

Abstract
The BubR1 gene encodes for a mitotic regulator that ensures accurate segregation of chromosomes through its role in the mitotic checkpoint and the establishment of proper microtubule-kinetochore attachments. Germline mutations that reduce BubR1 abundance cause aneuploidy, shorten lifespan, and induce premature aging phenotypes and cancer in both humans and mice. Reduced BubR1 expression is also a feature of chronological aging, but whether this age-related decline has biological consequences is unknown. Using a transgenic approach in mice, we show that sustained high expression of BubR1 preserves genomic integrity and reduces tumorigenesis, even in the presence of genetic alterations that strongly promote aneuplodization and cancer, such as oncogenic Ras. We find that BubR1 overabundance exerts its protective effect by correcting mitotic checkpoint impairment and microtubule-kinetochore attachment defects. Furthermore, sustained high expression of BubR1 extends lifespan and delays age-related deterioration and aneuploidy in several tissues. Collectively, these data uncover a generalized function for BubR1 in counteracting defects that cause whole chromosome instability and suggest that modulating BubR1 provides a unique opportunity to extend healthy lifespan.
During mitosis, duplicated chromosomes need to be separated equally amongst two identical cells. To safeguard that this process occurs without errors, mammalian cells have developed a surveillance mechanism, the mitotic checkpoint, which inhibits anaphase onset until chromosome bi-orientation has been achieved. BubR1 is a core mitotic checkpoint component that binds to and inhibits the Cdc20-activated anaphase-promoting complex (APC/CCdc20), a ubiquitin E3 ligase that initiates anaphase by orchestrating separase-mediated cleavage of cohesion rings that hold sister chromatids together1. BubR1 not only contributes to proper chromosome segregation through mitotic checkpoint activation but also by regulation of chromosome-spindle attachments2,3. Mutant mice carrying BubR1 hypomorphic alleles (BubR1H/H mice) that produce low amounts of the protein are prone to aneuploidy and develop various progeroid and age-related phenotypes, including short lifespan, growth retardation, cataracts, sarcopenia, subdermal fat loss, impaired wound healing, and reduced dermal thickness1,4–6. Mutations in BubR1 have been associated with mosaic variegated aneuploidy (MVA), a rare human syndrome characterized by aneuploidization, tumor predisposition, and several progeroid traits, including short lifespan, growth and mental retardation, cataracts, and facial dysmorphisms7–9. These data, together with the observation that BubR1 abundance declines with age in various mouse tissues1,4,5, led to the notion that BubR1 may contribute to chronological aging.
We reasoned that if depletion of BubR1 with age contributes to aging and age-related disorders, then increased expression of BubR1 might extend healthy lifespan. To test this hypothesis, we generated transgenic mice expressing BubR1 fused to a Flag tag under control of the ubiquitous CAGGS promoter (Fig. 1a). As a marker for transgene expression, we co-expressed enhanced green fluorescent protein (EGFP) from an internal ribosome entry site (IRES). Two independent BubR1 transgenic lines were obtained, henceforth referred to as T7 and T23. Western blot analysis of a broad spectrum of tissues indicated that T7 and T23 mice expressed moderate and high amounts of Flag-BubR1, respectively (Fig. 1b and Supplementary Fig. S1a–c). Expression of Flag-BubR1 corrected all premature aging phenotypes of BubR1H/H mice (Fig. 1c, and data not shown), confirming that Flag-BubR1 protein was functional and adequately expressed.
Fig. 1.
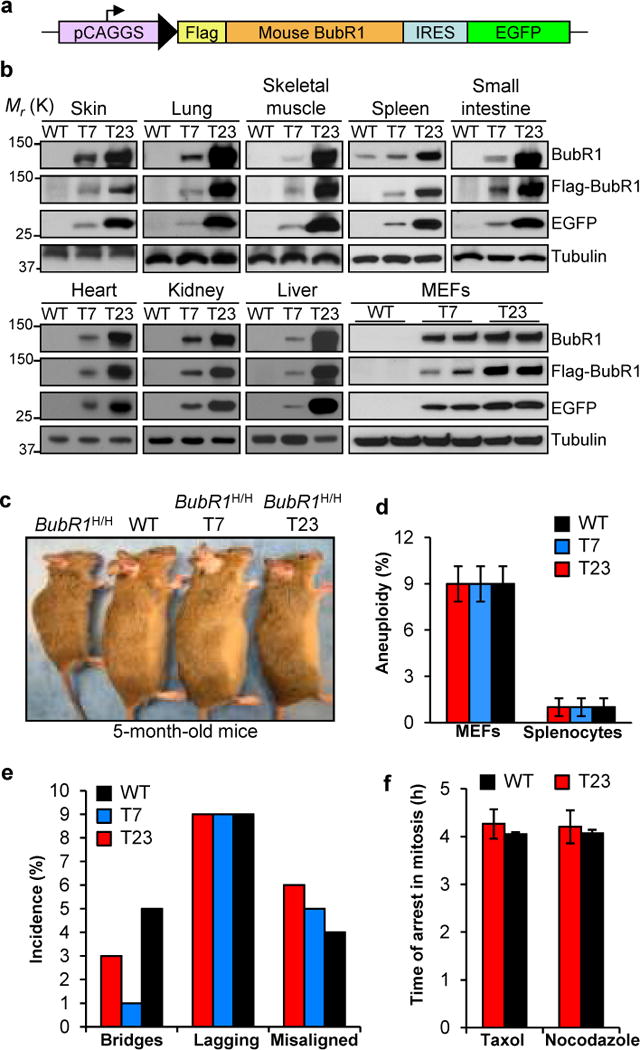
Transgenic mouse strains with moderate and high levels of Flag-BubR1 are chromosomally stable. (a) Flag-mBubR1 transgenic vector design. pCAGGS = promoter consisting of the CMV immediate enhancer and the chicken beta-actin promoter. (b) Western blots of tissue and MEF extracts from wildtype and Flag-BubR1 transgenic mice (strains T7 and T23). Tubulin was used as a loading control. (c) Both Flag-BubR1 transgenes correct the growth retardation and aging-associated phenotypes of BubR1H/H mice. (d) High BubR1 expression does not induce aneuploidy. Chromosome counts were done on splenocytes from 5-month-old mice and passage (P) 5 MEFs (n = 3 samples per genotype). Fifty spreads were counted per sample (150 total). Values represent means ± SD. (e) High BubR1 levels do not induce chromosome missegregation. (f) The mitotic checkpoint is not hyperactive at supranormal BubR1 levels. Three independent MEF lines per genotype were used in e and f. Values represent means ± SD in f.
Key mitotic regulators such as Bub1, Mad2 and UbcH10 cause aneuploidy and tumor formation when overexpressed in mice10–12. However, T7 and T23 splenocytes and mouse embryonic fibroblasts (MEFs) showed normal aneuploidy rates (Fig. 1d). Consistent with this, chromosome missegregation rates were not elevated in T7 and T23 MEFs (Fig. 1e). Transgenic MEFs showed increased BubR1-Cdc20 complex formation (Supplementary Fig. S1d), but this had no overt impact on mitotic checkpoint activity (Fig. 1f). Key mitotic regulators were expressed at normal levels in T23 MEFs (Supplementary Fig. S1e).
Tumor susceptibility of T7 and T23 transgenic mice was evaluated using 7,12-dimethylbenz(a)anthracene (DMBA), a carcinogen that causes lung and skin tumors13. Only 33% of T23 mice developed tumors in contrast to 100% of wildtype animals (Fig. 2a). Reductions in both skin and lung tumor incidence contributed to this decline (Fig. 2a). Tumor formation in T7 animals was not significantly reduced. Next, we examined the impact of moderate and high BubR1 overexpression on lung tumor formation in KrasLA1 mice14. KrasLA1 mice carry a conditional oncogenic Kras allele (KrasG12D) that becomes active upon intra-chromosomal homologous recombination. At 6 weeks, KrasLA1 mice on average had 17 lung tumors, whereas T23;KrasLA1 mice only had 8 (Fig. 2b). Again, the moderately expressed T7 transgene failed to reduce tumor formation (Supplementary Fig. S2a), indicating that tumor protection requires a threshold of BubR1 overexpression.
Fig. 2.
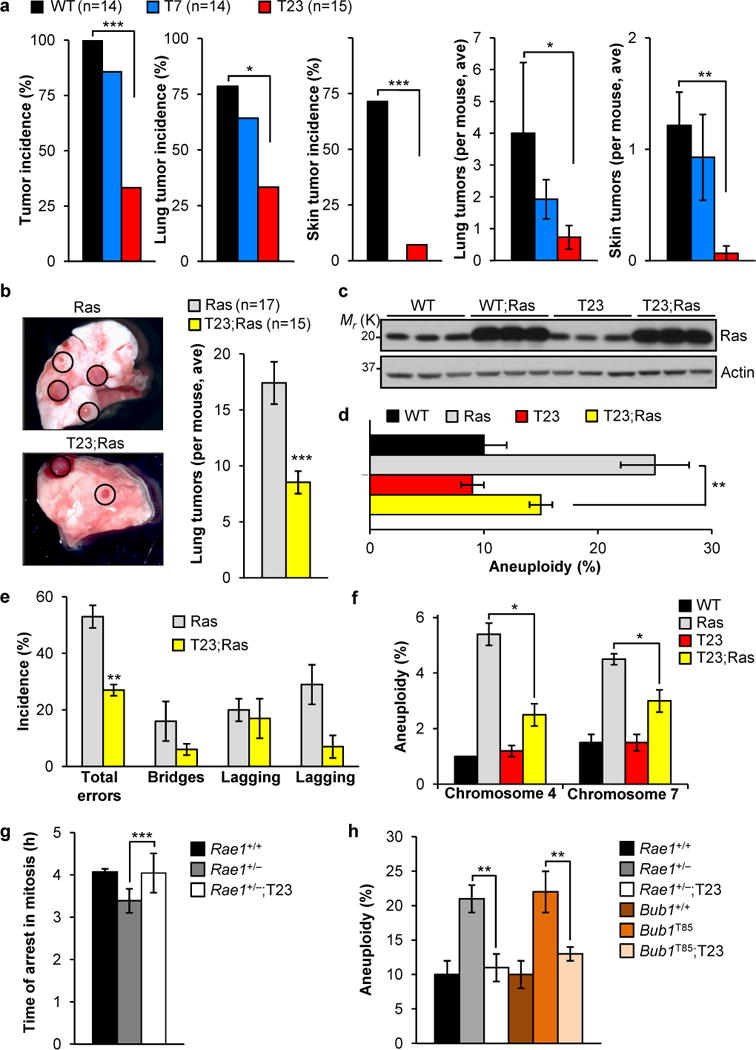
Resistance to tumorigenesis and chromosomal instability in BubR1 transgenic mice. (a) Tumor incidence and multiplicity in animals treated with DMBA on dorsal skin on post-natal day 5 and biopsied at 5 months of age. Values represent means ± SEM. (b) Resistance of BubR1 transgenic mice to lung tumors induced by oncogenic Kras (G12D). Cohorts of KrasLA1 and T23;KrasLA1 mice were killed at 6 weeks of age and lung tumors counted under a dissection microscope. Values represent means ± SD. (c) Western blots of cell extracts from wildtype and T23 MEFs with and without retrovirally expressed oncogenic Hras (G12V). Actin was used as a loading control. (d and e) Oncogenic Hras-induced aneuploidy (d) and chromosome missegregation (e) rates of MEF lines with and without BubR1 overexpression (n = 3 independent MEF lines each). Values represent means ± SD in d and means ± SEM in e. (f) Aneuploidy rates in lungs of wildtype and KrasLA1 mice with and without BubR1 overexpression. n = 3 mice for WT and T23, and n = 4 for Ras and T23;Ras. Values represent means ± SEM. (g) Nocodazole-challenge assay showing that elevated BubR1 expression restores normal mitotic checkpoint activity in MEFs with low amounts of Rae1. Values represent means ± SD. Three independent MEF lines per genotype were used. (h) Aneuploidy rates of mutant MEF lines with and without BubR1 overexpression (n = 5 MEF lines each). Values represent means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. n indicates the number of mice (mixed gender) used per genotype in a and b.
Aneuploidy is a hallmark of human cancers, and aneuploidy-prone mouse models indicate that this condition is causally implicated in tumor development15–17. This, combined with the observation that oncogenic Ras promotes chromosome missegregation18, led us to hypothesize that BubR1 overexpression suppresses tumorigenesis by counteracting Ras-mediated aneuploidization. To test this notion, we transduced wildtype and T23 MEFs with a retrovirus expressing oncogenic Ras (Fig. 2c) and performed chromosome counts on day 5-post infection. Indeed, BubR1 overexpression markedly decreased Ras-induced aneuploidy (Fig. 2d), which correlated with reduced chromosome missegregation (Fig. 2e). Next, we examined whether increased BubR1 also inhibits aneuploidization in lung tissue of KrasLA1 mice. Acquisition of oncogenic Ras in lung produced a hyperplastic epithelium (Supplementary Fig. S2b), which upon fluorescence in situ hybridization (FISH) analysis for chromosomes 4 and 7 showed increased aneuploidy (Fig. 2f). Aneuploidy was significantly reduced in T23;KrasLA1 mice (Fig. 2f), but not in T7;KrasLA1 mice (Supplementary Fig. S2c), indicating that protection against tumorigenesis by high levels of BubR1 tightly correlates with reduced aneuploidization.
To determine how high BubR1 levels counteract aneuploidization we focused on its known roles in mitotic checkpoint control and the establishment of proper microtubule-kinetochore attachments2,3,12,19. Mutant MEFs with low amounts of Rae1 have impaired mitotic checkpoint activity and are prone to chromosome segregation errors and aneuploidization20. On a T23 background, however, these MEFs had much lower missegregation and aneuploidy rates, which coincided with restoration of normal mitotic checkpoint activity (Fig. 2g,h and Supplementary Fig. S2d). Bub1T85 MEFs, which overexpress the mitotic checkpoint protein Bub1, have an intact mitotic checkpoint but are predisposed to chromosome missegregation and aneuploidy due to defective attachment error correction12. BubR1 overabundance markedly improved error correction in these MEFs, resulting in decreased aneuploidy (Fig. 2h and Supplementary Fig. S2d). Together, these findings indicate that increased BubR1 expression can preserve genomic integrity by ameliorating defects that perturb the mitotic checkpoint and/or kinetochore-microtubule attachment.
To analyze the impact of sustained high BubR1 expression on spontaneous tumorigenesis, we generated a cohort of T23 transgenic mice. As control cohorts, we used non-transgenic littermates and a transgenic strain expressing EGFP under the control of the CAGGS promoter, referred to as T-GFP mice. In accordance with conclusions from the DMBA and KrasLA1 studies, development of lethal tumors (i.e. malignant lymphomas, sarcomas, and carcinomas) was significantly delayed in T23, revealing a broad tumor protective effect of high BubR1 (Fig. 3a). The spontaneous tumors that developed in T23 animals contained low amounts of FLAG-BubR1 (Supplementary Fig. S3a,b), implying that tumorigenesis selected for loss of transgene expression. Consistent with attenuated tumor development, T23 mice showed increased longevity (Fig. 3b). Tumor protection and lifespan extension were observed in both genders, although more profoundly in males (Supplementary Fig. S3c–f).
Fig. 3.
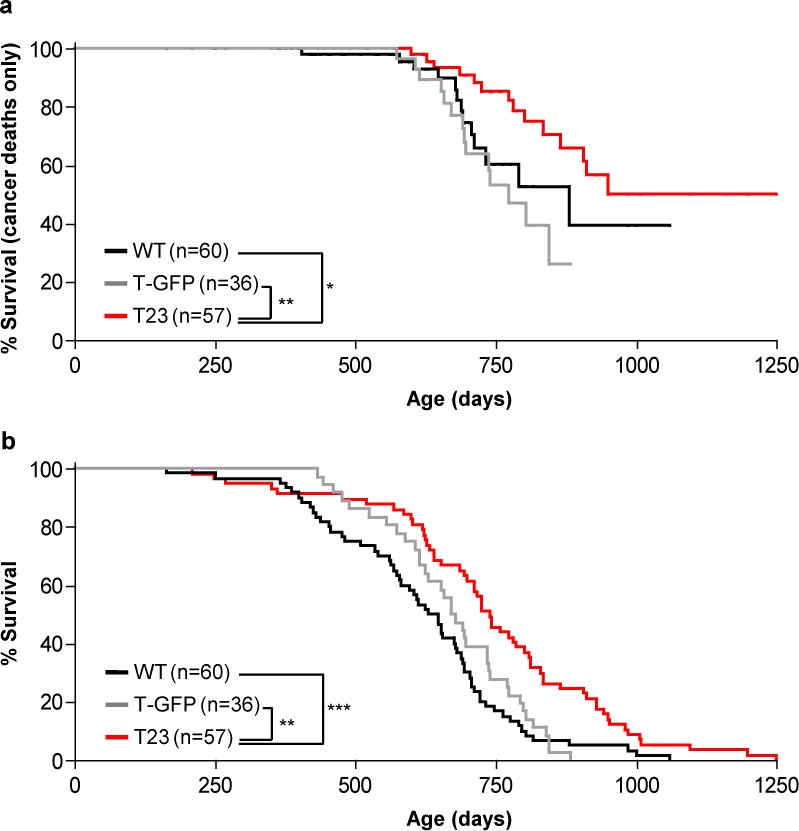
Increased BubR1 expression protects against spontaneous tumors and extends lifespan. (a) Survival curves of wildtype, T-GFP, and T23 mice dying of cancer. Only mice with malignant lymphomas, sarcomas and carcinomas are included. Statistical analysis of the survival curves is represented by the asterisks (log-rank test). (b) Overall survival curves of wildtype, T-GFP, and T23 mice. We note that maximum lifespan of T23 mice was also significantly extended compared to both wildtype and T-GFP control mice (p = 0.05 and 0.0056), respectively; one-sided Wang/Allison test referring to the proportion of mice alive at the 90th percentile survival point. Furthermore, we note that the median lifespan of our wildtype cohort is similar to that of an earlier, independent study performed at the same site40. *P < 0.05, **P < 0.01, ***P < 0.001. n indicates the number of mice (mixed gender) per genotype.
To explore whether high BubR1 expression has anti-aging effects independent of tumor-protection, we analyzed T23 and wildtype mice for distinct parameters of healthspan. Muscle loss is a hallmark of aging in both humans and rodents. In contrast to wildtype mice, which showed a 35% decrease in gastrocnemius muscle fiber diameter between 3 and 24 months of age, T23 mice were protected from muscle fiber atrophy (Fig. 4a). Relative gastrocnemius muscle weights of aged T23 mice were significantly larger than those of corresponding control mice (Supplementary Fig. S4a). Consistent with reduced muscle degeneration, aging induced upregulation of p16Ink4a and p19Arf, two biomarkers of senescence and aging21, was blunted in the gastrocnemius of T23 mice (Fig. 4b,c). T23 mice outperformed wildtype counterparts in treadmill exercise tests (Fig. 4d–f), validating preservation of muscle function. Additionally, renal sclerosis with glomerulosclerosis, interstitial fibrosis and tubular atrophy, which occurs with aging, was reduced in 24-month-old T23 animals (Fig. 4g,h). Blood urea nitrogen (BUN) levels of these animals were significantly lower than in age-matched wildtype animals (16.10 ± 1.95 mg/dl versus 23.02 ± 2.08 mg/dl, respectively, P = 0.0136, unpaired t test), indicating enhanced preservation of renal function. Aging of kidney is characterized by accumulation of cells with γ-H2AX foci22,23, which are thought to represent senescent cells29,30. The foci themselves represent sites of DNA damage, potentially resulting from aberrant sister chromatid segregation during mitosis24,25. Consistent with reduced age-related pathology, kidneys of 24-month-old T23 animals contained significantly fewer cells with γ-H2AX foci than those of age-matched control mice (Fig. 4i). Conversely, the incidence of cells with γ-H2AX foci in kidneys of progeroid BubR1 mice was markedly increased at 3 months of age (Fig. 4i), indicating that formation of DNA double strand breaks inversely correlates with level of BubR1 expression. Reactive oxygen species (ROS) have been linked to age-related DNA damage26, but we found no evidence for alterations in abundance of or tolerance to ROS in T23 mice (Supplementary Fig. S4b–d). Decreased accumulation of cells with γ-H2AX foci in kidney of 24-month-old T23 mice correlated with increased BrdU incorporation, further supporting the idea that elevated BubR1 attenuates senescence-associated replicative arrest (Supplementary Fig. S4e).
Fig. 4.
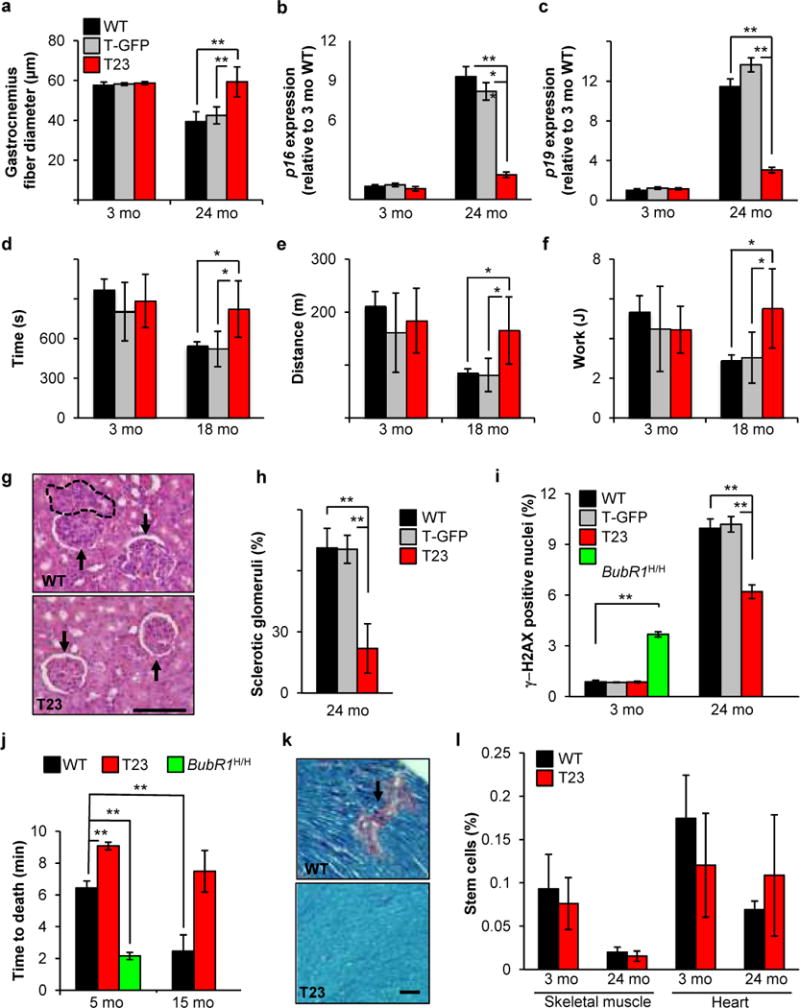
Increased BubR1 expression delays select age-related pathologies. (a) Sustained BubR1 expression attenuates muscle fiber atrophy. Gastrocnemius muscle fiber diameter declines with age in all genotypes but T23. (b) Quantitative RT-PCR analysis of p16Ink4a expression in gastrocnemius muscles 3 and 24-month-old transgenic and control mice relative to 3-month-old wildtype muscles. (c) Same as (b) but for p19Arf. (d–f) Exercise ability is enhanced in aged T23 mice. Time (d), distance (e), and work performed (f) are all improved in 18-month-old T23 animals compared to WT and T-GFP. (g) Hematoxylin-eosin stained kidney sections of 24-month-old mice. Dashed area depicts interstitial inflammation; arrows denote glomerular hypercellularity of WT mice (above) and normal glomeruli of T23 mice (below). Scale bar, 100 μm. (h) Percentage of sclerotic glomeruli from 24-month-old kidney sections. 40 glomeruli were scored for each animal. (i) DNA damage, as measured by γ-H2AX staining on cryosections of kidney tissue, is increased in BubR1H/H mice at a young age and reduced in transgenic mice at advanced age. (j) Cardiac stress tolerance correlates with BubR1 level of expression. (k) Ptah stained heart sections of 24-month-old wildtype and T23 mice. Interstitial fibrosis (pink area) is significantly reduced in T23 animals. Scale bar, 50 μm. For all analyses in a–k, n = 5 males per genotype per age group. Error bars represent SD. (l) Quantification of the percentage of stem cells isolated from 3 and 24-month-old wildtype and T23 mouse skeletal muscle (satellite cells) and heart (cardiac stem cells). n = 3 males per genotype per tissue. Values represent means ± SEM. We note that there are no significant differences between wildtype and T23. *P < 0.05, **P < 0.01, ***P < 0.001.
BubR1 progeroid mice are thought to succumb to early death due to heart failure27, prompting us to examine whether sustained BubR1 expression improves cardiac performance. In a cardiac stress tolerance test, in which mice are injected with a lethal dose of the β-adrenergic agonist isoproterenol, wildtype mice suffered cardiac arrest within 6.5 min (Fig. 4j). Consistent with reduced cardiac stress resistance, 5-month-old BubR1H/H mice died three times faster than control animals. In contrast, time to death was significantly extended in T23 animals. Furthermore, while cardiac stress tolerance of wildtype mice significantly declined between 5 and 15 months of age, T23 mice retained high cardiac performance. Age-related interstitial fibrosis in heart tissue was markedly lower in T23 mice (Fig. 4k), corroborating that BubR1 overexpression acts to preserve cardiac function and structural integrity. Attenuated deterioration of skeletal muscle and heart in T23 mice was not characterized by increased stem cell abundance within these tissues (Fig. 4l). T23 animals also showed resistance to age-related retinal atrophy (Supplementary Fig. S4f), which correlated well with increased BubR1 expression in eye (Supplementary Fig. S1c). In contrast, osteoporosis and cataractogenesis, two other age-related disorders that we screened for, were not delayed (Supplementary Fig. S4g–i), indicating that the anti-aging effect of increased BubR1 expression is confined to particular cell and tissue types.
Aneuploidy increases with aging in mouse splenocytes28. To determine whether age-related aneuplodization also occurs in other tissues and correlates with age-related tissue dysfunction, we performed interphase FISH for chromosomes 4 and 7 on tissues of 3- and 24-month-old wildtype and T23 mice. Aneuploidy rates for both chromosomes markedly increased with aging in wildtype lung tissue, but not in T23 lungs (Table 1). Similar data were obtained for tissues that exhibited characteristics of delayed aging in T23 mice, including skeletal muscle, kidney, eye, and heart (Table 1). The presence of age-related aneuploidy in wildtype heart was surprising, given that traditionally heart has been viewed as a terminally differentiated post-mitotic organ. However, recent studies indicate that the heart replaces its cells several times during its lifespan29. Inhibition of age-related aneuploidization was not universal as bone marrow and small intestine, two tissues with a relatively high mitotic index, showed similar age-related aneuploidy rates in T23 and wildtype mice (Table 1). Notably, stem cells in skeletal muscle, heart, and bone marrow showed resistance to age-related aneuploidization in both wildtype and T23 animals (Table 1), suggesting that these populations are well protected against whole chromosome instability. There are several plausible mechanisms for why aneuploidy rates of differentiated cells could be high with stem cell aneuploidy being low (Supplementary Fig. S5k). First, lineage-primed progenitors derived from stem cells undergo multiple rounds of division before differentiation occurs and aneuploidization might occur during this proliferative phase. Second, it is possible that aneuploidy does occur when stem cells divide, but that the daughter cell that inherits stemness dies while the progenitor daughter cell survives and continues to proliferate. A third possibility would be that aneuploidization of differentiated cells is due to polyploidy resulting from fusion of nuclei. Polyploid nuclei, however, were not observed or extremely rare in our samples (data not shown). Finally, age-related aneuploidization and tissue dysfunction were not attenuated in T7 mice (Supplementary Fig. S5), indicating that both beneficial effects require a certain threshold of BubR1 overexpression.
Table 1.
Improved healthspan of T23 tissues correlates with reduced aneuploidization with age. Interphase FISH analysis on 3 and 24-month-old wildtype and T23 mice. n = 3 males per genotype, per tissue, per age group. Values represent means ± SD.
| 3-mo-old mice
|
24-mo-old mice
|
|||||
|---|---|---|---|---|---|---|
| % Aneuploidy (SD) | % Aneuploidy (SD) | |||||
|
|
|
|||||
| Tissue or stem cell type | Chr. 4 | Chr. 7 | Chr. 4 | Chr. 7 | ||
| WT Lung | 1.7 (0.6) | 1.7 (0.6) | 3.7 (0.6) | * | 4.0 (0.0) | ** |
| T23 Lung | 1.3 (0.6) | 1.7 (0.6) | 1.3 (0.6) | 1.3 (0.6) | ||
| WT Skeletal muscle | 3.3 (0.6) | 2.7 (0.6) | 13.0 (1.2) | * | 14.0 (1.7) | * |
| T23 Skeletal muscle | 3.3 (0.6) | 2.7 (1.2) | 7.3 (1.5) | 8.0 (1.7) | ||
| WT Kidney | 1.7 (0.6) | 1.3 (0.6) | 5.7 (0.6) | * | 7.3 (1.5) | * |
| T23 Kidney | 1.7 (0.6) | 1.7 (0.6) | 2.7 (1.5) | 2.3 (2.0) | ||
| WT Heart | 2.3 (1.2) | 2.7 (0.6) | 11.3 (3.2) | * | 11.3 (1.2) | ** |
| T23 Heart | 2.3 (1.5) | 2.0 (1.0) | 5.3 (1.2) | 6.0 (1.0) | ||
| WT Eye | 2.3 (0.6) | 2.7 (0.6) | 9.3 (1.5) | ** | 8.0 (1.0) | ** |
| T23 Eye | 1.7 (0.6) | 2.3 (0.6) | 4.7 (0.6) | 4.7 (0.6) | ||
| WT Spleen | 1.3 (0.6) | 1.3 (0.6) | 3.3 (1.2) | 4.7 (0.6) | ||
| T23 Spleen | 1.7 (1.2) | 1.7 (1.2) | 2.7 (0.6) | 3.0 (1.0) | ||
| WT Bone marrow | 1.7 (0.6) | 2.0 (1.0) | 5.0 (1.0) | 3.0 (1.0) | ||
| T23 Bone marrow | 1.7 (1.2) | 2.0 (1.0) | 4.3 (0.6) | 4.0 (1.0) | ||
| WT Small intestine | 1.7 (0.6) | 1.7 (0.6) | 4.3 (0.6) | 4.7 (0.6) | ||
| T23 Small intestine | 2.0 (1.0) | 1.7 (0.6) | 5.0 (1.0) | 5.3 (1.2) | ||
| WT Satellite cells | 2.9 (0.1) | 3.3 (0.7) | 3.7 (1.1) | 4.8 (1.8) | ||
| T23 Satellite cells | 2.1 (0.2) | 3.5 (1.3) | 3.5 (1.4) | 5.1 (2.2) | ||
| WT Cardiac stem cells | 2.7 (0.6) | 2.3 (0.6) | 4.0 (1.0) | 3.3 (0.6) | ||
| T23 Cardiac stem cells | 2.3 (0.6) | 2.7 (0.6) | 3.3 (0.6) | 3.7 (1.2) | ||
| WT Hematopoietic stem cells | 1.3 (0.6) | 1.7 (0.6) | 2.3 (0.6) | 3.0 (1.0) | ||
| T23 Hematopoietic stem cells | 1.7 (0.6) | 2.0 (1.0) | 2.0 (1.0) | 2.3 (0.6) | ||
P < 0.05,
P < 0.01. Numbers indicated in red represent values at 24 months that were significantly higher than the same chromosome when analyzed at 3 months of age (P < 0.05).
Our data here show that sustained high expression of BubR1 in mice protects against cancer, attenuates age-related deterioration of select tissues, and extends median and maximum lifespan. We show that these beneficial effects tightly correlate with reduced oncogene-induced or age-related aneuploidization, and that high BubR1 levels act to reinforce mitotic checkpoint control and attachment error correction in the presence of genetic defects that cause mitotic stress. Based on these findings, it is tempting to speculate that high BubR1 expression extends health- and lifespan by attenuating chromosomal instability. Consistent with this hypothesis, many mouse models of accelerated aneuploidization are prone to tumorigenesis15,30,31. On the other hand, while BubR1 hypomorphic and Bub3/Rae1 double haploinsufficient mice exhibit overt premature aging phenotypes1,32, other aneuploidy prone mouse strains do not. One possibility is that aneuploidy is required, but not sufficient for induction of age-related pathologies. For instance, aneuploidy may contribute to the induction of age-related pathologies only in the presence of other age-associated damage, such as DNA double strand breaks (DSBs), mitochondrial DNA damage, and proteotoxic damage33,34. Our observation that DSBs, which can result from specific types of chromosome segregation errors26,27, were increased in kidney of BubR1 progeroid mice and decreased in kidney of T23 transgenics (Fig. 4i), support this notion.
Alternatively, the beneficial effects of sustained overexpression of BubR1 could be independent of chromosome segregation. BubR1 is normally present throughout the cell cycle and known to be implicated in roles outside mitosis, including DNA repair and ciliogenesis35,36. Furthermore, recent studies uncovered that APC/CCdc20, whose E3 ubiquitin ligase activity is controlled by BubR1, regulates important biological processes in post-mitotic cells such as morphogenesis and differentiation37–39. These findings raise the interesting possibility that BubR1 overexpression might exert its beneficial effects by controlling the stability of interphase APC/CCdc20 substrates implicated in the suppression of cellular stresses that engage cellular senescence or other aging-related pathways.
In summary, we provide the first experimental evidence that overexpression of a mitotic regulator can improve ploidy control, provide resistance to tumorigenesis and delay age-related decline in tissues and organs important to human health in the absence of apparent side effects. These findings identify BubR1 and its regulator(s) as unique and promising targets for treatment of a broad spectrum of aneuploid human cancers and key age-related disorders that dictate health.
METHODS
Generation of Flag-mBubR1 transgenic mice
T7 and T23 BubR1 transgenic mice overexpressing Flag-tagged murine BubR1 protein were generated according to previously described methods11,41. T-GFP mice, which overexpress EGFP using the same promoter sequence as was used for the BubR1 transgene, were obtained and used as a control (stock number 004178, strain C57BL/6J, Jackson Laboratories). We note that EGFP levels in T-GFP and T23 mice were similar. Protocols used for PCR genotyping of the above strains are available upon request. All mice were on a mixed C57BL/6-SV129 background and were housed in a pathogen-free barrier environment for the duration of the study. These animals were fed a 10% fat diet and maintained on a 12 h light: dark cycle and were inspected daily. Animals in survival curves were mice found dead or euthanized if unlikely to survive for more than 48 h. These mice were carefully screened for tumors. Tumors were collected and processed by standard procedures for histopathological evaluation. Malignant lymphomas, sarcomas and carcinomas were considered in our tumor-free survival analysis. We note that animals used for experiments, including healthspan analyses, were omitted from the survival analysis. For light and fluorescence imaging of pups and organs we used a fluorescent dissecting microscope mounted with a camera (Leica). All images were taken within an hour of harvest from an intra-cardially PBS-flushed mouse. Isoproterenol challenge assays were performed on male mice. A lethal dose (680 mg/kg) was given to all animals and time until death was measured. All animal procedures were reviewed and approved by the institutional animal care and use committee.
Statistical analyses
Prism software was used for the generation of all survival curves and statistical analysis. Log-rank tests were used to determine overall and pairwise significance for all survival curves (Fig. 3 and Supplementary Fig. 3c–f). Log-rank tests were also used in Fig. 2g, and Supplementary Fig. 4b and i. One-sided Wang-Allison tests were used to determine maximum lifespan changes42 in Fig. 3 and Supplementary Fig. 3c–f. Chi-square tests for overall and pairwise significance were used for overall, lung and skin tumor incidence (Fig. 2a, first three graphs). Mann-Whitney tests were used for pairwise significance analysis in the following figures: Fig. 2a (graphs for average number of tumors); Fig. 2b, f, and h; Fig. 4a–c and h–j; Supplementary Fig. 2a and c; Supplementary Fig. 4e–h; and Supplementary Fig. 5a–c and g–i. Kruskal-Wallis tests for overall significance were performed before conducting these Mann-Whitney tests. An unpaired t test was used for pairwise comparisons in the following figures: Fig. 1d–f; Fig. 2d and e; Fig. 4d–f and l; Table 1; Supplementary Fig. 2d; Supplementary Fig. 4a, c and d; and Supplementary Fig. 5d–f and j.
Western blot analysis, immunoprecipitations and immunohistochemistry
Immunoprecipitations and western blot analysis were performed as previously described43. Mitotic cell lysates were prepared as described3. Tissue lysates were prepared by first snap-freezing the tissue sample in liquid nitrogen and then grinding it to powder form in mortar and pestle. 10 mg of powder was suspended in 100 μl of 1X PBS and 100 μl of Laemmli lysis buffer and boiled for 10 min before loading into Tris-HCl polyacrylamide gel. Blots were probed with antibodies for P-H3Ser10 (Millipore; 06-570, 1:500), BubR11 (1:1000) Flag (Origene; TA100011, 1:1000), β-actin (Sigma; A5441, 1:40000), EGFP (Cell Signaling; 2956, 1:1000), Aurora B (BD Biosciences; 611083, 1:1000), Cdc20 (1:1000), Plk1 (1:500) and Hras (1:5000) (Santa Cruz Biotechnology, Inc.; SC-8358, SC-17783, and SC-520, respectively), Bub320 (1:1000), Mad211 (1:1000), and Bub144 (1:1000). All antibodies were detected with secondary HRP-conjugated goat anti-mouse or anti-rabbit antibodies (Jackson Immunoresearch; 1:10,000). Equal loading was confirmed by using α-tubulin (Sigma; T9026, 1:2000) or by Ponceau S staining. Immunohistochemistry for BubR1 (BD Biosciences; 612503, 1:500) was performed on formalin fixed, paraffin embedded lung tumor sections using the SignalStain Boost IHC detection reagent as per manufacturer’s instructions (Cell Signaling).
Generation and culture of MEFs and live cell imaging
Wildtype, T7 and T23 MEFs were generated and cultured as previously described1. Double mutant MEFs were produced by breeding T23 transgenic females to Rae1+/−20 and Bub1T85 males12. Live cell imaging-based analyses of chromosome segregation errors and mitotic checkpoint activity were as previously described11. Nocodazole and taxol challenge assays and mitotic timing experiments were performed as previously described3. Oncogenic Ras was expressed in passage 3 (P3) wildtype and T23 MEFs using pBabe puro HrasG12V (Addgene plasmid 9051). Puromycin-resistant MEFs (2 μg/ml puromycin) were analyzed by live cell imaging five days post-transduction. At least three independent MEF lines per genotype were used in all experiments.
Karyotype analyses
Splenocyte and MEF karyotype analyses were performed as described20. FISH analysis on single cells isolated from fresh lung tissue was performed as described6. Chromosome counts on MEFs expressing oncogenic Ras were done as follows: P3 wildtype and T23 MEFs were transduced with pBabe puro HrasG12V for two days, cultured in medium containing 2 μg/ml puromycin for two days and arrested in mitosis with colcemid on day 5. Interphase FISH analysis on cell suspensions prepared from various wildtype, T7 and T23 tissues was performed as previously described17 in the Mayo Clinic Cytogenetics Core Facility as described in detail by Bayani and Squire45. At least 100 cells were analyzed per sample, n ≥ 3 samples per genotype and tissue.
Tumor susceptibility studies
DMBA treatment of mice was performed as previously described13. K-rasLA1 mice14 were obtained from the MMHCC (NCI Frederick).
Age related phenotyping
Biweekly, mice were screened for overt cataracts32. Muscle fiber diameter measurements were taken on gastrocnemius muscles as previously described6. Exercise ability assessments were performed as described27. Individual gastrocnemius muscles were isolated from 18-month-old female mice. The relative weight is the weight of one gastrocnemius muscle divided by overall body mass. Bone mineral density and bone mineral content were determined using DEXA scanning. Formalin fixed, paraffin embedded kidney samples were stained using routine hematoxylin and eosin. Forty randomly selected glomeruli were scored for sclerosis. Glomeruli with > 50% sclerosis were determined to be sclerotic. Blood urea nitrogen (BUN) assays were performed on 24-month-old mice as described46. Ptah staining on formalin fixed, paraffin embedded heart samples were performed as described47. Retinal thickness was determined by measuring the thickness of the retinal layer in hematoxylin and eosin stained paraffin embedded eye tissue using a calibrated computer program (Olympus MicroSuite 5). Forty random measurements were taken for each sample. γ-H2AX staining was performed as described23.
Stem cell isolation and quantification
Satellite cells were labeled and purified as previously described48. Briefly, excised gastrocnemius muscles were washed in PBS and digested in DMEM containing collagenase type II solution (Worthington Biochemical Corp.) and 2% FCS for 45–60 min with agitation at 37°C. Cell suspensions were filtered through a 45 micron mesh and centrifuged for 5 min at 500 g. Cells were incubated in blocking buffer for 15 min on ice following antibody staining for CD45, CD34 and Sca1 (all from BD-Biosciences; 557659, 560230 and 553336, respectively). Cells with a CD45−, Sca-1− and CD34+ surface profile49 were collected using a FACS Aria Cell Sorter (BD Biosciences) running FACSDiva software. Enzymatic isolation of endogenous cardiac stem cells (CSC) was performed after Langendorf perfusion of the heart50 to obtain a viable single cell suspension. Following antibody staining for c-Kit, CD45 and CD34, cells with a c-Kit+, CD45− and CD34− surface profile were collected51 (c-Kit antibody was from BD-Biosciences; 561075). Identification and quantification of hematopoietic stem cells was performed as described52.
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) analysis was performed on cDNA generated from RNA extracted from gastrocnemius muscle of 3-month-old and 24-month-old mice as previously described6.
In vivo BrdU incorporation
Analyses for in vivo BrdU incorporation were performed on 24-month-old mice as previously described6.
Assessment of ROS resistance by paraquat
Paraquat (methyl viologen; Sigma) was dissolved in sterile saline and administered to 3 to 4 month-old mice by intraperitoneal injection at a dose of 60 mg/kg body weight. Animals were monitored for survival for 10 days following paraquat injection.
Hydrogen peroxide and DCFDA assay
Hydrogen peroxide (H2O2) levels in gastrocnemius muscle of 24-month-old mice were determined by using the Amplex Red Hydrogen Peroxide Assay Kit according to the protocol of the manufacturer (Invitrogen). Fluorescence was measured using an excitation wavelength of 545 nm and emission wavelength of 590 nm. The H2O2 concentration/mg protein was calculated by using a standard curve. H2O2 levels were presented as relative to 24-month-old wildtype mice. ROS levels in liver of 24-month-old mice were analyzed by using carboxy-DCFDA (2′,7′-dichlorodihydrofluorescein diacetate, Invitrogen). Tissue extracts were added to DCFDA (25 μM) and fluorescence was measured using an excitation wavelength of 485 nm and emission wavelength of 530 nm. For both experiments, the homogenization buffer consisted of 20 mM glycerolphosphate, 20 mM NaF, 2 mM sodium orthovanadate, 1 mM EDTA, 0.5 mM PMSF, 1 μM pepstatin, 100 mM Tris-HCl (pH 7.4) and freshly added mini EDTA-free cocktail tablet (Roche).
Supplementary Material
BubR1 expression analysis in T23 mouse tissues and analysis of mitotic checkpoint complex formation and levels of key mitotic regulators in T23 MEFs. (a) Western blot analysis of mitotic MEF extracts for BubR1. BubR1 protein levels in T23 MEFs were about 2-fold higher than in T7 MEFs and 10-fold higher than in wildtype MEFs. Actin served as a loading control. (b) Images of WT and transgenic pups obtained by light (left) and fluorescence microscopy (right). (c) The BubR1 transgene continues to be expressed late in life of transgenic animals and is not subject to silencing. Western blot analysis of tissues of 24-month-old WT and T23 mice. Blots were probed for BubR1. Ponceau S staining was used as a loading control. (d) Immunoblots of wildtype and T23 MEF extracts subjected to immunoprecipitation with the indicated antibodies. (e) Western blots of mitotic cell extracts of wildtype and T23 MEFs probed for key mitotic regulators. P-H3Ser10 was used as a control for equal loading of mitotic cells.
Protection against tumor formation and aneuploidization requires a threshold of BubR1 overexpression. (a) BubR1 T7 transgenic mice are not protected from lung tumors induced by oncogenic Kras (G12D). Cohorts of KrasLA1, and T7;KrasLA1 mice were killed at 6 weeks of age and lung tumors counted under a dissection microscope. Error bars indicate SEM. (b) Alveolar hyperplasia in K-rasLA1 and T23;K-rasLA1; mice is similar. Haematoxylin-eosin stained lung sections of mice of the indicated genotypes (at 6 weeks of age). Hyperplastic alveolar tissue (without tumors) of K-rasLA1 and T23;K-rasLA1 mice is shown. Note that lung tissue of T23 transgenic mice is histologically normal. (c) Aneuploidy rates in lungs of KrasLA1 mice with and without BubR1 clone T7 overexpression. (d) Chromosome missegregation rates of mutant MEF lines with and without BubR1 overexpression. Consistent with reduced aneuploidy rates, the fidelity of segregation is improved when BubR1 levels are high. *P < 0.05, ***P < 0.001. n indicates the number of mice (mixed gender) used per genotype in a and c, and the number of independent MEF lines used in d.
Overall and tumor-free survival of T23 animals is extended in both males and females. (a, b) Analysis of BubR1 protein levels in spontaneous tumors. (a) Western blot analysis of T23 liver tumors and flanking normal tissue for expression of transgenic BubR1 (FLAG-BubR1). Tubulin was used as a loading control. (b) Representative images of WT and T23 lung sections stained for BubR1. Note that BubR1 is present at high levels in normal lung tissue of T23 mice but not in neighboring tumor tissue. Scale bars: top, 1 mm; bottom, 100 μm. (c–f) Data presented are from the same mouse cohorts as in Fig. 4, but now separated by gender. (c) Overall survival of males and (d) females, and (e) tumor-free survival of males and (f) females. *P < 0.05, **P < 0.01, ***P < 0.001.
High levels of BubR1 delay age-related changes in a tissue specific manner. (a) BubR1 overexpression protects against loss of muscle mass. (b) Levels of BubR1 do not correlate with altered sensitivity to acute oxidative stress, as survival after paraquat injection in 3–4 month-old mice is unchanged. (c) H2O2 levels in gastrocnemius of 24-month-old mice in T23 is similar to wildtype. (d) Relative ROS levels are unchanged in livers of 24-month-old mice. (e) Proliferative capacity in kidney, as measured by BrdU incorporation, is increased in aged BubR1 transgenic mice. (f) Age-related retinal degeneration is prevented by high levels of BubR1. (g–h) High levels of BubR1 are unable to prevent osteoporosis. Bone mineral density (g) and bone mineral content (h) declines in mice with age. (i) Cataract onset and incidence are not prevented by overexpression of BubR1 in the eye. n = 5 females per genotype in a, and 5 males per genotype in c–h. n in b and i indicates the number of mice (mixed gender) used per genotype. Values in a and e–h are means ± SD, values in c and d are means ± SEM. *P < 0.05, **P < 0.01.
Age-related attenuation of tissue dysfunction requires a threshold level of BubR1 overexpression. (a) Gastrocnemius muscle fiber diameter measurements. (b) QRT-PCR analysis of p16Ink4a expression in young and old gastrocnemius muscles. (c) Same as (b) but for p19Arf. (d–f) Duration of exercise (d), distance travelled (e), and work performed (f) in treadmill exercise tests. (g) Analysis of glomerulosclerosis. (h) Age-related retinal degeneration is not attenuated in T7 mice. (i) Cardiac stress tolerance is not improved in T7 animals. For all analyses in a–i, n = 5 males per genotype per age group, values are means ± SD. (j) Interphase FISH analysis on various mouse tissues (n = 3 males per genotype per tissue). (k) Model for asymmetric aneuploidization of proliferating stems cells and their committed progeny: (top) stem cells segregate their chromosomes with high accuracy, but committed progenitors do not leading to aneuploid differentiated offspring; (bottom) stem cells undergoing inaccurate segregation die, while progenitor daughter cells do not, allowing them to produce aneuploid offspring.
Acknowledgments
We thank Paul Galardy, Robin Ricke, James Kirkland, Nathan LeBrasseur, and Richard Miller for feedback on the manuscript, and Karl Nath and Joseph Grande for help with the analysis of kidney phenotypes. This study was supported by NIH grant CA96985, the Ellison Medical Foundation, the Noaber Foundation and The Kogod Center on Aging.
Footnotes
AUTHOR CONTRIBUTIONS
D.J.B., M.M.D., T.W., K.B.J., L.M., J.H.v.R., R.C.D., S.R., A.B., A.T., L.S., V.S., and J.M.v.D. designed and performed experiments, B.v.d.S. helped supervise T.W., and D.J.B. and J.M.v.D. wrote the manuscript. All authors discussed results, made figures and edited the manuscript.
References
- 1.Baker DJ, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–749. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- 2.Lampson MA, Kapoor TM. The human mitotic checkpoint protein BubR1 regulates chromosome-spindle attachments. Nat Cell Biol. 2005;7:93–98. doi: 10.1038/ncb1208. [DOI] [PubMed] [Google Scholar]
- 3.Malureanu LA, et al. BubR1 N terminus acts as a soluble inhibitor of cyclin B degradation by APC/C(Cdc20) in interphase. Dev Cell. 2009;16:118–131. doi: 10.1016/j.devcel.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matsumoto T, et al. Aging-associated vascular phenotype in mutant mice with low levels of BubR1. Stroke. 2007;38:1050–1056. doi: 10.1161/01.STR.0000257967.86132.01. [DOI] [PubMed] [Google Scholar]
- 5.Hartman TK, Wengenack TM, Poduslo JF, van Deursen JM. Mutant mice with small amounts of BubR1 display accelerated age-related gliosis. Neurobiol Aging. 2007;28:921–927. doi: 10.1016/j.neurobiolaging.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 6.Baker DJ, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 2008;10:825–836. doi: 10.1038/ncb1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsuura S, et al. Monoallelic BUB1B mutations and defective mitotic-spindle checkpoint in seven families with premature chromatid separation (PCS) syndrome. Am J Med Genet A. 2006;140:358–367. doi: 10.1002/ajmg.a.31069. [DOI] [PubMed] [Google Scholar]
- 8.Hanks S, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet. 2004;36:1159–1161. doi: 10.1038/ng1449. [DOI] [PubMed] [Google Scholar]
- 9.Burtner CR, Kennedy BK. Progeria syndromes and ageing: what is the connection? Nat Rev Mol Cell Biol. 2010;11:567–578. doi: 10.1038/nrm2944. [DOI] [PubMed] [Google Scholar]
- 10.Sotillo R, et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Ree JH, Jeganathan KB, Malureanu L, van Deursen JM. Overexpression of the E2 ubiquitin-conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. J Cell Biol. 2010;188:83–100. doi: 10.1083/jcb.200906147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ricke RM, Jeganathan KB, van Deursen JM. Bub1 overexpression induces aneuploidy and tumor formation through Aurora B kinase hyperactivation. J Cell Biol. 2011;193:1049–1064. doi: 10.1083/jcb.201012035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serrano M, et al. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 14.Johnson L, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 15.Schvartzman JM, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer. 2010;10:102–115. doi: 10.1038/nrc2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10:478–487. doi: 10.1038/nrm2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baker DJ, Jin F, Jeganathan KB, van Deursen JM. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer cell. 2009;16:475–486. doi: 10.1016/j.ccr.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saavedra HI, et al. The RAS oncogene induces genomic instability in thyroid PCCL3 cells via the MAPK pathway. Oncogene. 2000;19:3948–3954. doi: 10.1038/sj.onc.1203723. [DOI] [PubMed] [Google Scholar]
- 19.Sudakin V, Chan GK, Yen TJ. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol. 2001;154:925–936. doi: 10.1083/jcb.200102093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Babu JR, et al. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J Cell Biol. 2003;160:341–353. doi: 10.1083/jcb.200211048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krishnamurthy J, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a) Mol Cell. 2004;14:501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 23.Matheu A, et al. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448:375–379. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- 24.Lukas C, et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol. 2011;13:243–253. doi: 10.1038/ncb2201. [DOI] [PubMed] [Google Scholar]
- 25.Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–1898. doi: 10.1126/science.1210214. [DOI] [PubMed] [Google Scholar]
- 26.Vijg J, Campisi J. Puzzles, promises and a cure for ageing. Nature. 2008;454:1065–1071. doi: 10.1038/nature07216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker DJ, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lushnikova T, Bouska A, Odvody J, Dupont WD, Eischen CM. Aging mice have increased chromosome instability that is exacerbated by elevated Mdm2 expression. Oncogene. 2011;30:4622–4631. doi: 10.1038/onc.2011.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bergmann O, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holland AJ, Cleveland DW. Losing balance: the origin and impact of aneuploidy in cancer. EMBO Rep. 2012;13:501–514. doi: 10.1038/embor.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfau SJ, Amon A. Chromosomal instability and aneuploidy in cancer: from yeast to man. EMBO Rep. 2012;13:515–527. doi: 10.1038/embor.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baker DJ, et al. Early aging-associated phenotypes in Bub3/Rae1 haploinsufficient mice. J Cell Biol. 2006;172:529–540. doi: 10.1083/jcb.200507081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, Mitchell JR, Hasty P. DNA double-strand breaks: a potential causative factor for mammalian aging? Mech Ageing Dev. 2008;129:416–424. doi: 10.1016/j.mad.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fang Y, et al. BubR1 is involved in regulation of DNA damage responses. Oncogene. 2006 doi: 10.1038/sj.onc.1209392. [DOI] [PubMed] [Google Scholar]
- 36.Miyamoto T, et al. Insufficiency of BUBR1, a mitotic spindle checkpoint regulator, causes impaired ciliogenesis in vertebrates. Hum Mol Genet. 2011;20:2058–2070. doi: 10.1093/hmg/ddr090. [DOI] [PubMed] [Google Scholar]
- 37.Malureanu L, et al. Cdc20 hypomorphic mice fail to counteract de novo synthesis of cyclin B1 in mitosis. J Cell Biol. 2010;191:313–329. doi: 10.1083/jcb.201003090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim AH, et al. A centrosomal Cdc20-APC pathway controls dendrite morphogenesis in postmitotic neurons. Cell. 2009;136:322–336. doi: 10.1016/j.cell.2008.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Y, et al. A Cdc20-APC ubiquitin signaling pathway regulates presynaptic differentiation. Science. 2009;326:575–578. doi: 10.1126/science.1177087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conover CA, Bale LK. Loss of pregnancy-associated plasma protein A extends lifespan in mice. Aging Cell. 2007;6:727–729. doi: 10.1111/j.1474-9726.2007.00328.x. [DOI] [PubMed] [Google Scholar]
- 41.van Ree J, Zhou W, Li M, van Deursen JM. Transgenesis in mouse embryonic stem cells. Methods Mol Biol. 2011;693:143–162. doi: 10.1007/978-1-60761-974-1_9. [DOI] [PubMed] [Google Scholar]
- 42.Wang C, Li Q, Redden DT, Weindruch R, Allison DB. Statistical methods for testing effects on “maximum lifespan”. Mech Ageing Dev. 2004;125:629–632. doi: 10.1016/j.mad.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 43.Kasper LH, et al. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol Cell Biol. 1999;19:764–776. doi: 10.1128/mcb.19.1.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeganathan K, Malureanu L, Baker DJ, Abraham SC, van Deursen JM. Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J Cell Biol. 2007;179:255–267. doi: 10.1083/jcb.200706015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bayani J, Squire JA. Fluorescence in situ Hybridization (FISH) Curr Protoc Cell Biol. 2004;Chapter 22 doi: 10.1002/0471143030.cb2204s23. [DOI] [PubMed] [Google Scholar]
- 46.Nath KA, Croatt AJ, Warner GM, Grande JP. Genetic deficiency of Smad3 protects against murine ischemic acute kidney injury. Am J Physiol Renal Physiol. 2011;301:F436–442. doi: 10.1152/ajprenal.00162.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ameri A, Kurachi S, Sueishi K, Kuwahara M, Kurachi K. Myocardial fibrosis in mice with overexpression of human blood coagulation factor IX. Blood. 2003;101:1871–1873. doi: 10.1182/blood-2002-05-1581. [DOI] [PubMed] [Google Scholar]
- 48.Conboy IM, Conboy MJ, Smythe GM, Rando TA. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003;302:1575–1577. doi: 10.1126/science.1087573. [DOI] [PubMed] [Google Scholar]
- 49.Sherwood RI, et al. Isolation of adult mouse myogenic progenitors: functional heterogeneity of cells within and engrafting skeletal muscle. Cell. 2004;119:543–554. doi: 10.1016/j.cell.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 50.Liu XK, et al. Genetic disruption of Kir6.2, the pore-forming subunit of ATP-sensitive K+ channel, predisposes to catecholamine-induced ventricular dysrhythmia. Diabetes. 2004;53(Suppl 3):S165–168. doi: 10.2337/diabetes.53.suppl_3.s165. [DOI] [PubMed] [Google Scholar]
- 51.Oh H, et al. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci USA. 2003;100:12313–12318. doi: 10.1073/pnas.2132126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pajerowski AG, et al. Adult hematopoietic stem cells require NKAP for maintenance and survival. Blood. 2010;116:2684–2693. doi: 10.1182/blood-2010-02-268391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
BubR1 expression analysis in T23 mouse tissues and analysis of mitotic checkpoint complex formation and levels of key mitotic regulators in T23 MEFs. (a) Western blot analysis of mitotic MEF extracts for BubR1. BubR1 protein levels in T23 MEFs were about 2-fold higher than in T7 MEFs and 10-fold higher than in wildtype MEFs. Actin served as a loading control. (b) Images of WT and transgenic pups obtained by light (left) and fluorescence microscopy (right). (c) The BubR1 transgene continues to be expressed late in life of transgenic animals and is not subject to silencing. Western blot analysis of tissues of 24-month-old WT and T23 mice. Blots were probed for BubR1. Ponceau S staining was used as a loading control. (d) Immunoblots of wildtype and T23 MEF extracts subjected to immunoprecipitation with the indicated antibodies. (e) Western blots of mitotic cell extracts of wildtype and T23 MEFs probed for key mitotic regulators. P-H3Ser10 was used as a control for equal loading of mitotic cells.
Protection against tumor formation and aneuploidization requires a threshold of BubR1 overexpression. (a) BubR1 T7 transgenic mice are not protected from lung tumors induced by oncogenic Kras (G12D). Cohorts of KrasLA1, and T7;KrasLA1 mice were killed at 6 weeks of age and lung tumors counted under a dissection microscope. Error bars indicate SEM. (b) Alveolar hyperplasia in K-rasLA1 and T23;K-rasLA1; mice is similar. Haematoxylin-eosin stained lung sections of mice of the indicated genotypes (at 6 weeks of age). Hyperplastic alveolar tissue (without tumors) of K-rasLA1 and T23;K-rasLA1 mice is shown. Note that lung tissue of T23 transgenic mice is histologically normal. (c) Aneuploidy rates in lungs of KrasLA1 mice with and without BubR1 clone T7 overexpression. (d) Chromosome missegregation rates of mutant MEF lines with and without BubR1 overexpression. Consistent with reduced aneuploidy rates, the fidelity of segregation is improved when BubR1 levels are high. *P < 0.05, ***P < 0.001. n indicates the number of mice (mixed gender) used per genotype in a and c, and the number of independent MEF lines used in d.
Overall and tumor-free survival of T23 animals is extended in both males and females. (a, b) Analysis of BubR1 protein levels in spontaneous tumors. (a) Western blot analysis of T23 liver tumors and flanking normal tissue for expression of transgenic BubR1 (FLAG-BubR1). Tubulin was used as a loading control. (b) Representative images of WT and T23 lung sections stained for BubR1. Note that BubR1 is present at high levels in normal lung tissue of T23 mice but not in neighboring tumor tissue. Scale bars: top, 1 mm; bottom, 100 μm. (c–f) Data presented are from the same mouse cohorts as in Fig. 4, but now separated by gender. (c) Overall survival of males and (d) females, and (e) tumor-free survival of males and (f) females. *P < 0.05, **P < 0.01, ***P < 0.001.
High levels of BubR1 delay age-related changes in a tissue specific manner. (a) BubR1 overexpression protects against loss of muscle mass. (b) Levels of BubR1 do not correlate with altered sensitivity to acute oxidative stress, as survival after paraquat injection in 3–4 month-old mice is unchanged. (c) H2O2 levels in gastrocnemius of 24-month-old mice in T23 is similar to wildtype. (d) Relative ROS levels are unchanged in livers of 24-month-old mice. (e) Proliferative capacity in kidney, as measured by BrdU incorporation, is increased in aged BubR1 transgenic mice. (f) Age-related retinal degeneration is prevented by high levels of BubR1. (g–h) High levels of BubR1 are unable to prevent osteoporosis. Bone mineral density (g) and bone mineral content (h) declines in mice with age. (i) Cataract onset and incidence are not prevented by overexpression of BubR1 in the eye. n = 5 females per genotype in a, and 5 males per genotype in c–h. n in b and i indicates the number of mice (mixed gender) used per genotype. Values in a and e–h are means ± SD, values in c and d are means ± SEM. *P < 0.05, **P < 0.01.
Age-related attenuation of tissue dysfunction requires a threshold level of BubR1 overexpression. (a) Gastrocnemius muscle fiber diameter measurements. (b) QRT-PCR analysis of p16Ink4a expression in young and old gastrocnemius muscles. (c) Same as (b) but for p19Arf. (d–f) Duration of exercise (d), distance travelled (e), and work performed (f) in treadmill exercise tests. (g) Analysis of glomerulosclerosis. (h) Age-related retinal degeneration is not attenuated in T7 mice. (i) Cardiac stress tolerance is not improved in T7 animals. For all analyses in a–i, n = 5 males per genotype per age group, values are means ± SD. (j) Interphase FISH analysis on various mouse tissues (n = 3 males per genotype per tissue). (k) Model for asymmetric aneuploidization of proliferating stems cells and their committed progeny: (top) stem cells segregate their chromosomes with high accuracy, but committed progenitors do not leading to aneuploid differentiated offspring; (bottom) stem cells undergoing inaccurate segregation die, while progenitor daughter cells do not, allowing them to produce aneuploid offspring.