

Abstract
Outbreaks of infection can be devastating for individuals and societies. In this review, we examine the applications of new high-throughput sequencing approaches to the identification and characterization of outbreaks, focusing on the application of whole-genome sequencing (WGS) to outbreaks of bacterial infection. We describe traditional epidemiological analysis and show how WGS can be informative at multiple steps in outbreak investigation, as evidenced by many recent studies. We conclude that high-throughput sequencing approaches can make a significant contribution to the investigation of outbreaks of bacterial infection and that the integration of WGS with epidemiological investigation, diagnostic assays and antimicrobial susceptibility testing will precipitate radical changes in clinical microbiology and infectious disease epidemiology in the near future. However, several challenges remain before WGS can be routinely used in outbreak investigation and clinical practice.
Outbreaks: definition and classification
Outbreaks of infection can be devastating for individuals and societies. In medieval times, the Black Death led to the death of up to a third of the inhabitants of Europe [1]. More recently, an outbreak of Shiga-toxin-producing Escherichia coli (STEC) struck Germany in May-June 2011, resulting in over 3,000 cases and over 50 deaths, and provided ample evidence of the harrowing effects of bacterial infection on a modern, industrialized society [2,3].
In its loosest sense, the term 'outbreak' can be used to refer to any increase in the incidence of a given infection, which can occur in response to local, societal or environmental changes: for example, one might see an increase in the prevalence of staphylococcal wound infections when hospital ward or operating theatre cleaning procedures change, or when there are changes in the use of antibiotics. However, in the strictest sense (which we adopt here), the term implies a series of infections caused by indistinguishable or closely linked isolates, which are sufficiently similar to justify talking about 'an outbreak strain'. Such outbreaks can range in size from a few individuals, for instance in a family outbreak or an outbreak on a hospital ward, to epidemics that rage across countries or continents.
Investigation of a suspected outbreak has two aims: termination of the cluster of disease and prevention of similar occurrences by understanding how such outbreaks originate. A key question surfaces at the start of any such investigation: is one really seeing an outbreak in the strictest sense, caused by a single strain, or is one merely seeing an increased incidence of infection, involving multiple unrelated strains? The answer to this question is of more than academic interest, as it dictates how the finite resources available for infection control are best deployed. For example, evidence of cross infection with a single methicillin-resistant Staphylococcus aureus (MRSA) strain on a ward might prompt an aggressive strategy of patient isolation and decolonization, whereas an increase in infections caused by diverse staphylococcal strains (presumably each derived from the patient's own microbiota) might prompt a look at policies for wound care or antibiotic usage. Similarly, identification and characterization of an outbreak strain or the discovery of its source or mode of transmission influences the behavior of the infection control team - potential responses include removal of the source, interruption of transmission or strengthening of host defenses.
In the past decade, many different kinds of outbreaks have hit the headlines (Table 1), with concern focused on the spread of multi-drug-resistant strains in hospitals (such as MRSA) [4] or in the community (such as multi-drug-resistant tuberculosis [5]); the threat of bioterrorism [6]; and 'emerging infections', caused by newly discovered pathogens, such as severe acute respiratory syndrome (SARS) or infection with the novel coronavirus 2012 (HCoV-EMC/2012) [7,8], or by novel variants of previously recognized species or strains, such as STEC O104:H4 [2,3]. Outbreaks are often linked to social factors, including mass travel, migration, conflict or societal breakdown, or to environmental threats, such as earthquakes or floods. They can arise from exposure to a common source in the environment (for example, legionellosis arising from a water source); when the period of exposure is brief, these events are termed 'point-source outbreaks'. Alternatively, outbreaks can be propagated by human-to-human spread or, in the case of zoonoses, such as swine or bird flu, can result from the spread to humans from animal reservoirs. Outbreaks can also be classified according to context, for example whether they occur in the community or in healthcare settings, or according to the mode of transmission, for example food-borne, waterborne, airborne or vector-borne.
Table 1.
A selection of recent outbreaks*
| Features | Disease or pathogen | When | Where | Scale | Comments |
|---|---|---|---|---|---|
| Airborne, point source | Legionnaire's disease | July 2012 | Stoke on Trent, UK | <10 cases | Likely source a hot tub |
| Airborne, propagated human-to-human | Measles | 2012 to now | South Wales, UK | >500 cases | Subsequent to poor take-up of measles, mumps and rubella (MMR) vaccine |
| Airborne, propagated human-to-human | Bordetella pertussis | 2011 to now | England and Wales, UK | >2,000 cases | Perhaps related to waning immunity in adults |
| Airborne, propagated human-to-human | Bovine tuberculosis | 2006 | Birmingham, UK | <10 cases | Spread through social links, including nightclub |
| Blood-borne | Hepatitis B | 2011 | Swansea, UK | ≥4 cases | Link between cases unclear |
| Bloodstream infection, common source | Anthrax | 2009 to 2012 | Europe, including UK | 100s of cases | Thought be associated with contaminated batch of heroin |
| Exposure to animal feces | E. coli O157 | September 2012 | Sutton Coldfield, UK | <10 cases | Contact between humans and animals in suburban park |
| Food-borne, point source | Salmonella Newport | Early 2012 | England, UK | >35 cases | Linked to consumption of watermelon |
| Hospital-acquired | Pseudomonas aeruginosa | Late 2011 to early 2012 | Northern Ireland, UK | 4 babies | Associated with contaminated hospital water supplies |
| Waterborne | Cholera | 2010 to now | Haiti | Occurred 10 months after powerful earthquake | |
| Waterborne | Cholera | 2008 to now | Zimbabwe | Exacerbated by consequences of economic collapse, including poor water sanitation | |
| Zoonotic, animal-to-human spread | Influenza H7N9 | April 2013 | China | >11 cases | Virus type known to be circulating in birds |
*This list is drawn largely from the BBC news website [58] and is illustrative rather than exhaustive.
Here, we examine the applications of new high-throughput sequencing approaches to the identification and characterization of outbreaks, focusing on the application of whole-genome sequencing (WGS) to outbreaks of bacterial infection. We describe how traditional epidemiological analysis works and show how WGS can be informative at multiple steps in outbreak investigation.
Epidemiological typing: progress and problems
Although traditional epidemiology can often track down the source of an outbreak (for example, a case-control study can identify the foodstuff responsible for a food-poisoning outbreak [9,10]), for several decades laboratory investigations have also had an important role in outbreak investigation and management [11]. Thus, when suspicion of an outbreak has been raised on clinical or epidemiological grounds, the laboratory can provide evidence to confirm or dismiss a common microbial cause. Alternatively, an increase in laboratory reports of a given pathogen may provide the first evidence that an outbreak is under way.
However, in addition to providing diagnostic information, the laboratory also offers epidemiological typing, which provides an assessment of how closely cases are related to each other. In broad terms, this means classifying isolates as unrelated (not part of an outbreak) or sufficiently closely related (in extremis, indistinguishable) to represent epidemic transmission.
Epidemiological typing requires the identification of stable distinguishing characteristics. Initially, this relied on analyses of useful phenotypic features (such as serological profiles, growth characteristics or susceptibilities to bacteriophage or antimicrobial agents) [11]. However, the arrival of molecular biology in general and specifically of the polymerase chain reaction (PCR) led to a profusion of genotypic approaches, largely documenting differences in patterns of bands seen on gels: examples include pulsed-field gel electrophoresis, ribotyping, variable number-tandem repeat typing, random amplification of polymorphic DNA, arbitrarily primed PCR and repetitive-element PCR [11].
This riotous proliferation of genotypic typing methods, often with complex and non-standardized workflows, led Achtman in the late 1990s to coin the phrase YATM for 'yet another typing method' [12] and to pioneer, with others, the adoption of sequence-based approaches, notably multilocus sequence typing (MLST) [13]. In this approach, differences in stretches of DNA sequence from conserved housekeeping genes are used to assign bacterial isolates to sequence types, which, in turn, often fall into larger clonal complexes. Sequence-based approaches bring the advantage of portability; in other words, results from one laboratory can be easily compared with those from others around the world. In addition, archiving of information in national or international datasets allows isolates and outbreaks to be placed in the wider context of pathogen population structure.
Yet, despite the advantages of sequence-based typing, drawbacks remain. For example, there is a lack of standardization, as evidenced by the existence of multiple MLST databases and even multiple competing MLST schemes for the same species [14,15]. In addition, costs and complex workflows mean that most pathogen typing is performed in batch mode, retrospectively, in reference laboratories that struggle to provide data with real-time impact - one possible exception is the near-real-time typing of Mycobacterium tuberculosis isolates in the UK [16]. Approaches such as MLST also lack the resolution needed to reconstruct chains of transmission within outbreaks, tending instead to lump together all isolates from an outbreak together as 'indistinguishable' members of the same sequence type.
The promise of whole-genome sequencing
WGS promises to deliver the ultimate high-resolution genotypic typing method [17-20]. Although we recognize that virologists pioneered the use of WGS for pathogen typing, targeting genomes small enough for WGS with traditional Sanger sequencing [21], here we will concentrate on the application of WGS to outbreaks of bacterial infection, catalyzed by the recent arrival in the marketplace of a range of technologies that fall under the umbrella term 'high-throughput sequencing' (sometimes called 'next-generation sequencing') [22,23].
High-throughput sequencing, especially with the arrival of bench-top sequencers [24,25], brings methodologies for bacterial WGS that are simple, quick and cheap enough to fall within the remit of an average-sized clinical or research laboratory. Through a single unified workflow, it becomes possible to identify all the features of interest of a bacterial isolate, speeding up the detection and investigation of outbreaks and delivering data in a portable digital format that can be shared internationally.
By delivering a definitive catalog of genetic polymorphisms (especially single-nucleotide polymorphisms or SNPs), WGS delivers far greater resolution than traditional methods. For instance, whereas MLST identified only a single sequence type for a collection of MRSA isolates, WGS identified several distinct clusters [26]. Two recent studies of tuberculosis transmission have shown that the resolution of WGS with SNP typing is much higher than that provided by the previous 'gold standard' typing method, mycobacterial interspersed repetitive unit variable number tandem repeat (MIRU-VNTR) typing [27,28]. WGS also links epidemiology to pathogen biology, delivering unprecedented insights into genome evolution, genome structure and gene content, including information on clinically important markers, such as resistance and virulence genes [11] (Figure 1).
Figure 1.
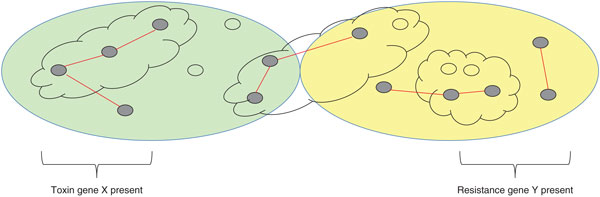
Whole-genome sequencing delivers high-resolution typing and insights into pathogen biology. In this hypothetical example, the two large ovals represent sets of isolates (small ovals) that have been assigned to genotypes using conventional laboratory typing. Clouds indicate clusters within those genotypes built using epidemiological data. Whole-genome sequencing provides a more detailed view of pathogen epidemiology, revealing previously unseen links (red lines) between genome-sequenced isolates (filled small ovals) within and between genotypes. Whole-genome sequencing also provides insights into pathogen biology, including the factors associated with virulence (represented here by toxin gene X) and drug resistance (represented here by resistance gene Y).
Applications of genome sequencing in outbreak investigation
Traditional outbreak investigation can be divided into discrete steps, although these often overlap. WGS has the potential to contribute to each of these steps (Table 2).
Table 2.
How whole-genome sequencing contributes to each step in outbreak investigation
| Step | Contribution of whole-genome sequencing (WGS) | References |
|---|---|---|
| Confirming the existence of an outbreak | Bench-top sequencing of whole bacterial genomes in near real time to confirm or refute the existence of outbreaks of MRSA or C. difficile | [25] |
| Open-ended diagnostic metagenomics to identify and characterize outbreak strain | [30] | |
| Case definition | WGS and/or metagenomics leads to the development of diagnostic reagents then used in defining cases within an outbreak | [3,31,32] |
| Descriptive study: collecting data and generating hypotheses | Integration of WGS with geographical data to uncover modes of spread of typhoid | [38] |
| Reconstruction of routes of transmission, including hidden transmission events | [25,45,59,60] | |
| Identification of virulence factors and antimicrobial resistance | [26,34,36] | |
| Analysis and hypothesis testing | Iterative refinements to assumptions and models | [25,27,36,41-47] |
| Institution and verification of control measures | Documenting effects of vaccination on pathogen populations | [48,49] |
| Confirmation that infections are imported rather than locally transmitted | [25,27,50] | |
| Communication | Need for user-friendly digital output easily transferred between laboratories and expert advice of clinical academics at home in research and clinical environments |
Confirming the existence of an outbreak
When pathogens are endemic, for example, MRSA or Clostridium difficile in healthcare facilities, it can be difficult to decide whether one or more outbreaks are under way or whether there has simply been a general rise in the incidence of infection. Eyre and colleagues [25] showed that bench-top sequencing of whole bacterial genomes could be used in near real time to confirm or refute the existence of outbreaks of MRSA or C. difficile in an acute hospital setting. In particular, they found that the genome sequences from an apparent cluster of C. difficile infections turned out to be unrelated and so did not represent an outbreak sensu stricto [25].
Metagenomics, that is, wholesale sequencing of DNA extracted from complex microbial communities without culture, capture or enrichment of pathogens or their sequences, provides an exciting new approach to the identification and characterization of outbreak strains that does away with the need for laboratory culture or target-specific amplification or enrichment. This approach has been used to identify the causes of outbreaks of viral infection [29]. Most recently, diagnostic metagenomics has been applied to stool samples collected during the German outbreak of STEC O104:H4, allowing recovery of draft genomes from the outbreak strain and several other pathogens and showing the applicability of diagnostic metagenomics to bacterial infections [30].
Case definition
Case definition within an outbreak usually involves a combination of clinical and laboratory criteria; for instance, a complex of symptoms and an associated organism. This definition can then be used for active case finding to identify additional patients in the cluster. During the German STEC outbreak, rapid genome sequencing together with crowd-sourced bioinformatics analyses led to the development of a set of diagnostic reagents that could then be used in defining cases within the outbreak [3]. Similarly, during new outbreaks of viral infection, genome-scale sequencing can act as a precursor to the development of simpler specific tests that can be used in case definition [31,32].
Descriptive study
During this phase of outbreak investigation, inferences from sequence data (such as on phylogeny, transmissibility, virulence or resistance) can be integrated with clinical and environmental metadata (such as geographical, temporal or anatomical data) to generate hypotheses and build and test models. For example, in a landmark study, Baker and colleagues [33] combined high-resolution genotyping and geospatial analysis to uncover the modes of transmission of endemic typhoid fever in an urban setting in Nepal.
During this phase of hypothesis generation, it may be possible to infer hidden transmission events. For instance, when faced with the recurrence of a strain of C. difficile in a hospital after more than 3 years of absence, Eyre and colleagues [25] concluded that unsuspected community transmission of C. difficile was the most likely explanation for their observations. They also noted that most of their C. difficile cases were unrelated to other recent cases in the hospital, from which they concluded that their hospital infection control policies were working as well as they could and that further reductions in the incidence of C. difficile infections would have to rely on additional and different interventions.
In some cases, it may be possible to hypothesize what determinants underlie the success of an outbreak strain. For example, the sasX gene (a mobile genetic element-encoded gene involved in nasal colonization and pathogenesis) appeared to be a key determinant of the successful spread of MRSA in China [34], and genes for the Panton-Valentine toxin were hypothesized to contribute to the spread of a novel MRSA genotype that caused an outbreak in a British special care baby unit [26].
Prediction of resistance phenotype from genotype has been applied routinely for years to viral pathogens such as human immunodeficiency virus, for which the cataloguing of resistance mutations in a publicly accessible database has greatly strengthened the utility of the approach [35]. Data are accumulating from S. aureus [36] and from E. coli strains that produce extended-spectrum beta-lactamases showing that WGS can be used to predict the resistance phenotype in bacteria (Nicole Stoesser, Department of Microbiology, John Radcliffe Hospital, Oxford, personal communication). Well-maintained databases documenting links between genotypes and resistance phenotypes are likely to add value to such ventures.
Host factors associated with disease may also be identified during data collection. Increasingly, whole-genome sequences of humans are available and being used to study population genetic risks for diseases, as reviewed recently by Chapman and Hill [37].
Analysis and hypothesis testing
During this stage, there is often a series of iterative refinements to assumptions and models. For example, in a detailed retrospective analysis of tuberculosis cases in the English Midlands, Walker and colleagues [27] first documented the diversity of M. tuberculosis genotypes in their collection and then explored how the patterns of genome diversity were reflected in contemporaneous and serial isolates from individual patients and among isolates from household outbreaks. This allowed them to define cut-offs in the number of SNPs that could be used to rule isolates in or out of a recent transmission event. In some instances, they could then allocate cases to clusters in which a link had been suspected, but had not been proven, by conventional epidemiological methods. In other cases, where a link had been suspected on grounds of ethnicity, they were able to exclude recent transmission within the West Midlands region.
Outbreaks of meningococcal disease caused by serogroup C have largely been eradicated in the UK by vaccination. However, a retrospective genomic analysis of strains from a meningococcal outbreak allowed chains of transmission to be identified [38]. This study pioneered the automated comparison of WGS data using a new public database, the Bacterial Isolate Genome Sequence Database (BIGSdb) [39]; the development of this kind of user-friendly, open-access tool is likely to underpin the adoption of WGS in epidemiological investigations in a clinical and public health environment.
Relatedness between isolates within an outbreak (and more widely) is often assessed by the construction of a phylogenetic tree [40]. Such phylogenetic inferences can enable the identification of sources or reservoirs of infection: examples include the acquisition of leprosy by humans from wild armadillos and the acquisition of Mycobacterium bovis in cattle from sympatric badger populations [41,42]. Integration of phylogeny with geography has allowed the origins and spread of pandemics and epidemics to be traced, including the Yersinia pestis pandemic [43] and, controversially, the 2010 cholera outbreak in Haiti, which has been traced to Nepalese peacekeepers [44].
Molecular phylogenies also make it possible to look back over years, decades, even centuries. For example, He and colleagues [45] showed that two distinct strains of fluoroquinolone-resistant C. difficile 027 emerged in the USA in 1993 to 1994, and that these showed different patterns of global spread. Genomic information, together with estimates from the sequence data of the time since isolates had diverged ('molecular clock' estimates) allowed them to reconstruct detailed routes of transmission within the UK. Similar studies have revealed patterns of the global spread of cholera, Shigella sonnei and MRSA [36,46,47].
Institution and verification of control measures
Vaccination provides a means of disrupting transmission by removing susceptible hosts from the population. For example, immunity to specific capsule types responsible for pneumococcal infection is targeted by their inclusion in a multivalent vaccine. High-throughput sequencing studies provide clear evidence that capsule switching is occurring in pneumococcal populations in response to vaccination, which has implications for disease control and vaccine design [48,49].
Viral illnesses have long been the target of successful vaccination programs. WGS analysis of rubella virus cases from the USA has confirmed that indigenous disease has been eradicated and that all the cases there are imported, with virus sequences matching those found elsewhere in the world [50].
Communication
To be useful to clinicians, whole-genome sequence data must be readily accessible in a portable, easily stored and searched, user-friendly format. However, data sharing even through established hospital informatics systems is a non-trivial task, particularly given the current diversity in sequencing platforms and analytical pipelines. Perhaps the answer here is to ensure the involvement of clinical academics with the relevant research credentials and accreditation to make clinical decisions, who might be best placed to pioneer the use of WGS data to manage outbreaks.
Conclusions and future perspectives
As we have seen, there is now ample evidence that WGS can make a significant contribution to the investigation of outbreaks of bacterial infection. It is therefore safe to conclude that once WGS has been integrated with epidemiological investigation, diagnostic assays and antimicrobial susceptibility testing, we will soon see large changes in the practice of clinical microbiology and infectious disease epidemiology. Nonetheless, several challenges remain before WGS can be routinely used in clinical practice (Table 3).
Table 3.
Whole-genome sequencing in outbreak investigations: opportunities and challenges
| Feature | Opportunities | Challenges |
|---|---|---|
| Sequence generation | Provision of data on a timescale that allows clinical interventions Costs now comparable to those of other clinically relevant expenditure (such as of antibiotic treatment or bed occupancy) Use now comparable to that of other automated laboratory systems Delivers far richer data than any previous method Potential for open-ended one-size-fits-all culture-independent workflow |
Chasing a moving target: difficult to devise stable and agreed standard operating procedures in the face of relentless technical innovation Proof needed that WGS cost-effective across a range of clinical applications Difficulties in predicting phenotype from genotype Still sufficiently technically demanding to require input of skilled staff Resistance to adoption of potentially disruptive technology |
| Data handling | Provides portable, digital, library-based approach | Large datasets require significant hardware for storage and analysis Need for standardized, robust, user-friendly analysis pipelines Issues over data storage, ownership and presentation need to be resolved Integration with healthcare informatics systems to allow easy communication with clinicians |
| Epidemiological analysis | WGS provides highest possible resolution Potential to link pathogen discovery, biology and evolution with phylogeny and epidemiology to facilitate iterative hypothesis generation, testing and refinement |
Need to move beyond SNP typing of draft genomes of colony-purified isolates to embrace full range of genome variation, including within-patient variation Better integration with conventional epidemiology required to place data in context and evaluate hypothesized routes of transmissions Acquiring clinical metadata often remains a bottleneck |
There is still a need for improved speed, ease of use, accuracy and longer read lengths. However, given the ongoing, relentless improvements in performance and cost-effectiveness of high-throughput sequencing, it is likely that these financial and technical challenges will be met relatively easily over the coming years [51]. Nonetheless, improvements in the analysis, archiving and sharing of WGS data need to occur before sequencing results can become trustworthy enough to guide clinical decision-making. Significant investment in establishing standards, databases and communication tools will be required to maximize the opportunities provided by WGS in epidemiology. There may also be organizational and ethical issues with data ownership and access [52].
Careful contextualization of WGS data will be needed before robust conclusions can be drawn, ideally within an agreed framework of standard operating procedures. Interpretation of genomic data requires a detailed knowledge of within-host and between-host genotypic diversity, whether defined at a single time point or longitudinally. Readings from the molecular clock provide the temporal information needed to reconstruct the emergence and evolution of lineages and transmission events within an outbreak. This means that extensive benchmarking will be needed to determine the rates of genomic change, which are likely to be species- and even lineage-specific. Only when WGS data have been obtained from a large number of epidemiologically linked and unlinked cases in a given lineage will it be possible to define cut-offs for the genomic differences that allow linked and unlinked cases to be accurately defined. This may also rely on comparisons with an 'outgroup', that is, a group of cases that clearly fall outside the outbreak cluster.
Estimates of rates of genetic change have been published for some organisms: for example, S. aureus mutates relatively rapidly, with 3 × 10-6 mutations per year, corresponding to 8.4 SNPs per genome per year [3,39], whereas M. tuberculosis evolves slowly, acquiring only 0.5 SNPs per genome per year [27,53-55]. However, such data are available for only a very limited number of other pathogens. This will need to be expanded significantly before routine use of WGS data becomes a reality. We suspect that there may be consistent differences in the mode and rate of genotypic change between organisms for which an asymptomatic carrier state (for example C. difficile) or a latent period (M. tuberculosis) exists and those, such as measles, for which there is no carrier state.
In conclusion, it is clear that WGS is already transforming the practice of outbreak investigation. However, the dizzyingly fast pace of change in this field, with steady improvements in high-throughput sequencing, make predictions about the future difficult, particularly now that nanopore sequencing technologies are poised to deliver a revolution in our ability to sequence macromolecules in clinical samples (not just DNA, but also RNA and even proteins) [56,57]. Portable nanopore technologies might provide a route to real-time near-patient testing and environmental sampling, as well as delivering a combined read-out of genotype and phenotype in bacterial cells (perhaps even allowing direct detection of the expression of resistance determinants). It also seems likely that clinical diagnostic metagenomics [30], perhaps equipped with target-specific enhancements such as sorting or capture of cells or DNA, will deliver improved genomic epidemiological information, including insights into within-patient pathogen population genetics and identification and typing of non-culturable or difficult-to-culture organisms.
One thing is certain: the future of bacterial outbreak investigation will rely on a new paradigm of genomics and metagenomics. Therefore, it is up to all clinical and epidemiological researchers to embrace the opportunities and meet the challenges of this new way of working
Abbreviations
MLST: multilocus sequence typing; MRSA: methicillin-resistant Staphylococcus aureus; SNP: single-nucleotide polymorphism; STEC: Shiga-toxin-producing Escherichia coli; WGS: whole-genome sequencing.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Esther R Robinson, Email: estherrobinson@doctors.org.uk.
Timothy M Walker, Email: timothy.walker@ndm.ox.ac.uk.
Mark J Pallen, Email: m.pallen@warwick.ac.uk.
Acknowledgements
TMW is an MRC Research Training Fellow.
References
- Ziegler P. The Black Death. London: Penguin; 1998. [Google Scholar]
- Frank C, Werber D, Cramer JP, Askar M, Faber M, an der Heiden M, Bernard H, Fruth A, Prager R, Spode A, Wadl M, Zoufaly A, Jordan S, Kemper MJ, Follin P, Müller L, King LA, Rosner B, Buchholz U, Stark K, Krause G. HUS Investigation Team. Epidemic profile of shiga-toxin-producing Escherichia coli O104:H4 outbreak in germany - preliminary report. N Engl J Med. 2011;365:1771–1780. doi: 10.1056/NEJMoa1106483. [DOI] [PubMed] [Google Scholar]
- Rohde H, Qin J, Cui Y, Li D, Loman NJ, Hentschke M, Chen W, Pu F, Peng Y, Li J, Xi F, Li S, Li Y, Zhang Z, Yang X, Zhao M, Wang P, Guan Y, Cen Z, Zhao X, Christner M, Kobbe R, Loos S, Oh J, Yang L, Danchin A, Gao GF, Song Y, Li Y, Yang H. et al. Open-source genomic analysis of Shiga-toxin-producing E. coli O104:H4. N Engl J Med. 2011;365:718–724. doi: 10.1056/NEJMoa1107643. [DOI] [PubMed] [Google Scholar]
- Stefani S, Chung DR, Lindsay JA, Friedrich AW, Kearns AM, Westh H, Mackenzie FM. Meticillin-resistant Staphylococcus aureus (MRSA): global epidemiology and harmonisation of typing methods. Int J Antimicrob Agents. 2012;39:273–282. doi: 10.1016/j.ijantimicag.2011.09.030. [DOI] [PubMed] [Google Scholar]
- Pontali E, Matteelli A, Migliori GB. Drug-resistant tuberculosis. Curr Opin Pulm Med. 2013;19:266–272. doi: 10.1097/MCP.0b013e32835f1bf3. [DOI] [PubMed] [Google Scholar]
- Nordmann BD. Issues in biosecurity and biosafety. Int J Antimicrob Agents. 2010;36(Suppl 1):S66–S69. doi: 10.1016/j.ijantimicag.2010.06.025. [DOI] [PubMed] [Google Scholar]
- Cleri DJ, Ricketti AJ, Vernaleo JR. Severe acute respiratory syndrome (SARS). Infect Dis Clin North Am. 2010;24:175–202. doi: 10.1016/j.idc.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Osterhaus AD, Haagmans BL, Gorbalenya AE, Snijder EJ, Fouchier RA. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio. 2012;3:e00473–12. doi: 10.1128/mBio.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz U, Bernard H, Werber D, Bohmer MM, Remschmidt C, Wilking H, Delere Y, an der Heiden M, Adlhoch C, Dreesman J, Ehlers J, Ethelberg S, Faber M, Frank C, Fricke G, Greiner M, Hohle M, Ivarsson S, Jark U, Kirchner M, Koch J, Krause G, Luber P, Rosner B, Stark K, Kuhne M. German outbreak of Escherichia coli O104:H4 associated with sprouts. N Engl J Med. 2011;365:1763–1770. doi: 10.1056/NEJMoa1106482. [DOI] [PubMed] [Google Scholar]
- King LA, Nogareda F, Weill FX, Mariani-Kurkdjian P, Loukiadis E, Gault G, Jourdan-DaSilva N, Bingen E, Mace M, Thevenot D, Ong N, Castor C, Noel H, Van Cauteren D, Charron M, Vaillant V, Aldabe B, Goulet V, Delmas G, Couturier E, Le Strat Y, Combe C, Delmas Y, Terrier F, Vendrely B, Rolland P, de Valk H. Outbreak of Shiga toxin-producing Escherichia coli O104:H4 associated with organic fenugreek sprouts, France, June 2011. Clin Infect Dis. 2012;54:1588–1594. doi: 10.1093/cid/cis255. [DOI] [PubMed] [Google Scholar]
- Sabat AJ, Budimir A, Nashev D, Sa-Leao R, van DJ, Laurent F, Grundmann H, Friedrich AW. Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Euro Surveill. 2013;18:20380. doi: 10.2807/ese.18.04.20380-en. [DOI] [PubMed] [Google Scholar]
- Achtman M. A surfeit of YATMs?. J Clin Microbiol. 1996;34:1870. doi: 10.1128/jcm.34.7.1870-1870.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, Feavers IM, Achtman M, Spratt BG. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci USA. 1998;95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Losada M, Cabezas P, Castro-Nallar E, Crandall KA. Pathogen typing in the genomics era: MLST and the future of molecular epidemiology. Infect Genet Evol. 2013;16C:38–53. doi: 10.1016/j.meegid.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Chaudhuri RR, Henderson IR. The evolution of the Escherichia coli phylogeny. Infect Genet Evol. 2012;12:214–226. doi: 10.1016/j.meegid.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Hawkey PM, Smith EG, Evans JT, Monk P, Bryan G, Mohamed HH, Bardhan M, Pugh RN. Mycobacterial interspersed repetitive unit typing of Mycobacterium tuberculosis compared to IS6110-based restriction fragment length polymorphism analysis for investigation of apparently clustered cases of tuberculosis. J Clin Microbiol. 2003;41:3514–3520. doi: 10.1128/JCM.41.8.3514-3520.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JZ, Pallen MJ, Oppenheim B, Constantinidou C. Genome sequencing in clinical microbiology. Nat Biotechnol. 2012;30:1068–1071. doi: 10.1038/nbt.2410. [DOI] [PubMed] [Google Scholar]
- Loman NJ, Constantinidou C, Chan JZ, Halachev M, Sergeant M, Penn CW, Robinson ER, Pallen MJ. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat Rev Microbiol. 2012;10:599–606. doi: 10.1038/nrmicro2850. [DOI] [PubMed] [Google Scholar]
- Pallen MJ, Loman NJ, Penn CW. High-throughput sequencing and clinical microbiology: progress, opportunities and challenges. Curr Opin Microbiol. 2010;13:625–631. doi: 10.1016/j.mib.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Pallen MJ, Loman NJ. Are diagnostic and public health bacteriology ready to become branches of genomic medicine?. Genome Med. 2011;3:53. doi: 10.1186/gm269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arens M. Methods for subtyping and molecular comparison of human viral genomes. Clin Microbiol Rev. 1999;12:612–626. doi: 10.1128/cmr.12.4.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Stranneheim H, Lundeberg J. Stepping stones in DNA sequencing. Biotechnol J. 2012;7:1063–1073. doi: 10.1002/biot.201200153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012;30:434–439. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- Eyre DW, Golubchik T, Gordon NC, Bowden R, Piazza P, Batty EM, Ip CL, Wilson DJ, Didelot X, O'Connor L, Lay R, Buck D, Kearns AM, Shaw A, Paul J, Wilcox MH, Donnelly PJ, Peto TE, Walker AS, Crook DW. A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance. BMJ Open. 2012;2 doi: 10.1136/bmjopen-2012-001124. pii: e001124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SR, Cartwright EJ, Torok ME, Holden MT, Brown NM, Ogilvy-Stuart AL, Ellington MJ, Quail MA, Bentley SD, Parkhill J, Peacock SJ. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect Dis. 2013;13:130–136. doi: 10.1016/S1473-3099(12)70268-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, Eyre DW, Wilson DJ, Hawkey PM, Crook DW, Parkhill J, Harris D, Walker AS, Bowden R, Monk P, Smith EG, Peto TE. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013;13:137–146. doi: 10.1016/S1473-3099(12)70277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardy JL, Johnston JC, Ho Sui SJ, Cook VJ, Shah L, Brodkin E, Rempel S, Moore R, Zhao Y, Holt R, Varhol R, Birol I, Lem M, Sharma MK, Elwood K, Jones SJ, Brinkman FS, Brunham RC, Tang P. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. 2011;364:730–739. doi: 10.1056/NEJMoa1003176. [DOI] [PubMed] [Google Scholar]
- Capobianchi MR, Giombini E, Rozera G. Next-generation sequencing technology in clinical virology. Clin Microbiol Infect. 2013;19:15–22. doi: 10.1111/1469-0691.12056. [DOI] [PubMed] [Google Scholar]
- Loman NJ, Constantinidou C, Christner M, Rohde H, Chan JZ-M, Quick J, Weir JC, Quince C, Smith GP, Betley JR, Aepfelbacher M, Pallen MJ. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli {O104:H4}. JAMA. 2013;309:1502–1510. doi: 10.1001/jama.2013.3231. [DOI] [PubMed] [Google Scholar]
- Palacios G, Druce J, Du L, Tran T, Birch C, Briese T, Conlan S, Quan PL, Hui J, Marshall J, Simons JF, Egholm M, Paddock CD, Shieh WJ, Goldsmith CS, Zaki SR, Catton M, Lipkin WI. A new arenavirus in a cluster of fatal transplant-associated diseases. N Engl J Med. 2008;358:991–998. doi: 10.1056/NEJMoa073785. [DOI] [PubMed] [Google Scholar]
- Towner JS, Sealy TK, Khristova ML, Albarino CG, Conlan S, Reeder SA, Quan PL, Lipkin WI, Downing R, Tappero JW, Okware S, Lutwama J, Bakamutumaho B, Kayiwa J, Comer JA, Rollin PE, Ksiazek TG, Nichol ST. Newly discovered Ebola virus associated with hemorrhagic fever outbreak in Uganda. PLoS Pathog. 2008;4:e1000212. doi: 10.1371/journal.ppat.1000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker S, Holt KE, Clements AC, Karkey A, Arjyal A, Boni MF, Dongol S, Hammond N, Koirala S, Duy PT, Nga TV, Campbell JI, Dolecek C, Basnyat B, Dougan G, Farrar JJ. Combined high-resolution genotyping and geospatial analysis reveals modes of endemic urban typhoid fever transmission. Open Biol. 2011;1:110008. doi: 10.1098/rsob.110008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Du X, Villaruz AE, Diep BA, Wang D, Song Y, Tian Y, Hu J, Yu F, Lu Y, Otto M. MRSA epidemic linked to a quickly spreading colonization and virulence determinant. Nat Med. 2012;18:816–819. doi: 10.1038/nm.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer RW. Rationale and uses of a public HIV drug-resistance database. J Infect Dis. 2006;194(Suppl 1):S51–S58. doi: 10.1086/505356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden MT, Hsu LY, Kurt K, Weinert LA, Mather AE, Harris SR, Strommenger B, Layer F, Witte W, de Lencastre H, Skov R, Westh H, Zemlickova H, Coombs G, Kearns AM, Hill RL, Edgeworth J, Gould I, Gant V, Cooke J, Edwards GF, McAdam PR, Templeton KE, McCann A, Zhou Z, Castillo-Ramirez S, Feil EJ, Hudson LO, Enright MC, Balloux F. et al. A genomic portrait of the emergence, evolution, and global spread of a methicillin-resistant Staphylococcus aureus pandemic. Genome Res. 2013;23:653–664. doi: 10.1101/gr.147710.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman SJ, Hill AV. Human genetic susceptibility to infectious disease. Nat Rev Genet. 2012;13:175–188. doi: 10.1038/nrg3114. [DOI] [PubMed] [Google Scholar]
- Jolley KA, Hill DM, Bratcher HB, Harrison OB, Feavers IM, Parkhill J, Maiden MC. Resolution of a meningococcal disease outbreak from whole-genome sequence data with rapid Web-based analysis methods. J Clin Microbiol. 2012;50:3046–3053. doi: 10.1128/JCM.01312-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley KA, Maiden MC. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Rannala B. Molecular phylogenetics: principles and practice. Nat Rev Genet. 2012;13:303–314. doi: 10.1038/nrg3186. [DOI] [PubMed] [Google Scholar]
- Truman RW, Singh P, Sharma R, Busso P, Rougemont J, Paniz-Mondolfi A, Kapopoulou A, Brisse S, Scollard DM, Gillis TP, Cole ST. Probable zoonotic leprosy in the southern United States. N Engl J Med. 2011;364:1626–1633. doi: 10.1056/NEJMoa1010536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biek R, O'Hare A, Wright D, Mallon T, McCormick C, Orton RJ, McDowell S, Trewby H, Skuce RA, Kao RR. Whole genome sequencing reveals local transmission patterns of Mycobacterium bovis in sympatric cattle and badger populations. PLoS Pathog. 2012;8:e1003008. doi: 10.1371/journal.ppat.1003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli G, Song Y, Mazzoni CJ, Eppinger M, Roumagnac P, Wagner DM, Feldkamp M, Kusecek B, Vogler AJ, Li Y, Cui Y, Thomson NR, Jombart T, Leblois R, Lichtner P, Rahalison L, Petersen JM, Balloux F, Keim P, Wirth T, Ravel J, Yang R, Carniel E, Achtman M. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat Genet. 2010;42:1140–1143. doi: 10.1038/ng.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriksen RS, Price LB, Schupp JM, Gillece JD, Kaas RS, Engelthaler DM, Bortolaia V, Pearson T, Waters AE, Upadhyay BP, Shrestha SD, Adhikari S, Shakya G, Keim PS, Aarestrup FM. Population genetics of Vibrio cholerae from Nepal in 2010: evidence on the origin of the Haitian outbreak. mBio. 2011;2:e00157–11. doi: 10.1128/mBio.00157-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Miyajima F, Roberts P, Ellison L, Pickard DJ, Martin MJ, Connor TR, Harris SR, Fairley D, Bamford KB, D'Arc S, Brazier J, Brown D, Coia JE, Douce G, Gerding D, Kim HJ, Koh TH, Kato H, Senoh M, Louie T, Michell S, Butt E, Peacock SJ, Brown NM, Riley T, Songer G, Wilcox M, Pirmohamed M, Kuijper E. et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet. 2013;45:109–113. doi: 10.1038/ng.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt KE, Baker S, Weill FX, Holmes EC, Kitchen A, Yu J, Sangal V, Brown DJ, Coia JE, Kim DW, Choi SY, Kim SH, da Silveira WD, Pickard DJ, Farrar JJ, Parkhill J, Dougan G, Thomson NR. Shigella sonnei genome sequencing and phylogenetic analysis indicate recent global dissemination from Europe. Nat Genet. 2012;44:1056–1059. doi: 10.1038/ng.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutreja A, Kim DW, Thomson NR, Connor TR, Lee JH, Kariuki S, Croucher NJ, Choi SY, Harris SR, Lebens M, Niyogi SK, Kim EJ, Ramamurthy T, Chun J, Wood JL, Clemens JD, Czerkinsky C, Nair GB, Holmgren J, Parkhill J, Dougan G. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature. 2011;477:462–465. doi: 10.1038/nature10392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, van der Linden M, McGee L, von Gottberg A, Song JH, Ko KS, Pichon B, Baker S, Parry CM, Lambertsen LM, Shahinas D, Pillai DR, Mitchell TJ, Dougan G, Tomasz A, Klugman KP, Parkhill J, Hanage WP, Bentley SD. Rapid pneumococcal evolution in response to clinical interventions. Science. 2011;331:430–434. doi: 10.1126/science.1198545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu FZ, Eutsey R, Ahmed A, Frazao N, Powell E, Hiller NL, Hillman T, Buchinsky FJ, Boissy R, Janto B, Kress-Bennett J, Longwell M, Ezzo S, Post JC, Nesin M, Tomasz A, Ehrlich GD. In vivo capsular switch in Streptococcus pneumoniae - analysis by whole genome sequencing. PLoS One. 2012;7:e47983. doi: 10.1371/journal.pone.0047983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abernathy E, Chen MH, Bera J, Shrivastava S, Kirkness E, Zheng Q, Bellini W, Icenogle J. Analysis of whole genome sequences of 16 strains of rubella virus from the United States, 1961--2009. Virol J. 2013;10:32. doi: 10.1186/1743-422X-10-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson E, Lundeberg J, Ahmadian A. Generations of sequencing technologies. Genomics. 2009;93:105–111. doi: 10.1016/j.ygeno.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Rump B, Cornelis C, Woonink F, Verwieij M. The need for ethical reflection on the use of molecular microbial characterisation in outbreak management. Eurosurveillance. 2013;18:9. doi: 10.2807/ese.18.04.20384-en. [DOI] [PubMed] [Google Scholar]
- Didelot X, Bowden R, Wilson DJ, Peto TE, Crook DW. Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet. 2012;13:601–612. doi: 10.1038/nrg3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young BC, Golubchik T, Batty EM, Fung R, Larner-Svensson H, Votintseva AA, Miller RR, Godwin H, Knox K, Everitt RG, Iqbal Z, Rimmer AJ, Cule M, Ip CL, Didelot X, Harding RM, Donnelly P, Peto TE, Crook DW, Bowden R, Wilson DJ. Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease. Proc Natl Acad Sci USA. 2012;109:4550–4555. doi: 10.1073/pnas.1113219109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CB, Lin PL, Chase MR, Shah RR, Iartchouk O, Galagan J, Mohaideen N, Ioerger TR, Sacchettini JC, Lipsitch M, Flynn JL, Fortune SM. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat Genet. 2011;43:482–486. doi: 10.1038/ng.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra RD, Kim J, Dunbar WB. Recent advances in nanopore sequencing. Electrophoresis. 2012;33:3418–3428. doi: 10.1002/elps.201200272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivala J, Marks DB, Akeson M. Unfoldase-mediated protein translocation through an alpha-hemolysin nanopore. Nat Biotechnol. 2013;31:247–250. doi: 10.1038/nbt.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BBC News. http://www.bbc.co.uk/news
- Didelot X, Eyre DW, Cule M, Ip CL, Ansari MA, Griffiths D, Vaughan A, O'Connor L, Golubchik T, Batty EM, Piazza P, Wilson DJ, Bowden R, Donnelly PJ, Dingle KE, Wilcox M, Walker AS, Crook DW, A Peto TE, Harding RM. Microevolutionary analysis of Clostridium difficile genomes to investigate transmission. Genome Biol. 2012;13:R118. doi: 10.1186/gb-2012-13-12-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis T, Loman NJ, Bingle L, Jumaa P, Weinstock GM, Mortiboy D, Pallen MJ. High-throughput whole-genome sequencing to dissect the epidemiology of Acinetobacter baumannii isolates from a hospital outbreak. J Hosp Infect. 2010;75:37–41. doi: 10.1016/j.jhin.2010.01.012. [DOI] [PubMed] [Google Scholar]