

Abstract
Progranulin (PGRN) is a secreted glycoprotein expressed in neurons and glia that is implicated in neuronal survival on the basis that mutations in the GRN gene causing haploinsufficiency result in a familial form of frontotemporal dementia (FTD). Recently, a direct interaction between PGRN and tumor necrosis factor receptors (TNFR I/II) was reported and proposed to be a mechanism by which PGRN exerts anti-inflammatory activity, raising the possibility that aberrant PGRN–TNFR interactions underlie the molecular basis for neuroinflammation in frontotemporal lobar degeneration pathogenesis. Here, we report that we find no evidence for a direct physical or functional interaction between PGRN and TNFRs. Using coimmunoprecipitation and surface plasmon resonance (SPR) we replicated the interaction between PGRN and sortilin and that between TNF and TNFRI/II, but not the interaction between PGRN and TNFRs. Recombinant PGRN or transfection of a cDNA encoding PGRN did not antagonize TNF-dependent NFκB, Akt, and Erk1/2 pathway activation; inflammatory gene expression; or secretion of inflammatory factors in BV2 microglia and bone marrow-derived macrophages (BMDMs). Moreover, PGRN did not antagonize TNF-induced cytotoxicity on dopaminergic neuroblastoma cells. Last, co-addition or pre-incubation with various N- or C-terminal-tagged recombinant PGRNs did not alter lipopolysaccharide-induced inflammatory gene expression or cytokine secretion in any cell type examined, including BMDMs from Grn+/− or Grn−/− mice. Therefore, the neuroinflammatory phenotype associated with PGRN deficiency in the CNS is not a direct consequence of the loss of TNF antagonism by PGRN, but may be a secondary response by glia to disrupted interactions between PGRN and Sortilin and/or other binding partners yet to be identified.
Introduction
Progranulin (PGRN) is a secreted glycoprotein expressed in both neurons and microglia but whose exact physiological function in the brain is not well understood. PGRN function has been implicated in embryonic development, cell motility, tissue repair, and tumor growth (Toh et al., 2011; Cenik et al., 2012). PGRN is composed of 7.5 repeats of the granulin (GRN) domain that can be proteolytically processed into 6 kDa GRN peptides; full-length PGRN is thought to have anti-inflammatory properties, while the GRNs are pro-inflammatory (Cenik et al., 2012). PGRN has been hypothesized to promote neuronal survival on the basis that mutations in the GRN gene, which result in haploinsufficiency and reduced levels of PGRN in the brain, cause a familial form of frontotemporal dementia (FTD) (Ward and Miller, 2011), the most common form of early onset dementia. PGRN has also been described to increase neurite outgrowth, branching, and survival in primary neuronal cultures and neuron-like cell lines (Van Damme et al., 2008; Ryan et al., 2009; Gao et al., 2010; Tapia et al., 2011; Xu et al., 2011; Gass et al., 2012). In support of this idea, Grn−/− mice display dysregulated microglia activation in the CNS and increased sensitivity to neurotoxin-induced degeneration (Martens et al., 2012). However, the precise mechanism by which PGRN may be protective to neurons remains to be defined. Interactions between PGRN and the receptor sortilin have been reported and proposed to be important for internalization of full-length PGRN and its neurotrophic activity (Hu et al., 2010; Zheng et al., 2011). Studies on Grn−/− mice suggest that PGRN is essential for healthy aging and PGRN-related neurodegenerative diseases may result from impaired synaptic plasticity and lifetime depletion of neurotrophic support in association with dysregulated inflammation (Kayasuga et al., 2007; Ahmed et al., 2010; Yin et al., 2010a,b; Petkau et al., 2012), thus highlighting possible new molecular targets for PGRN-related FTD treatment. Direct antagonism of TNFRI and TNFRII binding to TNF by PGRN was recently reported as a mechanism via which PGRN exerts anti-inflammatory activities in a mouse model of rheumatoid arthritis (Tang et al., 2011), raising the interesting possibility that disruption of PGRN-TNFRI/II interactions may also underlie FTD pathogenesis. TNF has a well recognized role in immune function (Liu, 2005; Garcia et al., 2011) and a less well understood role in the CNS but has been implicated in synaptic plasticity and neuroinflammation-induced cell death (Stellwagen and Malenka, 2006; McCoy and Tansey, 2008). The primary goal of these studies was to confirm the reported interaction between PGRN and TNFRs and to further investigate the functional significance of this reported interaction in neuronal cells that express TNFRs and are sensitive to TNF-induced death.
Materials and Methods
Animals.
Male mice from two different Grn knock-out mouse models were used in these studies as indicated below. Grn+/+, Grn−/−, and Grn+/− mice were generated [Robert Farese Jr., University of California, San Francisco (UCSF)] on a C57BL/6 background as described previously (Martens et al., 2012) and a different Grn knock-out mouse generated and characterized by Aihao Ding et al. on a C57BL/6 background as described previously (Yin et al., 2010a,b) was purchased from The Jackson Laboratory (B6.Cg-Grntm1.1Aidi/J catalog #013175). All mice were housed in a pathogen-free, climate-controlled facility in the Division of Animal Resources at Emory University School of Medicine or UCSF and given food and water ad libitum. All animal studies were reviewed and approved by the Institutional Animal Care and Use Committees at Emory or UCSF in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Reagents.
A number of different recombinant PGRN proteins were used in our studies. hPGRN-FLAG, the human PGRN gene, was amplified using cDNA obtained from HEK293T cells using the Phusion DNA polymerase kit (New England BioLabs). The forward and reverse primers used are CGTACGAATTCATGTGGACCCTGGTGAGCTGGGT and GCTACGCGGCCGCCAGCAGCTGTCTCAAGGCTGG, respectively. The PCR product was gel purified, subjected to restriction digestion with EcoRI and NotI, and subcloned into the multiple cloning site of pcDNA3, upstream of three tandem copies of the FLAG epitope tag. The integrity of the plasmid (pcDNA-human progranulin-3×FLAG) was confirmed by DNA sequencing. Recombinant FLAG-tagged hPGRN was purified from the conditioned media of transfected HEK293T cells using anti-FLAG M2 magnetic beads. Briefly, HEK293T cells were transfected with pcDNA-human progranulin-3×FLAG. After 3 d, the conditioned media was collected, cleared at 1500 × g at 4°C for 10 min, concentrated using Amicon Ultra 10 kDa columns, and incubated with anti-FLAG M2 magnetic beads (Sigma) at 4°C on an end-over-end rotator for 4 h. Then the magnetic beads were washed three times with PBS for 5 min. The recombinant PGRN was eluted from the magnetic beads by competition with 500 ng/μl FLAG peptide (Sigma). The eluate was applied to an Amicon Ultra 10 kDa column to remove the FLAG peptide. Aliquots of the recombinant PGRN were stored at −20°C. mPGRN6×His on C terminus, mouse recombinant PGRN with a C-terminal 6-His tag, was purchased (R&D Systems; catalog #2557-PG). hPGRN, human recombinant PGRN, was obtained from Abnova. hPGRN, human recombinant untagged PGRN, was obtained from Five Prime Therapeutics. hPGRN-TAP, tandem affinity purification (TAP), tags were cloned onto the carboxyl (C) terminus or amino (N) terminus of full-length hPGRN to generate C-TAP PGRN and N-TAP PGRN, respectively. The C-TAP PGRN construct consists of a tandem Strep-II (also known as One-Strep; IBA) tag followed by the FLAG epitope as described previously (Gloeckner et al., 2009). N-TAP PGRN has a tandem Strep-II tag followed by a V5 epitope tag that was inserted following the PGRN signal peptide sequence to allow proper processing and secretion of the protein while retaining the TAP tag. Stable HEK293T cell lines expressing C-TAP PGRN and N-TAP PGRN were generated and secreted PGRN proteins were purified from conditioned media over Strep-Tactin superflow columns from IBA Life Sciences as described by the manufacturer (IBA). PGRN-Fc, a cDNA-encoding mouse PGRN obtained from the IMAGE consortium, and a plasmid encoding part of the Fc portion of human Ig (obtained as a kind gift from Dr. Thomas Sudhof) were used to subclone into pcDNA4 (Invitrogen) and generate an Fc-tagged mouse PGRN using standard molecular cloning techniques. A clone of HEK293 cells stably overexpressing PGRN-Fc was generated using Zeocin (Invitrogen) selection. PGRN-Fc was purified from conditioned medium by affinity chromatography using Protein A Agarose beads (Roche). Briefly, conditioned medium was adjusted to pH 8 with 1 m Tris-HCl; PGRN-Fc-tagged protein was bound to the beads overnight at 4°C; the beads were collected by passing through a chromatography column and washed with buffer containing 100 mm Tris-HCl, pH 8, 0.5 m NaCl, 0.1% Triton X-100; the protein was eluted with 100 mm glycine, pH 3, immediately neutralized with Tris buffer-exchanged into PBS supplemented with 10% glycerol and 0.5 mm EDTA; and stored at −80°C until use in biochemical experiments. All batches of mouse or human PGRN obtained from commercial sources or purified from HEK193 cells in-house were confirmed to be active as measured by phospho-Erk1/2 and p-Akt activity in immunoblots of lysates from PGRN-treated H4 glioma cells.
Coimmunoprecipitation.
Proteins (N-TAP PGRN, C-TAP PGRN, or human TNF;R&D Systems) were tested separately for interaction with purified recombinant receptors that contained a human Fc tag (human TNFRI-Fc or TNFRII-Fc; 250 ng; R&D Systems) in coimmunoprecipitation (Co-IP) buffer (TBS plus 0.05% Tween 20 and 1% bovine serum albumin) along with 10 μl of magnetic protein A beads (Invitrogen) and allowed to incubate overnight at 4°C with rotation. Protein A beads were used to pull down the Fc-tagged recombinant TNFR complexes and were collected on a magnetic rack (Invitrogen), washed three times (200 μl each) with Co-IP buffer, then eluted in SDS-PAGE sample buffer at 70°C. An anti-PGRN antibody (α-PCDGF, rabbit; Invitrogen 40–3400) was used as a positive control to confirm stoichiometric pull down of PGRN. Samples were analyzed by dual-color infrared Western blots as previously described (Kukar et al., 2008). Briefly, samples were separated on Criterion gels (Bio-Rad), transferred to nitrocellulose membranes, blocked with Odyssey blocker (LICOR) for ∼2 h, and incubated with primary antibodies overnight. TNFR I/II proteins were visualized by immunoblotting with an anti-human IgG antibody conjugated to IR700 (Rockland). N-TAP and C-TAP PGRN were visualized by immunoblotting with anti-Strep II MAB Classic conjugated to Oyster 800 (IBA). TNF was detected with a mouse anti-TNF antibody (MAB610; R&D Systems) followed by incubation with an anti-mouse IgG secondary antibody conjugated to Alexa 680 (Invitrogen).
Surface plasmon resonance.
The following reagents were used in surface plasmon resonance (SPR) studies: 25 μg/ml anti-human IgG Fc in pH 5 acetate buffer, 25 μg/ml anti-Strep II in pH 5 acetate buffer, 10 μg/ml TNFRI in HBS-P buffer, 10 μg/ml TNFRII in HBS-P buffer, 10 μg/ml PGRN in HBS-P buffer, 10 μg/ml TNF-α in HBS-P buffer, and 10 μg/ml sortilin in HBS-P buffer. The regeneration buffer for anti-human IgG Fc and Fc-tagged proteins consisted of 3 m MgCl2 and the regeneration buffer for anti-Strep II- and Strep II-tagged protein was pH 1.5 glycine. The following steps were performed for TNFR (I and II) binding analyses: (1) anti-human IgG FC was immobilized on a new CM5 chip through the EDC/NHS chemistry; (2) TNFR (I or II) was injected over the anti-human IgG Fc-coated channel for 2 min at a flow rate of 10 μl/min and A response with ∼872 RU was obtained; (3) a blank sample (HBS-P) was injected over TNFR (I or II) layer first to obtain the background sensor response; (4) next, the regeneration buffer was flowed over the sensor surface for 180 s at a flow rate of 30 μl/min; (5) step (2) was repeated and followed with a PGRN solution injection over the TNFR (I and II) layer to obtain the binding sensorgram between TNFR (I or II) and PGRN; and (6) step (4) and (3) were repeated and followed with a TNF injection as the positive control. The sensorgrams were shown in Figure 2A,B. Similar procedures were followed to capture TNFRII on the sensor surface to study the binding between TNFRII and PGRN. Again, blank and TNF were used as the negative control and positive control, respectively. For PGRN binding analysis, the following steps were performed: (1) anti-Strep II antibody was first immobilized on a CM5 chip through the EDC/NHS chemistry; (2) Strep II-tagged PGRN was injected over the anti-Strep II-coated channel for 2 min at flow rate of 10 μl/min, and a binding response with ∼340 RU was obtained; (3) a blank sample (HBS-P) was injected over Strep II-tagged protein layer first to obtain the background sensor response; (4) next, regeneration buffer was flowed over the sensor surface for 180 s at the flow rate of 30 μl/min; (5) step (2) was repeated and followed with a sample solution injection (TNFRI) over the PGRN layer; and (6) step (4) and (5) were repeated and followed for each samples (TNFRII and sortilin) binding analyses. The sensorgrams were shown in Figure 2C.
Figure 2.
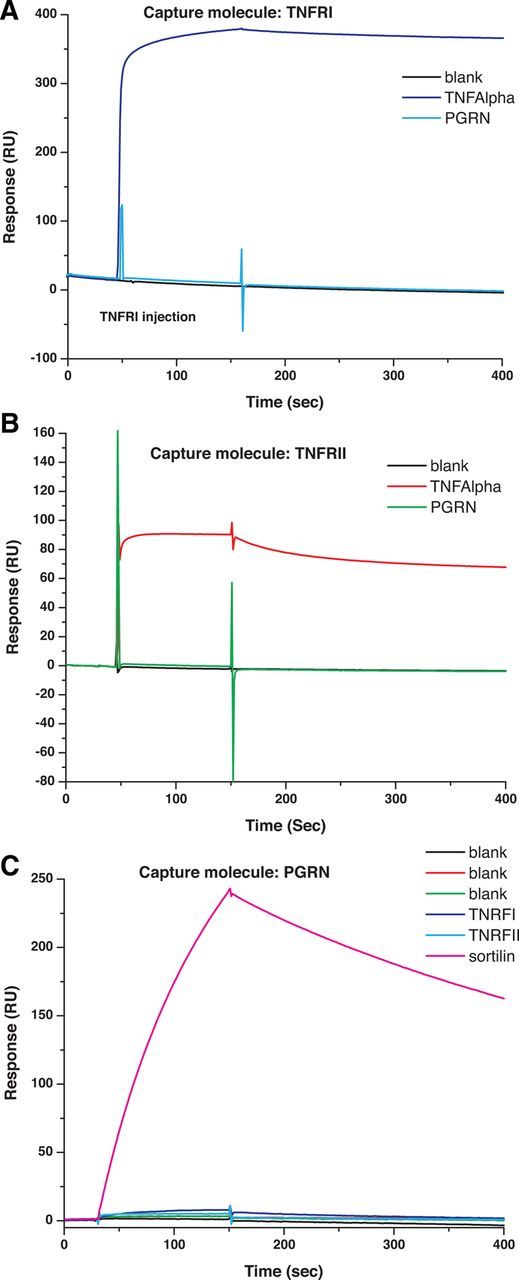
Purified recombinant PGRN does not bind purified TNFRI or II. Sensorgrams of the binding analysis between purified recombinant PGRN and TNF with TNFRI (A) or TNFRII (B) and between PGRN and sortilin (C). The binding between TNFRI and TNF, between TNFRII and TNF (red curves), and the binding between PGRN and sortilin have a fast on rate and a slow off rate, indicating a stable binding, whereas the binding between TNFRI and PGRN or TNFRII and PGRN (green curves) is indistinguishable from the blank, suggesting the binding is negligible. Affinity measurements for TNF–TNFRI and TNF-TNFRII are summarized in Table 1.
Cell culture.
Culture conditions for the BV2 microglia cell line consisted of DMEM/F-12 medium supplemented with 5% heat-inactivated fetal bovine serum (FBS). The MN9D dopaminergic neuroblastoma cell line was developed by Dr. Alfred Heller (Choi et al., 1991) and was a generous gift from Dr. Michael Zigmond at the University of Pittsburgh. MN9D cells were grown in culture in sterile complete media (CM), which consisted of high glucose (4500 mg/L) DMEM (Sigma, D5648) dissolved in sterile tissue culture tested water (Sigma) supplemented with 10% FBS (Hyclone Fetal Clone III), sodium bicarbonate (3.7 g/L; Sigma), 25 mm HEPES (Sigma), and 1% penicillin/streptomycin (Sigma) at a final pH of 7.3 in a humidified 5% CO2 incubator at 37°C. MN9D cell cultures were seeded in 75 cm2 tissue culture flasks (Costar) and plated at a density of 7500 cells per well for 96-well plates (100 μl CM per well). After plating and allowing attachment of cells overnight in CM, MN9D cells were differentiated for 72 h via a complete media change to differentiation media (DM), which contained serum-free DMEM (same CM as above, except FBS was excluded) supplemented with 5 mm 2-propylpentanoic acid (valproic acid; Sigma, P6273) and 1× N2 supplement (Invitrogen) as published previously (J. K. Lee et al., 2008). H4 glioma cells were plated in DMEM supplemented with 10% FBS into 10 cm dishes. For experiments, cells were serum deprived in DMEM and treated for the times indicated with N-TAP-PGRN or C-TAP-PGRN at 500 ng/ml. Cells were washed in PBS, lysed in PBS + 0.1% Triton X-100 (with protease and phosphatase inhibitors), and after centrifugation the supernatant was diluted into SDS-PAGE buffer for immunoblot analyses with antibodies specific for phospho-Akt, Akt, phospho-Erk1/2, or total Erk.
SDS-PAGE and Western blot analysis.
Cells were lysed in a buffer containing 1% NP-40, 10 mm Tris, pH 7.4, 150 mm NaCl, 100 μg/ml PMSF, and protease inhibitor mix (Sigma) for 30 min on ice. Lysates were resuspended in 2× Laemmli sample buffer and loaded on precast 12% SDS-PAGE gels (Bio-Rad), transferred onto PDVF membranes (Millipore), and probed with phospho-p38MAPK, phospho-Erk1/2, phospho-IKB, phospho-p65, and p65 antibodies (Cell Signaling Technology) or anti-GAPDH (1:1000) antibody (Santa Cruz Biotechnology) plus the appropriate horseradish peroxidase-conjugated secondary antibody (1:5000; Jackson ImmunoResearch). Immunoreactive bands were visualized with SuperSignal West Femto HRP substrate (Thermo Fisher Scientific) according to the manufacturer's instructions and imaged on a Syngene G:Box Chemi gel documentation station. Membranes were stripped with 0.2 m glycine, 1% SDS, and 0.1% Tween 20, pH 2.2, and reprobed as necessary.
NFκB-luciferase reporter assays.
NFκB-luciferase reporter plasmid (NFκB-Luc) was a gift from Dr. James Chen (Seth et al., 2005). A mammalian expression plasmid encoding β-galactosidase (β-gal) was kindly provided by Dr. Paul Dutchak. These plasmids were prepared using NucleoBond (Takara) endotoxin-free maxi-prep kits. The plasmid encoding mouse GRN was generated by inserting full-length GRN cDNA into pcDNA4 (Invitrogen) by standard molecular cloning techniques. The plasmid was fully sequenced, and is available from the Herz and Yu labs. rhPGRN and rhTNF-α were obtained from R&D Systems; stock solutions were prepared in sterile, cell culture grade PBS. HEK293A cells were cultured as described previously (Sephton et al., 2010) in DMEM/10% FBS (Invitrogen) and transiently transfected with FUGENE 6 (Roche) reagent, 48 h before NFκB-luciferase reporter assays. In the experiments summarized in Figure 4B, NFκB-Luc, β-gal, and test plasmids were transfected in 1:1:3 ratio. The cells were plated on 96-well plates and serum starved for 12 h. Etanercept or rhPGRN was applied 30 min before TNF-α treatment. All reagents were diluted and applied in serum-free DMEM. Five to six hours after TNF-α treatment, cells were lysed in luciferase assay buffer (Promega) + 1% Triton X-100 (100 μl/well). Luciferase activity was measured with Victor3 Multi-label Counter (PerkinElmer). Then, 120 μl/well β-gal buffer (60 mm Na2HPO4, 40 mm NaH2PO4, 10 mm KCl, 1 mm MgCl2, 0.4 mg/ml ONPG, 0.28% 2-mercaptoethanol) was added and OD420 was measured ∼30 min later with a spectrophotometer. Western blotting was performed as described previously (Cenik et al., 2012).
Figure 4.

Co-addition of recombinant PGRN and transfected PGRN fail to diminish NFκB reporter activation in response to TNF. A, HEK293A cells were transfected with NFκB-luciferase reporter and β-gal expression plasmids. Pretreatments and recombinant human TNF were applied at the indicated concentrations. Luciferase reporter activity in relative light units (RLU) was normalized to β-gal activity (measured by optical density at 420 nm, OD420) and expressed as percentage of activity observed in the untreated wells (leftmost column). Error bars indicate SEM, n = 4–5. *p < 0.05 versus no pretreatment for the same TNF concentration. NS indicates p > 0.05 for the same TNF concentration. B, HEK293A cells were transfected with NFκB-luciferase reporter and β-gal expression plasmids, plus either control plasmid (empty vector) or a plasmid encoding full-length mouse progranulin (GRN), as indicated. NFκB reporter activity was measured and analyzed as in A. Error bars indicate SEM, n = 5. C, Western blot analysis was performed on cells transfected in parallel to cells used in B to confirm PGRN expression.
Cell viability assays.
Treated diff-MN9D cells in 96-well plates were evaluated for overall viability using the MTS assay (Promega; CellTiter 96 Aqueous One Solution Cell Proliferation Assay) according to the manufacturer's instructions. Twenty microliters of the MTS reagent was added to cell cultures with DM-containing treatments and/or inhibitors. The cells were incubated with the MTS reagent at 37°C, 5% CO2 for 3 h before colorimetric quantification of MTS reduction into a blue formazan byproduct by metabolically active cells. The absorbance of blue formazan was measured at 492 nm wavelength using a Multiskan Ascent absorbance plate reader (Thermo LabSystems). Each experimental condition was performed in quadruplicate and three to four independent experiments were conducted.
Bone marrow-derived macrophages culture.
For recombinant PGRN co-addition experiments (see Figs. 8, 9), bone marrow-derived macrophages (BMDMs) from C57BL/6 Grn+/+ or Grn+/− (heterozygous) male mice, generated as described previously (Martens et al., 2012), were harvested and plated at a density of 0.5 million cells per well in a 12-well tissue culture plate in CM, which consisted of DMEM/F12 (D6421; Sigma), 20% heat-inactivated FBS (S11150; Atlanta Biologicals), 1% l-glutamine (Sigma), 1% penicillin/streptomycin (Sigma), and 20% CM from L929 (ATCC catalog #CCL1) mouse fibroblast cell line [DMEM (D5796, Invitrogen), 10% FBS (Sigma), 1% l-glutamine, 1% penicillin/streptomycin]. BMDMs were incubated with 10 ng/ml TNF, 1 μg/ml lipopolysaccharide (LPS), or 5 nm mouse recombinant PGRN (R&D Systems) singly or in combination for 24 h. For recombinant PGRN pre-incubation experiments (see Fig. 10), BMDMs were harvested from C57BL/6 Grn+/+ or Grn−/− mice generated and characterized by Aihao Ding et al. on a C57BL/6 background as described previously (Yin al., 2010a,b). BMDMs were plated at a density of 0.5 million cells per well in a 12-well tissue culture plate in CM. Before pre-incubation experiments with recombinant PGRNs, cells were switched to treatment media, which consisted of DMEM/F12, 1% l-glutamine, 1% penicillin/streptomycin, and 20% conditioned media from L929 mouse fibroblast cell line. Cells were pretreated in triplicate with 325 ng/ml (∼5 nm) PGRN for 16 h followed by stimulation with 100 ng/ml LPS (Sigma) for 24 h. Conditioned media were collected for multiplexed immunoassay analysis using a 7-plex mouse inflammatory factor array (Meso Scale Discovery). Cells were harvested and RNA was extracted using RNeasy mini kit (Qiagen) and processed for quantitative real-time PCR (qPCR) analysis. hPGRN flag denotes C-term FLAG-tagged human PGRN purified in-house at UCSF, 325 ng/ml; mPGRN obtained from R&D Systems, 325 ng/ml; and hPGRN denotes untagged PGRN obtained by Five Prime Therapeutics, 425 ng/ml.
Figure 8.

Co-addition of recombinant PGRNs does not directly antagonize TNF- or LPS-induced inflammatory gene expression in BV2 microglia or wild-type BMDMs. BV2 microglia cells (A) or BMDMs from wild-type C57/B6 mouse (B) were incubated with 10 ng/ml TNF, 1 μg/ml LPS, or 5 nm mouse recombinant PGRN (R&D Systems) singly or in combination for 24 h. Cells were harvested for RNA extraction and real-time qPCR analyses of TNF and IL-1β mRNA expression normalized to GAPDH mRNA expression (see Materials and Methods). One-way ANOVA followed by Tukey's post hoc test; *p < 0.05. The data shown are representative of 3–4 independent experiments.
Figure 9.

Co-addition of recombinant mPGRN does not directly antagonize TNF- or LPS- induced cytokine secretion by BMDMs from Grn+/− mice. Mouse BMDMs from C57BL/6 Grn+/+ or Grn+/− (heterozygous) mice generated as described previously (Martens et al., 2012) were harvested and plated at a density of 0.5 million cells per well in a 12-well tissue culture plate. Cells were incubated with 10 ng/ml TNF (A), 1 μg/ml LPS (B), or 5 nm mouse recombinant PGRN (R&D Systems) singly or in combination for 24 h. Conditioned media were collected and subjected to multiplexed immunoassay analysis using a 7-plex mouse inflammatory factor array (Meso Scale Discovery; see Materials and Methods). One-way ANOVA revealed no significant differences. The data shown are representative of two independent experiments.
Figure 10.

Prolonged pre-incubation with recombinant mouse or human PGRN has no modulatory effects on LPS-induced inflammatory cytokine secretion in BMDMs from Grn−/− mice. Mouse BMDMs from C57BL/6 Grn−/− (homozygous) mice (purchased from The Jackson Laboratory) were harvested and plated at a density of 0.5 million cells per well in a 12-well tissue culture plate. Before experiments, cells were switched to treatment (see Materials and Methods) media and incubated with 5 nm mouse recombinant PGRN with a C-terminal 6-His tag (R&D Systems), or N- or C-TAP-hPGRN for 16 h. Cells were then treated with 100 ng/ml LPS or saline for 24 h for collection of conditioned media for 7-plex mouse immunoassay analysis of inflammatory factors (Meso Scale Discovery; see Materials and Methods). One-way ANOVA revealed no significant differences. The data shown are representative of four independent experiments performed with two different Grn−/− mouse lines.
qPCR.
QPCR was performed as previously described (Kurrasch et al., 2004). Total RNA was isolated from cells in culture using the RNeasy isolation kit (Qiagen), treated with DNaseI, and reverse transcribed using Superscript II RNase H reverse transcriptase (Invitrogen). qPCR was performed using SYBR Green in 384-well format using an ABI Prism 7900HT Fast Detection System (Applied Biosystems). Oligonucleotide primers for qPCR were obtained from Integrated DNA Technologies. Primer sequences (available upon request) for the genes of interest were validated using tissues with high expression listed in www.biogps.org and used for gene amplification. Levels of mRNA expression were normalized to those of the mouse housekeeping genes cyclophilin B and GAPDH. Values represent the mean value of triplicate samples ± SEM. Data are representative of at least four independent experiments.
Multiplexed immunoassays for inflammatory factor secretion.
For mouse inflammatory cytokine multiplexed assay murine microglial BV2 cells or BMDM cultures were grown as indicated. Conditioned media from those cultures were collected to measure the production of cytokines including murine interferon-γ, IL-1β, IL-6, IL-10, IL-12p70, KC, and TNF using a multiplex assay per the manufacturer's instructions (Meso Scale Discovery).
qPCR array.
BV2 microglia were plated at 150,000 cells per well in a 12-well plate in DMEM/F-12 with 5% FBS. Cells were pretreated with 5 nm mouse recombinant PGRN (R&D Systems) for 16 h, followed by 1 μg/ml LPS for 24 h. RNA was extracted using Qiagen RNeasy mini kit and converted into first-strand cDNA using RT2 First Strand Kit (SABiosciences). qPCR was performed using an ABI Prism 7900HT Fast Detection System (Applied Biosystems). Each 10 μl reaction was performed in 384-well format of mouse inflammatory cytokines and receptors RT2 Profiler PCR Array (SABiosciences catalog #PAMM-011). The PCR mix was denatured at 95°C for 10 min before the first PCR cycle. The thermal cycle profile was denaturation for 15 s at 95°C and annealing for 60 s at 60°C. A total of 40 PCR cycles were performed. The resulting threshold cycle values for all wells were analyzed using the Data Analysis Template Excel file provided by SABiosciences.
Statistical analyses.
Differences treatments among the different groups were analyzed by two-way ANOVA followed by the Bonferroni post hoc test for p values significance. Differences treatments within the group were analyzed by one-way ANOVA followed by the Tukey's post hoc test for p values significance. Values expressed are the group mean +/− SEM.
Results
Purified PGRN does not bind purified TNFRI or II
To examine the interaction of PGRN and TNFR in cellular systems relevant to the CNS, we performed Co-IP experiments in neuroblastoma and neuroglioma cells. Unexpectedly, we were unable to detect Co-IP of endogenous TNFRI and PGRN in either cell line (data not shown). To eliminate the possibility that this negative result was due to low protein expression levels, we performed Co-IPs using purified recombinant PGRN and TNFR (Fig. 1). Neither TNFRI nor TNFRII was able to pull down PGRN tagged at the C terminus with a TAP tag under buffer conditions containing 0.05% Tween 20. Sortilin has also been described as a receptor for PGRN (Carrasquillo et al., 2010; Hu et al., 2010) and this interaction is disrupted by C-terminal tags (Zheng et al., 2011). Therefore, we asked if PGRN tagged on the N terminus interacted with TNFRI or TNFRII, but were unable to detect pull down. However, TNFRI and TNFRII were able to Co-IP the known ligand soluble TNF.
Figure 1.

Purified recombinant PGRN does not interact with purified recombinant TNFR I or II. Purified recombinant N-TAP PGRN, C-TAP PGRN, and soluble TNF were tested separately for interaction with purified recombinant TNFRs that contained a human Fc tag (human TNFRI-Fc or TNFRII-Fc) as described (see Materials and Methods). In a separate reaction, an anti-PGRN antibody (α-PCDGF) was incubated with N- and C-TAP PGRN to confirm stoichiometric pull down of PGRN. After SDS-PAGE and immunoblotting, the samples were analyzed by dual-color infrared (LI-COR Odyssey Fc) Western blots as previously described (Kukar et al., 2008; see Materials and Methods).
We next used SPR to perform a more detailed analysis of the binding of PGRN to recombinant TNFRI, TNFRII, or sortilin in comparison with the affinity of TNF to its receptors. Two experimental approaches were designed and conducted. In the first approach, TNFR proteins were immobilized on the SPR chip surface and PGRN diluted into buffer was flowed over the SPR chip to examine binding. In the second approach, PGRN was immobilized on the SPR chip surface and TNFR solutions were flowed over the SPR surface to detect binding between the PGRN and TNFR proteins. The binding between TNFRI and TNF or between TNFRII and TNF (red curves) has a fast on rate and a slow off rate, indicating stable binding between TNFRI and TNF or TNFRII and TNF, respectively (Fig. 2A,B). The binding between TNFRI and PGRN or TNFRII and PGRN (green curves) is indistinguishable from the blank, suggesting the binding is negligible (Fig. 2A,B). The same conclusion is reached based on the second experimental approach, as shown in Figure 2C. Next, we repeated the experiments with varying amounts of soluble TNF bound and TNFRI or TNFRII as the capture molecules to calculate the binding affinities. To extract the Kd values, a 1:1 steady-state model was used for curve fitting: (Req= CRmaxKd + C + RI) where C is the concentration, Req is the steady-state binding levels, and RI is the bulk refractive index contributions. The fitted Kd for TNFRI and TNF or TNFRII and TNF are 8.03 × 10−8 m and 1.23 × 10−7 m, respectively. The Kd for PGRN binding to sortilin was 3.07 10−8 m (Table 1). In summary, the binding affinities derived from our studies were in line with the published affinities for TNF–TNFRs and PGRN–Sortilin but we found no evidence of direct interactions between PGRN and TNFRs.
Table 1.
Binding constants derived from SPR studies
| Protein | Kd |
|---|---|
| TNF–TNFRI | 8.03 × 10−8 m |
| TNF–TNFRII | 1.23 × 10−7 m |
| PGRN–Sortilin | 3.07 × 10−8 m |
Recombinant PRGN does not directly antagonize TNF-induced signaling
Based on the lack of detectable physical interactions between purified PGRN and TNFRs or in cell lysates after transfection of human PGRN cDNA and exogenously added TNFRs, we predicted that addition of mPGRN to cells would not antagonize TNF-dependent signaling. To test this directly, we first stimulated the mouse microglia BV2 cell line with soluble TNF for up to 60 min and found the expected transient phosphorylation of the p65RelA subunit indicative of activation of the NFκB complex and transient phosphorylation of p42/p44MAPKs (p-erk1/2) (Fig. 3A). However, co-addition of mPGRN and TNF did not block or attenuate the TNF-dependent activation of either signaling pathway. Notably, addition of mPGRN alone to BV2 cells did not induce activation of NFκ signaling but transiently increased p-erk (Figs. 3A, 6A). To extend and confirm these findings we harvested BMDMs from wild-type mice and also transient activation of p-erk signaling by PGRN alone but no effect of PGRN on TNF-dependent p65 phosphorylation (Fig. 3B).
Figure 3.

Co-addition of recombinant PGRN does not directly antagonize TNF-induced signaling in microglia or BMDMs. BV2 microglia cells (A) or wild-type C57/B6 mouse BMDMs (B) were incubated with 10 ng/ml TNF, or 5 nm mouse recombinant PGRN (R&D Systems), or both for the times indicated. Cell lysates were harvested for SDS-PAGE and were analyzed by immunoblot using antibodies specific for phospho-p65, p65, phospho-Erk1/2, phospho-IKB, or GAPDH for normalization (see Materials and Methods). The data shown are representative of 3–4 independent experiments.
Figure 6.

Co-addition of recombinant mPGRN does not antagonize LPS-induced signaling in BV2 and BMDM. BV2 microglia cells (A) or mouse BMDMs (B) were incubated with 1 μg/ml LPS, or 5 nm mouse recombinant PGRN (R&D Systems) singly or in combination for the times indicated. Cell lysates were harvested for SDS-PAGE and were analyzed by immunoblot using antibodies against phospho-p65, p65, phospho-IKB, phospho-Erk1/2, phospho-p38MAPK, or GAPDH for normalization. The data shown are representative of 3–4 independent experiments.
To extend and confirm these findings, we investigated the ability of PGRN to antagonize TNF-induced NFκB activation using luciferase reporter assays in HEK293A cells. First, HEK293A cells were transfected with an NFκB-luciferase reporter and β-gal expression plasmids to measure TNF-dependent NFκB transcriptional activity in cells treated with TNF in the presence or absence of rhPGRN or the TNFRII decoy receptor etanercept as a positive control for inhibition. We found that while etanercept completely abolished the response to TNF, rhPGRN had no effect on NFκB activation (Fig. 4A). Next, HEK293A cells were transfected with NFκB-luciferase reporter and β-gal expression plasmids, plus either a control plasmid (empty vector) or a plasmid encoding full-length mouse progranulin (mPGRN), as indicated. In the absence of TNF, there was a slight trend toward lesser reporter activation in the cells transfected with the mPGRN plasmid (84.5 ± 7.4%); however, the effect was not statistically significant (NS) either in the presence or absence of TNF (Fig. 4B). To confirm that the cells transfected with mPGRN were in fact expressing PGRN protein, Western blot analysis was performed on cells transfected in parallel to untransfected HEK293A cells. Transfection with the mPGRN plasmid greatly enhanced mPGRN immunoreactivity in cell lysates and CM (Fig. 4C). While low levels of endogenous human PGRN expression are also evident in control cell lysates, the level of endogenous human PGRN in the CM is undetectable by this assay. In summary, TNF-induced NFκB signaling is not antagonized by recombinant human PGRN or overexpression of mPGRN in mammalian cells.
PRGN does not antagonize TNF-induced neurotoxicity in dopaminergic neuron-like cells
Next, we investigated whether PGRN could antagonize TNF-induced neurotoxicity in the neurally differentiated MN9D mouse ventral mesencephalon neuroblastoma cell line (Choi et al., 1991). Differentiated MN9D (diff-MN9D) cells were treated with soluble TNF in the presence or absence of recombinant Fc-fused human PGRN or C-terminal 6-His-tagged mouse PGRN at the concentrations indicated or with etanercept as a positive control for TNF inhibition. As described previously (McCoy et al., 2006, 2008), we found that diff-MN9D cells displayed reduced mitochondrial metabolism and viability after 48 h of exposure to soluble TNF (5 ng/ml) and coincubation with the TNF inhibitor etanercept significantly attenuated the loss of viability (Fig. 5A). Consistent with the observations that PGRN did not directly bind to or associate with TNFRs and did not antagonize TNF-induced signal transduction, co-addition of either mouse or human PGRN with soluble TNF did not inhibit TNF-induced cytotoxicity and had no effect on its own either. Identical results were obtained with recombinant N- and C-TAP-tagged PGRN and with PGRN from a number of commercial vendors (Alexis and Abnova; data not shown). N- and C-TAP-hPGRN did, however, induce time-dependent phosphorylation of AKT in H4 neuroglioma cells (Fig. 5B), confirming that our recombinant PGRN is active in previously reported signaling pathways (Zanocco-Marani, Bateman et al., 1999). Together, our observations derived from binding, signaling, gene reporter, and cytotoxicity assays are internally consistent and demonstrate there is no functional interaction between PGRN and TNF-dependent signaling in microglia, BMDMs, and neuroblastoma cells.
Figure 5.

Co-addition of recombinant mPGRN or hPGRN does not directly antagonize TNF-induced neurotoxicity in dopaminergic neuron-like cells. A, Neuronally differentiated MN9D dopaminergic neuroblastoma cells plated in 96-well plates were treated for 24 h with 5 ng/ml TNF, mouse recombinant PGRN, or Fc-tagged human PGRN singly or in combination at the concentrations (in ng/ml) indicated. Etanercept was used at 200 ng/ml as a positive control for TNF inhibition. Cell viability was measured using the MTS assay (Promega, CellTiter 96 Aqueous One Solution Cell Proliferation Assay) according to the manufacturer's instructions. One-way ANOVA followed by Tukey's post hoc test; ##p < 0.01, *p < 0.05. The data shown are representative of 4–5 independent experiments. B, H4 glioma cells plated on 10 cm dishes were treated for the times indicated with N-TAP-PGRN or C-TAP-PGRN at 500 ng/ml. Cells were washed in PBS, lysed in PBS + 0.1% Triton X-100 (with protease and phosphatase inhibitors), and resuspended after centrifugation into SDS-PAGE buffer for immunoblot analyses with antibodies specific for phospho-Akt, Akt, phospho-Erk1/2, or total Erk (see Materials and Methods). The data shown are representative of three independent experiments.
PRGN does not antagonize LPS-induced signal transduction in BV2 microglia or wild-type BMDMs
Next, we sought to investigate whether recombinant PGRN co-addition with LPS resulted in any detectable antagonism of LPS-induced inflammatory signaling in mouse BV2 microglia or BMDM from wild-type mice. As described previously (J. K. Lee et al., 2011), LPS induced robust but transient activation of the NFκB pathway in BV2 microglia cells as evidenced by phosphorylation of p65RelA and IKB and in activation of p42/p44MAPK pathway as measured by phosphorylation of Erk1/2 (Fig. 6A); although, however, PGRN alone elicited transient p-erk activation, co-addition of PGRN with LPS did not attenuate LPS-induced activation of these signaling pathways. Similar results were obtained with BMDMs (Fig. 6B). Together these data indicate recombinant PGRN does not exert anti-inflammatory activity to antagonize LPS-induced signaling in microglia or macrophages.
Pre-incubation with recombinant PRGN does not significantly change the network of inflammatory gene expression regulated by LPS in BV2 microglia
In an attempt to expand the survey of potential anti-inflammatory properties by PGRN, we treated BV2 microglia cells with LPS, PGRN alone, or PGRN plus LPS together for 24 h then harvested the cells to extract RNA for gene expression analysis. Using an inflammatory gene PCR array, we found that the inflammatory gene profile induced by LPS alone relative to control (saline-treated cells) (Fig. 7A) was nearly identical to that induced by PGRN + LPS relative to control-treated cells (Fig. 7B), suggesting that PGRN pre-incubation was not able to significantly change the inflammatory profile induced by LPS. Last, analysis of the inflammatory gene profile induced by PGRN alone (Fig. 7C) revealed that several inflammatory factors and receptors (including IL-11, CCL6, CCL17, CCR6, and CXCR3) were upregulated by PGRN itself while only two inflammatory ligands (IL-17 and CD40L) were downregulated by PGRN.
Figure 7.
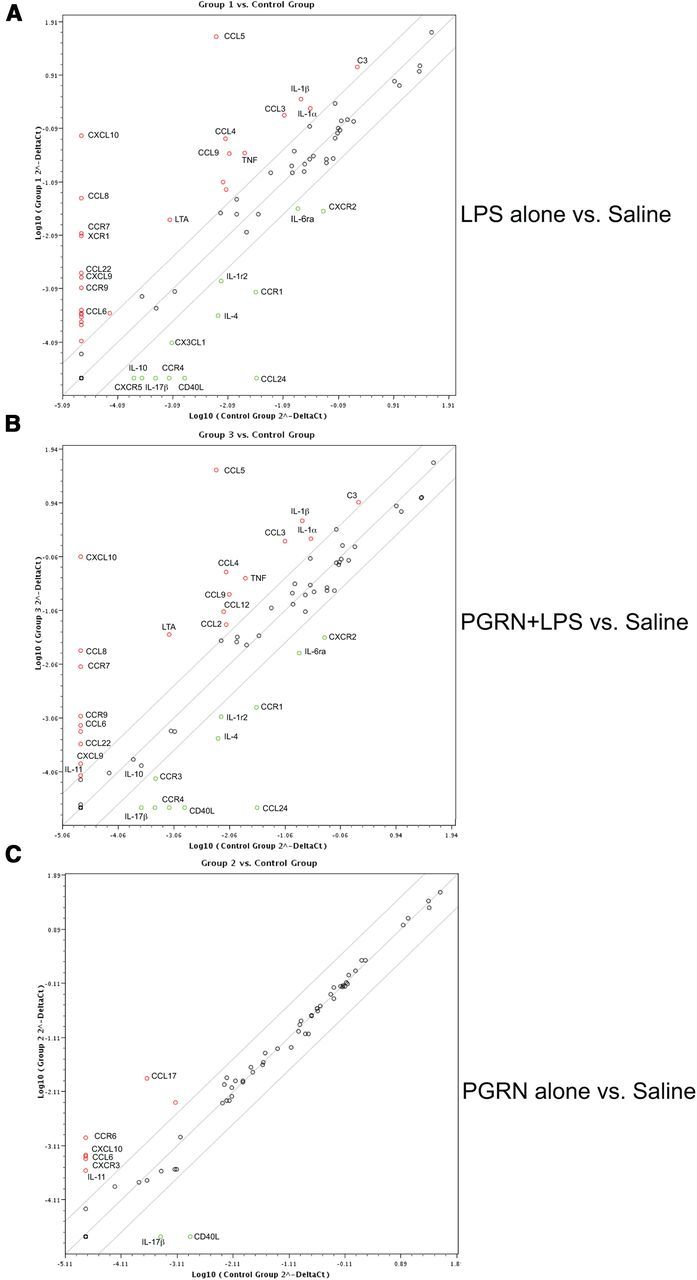
Inflammatory gene expression profile in PGRN pretreated microglia. BV2 microglia were plated at 150,000 cells per well in a 12-well plate in DMEM/F-12 with 5% FBS. Cells were treated for 16 h with saline followed by 24 h of saline (Control), or saline for 16 h + 1 μg/ml LPS for 24 h (Group 1), or 5 nm mouse recombinant PGRN (R&D Systems) for 16 h + saline for 24 h (Group 2), or 5 nm mouse PGRN for 16 h + 1 μg/ml LPS for 24 h (Group 3). RNA was extracted using Qiagen RNeasy mini kit and converted into first-strand cDNA using RT2 First Strand Kit (SA Biosciences). qPCR was performed using an ABI Prism 7900HT Fast Detection System (Applied Biosystems). Each 10 μl reaction was performed in a 384-well format of mouse inflammatory cytokines and receptors RT2 Profiler PCR Array (SABiosciences). The resulting threshold cycle values for all wells were analyzed using the Scatter Plot Data Analysis Tool provided by SA Biosciences. Scatter plot of gene expression of (A) LPS-stimulated BV2 microglia (Group 1) versus saline-treated (Control) BV2 microglia, (B) PGRN + LPS-stimulated BV2 microglia (Group 3) versus saline-treated (Control) BV2 microglia, and (C) PGRN-stimulated BV2 microglia cells versus saline-treated (Control) BV2 microglia. Genes shown in red were upregulated and genes shown with in green were downregulated in the treatment condition, which is plotted on the y-axis relative to the control condition plotted on the x-axis.
Co-addition of recombinant mPGRN does not directly antagonize TNF- or LPS-induced inflammatory gene expression in BV2 microglia or wild-type BMDMs
Next, we sought to investigate the specific effects of co-addition of various different types of recombinant PGRN proteins (C-terminal 6-His-mPGRN, N-TAP-hPGRN, or C-TAP-hPGRN) on BV2 microglia cells and wild-type BMDMs. Cells were treated with TNF or LPS for 24 h after which cells were harvested to measure mRNA levels of TNF and IL-1β as transcripts for two key pro-inflammatory cytokines. We found no inhibitory effects by any recombinant PGRN on the TNF- or LPS-induced increases of IL-1β or TNF mRNAs in either BV2 (Fig. 8A) or BMDMs (Fig. 8B) and an unexpected yet very modest pro-inflammatory effect by C-TAP-hPGRN in BV2 microglia. In agreement with other assays, addition of recombinant mPGRN alone had no effect on TNF and IL-1β mRNAs.
Co-addition of recombinant mPGRN does not directly antagonize TNF- or LPS- induced cytokine secretion by BMDMs derived from Grn+/− mice
To rule out the possibility that endogenous PGRN levels were masking our ability to detect anti-inflammatory activity by exogenously added recombinant PGRNs, we measured the levels of TNF- and LPS-induced IL-6, IL-10, or TNF secretion into the media by BMDMs harvested from Grn+/− mice generated as described previously (Martens et al., 2012) and compared them to those from Grn+/+. We found that both TNF and LPS induced robust and comparable secretion of these three cytokines in BMDMs from both Grn+/+ and Grn+/− mice and co-addition of recombinant mouse PGRN did not attenuate these responses (Fig. 9). These findings indicate that endogenous PGRN is not interfering with our ability to detect anti-inflammatory effects of recombinant PGRN because reduction of endogenous PGRN levels by 50% in BMDMs does not result in enhanced baseline cytokine secretion from BMDMs and exogenously added recombinant mouse PGRN does not alter the TNF- or LPS-evoked responses.
Prolonged pre-incubation with recombinant mouse or human PRGN does not alter inflammatory factor production by BMDMs from Grn−/− mice
Last, we investigated whether further reduction of endogenous PGRN levels and a more extended pre-incubation with recombinant PGRN could elicit more robust anti-inflammatory gene expression or attenuate a response to a strong inflammatory challenge. We performed a set of four independent pre-incubation experiments in BMDMs from Grn+/+ and Grn−/− mice generated as described previously (Yin et al., 2010a,b) as well as in Grn+/+ and Grn−/− mice generated as described previously (Martens et al., 2012). Cells were exposed to recombinant mPGRN or human N- or C-TAP-PGRN for 16 h followed by addition of LPS (100 ng/ml) or saline for 6 h (after which time cells were harvested for inflammatory gene expression by qPCR) or for 24 h (after which time Conditioned media were collected for multiplexed immunoassay analysis of inflammatory factor expression). Consistent with other gene expression results, we found no significant anti-inflammatory effects by any recombinant PGRN on LPS-induced secretion of IL-1β, TNF, IFN-γ, IL-6, IL-10, or IL-12 proteins in BMDMs from Grn−/− mice (Fig. 10). It should be noted that on occasion, we observed detectable but very modest and inconsistent inhibitory (sometimes stimulatory) effects of recombinant mPGRN and N- and C-TAP-hPGRN on cytokine secretion that were not reproducible from experiment to experiment (data not shown).
Discussion
We sought to confirm the physical interaction reported by Tang et al. (2011) between PGRN and TNFRs to subsequently investigate the functional significance of this interaction within the context of microglia, macrophage activation, and dopamine neuron survival. Surprisingly, we found no evidence from immunoprecipitation and SPR studies to support a direct physical interaction between PGRN and either TNFR. Given that TNFRI is ubiquitously expressed in detectable amounts in most cell types (Tang et al., 2011), we first attempted to detect the interaction between endogenous TNFRI and PGRN in neuroblastoma and neuroglioma cells but were unable to do so. Next we used purified recombinant epitope-tagged proteins and performed reciprocal immunoprecipitations in vitro, using recombinant soluble TNF as a positive control for interactions with TNFRI and II, but again found no evidence of any direct association between PGRN and either TNFR.
Moreover, using state-of-the art SPR instrumentation and conditions comparable to those used by Tang et al. (2011) we were able to detect the previously reported interaction of TNF–TNFRI, TNF–TNFRII, and PGRN–Sortilin at affinities similar to those reported in the literature, but were unable to detect any interaction between PGRN and either TNFR regardless of whether recombinant PGRN or TNFR was used as the capture protein on the SPR chip. It is currently unclear why our results differ with the interaction data reported by Tang et al. (2011) that PGRN binds to TNFR. Some possibilities include the following: (1) the initial PGRN–TNFR was a false positive arising from the yeast-two hybrid assay or (2) the Co-IP of PGRN and TNFR was a result of nonspecific interactions due to high concentration of antibody (25 μg/ml), overnight incubation with Protein A Agarose beads and/or cross-linking of primary antibodies to Protein A-agarose, and low stringency washes (50 mm Tris HCl, pH 7.4, with 150 m NaCl) (Tang et al., 2011). In support of this idea, we detected the expected nonspecific interactions between recombinant TNFRs, PGRN, Sortilin, and beads under low-detergent buffer conditions (data not shown).
Consistent with a lack of physical interactions between PGRN and TNFRs, we observed no functional antagonism by recombinant PGRN against soluble TNF-dependent signaling, gene expression, or cytotoxicity in a variety of cell types using multiple assays when both ligands were added to cells in culture simultaneously. To rule out that the negative results could be due to a particular source of PGRN, we tested the ability of several different sources and batches of recombinant mouse and human PGRN (with N- or C-terminal tags) to antagonize soluble TNF-dependent neurotoxicity in mouse ventral mesencephalon MN9D neuroblastoma cells, which express high levels of TNFRI and lose viability when exposed to soluble TNF (Tran et al., 2008; Harms et al., 2011, 2012), and found neither mouse nor human PGRN was able to rescue from TNF-dependent toxicity. In fact, in several experimental paradigms using wild-type cells, we observed detectable but inconsistent potentiation of TNF- and LPS-dependent inflammatory signaling that we could not attribute to PGRN cleavage. Specifically, we considered that lack of antagonism by PGRN during co-addition with soluble TNF might be due to proteolytic processing of PGRN to GRNs, which are known to exert pro-inflammatory activity (Zhu et al., 2002; He and Bateman, 2003; He et al., 2003). However, the addition of secretory leukocyte protease inhibitor to protect PGRN from cleavage did not change the TNF-induced neurotoxicity in MN9D cells (data not shown) and led us to conclude PGRN cleavage was not the reason for it. We also considered the possibility that endogenous production of PGRN by the cells and secretion into the medium could obscure our ability to observe functional antagonism of TNF-dependent signaling. To rule out this possibility, we used BMDMs from Grn+/− or Grn−/− mice in some of our experiments and all cells were treated in freshly replenished media when stimulated in the presence or absence of the different recombinant PGRN batches obtained commercially or purified in-house using affinity tags. On the basis of these internally consistent data, we conclude that the inflammatory phenotype observed after deletion of Grn in mice is not a direct consequence of the loss of TNF antagonism by PGRN, but instead reflects an as yet unidentified downstream PGRN target.
Given that dysregulated neuroinflammation in Grn−/− mice could not be attributed to lack of direct antagonism of TNF–TNFR binding interactions, we considered that perhaps the effects of PGRN on inflammatory responses could be mediated through changes in basal levels of inflammatory gene expression. To address this possibility, we investigated whether pre-incubation with exogenously added recombinant PGRN could change the outcome of LPS-induced inflammatory gene transcription or cytokine secretion in BV2 microglia or BMDMs from mice with various Grn gene dosages. We found that co-addition of recombinant PGRN had no significant effects on cytokine mRNA expression in BMDMs from Grn+/− or Grn−/− mice stimulated with LPS. Curiously, we also observed detectable but weak and not strictly anti-inflammatory effects on inflammatory gene expression in BV2s and in wild-type BMDMs N- and C-tagged recombinant PGRNs after prolonged incubation. These findings raise the interesting possibility that perhaps internalization of PGRN through some unknown mechanism may be able to suppress LPS-induced inflammation in a cell-autonomous manner. In support of this idea, our recent work demonstrated that lentiviral transduction of murine PGRN can rescue the heightened LPS-induced expression of inflammatory cytokine mRNAs in Grn−/− microglia (Martens et al., 2012). Nevertheless, the current results fail to find any role for TNFRs.
As reported by several groups, genetic ablation of Grn results in increased microglia activation in the CNS in vivo with age (Ahmed et al., 2010; Yin et al., 2010a,b; Ghoshal et al., 2012; Wils et al., 2012). Our findings indicate that enhanced neuroinflammation is not caused by disruption of PGRN–TNFR interactions. Although exogenous addition of recombinant PGRN had very modest and variable effects on LPS-induced inflammatory signaling and cytokine secretion, these studies do not exclude the possibility that PGRN could modulate inflammatory responses in a cell-autonomous manner. However, another equally plausible explanation may be that the neuroinflammation in Grn-null mice is an indirect result of neuronal dysfunction due to decreased neurotrophic support by PGRN, which in turn leads to release of signals from neurons that activate microglia and astrocytes, possibly chemokines detectably elevated by recombinant PGRN in our studies (CCL6, CCL17, CXCL10, and IL-11). Alternatively, complete loss of PGRN has been recently described in human patients with neuronal ceroid lipofuscinosis (NCL) (Smith et al., 2012), suggesting that loss of PGRN leads to aberrant lysosomal function and defective lipid metabolism, which may trigger neuroinflammation and accelerate neurodegeneration (Cenik et al., 2012).
In summary, we find no evidence that PGRN and TNFR I/II interact either physically or through intracellular signaling pathways. Thus, the pleiotropic functions of PGRN are likely mediated through alternative receptors, such as sortilin (or an unknown receptor, or other biological pathways inside the cell). The implication of our findings is that recombinant PGRN therapy in frontotemporal lobar degeneration (FTLD) patients with PGRN haploinsufficiency is unlikely to have direct anti-inflammatory effects in FTLD. Further work to define the mechanisms of PGRN activity may ultimately lead to new treatments for diseases linked to decreased PGRN, which include not only FTD but now extend to NCL (Smith 2012), hippocampal sclerosis (Dickson et al., 2010), and Alzheimer's disease (Fenoglio et al., 2009; M. J. Lee et al., 2011; Kamalainen et al., 2013).
Footnotes
This work was supported in part by grants from NIH-NINDS 5RO1NS049433-05 (M.G.T.); NIH-NIA 4R00AG032362-03, 5P50AG016574-14, and Alzheimer's Association New Investigator Research Grant (12-242771) (T.K.); NIH-HL20948 and HL63762 (J.H.), and the Consortium for Frontotemporal Dementia Research (L.H.M., J.H., R.V.F.). We would like to thank members of our laboratories for helpful discussions.
References
- Ahmed Z, Sheng H, Xu YF, Lin WL, Innes AE, Gass J, Yu X, Wuertzer CA, Hou H, Chiba S, Yamanouchi K, Leissring M, Petrucelli L, Nishihara M, Hutton ML, McGowan E, Dickson DW, Lewis J. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am J Pathol. 2010;177:311–324. doi: 10.2353/ajpath.2010.090915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo MM, Nicholson AM, Finch N, Gibbs JR, Baker M, Rutherford NJ, Hunter TA, DeJesus-Hernandez M, Bisceglio GD, Mackenzie IR, Singleton A, Cookson MR, Crook JE, Dillman A, Hernandez D, Petersen RC, Graff-Radford NR, Younkin SG, Rademakers R. Genome-wide screen identifies rs646776 near sortilin as a regulator of progranulin levels in human plasma. Am J Hum Genet. 2010;87:890–897. doi: 10.1016/j.ajhg.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenik B, Sephton CF, Kutluk Cenik B, Herz J, Yu G. Progranulin: a proteolytically processed protein at the crossroads of inflammation and neurodegeneration. J Biol Chem. 2012;287:32298–32306. doi: 10.1074/jbc.R112.399170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HK, Won LA, Kontur PJ, Hammond DN, Fox AP, Wainer BH, Hoffmann PC, Heller A. Immortalization of embryonic mesencephalic dopaminergic neurons by somatic cell fusion. Brain Res. 1991;552:67–76. doi: 10.1016/0006-8993(91)90661-E. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Baker M, Rademakers R. Common variant in GRN is a genetic risk factor for hippocampal sclerosis in the elderly. Neurodegener Dis. 2010;7:170–174. doi: 10.1159/000289231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenoglio C, Galimberti D, Cortini F, Kauwe JS, Cruchaga C, Venturelli E, Villa C, Serpente M, Scalabrini D, Mayo K, Piccio LM, Clerici F, Albani D, Mariani C, Forloni G, Bresolin N, Goate AM, Scarpini E. Rs5848 variant influences GRN mRNA levels in brain and peripheral mononuclear cells in patients with Alzheimer's disease. J Alzheimers Dis. 2009;18:603–612. doi: 10.3233/JAD-2009-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Joselin AP, Wang L, Kar A, Ray P, Bateman A, Goate AM, Wu JY. Progranulin promotes neurite outgrowth and neuronal differentiation by regulating GSK-3beta. Protein Cell. 2010;1:552–562. doi: 10.1007/s13238-010-0067-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia I, Olleros ML, Quesniaux VF, Jacobs M, Allie N, Nedospasov SA, Szymkowski DE, Ryffel B. Roles of Soluble and Membrane TNF and Related Ligands in Mycobacterial Infections: effects of Selective and Non-selective TNF Inhibitors During Infection. Adv Exp Med Biol. 2011;691:187–201. doi: 10.1007/978-1-4419-6612-4_20. [DOI] [PubMed] [Google Scholar]
- Gass J, Lee WC, Cook C, Finch N, Stetler C, Jansen-West K, Lewis J, Link CD, Rademakers R, Nykjær A, Petrucelli L. Progranulin regulates neuronal outgrowth independent of Sortilin. Mol Neurodegener. 2012;7:33. doi: 10.1186/1750-1326-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshal N, Dearborn JT, Wozniak DF, Cairns NJ. Core features of frontotemporal dementia recapitulated in progranulin knockout mice. Neurobiol Dis. 2012;45:395–408. doi: 10.1016/j.nbd.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloeckner CJ, Boldt K, Schumacher A, Ueffing M. Tandem affinity purification of protein complexes from mammalian cells by the Strep/FLAG (SF)-TAP tag. Methods Mol Biol. 2009;564:359–372. doi: 10.1007/978-1-60761-157-8_21. [DOI] [PubMed] [Google Scholar]
- Harms AS, Barnum CJ, Ruhn KA, Varghese S, Treviño I, Blesch A, Tansey MG. Delayed dominant-negative TNF gene therapy halts progressive loss of nigral dopaminergic neurons in a rat model of Parkinson's disease. Mol Ther. 2011;19:46–52. doi: 10.1038/mt.2010.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms AS, Lee JK, Nguyen TA, Chang J, Ruhn KM, Treviño I, Tansey MG. Regulation of microglia effector functions by tumor necrosis factor signaling. Glia. 2012;60:189–202. doi: 10.1002/glia.21254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Bateman A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med. 2003;81:600–612. doi: 10.1007/s00109-003-0474-3. [DOI] [PubMed] [Google Scholar]
- He Z, Ong CH, Halper J, Bateman A. Progranulin is a mediator of the wound response. Nat Med. 2003;9:225–229. doi: 10.1038/nm816. [DOI] [PubMed] [Google Scholar]
- Hu F, Padukkavidana T, Vægter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68:654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamalainen A., Viswanathan, Natunen T, Helisalmi S, Kauppinen T, Pikkarainen M, Pursiheimo JP, Alafuzoff I, Kivipelto M, Haapasalo A, Soininen H, Herukka SK, Hiltunen MJ. GRN Variant rs5848 Reduces Plasma and Brain Levels of Granulin in Alzheimer's Disease Patients. J Alzheimers Dis. 2013;33:23–27. doi: 10.3233/JAD-2012-120946. [DOI] [PubMed] [Google Scholar]
- Kayasuga Y, Chiba S, Suzuki M, Kikusui T, Matsuwaki T, Yamanouchi K, Kotaki H, Horai R, Iwakura Y, Nishihara M. Alteration of behavioural phenotype in mice by targeted disruption of the progranulin gene. Behav Brain Res. 2007;185:110–118. doi: 10.1016/j.bbr.2007.07.020. [DOI] [PubMed] [Google Scholar]
- Kukar TL, Ladd TB, Bann MA, Fraering PC, Narlawar R, Maharvi GM, Healy B, Chapman R, Welzel AT, Price RW, Moore B, Rangachari V, Cusack B, Eriksen J, Jansen-West K, Verbeeck C, Yager D, Eckman C, Ye W, Sagi S, et al. Substrate-targeting gamma-secretase modulators. Nature. 2008;453:925–929. doi: 10.1038/nature07055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurrasch DM, Huang J, Wilkie TM, Repa JJ. Quantitative real-time PCR measurement of regulators of G-protein signaling (RGS) mRNA levels in mouse tissues. Methods Enzymol. 2004;389:3–15. doi: 10.1016/S0076-6879(04)89001-3. [DOI] [PubMed] [Google Scholar]
- Lee JK, McCoy MK, Harms AS, Ruhn KA, Gold SJ, Tansey MG. Regulator of G-protein signaling 10 promotes dopaminergic neuron survival via regulation of the microglial inflammatory response. J Neurosci. 2008;28:8517–8528. doi: 10.1523/JNEUROSCI.1806-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Chung J, McAlpine FE, Tansey MG. Regulator of G-protein signaling-10 negatively regulates NF-{kappa}B in microglia and neuroprotects dopaminergic neurons in hemiparkinsonian rats. J Neurosci. 2011;31:11879–11888. doi: 10.1523/JNEUROSCI.1002-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Chen TF, Cheng TW, Chiu MJ. rs5848 variant of progranulin gene is a risk of Alzheimer's disease in the Taiwanese population. Neurodegener Dis. 2011;8:216–220. doi: 10.1159/000322538. [DOI] [PubMed] [Google Scholar]
- Liu ZG. Molecular mechanism of TNF signaling and beyond. Cell Res. 2005;15:24–27. doi: 10.1038/sj.cr.7290259. [DOI] [PubMed] [Google Scholar]
- Martens LH, Zhang J, Barmada SJ, Zhou P, Kamiya S, Sun B, Min SW, Gan L, Finkbeiner S, Huang EJ, Farese RV., Jr Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J Clin Invest. 2012;122:3955–3959. doi: 10.1172/JCI63113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008;5:45. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MK, Martinez TN, Ruhn KA, Szymkowski DE, Smith CG, Botterman BR, Tansey KE, Tansey MG. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson's disease. J Neurosci. 2006;26:9365–9375. doi: 10.1523/JNEUROSCI.1504-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MK, Martinez TN, Ruhn KA, Wrage PC, Keefer EW, Botterman BR, Tansey KE, Tansey MG. Autologous transplants of Adipose-Derived Adult Stromal (ADAS) cells afford dopaminergic neuroprotection in a model of Parkinson's disease. Exp Neurol. 2008;210:14–29. doi: 10.1016/j.expneurol.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkau TL, Neal SJ, Milnerwood A, Mew A, Hill AM, Orban P, Gregg J, Lu G, Feldman HH, Mackenzie IR, Raymond LA, Leavitt BR. Synaptic dysfunction in progranulin-deficient mice. Neurobiol Dis. 2012;45:711–722. doi: 10.1016/j.nbd.2011.10.016. [DOI] [PubMed] [Google Scholar]
- Ryan CL, Baranowski DC, Chitramuthu BP, Malik S, Li Z, Cao M, Minotti S, Durham HD, Kay DG, Shaw CA, Bennett HP, Bateman A. Progranulin is expressed within motor neurons and promotes neuronal cell survival. BMC Neurosci. 2009;10:130. doi: 10.1186/1471-2202-10-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, Good SK, Dewey CM, Mayer P, 3rd, Herz J, Yu G. TDP-43 is a developmentally regulated protein essential for early embryonic development. J Biol Chem. 2010;285:6826–6834. doi: 10.1074/jbc.M109.061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, Sims KB, Lewis J, Lin WL, Dickson DW, Dahl HH, Bahlo M, Berkovic SF. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. 2012;90:1102–1107. doi: 10.1016/j.ajhg.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Tang W, Lu Y, Tian QY, Zhang Y, Guo FJ, Liu GY, Syed NM, Lai Y, Lin EA, Kong L, Su J, Yin F, Ding AH, Zanin-Zhorov A, Dustin ML, Tao J, Craft J, Yin Z, Feng JQ, Abramson SB, et al. The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science. 2011;332:478–484. doi: 10.1126/science.1199214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia L, Milnerwood A, Guo A, Mills F, Yoshida E, Vasuta C, Mackenzie IR, Raymond L, Cynader M, Jia W, Bamji SX. Progranulin deficiency decreases gross neural connectivity but enhances transmission at individual synapses. J Neurosci. 2011;31:11126–11132. doi: 10.1523/JNEUROSCI.6244-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh H, Chitramuthu BP, Bennett HP, Bateman A. Structure, function, and mechanism of progranulin; the brain and beyond. J Mol Neurosci. 2011;45:538–548. doi: 10.1007/s12031-011-9569-4. [DOI] [PubMed] [Google Scholar]
- Tran TA, McCoy MK, Sporn MB, Tansey MG. The synthetic triterpenoid CDDO-methyl ester modulates microglial activities, inhibits TNF production, and provides dopaminergic neuroprotection. J Neuroinflammation. 2008;5:14. doi: 10.1186/1742-2094-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Van Hoecke A, Lambrechts D, Vanacker P, Bogaert E, van Swieten J, Carmeliet P, Van Den Bosch L, Robberecht W. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181:37–41. doi: 10.1083/jcb.200712039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward ME, Miller BL. Potential mechanisms of progranulin-deficient FTLD. J Mol Neurosci. 2011;45:574–582. doi: 10.1007/s12031-011-9622-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wils H, Kleinberger G, Pereson S, Janssens J, Capell A, Van Dam D, Cuijt I, Joris G, De Deyn PP, Haass C, Van Broeckhoven C, Kumar-Singh S. Cellular ageing, increased mortality and FTLD-TDP-associated neuropathology in progranulin knockout mice. J Pathol. 2012;228:67–76. doi: 10.1002/path.4043. [DOI] [PubMed] [Google Scholar]
- Xu J, Xilouri M, Bruban J, Shioi J, Shao Z, Papazoglou I, Vekrellis K, Robakis NK. Extracellular progranulin protects cortical neurons from toxic insults by activating survival signaling. Neurobiol Aging. 2011;32:2326, e5–e16. doi: 10.1016/j.neurobiolaging.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Nathan C, Ding A. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010a;207:117–128. doi: 10.1084/jem.20091568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Dumont M, Banerjee R, Ma Y, Li H, Lin MT, Beal MF, Nathan C, Thomas B, Ding A. Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASEB J. 2010b;24:4639–4647. doi: 10.1096/fj.10-161471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanocco-Marani T, Bateman A, Romano G, Valentinis B, He ZH, Baserga R. Biological activities and signaling pathways of the granulin/epithelin precursor. Cancer Res. 1999;59:5331–5340. [PubMed] [Google Scholar]
- Zheng Y, Brady OA, Meng PS, Mao Y, Hu F. C-terminus of progranulin interacts with the beta-propeller region of sortilin to regulate progranulin trafficking. PLoS One. 2011;6:e21023. doi: 10.1371/journal.pone.0021023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, Lacomis L, Erdjument-Bromage H, Tempst P, Wright CD, Ding A. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 2002;111:867–878. doi: 10.1016/S0092-8674(02)01141-8. [DOI] [PubMed] [Google Scholar]