

Abstract
A simple and rapid multiplex substrate profiling method has been developed to reveal the substrate specificity of any endo- or exo-peptidase using LC-MS/MS sequencing. A physicochemically diverse library of peptides was generated by incorporating all combinations of neighbor and near-neighbor amino acid pairs into decapeptide sequences that are flanked by unique dipeptides at each terminus. Addition of a panel of evolutionarily diverse peptidases to a mixture of these tetradecapeptides generated prime and non-prime site information and substrate specificity matched or expanded upon previous substrate motifs. This method biochemically confirmed the activity of the klassevirus 3C gene responsible for polypeptide processing and allowed Granzyme B substrates to be ranked by enzymatic turnover efficiency using label-free quantitation of precursor ion abundance. Furthermore, the proteolytic secretions from a parasitic flatworm larvae and a pancreatic cancer cell line were deconvoluted in a subtractive strategy using class-specific peptidase inhibitors.
Peptidases represent the largest class of post-translational modifying enzymes in the human proteome. An estimated 2% of human genes encode for 687 peptidases or peptidase-like homolog transcripts that result in ~550 predicted active enzymes1. The human proteome represents the potential substrate library for these enzymes and all proteins are proteolytically modified, either by limited proteolysis or final degradation. Uncovering the substrate specificity of these peptidases is central to understanding their physiological role in homeostasis and disease.
While it is still common practice to use generic protein or peptide substrates to test for proteolytic activity, the field has been continuously developing biological and chemical tools to characterize the substrate specificity in greater detail2. Diverse peptide sequences expressed on the surface of phage or bacteria have revealed the substrate specificity of Factor Xa3 and Caspase-34, while N-terminal sequencing of peptide mixtures has been used to profile matrix metallopeptidases5. In addition, positionally arranged fluorogenic substrate libraries have been used to rapidly uncover the non-prime side (N-terminal to the scissile bond) specificity of many enzymes6,7. More recently, proteome-derived substrate libraries have been combined with mass spectrometry to identify cleavage products within a protein extract8. These ‘degradomic’ methods require complex labeling strategies to differentiate between specific cleavages produced by the peptidase of interest and other peptidases within the sample9,10.
There remains a need for a rapid, quantitative and highly reproducible assay that can provide specificity profiles and kinetic constants for any endo- or exo-acting peptidase. Presented here is a direct cleavage assay that uses mass spectrometry-based peptide sequencing for detection of degradation products in a mixture of synthetic peptides. Design of the sequences is based on our hypothesis that cleavage by a peptidase requires no more than two amino acids suitably positioned within a peptide substrate. Therefore, the library contains all combinations of neighbor and near neighbor amino acid pairs. A peptide length of 14 residues was selected to allow sufficient substrate length for binding of endopeptidases and to minimize tertiary structure that could limit side-chain accessibility. In this study we examined whether the depth of information obtained from a chemically defined pool of peptides could be sufficient to determine a specificity signature of a peptidase. We demonstrate that Multiplex Substrate Profiling by Mass Spectrometry (MSP-MS) can generate prime and non-prime side substrate specificity data for representative endo- and exo-peptidases from multiple families. Due to the high information content relative to low background, this method requires no additional labeling or sample fractionation and is therefore highly reproducible. Peptide degradation kinetics can be monitored by label-free quantitation of parent ion MS peaks and we deconvolute biological samples containing multiple peptidases through the use of class-specific inhibitors.
Results
To profile the substrate specificity of all peptidase families, a defined library of 124 peptides with extensive physicochemical diversity was synthesized. Within the central decapeptide region of each sequence, two copies of every amino acid pair (XY) and one copy of every X*Y and X**Y pair were accommodated, where X and Y represent defined amino acids and * indicates a random amino acid. In order to create diversity at each terminus for exo-acting enzymes, a unique dipeptide sequence was placed at both the N-terminus and C-terminus of each decapeptide core to produce a library of 124 tetra-decapeptides resulting in 1612 potential cleavage sites (Fig. 1a). The abundance of each amino acid within the library ranges from 4.2 to 6.8%. By comparison, vertebrate protein sequences in the SwissProt database show significant bias in amino acid usage, ranging from 1.3% (Trp) to 8.1% (Leu) (Supplementary Fig. 1). For the MSP-MS assay, peptides were pooled at equimolar concentration and combined with a peptidase in assay buffer. At defined time intervals, samples were quenched and injected into a LC-MS/MS system (Fig. 1b).
Figure 1. Design of a physiochemically diverse peptide library and development of a multiplex substrate assay.
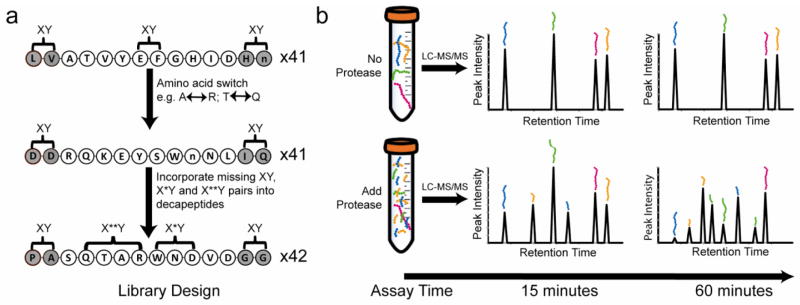
(a) Design of a library of 14-mer peptides by accommodating all neighbor (XY) and near neighbor pairs (X*Y and X**Y) into a core decapeptide (unshaded residues). X and Y correspond to defined amino acids while * corresponds to a random amino acid. The termini (shaded residues) were generated using amino acid pairs (XY) selected from 11 pools of amino acids. ‘n’ corresponds to norleucine. (b) Illustration of the multiplex substrate profiling assay. A peptidase or mixture of peptidases is added to the peptides and aliquots are removed at multiple time intervals and quenched. Samples are injected into a LC-MS/MS system to detect time dependant appearance of cleavage products.
Validation of the MSP-MS assay using the endopeptidase Cathepsin E
The aspartyl peptidase, cathepsin E was selected to validate the MSP-MS assay because it has been thoroughly characterized using proteome-derived peptide libraries11. Cathepsin E was incubated with the peptide library and a sample was removed at eight time intervals and quenched with pepstatin. After five minutes incubation, LC-MS/MS sequencing uncovered 114 cathepsin E cleavage sites and by 1200 minutes 14.5% of all peptide bonds in the library were hydrolyzed (Fig. 2a). For each cleaved bond observed at five minutes, the residues in P4 to P4′ were identified and a substrate signature was generated using iceLogo12 (Fig. 2b). Cathepsin E favored Phe and norleucine (Nle) at the P1 position and Nle and Val at P1′ while Gly at P1 and His at P1′ were disfavored. After incubation for 1200 minutes the specificity at both P1 and P1′ broadened to include Leu and Trp at P1 and Ile and Phe at P1′ (Fig. 2c) which strongly correlated (Pearson score >0.75) with cathepsin E specificity obtained from proteome derived sequences11 (Supplementary Table 1).
Figure 2. Validation of the multiplex substrate profiling technique using the aspartyl peptidase, Cathepsin E.

(a) Quantitative assessment of cleaved bonds at increasing time intervals. (b–c) iceLogos generated from amino acids that are enriched or de-enriched in the P4 to P4′ position Cathepsin E cleavage sites after incubation for 5 minutes (b) and 20 hours (c). Percent difference corresponds to the difference of amino acid frequency surrounding the Cathepsin E cleavage sites relative to the frequency of amino acids surrounding all peptide bonds in the library (n= 1612). In the iceLogo, “M” corresponds to norleucine. (d) A bar chart representing tetrapeptide sequences containing Phe in the third position (**F↓*) and the time that cleavage is first observed. All **F* motifs in the library are listed in Supplementary Table 4.
The MSP-MS assay uncovered a previously, uncharacterized carboxypeptidase function of cathepsin E as 12.9% of cleavage sites occurred at the carboxy terminus of substrates. Furthermore, in 98.3% of cleavage sites the S3 to S1′ subsites were occupied, suggesting these subsites were most important for substrate recognition. To examine the neighboring effects within the P3 to P1′ sites, substrates containing with the **F↓* motif were analyzed in detail as Phe in the P1 position is the single most preferred residue in the cathepsin E substrate binding pocket (Fig. 2d). When paired with a hydrophobic residue in the P1′ position, cleavage was generally observed within 30 minutes while Pro and polar residues were disfavored. In certain peptide sequences P1 Phe is surrounded by non-favorable residues however cleavage still occurs rapidly indicating flexibility in the substrate binding pocket.
Profiling exopeptidase substrate specificity with MSP-MS
Exopeptidases, such as prolycarboxypeptidase (PRCP) process peptide hormones by removing residues from the carboxy termini. Known PRCP substrates have a strict specificity for Pro-X bonds13, however the MSP-MS assay revealed that the enzyme readily accepted Ala or Nle in the S1 pocket (Fig. 3a). Cleavage occurred rapidly when P1 Pro, Ala or Nle were paired with hydrophobic residues such as Nle, Val, Leu, Ala and Pro in the P1′ position. PRCP had no tolerance for Arg or Lys in the S1′ pocket but accepted His, Asp and Glu in certain cases. In several substrates, time dependent trimming was observed. In one example, cleavage at P-V, AP, A-A and K-A bonds in the substrate RnENYnVLTKAAPV was evident at 5, 10, 60 and 1200 minutes, respectively.
Figure 3. Substrate profiling of an exopeptidase PRCP.
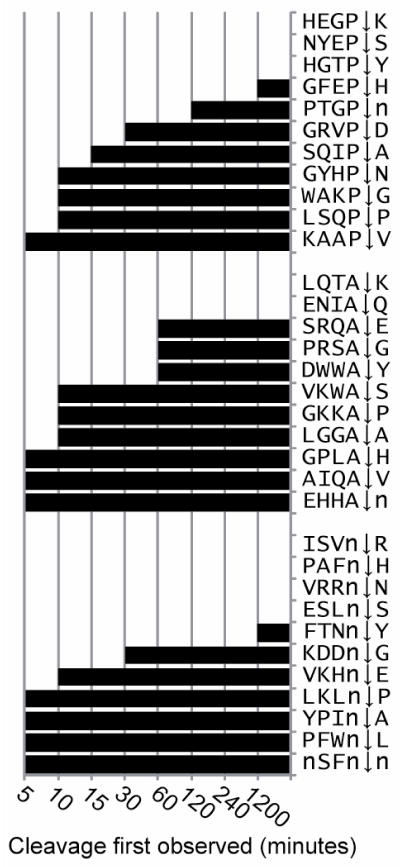
PRCP releases amino acids from the C-terminus of peptides when Pro, Ala or Nle are in the P1 position. The time, if any, that cleavage was first observed is illustrated by the length of each bar.
Quantification of cleavage efficiency (kcat/KM) for individual substrates
Granzyme B and human rhinovirus 14 (HRV14) 3C are highly specific peptidases that are involved in apoptosis and viral polypeptide processing, respectively. Unlike other enzymes with broader specificity, cleavage occurred in the MSP-MS assay at a single site within a substrate and therefore products were not subsequently degraded at secondary sites. This allowed for the quantification of the extracted ion chromatogram for each product as it accumulated over time and calculation of catalytic efficiency. The best substrate for Granzyme B (KHPLETVYAD↓SSEW) had a catalytic efficiency of 127,000 ± 13,000 M−1 s−1 (Fig. 4a–b) which closely matched previous studies (116,000 M−1 s−1) using a fluorescent substrate containing the sequence, VVAD↓SSMES14. HRV14 3C cleaved at only one site (YnDSIRHQ↓GPFWnL) therefore this substrate was pooled with other peptides containing Gln-Gly and Gln-Ser pairs and a selection of peptides of known or putative viral polypeptide processing sites (Supplementary Table 2). The catalytic efficiency of cleavage in YnDSIRHQ↓GPFWnL was comparable with the HRV14 2C-3A polypeptide release site and >100-fold superior to the HRV14 3C-3D release site (Supplementary Fig. 2), as has been observed previously15.
Figure 4. Specificity constants of individual substrates can be calculated from the MSP-MS assay.

(a) The precursor ion abundance of KARSAFAEnWPDHN (black) and cleaved product (grey) at four time intervals after addition of Granzyme B to the MSP-MS assay. The precursor ion abundance was calculated using Xcalibur. (b) Progress curves of product formation for peptides KHPLETVYAD and KARSAFAEnWPD. In each case, the parent substrates were completely hydrolyzed by Granzyme B within 1200 minutes, therefore the concentration of each product is known and kinetic values were subsequently calculated.
Screening peptidase open reading frames for proteolytic activity
We have validated that the peptide library is broadly applicable to profile purified peptidases from multiple families that include cruzain, matriptase, DPP-IV, MMP2, eqolisin, aspergillopepsin, HIV-1 and HIV-2 protease (Supplementary Fig. 3). However, isolation of pure, stable peptidases from native or recombinant sources is time consuming and labor intensive; therefore, we investigated if sufficient enzymatic activity could be generated from an in-vitro transcription/translation (IVTT) system to perform an MSP-MS assay. Using a bacterial IVTT system, a selection of highly specific 3C cysteine peptidases from picornaviruses were expressed, partially purified (Supplementary Fig. 4) and assayed with the same pool of peptides as outlined above for commercial grade HRV14 3C (Supplementary Table 2). Cleavage of YnDSIRHQ↓GPFWnL was evident by IVTT-derived HRV14 3C, poliovirus 3C, enterovirus 71, hepatitis A virus and a previously uncharacterized klassevirus 3C peptidase. The HRV14 2C-3A substrate was cleaved by HRV14, enterovirus 71 and poliovirus 3C peptidases but not by the others while the predicted klassevirus polypeptide release sites were not cleaved, even by the klassevirus enzyme. Taken together, these data indicate that rapid expression and partial purification of peptidases (<2 hours) can be combined with a highly sensitive peptidase assay to distinguish active from inactive enzymes.
Identifying the proteolytic signatures of a complex biological system
Schistosomiasis affects upwards of 200 million people worldwide and is ranked second to malaria in overall morbidity caused by a parasitic organism. The transition by larvae from the intermediate snail host and penetration of human skin is facilitated by a set of secreted peptidases. The process by which Schistosoma mansoni larvae, termed cercaria are “shed” from the snail host can be recapitulated in vitro16. Using the MSP-MS technique, non-infected snail shed was shown to consist of peptidases with a P1-Arg specificity, most likely due to tryptase17 (Fig. 5a). Infection with S. mansoni released peptidases with a preference for Tyr, Phe and Nle in P1 position, Pro in P2 and Nle in P1′. (Fig. 5b). Proteomic analysis determined that S. mansoni secretions contained serine, cysteine and metallo-peptidases17,18 therefore sheds were incubated with the metal chelator, EDTA, cysteine peptidase inhibitors E-64 and CAO74 or a peptide linked chloromethylketone known to be selective for elastase-type peptidases16. No significant changes were observed following treatment with EDTA or E-64/CAO74 (Supplementary Fig. 5), while the elastase inhibitor resulted in a 36% reduction in cleaved bonds and a de-enrichment of Tyr, Phe and Nle at the P1 position (Fig. 5c). MSP-MS profiling of a mixture of S. mansoni cercarial elastase isoforms confirmed the source of the peptidase activity in the parasite sheds (Fig. 5d).
Figure 5. Class specific peptidase inhibitors can dissect the proteolytic signatures of biological samples.

Substrate signature of material from (a) uninfected and (b) Schistosoma mansoni infected fresh water snails after 240 minutes incubation with the substrate library. (c) Infected snail material was pre-treated with AlaAlaProPhe-CMK inhibitor prior to multiplex substrate profile assay. (d) Cleavage site dataset of two semi-pure preparations of S. mansonii cercarial elastases were combined to generate a substrate signature for this peptidase group. (e) Substrate signature of all cleavage sites observed within 1200 minutes using conditioned media from pancreatic ductal adenocarcinoma cells as a source of proteases. Media was pre-treated with (f) E-64, (g) 1,10 phenanthroline and (h) pepstatin and a substrate signature was generated of the remaining cleavage events. In all cases, only amino acids that are enriched or de-enriched are illustrated in the substrate signature and ‘M’ corresponds to norleucine.
In many human cancers, dysregulation of protease activity can lead to degradation of extracellular matrices, thereby facilitating neoplastic progression19. Pancreatic ductal adenocarcinoma (PDAC) is an aggressive form of cancer with limited response to treatment leading to an average survival rate of six months. In order to interrogate the role of extracellular proteases in PDAC, proteomic analysis and MSP-MS was performed on conditioned media from a primary mouse PDAC cell line. The proteomic study identified six peptidases in the conditioned media (Supplementary Table 3), most of which were optimally active between pH 4.5 and 620–23. The extracellular environment within pancreatic tumors is known to be acidic so the MSP-MS assay was performed at pH 5.2. Under these conditions, a total of 98 unique cleavage sites were identified and the substrate signature revealed a preference for hydrophobic residues in the P1 and P1′ positions (Fig. 5e). When conditioned media was pre-treated with either E-64 or the metallopeptidase inhibitor, 1,10-phenanthroline, the majority of cleavage sites remained unchanged (Fig. 5f–g). However, treatment with pepstatin completely altered the cleavage signature (Fig. 5h). As cathepsin E is the only pepstatin-sensitive peptidase detected in media and the substrate signature is similar to that obtained from the recombinant enzyme (Fig. 2c), we concluded that cathepsin E is the major proteolytic activity secreted by PDAC cells.
Discussion
Fluorescent and colorimetric substrates have been the standard reagent for detecting and characterizing peptidases for decades. However, with the recent advances in mass spectrometry, substrates containing reporter groups are no longer required to obtain subsite specificity. Zhu and co-workers used mass spectrometry to identify candidate DPP-IV substrates from mixtures of peptide hormones and neuropeptides24 and several groups have obtained peptidase specificity data from protein or peptide substrates derived from bacterial or mammalian proteomes. These techniques utilize a variety of chemical or enzymatic strategies to enrich for peptidase digestion products that are subsequently sequenced by mass spectrometry25. We have rationally designed a synthetic peptide library composed of diverse yet defined sequences that can be used to obtain the prime and non-prime side specificity of any endo- or exo- peptidase. In a single kinetic assay, hydrolysis of any of the 1612 peptide bonds can be readily monitored using LC-MS/MS sequencing without any enrichment of substrates or products.
We first hypothesized that substrate recognition by peptidases requires no more than two amino acids suitably positioned within a peptide. This hypothesis was generated from the wealth of data derived from synthetic and proteome-derived peptide libraries26 and examples include P2 and P1 of cathepsin L, K, S and F7, P4 and P1 of Granzyme B27, and P3 and P1′ of MMP2 and MMP928. Diversity of the library was subsequently generated by designing peptides that encompass all neighbor and near-neighbor amino acid pairs.
To validate the library content and the MSP-MS technique we profiled the endopeptidases cathepsin E, a pancreatic tumor marker29 and PRCP, a key exopeptidase involved in the renin-angiotensin system and body weight regulation. Analysis revealed that cathepsin E requires P3 to P1′ residues for substrate recognition and therefore has carboxypeptidase-type activity. Early in the assay, primary cleavage sites occurred between P1 Phe and Nle and P1′ Nle and Val but after extended incubation, secondary cleavage occurred between other hydrophobic pairs. Interestingly, cathepsin E specificity from previous studies11 had strongest correlation with the 1200 minute MSP-MS assay data, indicating that primary cleavage sites may been overlooked. This primary cleavage site data will be used in future studies to generate improved fluorescent substrate probes to selectively image Cathepsin E activity in pancreatic tumors.
Prolylcarboxypeptidase releases amino acids from the C-terminus of substrates such as α-melanocyte stimulating hormone, plasma prekallikrein and angiotensin II and III all of which have Pro in the penultimate position30. Using MSP-MS, PRCP cleaved single amino acids from the C-terminus but not exclusively after Pro as its name suggests. In fact, cleavage after Ala or Nle was often identified at earlier time points compared to Pro containing peptides. Inhibition of PRCP in mice causes a reduction in body weight making the enzyme a target for treatment of obesity; however until now the substrate specificity has not been thoroughly investigated. Norleucine is an isosteric analog of methionine and therefore proteins or peptides containing either Met or Ala in the penultimate position should now be assessed as potential physiological substrates for PRCP.
Using the MSP-MS assay, individual substrates within a peptide mixture can be ranked using kinetic values extracted from progress curves. In one example, eleven substrates of Granzyme B, a peptidases that initiates natural killer cell-mediated apoptosis, were identified and kcat/KM values were calculated directly from the multiplex assay for the most efficient reactions. All substrates have Asp in the P1 position which correlates with previous studies31 however not all Asp containing substrates are cleaved. Therefore the MSP-MS assay can generate a list of true positive and true negative substrates which will be used to strengthen bioinformatic methods for predicting natural substrates31.
Our studies have generated comprehensive substrate signatures for a panel of enzymes representing five families (Supplementary Fig. 2). Furthermore, we have used the MSP-MS assay to profile the specificity of a gut associated hemoglobinase from the Lyme disease tick vector Ixodes ricinus32. To highlight the sensitivity of the assay, a set of highly specific viral peptidase genes were subjected to in vitro transcription/translation and the resulting proteins were assayed with a mixture of library peptides and known or putative substrate sequences. We determined that these enzymes exclusively hydrolyse Gln-Gly bonds and confirmed that the 3C gene of klassevirus33, a novel picornavirus associated with pediatric gastroenteritis is a functional peptidase.
Functional characterization of peptidases within a biological system has traditionally involved identifying a candidate protein, determining the substrate specificity, and generating a specific probe or inhibitor. While the candidate approach has a proven track record, we sought to characterize proteolysis using an unbiased global approach. In this study, peptidase substrate profiling using material shed from healthy snails had trypsin-like specificity (P1-Arg) while shed from Schistosoma mansoni infected snails had a mixture of trypsin and elastase-like activity. A previous study has shown that topical application of AlaAlaProPhe-CMK on human skin prevents invasion by the parasite34. In our study, we demonstrate that this inhibitor reduces the overall proteolytic activity in parasite secretions by targeting the elastase-like peptidases which dominate in the larvae.
The secretome of a primary pancreatic adenocarcinoma cell line revealed multiple secreted peptidases in the media. Using the MSP-MS assay, robust cleavage between pairs of hydrophobic residues was observed in the same media and this specificity was altered in the presence of an aspartic peptidase inhibitor. The sole aspartic peptidase, cathepsin E, appears to be a low abundant protein but represents the major proteolytic activity. Analysis of the proteome identified cysteine and metalloproteinase inhibitors but no endogenous aspartic peptidase inhibitor was evident. These data provide functional support for a recent study that uses a fluorescent cathepsin E substrate to detect pancreatic cancer in a mouse model29. Taken together, proteomic studies alone cannot be used to predict the role of active proteolytic enzymes. However, substrate profiling with MSP-MS provides a complementary, functional profile of peptidase activity in complex biological samples.
In this study, we report the development and validation of a simple and direct substrate profiling assay that uses a rationally designed set of peptides to simultaneous monitor amino-, carboxy- and endo peptidase activity in complex mixtures of peptidases. While the current set of peptides is sufficient to identify cleavage site specificity, we anticipate that greater sequence resolution can be achieved by synthesizing secondary libraries that iteratively explore the preferred sequence space for a peptidase. In certain cases the preferred peptide substrate can be a powerful probe for identifying endogenous substrates35. However, substrates determined in vitro must take into account the complex biology associated with the enzyme. Detailed knowledge of the substrate specificities of individual peptidases and those in complex biological systems affords new opportunities to understand their role in homeostasis and disease and will aid the development of chemical tools for detection or inhibition.
ONLINE METHODS
Peptide Library Design
For the MSP-MS assay, a library of 124 peptides with extensive physicochemical diversity was developed. Cysteine was omitted due to potential disulfide bond formation, and norleucine (Nle) replaced oxidation prone methionine. Peptide sequence diversity was designed in a two-part strategy.
For the endopeptidase component, a novel algorithm was developed to arrange amino acids pairs into the minimal number of decapeptide sequences, such that no decapeptide had more than two identical residues or more than one pair. All but one pair is incorporated into 41 sequences. In order to generate diversity surrounding each pair, a second set of 41 decapeptides was designed by substituting all amino acids with a physicochemically distinct counterpart, e.g. Ala residues were substituted for Arg and vice versa. These 82 decapeptide are defined as the “XY decapeptide sublibraries” where X and Y represent defined amino acids. Next, all near-neighbor amino acid pairs separated by one (X*Y) or two (X**Y) random amino acids were identified in the XY decapeptide sublibraries. Any pair not present (including the missing XY pair) was manually assembled into 42 additional decapeptides to generate a final set of 124 decapeptide sequences. For generating diversity for exopeptidases, amino acids were first combined into eleven groups of distinct physicochemical characteristics, specifically (Ile/Leu/Val), (Ser/Thr), (Glu/Asp), (Lys/Arg), (Tyr/Trp/Phe), (Gln/Asn), (Gly), (Pro), (Ala), (His) and (Nle). Next, 121 dipeptide sequences were generated by pairing a single amino acid, chosen at random, from each group. Each pair represents the amino terminal dipeptide of the final 14-mer sequence. This procedure was repeated to generate an additional 121 pairings for positioning at the carboxy terminus.
Finally, 14-mer sequences were assembled by combining an N-terminal and C-terminal dipeptide with each core decapeptide sequence such that no more than three identical residues are present in the entire sequence. Three additional N-terminal and C-terminal pairs were generated manually to ensure that the number of exo-sequences (121) match the number of decapeptides (124).
Peptide Synthesis
Peptide synthesis was performed on an automated peptide synthesizer (Protein Systems, model 433A), using solid phase conditions, rink amide AM resin (Novabiochem), and Fmoc main-chain protecting group chemistry. For the coupling of Fmoc-protected amino acids (Novabiochem), 10 equivalents of amino acid and a 1:1:2 molar ratio of coupling reagents HBTU/HOBt (Novabiochem)/DIEA were employed. Isolation of desired peptides was achieved by trifluoroacetic acid-mediated deprotection and cleavage, ether precipitation to yield the crude product, and high performance liquid chromatography (HPLC) (Varian ProStar) on a reverse phase C18 column (Varian) to yield the pure compounds. Chemical composition of the pure products was confirmed by LC-MS mass spectrometry (Waters Micromass ZQ) and 5 mM stock solutions of each peptide were dissolved in DMSO. The average cost of synthesis and purification was ~$10/amino acid. A subset of peptides were synthesized and purified by AnaSpec (Fremont, CA). In all cases, the purity of each peptide was 90% or greater.
Enzymes
Proteases were either purchased or obtained through kind gifts, and include mouse cathepsin E (R&D Systems), HRV 3C (EMD Chemicals), rat granzyme B (Cheryl Tajon, UCSF), human matriptase-1 (Christopher Brown, UCSF), matrix metalloprotease 2 (AnaSpec), Talaromyces emersonii eqolisin (Maria Tuohy, NUI Galway, Ireland), aspergillopepsin I (Sigma), cruzain (Greg Lee, UCSF), human dipeptidyl peptidase IV (Sigma), human prolylcarboxypeptidase (Wayne Geissler, Merck) and HIV-1 and HIV-2 Protease (Starlynn Clarke, UCSF).
A selection of viral proteases were amplified using the oligonucleotides listed in Supplementary Table 5 were cloned using InFusion Advantage (Clontech) into a pET23b vector linearized with NdeI and XhoI. 1 microgram of sequence-confirmed plasmid was used as input for the S30 T7 High Yield Protein Expression System (Promega), and purified using MagneHis Protein purification system (Promega), following manufacturer’s suggested protocols. 10 μL of the 50 μL eluate was run on a 4-12% Bis-Tris NuPage acrylamide gel (Life Technologies) and silver stained.
Isoforms of cercarial elastase were partially purified from Schistosoma mansoni cercariae after sonication in 300 mM sodium acetate, pH 6.5, 0.1% Triton X-100, 0.1% Tween-20, 0.05% NP40. Soluble protein was harvested by centrifugation for 15 minutes at 7,500 g, followed by 0.2 μm filtration. The supernatant was loaded onto a SR 16/100 column packed with Sephacryl 200 (GE Healthcare) and 4 ml fractions were collected at 4°C. Fractions were assayed using 10 μl of sample and 100 μl of assay buffer (100 mM glycine, pH 9.0, 100 μM succinyl-ala-ala-pro-phe-p-nitroanilide (AAPF-pNA). Proteolytically active fractions were used in the MSP-MS assay. These fractions were also size separated on a 10% Bis-Tris polyacrylamide gel (Life Technologies) and silver stained. Protein bands corresponding to the correct molecular weight of cercarial elastases were excised from the gel, digested with trypsin and analyzed by LC-MS/MS to determine the protein composition.
Schistosoma mansoni secretions
Schistosoma mansoni infected Biomphalaria glabrata were a kind gift from Fred A. Lewis at the NIH-NIAID Schistosomiasis Resource Center. Cercariae were shed from several hundred infected B. glabrata and secretions were collected as previously described by Salter and colleagues36. Isolated secretions were lyophilized and re-suspended in 50mM Tris-HCl, pH 7.5 and sonicated for 1 minute. The soluble fraction was isolated by centrifugation at 16,000 g at 4°C. For inhibition studies the soluble fraction was pre-treated 30 mins at room temperature with either 25mM EDTA, 500nM succinyl ala-ala-pro-phe-chloromethyl ketone or a mixture of 250nM CAO74 and 250nM E-64 prior to addition to MSP-MS assay. The assay was performed in 50 mM Tris-HCl pH 7.5.
Pancreatic cancer secretions
The p48-Cre/+; LSL-Kras/+; Trp53F/+ mouse cell line was a kind gift from the laboratory of Douglas Hanahan. Cells were maintained in DMEM containing 10% FBS and 1X Penn/Strep and grown to ~80% confluency in triplicate T75 flasks. Media was removed and cells were washed five times with Opti-MEM (Invitrogen) and incubated with the same media. After 20 hours, the conditioned media was removed and sterile filtered (0.22 um). The cells were treated with trypsin and viability was calculated using trypane blue staining. The conditioned media was buffer-exchanged into PBS and concentrated 50-fold in an Amicon Ultra centrifugal filter with 10 kDa cut-off. An aliquot of each triplicate sample was digested with trypsin and subjected to LC-MS/MS sequencing (described below). The remaining conditioned media was pooled and acidified to pH 5.2 with 200 mM ammonium acetate. The sample was split into four tubes and treated with ethanol (vehicle control), 1 μM E64, 1 μM 1,10-Phenanthroline or 400 nM pepstatin for 30 minutes at room temperature prior to addition to the MSP-MS assay. The assay was performed in PBS acidified to pH 5.2 with 200 mM ammonium acetate.
Protein Identification by Mass Spectrometry
Protein identification in pancreatic cancer secretion samples was performed using peptide sequencing by mass spectrometry. Secretion samples were digested with trypsin in solution as follows. Secretion sample (20-50 μg total protein) was incubated with urea (5 M final) and 10 mM DTT for 10 min at 56 °C. Following reduction, the sample was alkylated with 15 mM iodoacetamide (45 min, dark, 21 °C), then quenched with 10 mM additional DTT, and the final volume was diluted 5X in 100 mM ammonium bicarbonate. Trypsin (sequencing grade, Promega) was added at 1:50 trypsin: total protein for digestion overnight at 37 °C. The sample was then acidified with 10% formic acid to pH 2-3 and desalted using C18 Zip-tips (Millipore). Extracted peptides were sequenced using an LTQ-Orbitrap Velos (Thermo) mass spectrometer, equipped with 10,000 psi system nanoACUITY (Waters) UPLC instrument for reversed phase chromatography with a C18 column (BEH130, 1.7 μm bead size, 100 μm x 100 mm). The LC was operated at 600 nL/min flow rate, and peptides were separated using a linear gradient over 42 min from 2% B to 30% B, with solvent A: 0.1% formic acid in water and solvent B: 0.1% formic acid in 70% acetonitrile. Survey scans were recorded over 350–1900 m/z range, and MS/MS was performed in data dependent acquisition mode with HCD fragmentation on the seven most intense precursor ions.
Mass spectrometry peak lists were generated using in-house software called PAVA, and data were searched using Protein Prospector software v. 5.10.0 37. Database searches were performed against the SwissProt M. musculus database (downloaded March 21, 2012), containing 16,520 entries. For estimation of false discovery rate, this database was concatenated with a fully randomized set of sequence entries38. Data were searched with mass tolerances of 20 ppm for parent and 30 ppm for fragment ions.
For database searching, peptide sequences were matched as tryptic peptides with no missed cleavages, and carbamidomethylated cysteines as a fixed modification. Variable modifications included oxidation of methionine, N-terminal pyroglutamate from glutamine, loss of methionine and N-terminal acetylation. Protein Prospector score parameters were: minimum protein score of 22, minimum peptide score of 15, and maximum expectation values of 0.01 for protein and 0.001 for peptide matches, resulting in a protein false discovery rate of 1.5%. Protein identification results from three biological replicates are reported with a spectral count as an approximation of protein abundance, along with percent sequence coverage and an expectation value for the probability of the protein identification39,40.
Multiplex Peptide Cleavage Assay
Purified peptidase concentration ranged from 10–100 nM while biological samples were assayed at 20 μg/ml of total protein. In order to reproducibly detect all 124 intact peptides, three pools containing 52, 52 and 20 peptides were prepared and the concentration of each peptide in the assay was 500 nM which is 10-fold below the typical KM for peptidases. Whenever enzymatic activity parameters such as specific activity, pH optima, or cofactor requirements were known for a given peptidase, this information was used in the assay preparation. Each peptide pool was incubated at room temperature with peptidase and aliquots were removed and acid quenched to pH 3 or less with formic acid (4% final) at defined time intervals. A control sample lacking enzyme was also prepared under identical conditions and quenched at the first and last time point of the assay to account for non-enzymatic degradation of the substrates. For enzymes with low pH optima, specific inhibitors such as pepstatin for aspartic acid peptidases and TA1 for eqolisin were used41. All of the peptides could be cleaved by one of four enzymes (cathepsin E, eqolisin, cruzain and matriptase) suggesting that any potential secondary structure of the peptides does not appear to limit access of the sequence to proteolysis.
Peptide Cleavage Site Identification by Mass Spectrometry
Cleavage site identification was performed using peptide sequencing by mass spectrometry. Samples containing 1–3 μg of total peptide (calculated as 60 μl of enzyme reaction containing peptide pools at 500 nM) were desalted using C18 zip tips (Millipore) and rehydrated in 0.1% formic acid. Total peptide corresponding to 0.1 μg was injected on the column. For LC-MS/MS, a linear ion trap LTQ mass spectrometer (Thermo) equipped with an Ultimate HPLC and Famos autoinjector (LC Packings) was used, with a C18 “Magic” column (Michrom Bioresources, Inc., 5 μm bead size, 0.3 x 150 mm Magic, 200 Å). The LC system was operated at 5 μL/min flow rate, and peptides were separated using a linear gradient over 42 min from 2% B to 40% B, with solvent A: 0.1% formic acid in water and solvent B: 0.1% formic acid in 70% acetonitrile, 30% water. Survey scans were taken over 300–1500 m/z, and the top three ions in the survey scan were subjected to a high resolution MS “zoom” scan of the precursor and then a CID fragmentation MS/MS scan. Each sample requires 1 hour of mass spectrometry time and therefore a typical assay with four timepoints requires 12 hours of instrument time.
Mass spectrometry peak lists were generated using in-house software called PAVA based on the Raw_Extract script from Xcalibur v2.4 (Thermo Scientific), and data were searched using Protein Prospector software v. 5.10.0 (UCSF). Database searches were performed against a defined library of 124 sequences. Data were searched with parent mass and fragment mass tolerances of 0.8 Da. For database searching, peptide sequences were matched with no enzyme specificity requirement, and variable modifications including oxidation of Trp, Pro and Phe, and N-terminal pyroglutamate from glutamine. For estimation of false discovery rate with this small database size, four different decoy databases containing the randomized sequences of the same 124 entries were concatenated to the original 124 entries to create a final database of 620 sequences. The data for replicate no enzyme control samples were searched using this large random concatenated database. False discovery rate was calculated using the formula: FDR = 0.25 x FP/(TP), where FP = false positives and TP = true positive peptides identified in the search38. Protein Prospector score thresholds were selected to be minimum protein score of 20, minimum peptide score of 15, and maximum expectation values of 0.1 for “protein” and 0.05 for peptide matches, and resulted in a peptide false discovery rate of <0.17%. Cleavage site data was extracted from Protein Prospector using “MSP extractor” software which will be made available at www.craiklab.ucsf.edu. Only amino acids that differ significantly (p ≤ 0.05) from the total amino acid frequency in the library are highlighted in the iceLogos. Starting with a set of raw data files from multiple timepoints, a substrate signature can be generated for an enzyme or biological sample in ~30 minutes. Cleavage sequences for all enzyme assays are listed in the Supplementary MSP-MS Cleavages file.
Kinetics Calculations
MS acquisition, peak integration, and data analysis were performed using Xcalibur software version 1.2 (Thermo Finnigan). Kinetics were fit to the formula Y = e(−t*kcat/KM*[E0]) and fitted using non-linear least squares fitting to the enzyme kinetics model in GraphPad Prism v. 5. Catalytic efficiency was solved from the overall rate by estimating enzyme concentration, and is reported as kcat/KM with a χ2 value for the quality of the data fit.
Supplementary Material
Acknowledgments
We thank K.C. Lim of the Sandler Center for Drug Discovery at UCSF for maintenance of Biomphalaria glabrata. This project was supported by grants from the Sandler Center (CSC) and the NIH; P50 GM082250 (CSC), RO1 CA128765 (CSC), P41 RR001614 (ALB), P41 GM103481 (ALB) and K12 GM081266 (AAER).
Footnotes
Author contributions
AJO, MZ and CSC conceptualized the MSP-MS library and assay. CSC directed and coordinated the project. MOA and GQ wrote the pair fitting script and JBS wrote the MSP-MS extractor script. AJO, AAER and MZ synthesized and purified the peptides. AJO, AAER and GMK performed the MSP-MS assays and analyzed the data. GMK, DAM and ALB developed the mass spectrometry protocol. JI and JHM generated S. mansoni infected snail sample. AJO and DRH generated conditioned PDAC media. ALG and JLD provided viral peptidases. AJO, GMK, AAER and CSC wrote the manuscript and all authors participated in editing it.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Puente S, Guti A, Velasco G. Proteases and their Inhibitors in Neurodegenerative Disease A genomic view of the complexity of mammalian proteolytic systems. 2005:331–334. doi: 10.1042/BST0330331. [DOI] [PubMed] [Google Scholar]
- 2.Van Damme P, Vandekerckhove J, Gevaert K. Disentanglement of protease substrate repertoires. Biological chemistry. 2008;389:371–81. doi: 10.1515/BC.2008.043. [DOI] [PubMed] [Google Scholar]
- 3.Matthews DJ, Wells JA. Substrate phage: selection of protease substrates by monovalent phage display. Science (New York, NY) 1993;260:1113–7. doi: 10.1126/science.8493554. [DOI] [PubMed] [Google Scholar]
- 4.Boulware KKT, Daugherty PS. Protease specificity determination by using cellular libraries of peptide substrates (CLiPS) Proc Natl Acad Sci USA. 2006;103:7583–7588. doi: 10.1073/pnas.0511108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turk BE, Huang LL, Piro ET, Cantley LC. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nature Biotechnology. 2001;19:661–667. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- 6.Harris JL, et al. Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:7754–9. doi: 10.1073/pnas.140132697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choe Y, et al. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. The Journal of biological chemistry. 2006;281:12824–32. doi: 10.1074/jbc.M513331200. [DOI] [PubMed] [Google Scholar]
- 8.auf dem Keller U, Schilling O. Proteomic techniques and activity-based probes for the system-wide study of proteolysis. Biochimie. 2010;92:1705–14. doi: 10.1016/j.biochi.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 9.Mahrus S, et al. Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell. 2008;134:866–76. doi: 10.1016/j.cell.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Staes A, et al. Improved recovery of proteome-informative, protein N-terminal peptides by combined fractional diagonal chromatography (COFRADIC) Proteomics. 2008;8:1362–70. doi: 10.1002/pmic.200700950. [DOI] [PubMed] [Google Scholar]
- 11.Impens F, et al. A quantitative proteomics design for systematic identification of protease cleavage events. Molecular & cellular proteomics: MCP. 2010;9:2327–33. doi: 10.1074/mcp.M110.001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colaert N, Helsens K, Martens L, Vandekerckhove J, Gevaert K. Improved visualization of protein consensus sequences by iceLogo. Nature methods. 2009;6:786–7. doi: 10.1038/nmeth1109-786. [DOI] [PubMed] [Google Scholar]
- 13.Chajkowski SM, et al. Highly selective hydrolysis of kinins by recombinant prolylcarboxypeptidase. Biochemical and biophysical research communications. 2011;405:338–43. doi: 10.1016/j.bbrc.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun J, et al. Importance of the P4′ residue in human granzyme B inhibitors and substrates revealed by scanning mutagenesis of the proteinase inhibitor 9 reactive center loop. The Journal of biological chemistry. 2001;276:15177–84. doi: 10.1074/jbc.M006645200. [DOI] [PubMed] [Google Scholar]
- 15.Cordingley MG, Callahan PL, Sardana VV, Garsky VM, Colonno RJ. Substrate requirements of human rhinovirus 3C protease for peptide cleavage in vitro. The Journal of biological chemistry. 1990;265:9062–5. [PubMed] [Google Scholar]
- 16.Ingram JR, et al. Investigation of the Proteolytic Functions of an Expanded Cercarial Elastase Gene Family in Schistosoma mansoni. PLoS neglected tropical diseases. 2012;6:e1589. doi: 10.1371/journal.pntd.0001589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knudsen GM, Medzihradszky KF, Lim KC, Hansell E, McKerrow JH. Proteomic analysis of Schistosoma mansoni cercarial secretions. Molecular & cellular proteomics: MCP. 2005;4:1862–75. doi: 10.1074/mcp.M500097-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.Curwen RS, Ashton PD, Sundaralingam S, Wilson RA. Identification of novel proteases and immunomodulators in the secretions of schistosome cercariae that facilitate host entry. Molecular & cellular proteomics_: MCP. 2006;5:835–44. doi: 10.1074/mcp.M500313-MCP200. [DOI] [PubMed] [Google Scholar]
- 19.Nolan-Stevaux O, et al. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes & development. 2009;23:24–36. doi: 10.1101/gad.1753809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fricker L. Carboxypeptidase E. Handbook of Proteolytic Enzymes. 2004:840–844. [Google Scholar]
- 21.Caglic D, et al. Murine and human cathepsin B exhibit similar properties: possible implications for drug discovery. Biological chemistry. 2009;390:175–9. doi: 10.1515/BC.2009.021. [DOI] [PubMed] [Google Scholar]
- 22.Mason RW, Johnson Da, Barrett aJ, Chapman Ha. Elastinolytic activity of human cathepsin L. The Biochemical journal. 1986;233:925–7. doi: 10.1042/bj2330925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaidi N, Herrmann T, Voelter W, Kalbacher H. Recombinant cathepsin E has no proteolytic activity at neutral pH. Biochemical and biophysical research communications. 2007;360:51–5. doi: 10.1016/j.bbrc.2007.05.187. [DOI] [PubMed] [Google Scholar]
- 24.Zhu L, et al. The role of dipeptidyl peptidase IV in the cleavage of glucagon family peptides: in vivo metabolism of pituitary adenylate cyclase activating polypeptide-(1–38) The Journal of biological chemistry. 2003;278:22418–23. doi: 10.1074/jbc.M212355200. [DOI] [PubMed] [Google Scholar]
- 25.Impens F, et al. MS-driven protease substrate degradomics. Proteomics. 2010;10:1284–96. doi: 10.1002/pmic.200900418. [DOI] [PubMed] [Google Scholar]
- 26.Rawlings ND, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic acids research. 2012;40:D343–50. doi: 10.1093/nar/gkr987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruggles SW, Fletterick RJ, Craik CS. Characterization of structural determinants of granzyme B reveals potent mediators of extended substrate specificity. The Journal of biological chemistry. 2004;279:30751–9. doi: 10.1074/jbc.M400949200. [DOI] [PubMed] [Google Scholar]
- 28.Prudova A, auf dem Keller U, Butler GS, Overall CM. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Molecular & cellular proteomics: MCP. 2010;9:894–911. doi: 10.1074/mcp.M000050-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cruz-Monserrate Z, et al. Detection of pancreatic cancer tumours and precursor lesions by cathepsin E activity in mouse models. Gut. 2011 doi: 10.1136/gutjnl-2011-300544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou C, et al. Design and synthesis of prolylcarboxypeptidase (PrCP) inhibitors to validate PrCP as a potential target for obesity. Journal of medicinal chemistry. 2010;53:7251–63. doi: 10.1021/jm101013m. [DOI] [PubMed] [Google Scholar]
- 31.Barkan DT, et al. Prediction of protease substrates using sequence and structure features. Bioinformatics (Oxford, England) 2010;26:1714–22. doi: 10.1093/bioinformatics/btq267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sojka D, et al. Characterization of the gut-associated cathepsin D hemoglobinase from the tick Ixodes ricinus (IrCD1) The Journal of biological chemistry. 2012:1–21. doi: 10.1074/jbc.M112.347922. at < http://www.ncbi.nlm.nih.gov/pubmed/22539347>. [DOI] [PMC free article] [PubMed]
- 33.Greninger AL, et al. The complete genome of klassevirus - a novel picornavirus in pediatric stool. Virology journal. 2009;6:82. doi: 10.1186/1743-422X-6-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cohen FE, et al. Arresting tissue invasion of a parasite by protease inhibitors chosen with the aid of computer modeling. Biochemistry. 1991;30:11221–9. doi: 10.1021/bi00111a005. [DOI] [PubMed] [Google Scholar]
- 35.Harris JL, Peterson EP, Hudig D, Thornberry Na, Craik CS. Definition and redesign of the extended substrate specificity of granzyme B. The Journal of biological chemistry. 1998;273:27364–73. doi: 10.1074/jbc.273.42.27364. [DOI] [PubMed] [Google Scholar]
- 36.Salter JP, et al. Cercarial elastase is encoded by a functionally conserved gene family across multiple species of schistosomes. The Journal of biological chemistry. 2002;277:24618–24. doi: 10.1074/jbc.M202364200. [DOI] [PubMed] [Google Scholar]
- 37.Chalkley RJ, Baker PR, Medzihradszky KF, Lynn AJ, Burlingame aL. In-depth analysis of tandem mass spectrometry data from disparate instrument types. Molecular & cellular proteomics_: MCP. 2008;7:2386–98. doi: 10.1074/mcp.M800021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature methods. 2007;4:207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 39.Liu H, Sadygov RG, Yates JR. A Model for Random Sampling and Estimation of Relative Protein Abundance in Shotgun Proteomics proteolytic digestion and liquid chromatography in com. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 40.Choi H, Fermin D, Nesvizhskii AI. Significance analysis of spectral count data in label-free shotgun proteomics. Molecular & cellular proteomics_: MCP. 2008;7:2373–85. doi: 10.1074/mcp.M800203-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Donoghue AJ, et al. Inhibition of a secreted glutamic peptidase prevents growth of the fungus Talaromyces emersonii. The Journal of biological chemistry. 2008;283:29186–95. doi: 10.1074/jbc.M802366200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.