

Abstract
The frequency of biaryl substructures in a database of approved oral drugs has been analyzed. This led to designation of 20 prototypical biaryls plus 10 arylpyridinones for parameterization in the OPLS all-atom force fields. Bond stretching, angle-bending, and torsional parameters were developed to reproduce the MP2 geometries and torsional energy profiles. The transferability of the new parameters was tested through their application to three additional biaryls. The torsional energetics for the 33 biaryl molecules are analyzed and factors leading to preferences for planar and non-planar geometries are identified. For liquid biphenyl, the computed density and heat of vaporization at the boiling point (255 C) are also reported.
1. INTRODUCTION
Biaryls, compounds with two aryl rings connected via one single bond, are chemical fragments that are often found in drugs and drug-like compounds. Many top selling drugs contain biaryl fragments, including Lipitor, Crestor, Diovan, and Celebrex. Brameld et al. analyzed the Prous database and found that biphenyl and 5-phenyl-1H-tetrazole were the third and seventh most frequently observed cores in compounds entering phase I clinical trials.1 Our laboratory has also had high interest in biaryls since they arise regularly in our inhibitor design efforts, e.g., for fibroblast growth factor receptor 1 (FGFR1) kinase,2 the von Hippel-Lindau E3 ubiquitin ligase/HIF-1α interaction,3 and Plasmodium falciparum macrophage migration inhibitory factor (MIF).4 Biaryl cores also emerged in the development of agonists of human MIF-CD74 binding.5 The effectiveness of synthetic methods to produce biaryls, such as the Suzuki6 and Ullmann couplings,7 facilitates inclusion of biaryl substructures in small molecule screening libraries, which over time can be expected to lead to more biaryls being reported as core structures in biologically active agents.
The molecular geometry of the prototypical case, biphenyl, has been studied extensively, both theoretically8,9 and via electron diffraction.10 Theory and experiment concur on the location of the energy minimum; however, agreement on the torsional energy barriers has been challenging.8 Geometries and rotational barriers for some substituted biphenyls have also been studied through electron diffraction,11–14 NMR,15 and computation.16 For biaryls incorporating heterocycles, little experimental data on torsional energetics is available, while there have been some computational studies, e.g., for 2,2′-bipyridine,17 2-phenylpyridine,17 and 4,4′-bipyridine.18 Though X-ray single crystal data can provide comparisons with computed molecular geometries, crystal packing effects can be considerable.19 This causes dihedral distributions of biaryls, such as substituted biphenyls, to be shifted towards planar structures in comparison to the gas phase.
Accurate description of biaryl geometries and torsional energetics is important for molecular modeling since it is frequently employed in the identification and optimization of biologically active compounds.20 For calculations on biomolecular systems including protein-ligand complexes, the modeling typically uses molecular mechanics force fields, which require parameterization to reproduce observed structures and properties for both isolated molecules and condensed-phase systems.21 Clearly the preferred planarity or non-planarity of a biaryl system and the associated torsional barriers are of great importance in gauging the binding affinity of a biaryl-containing ligand. The present study was undertaken to improve the accuracy of the OPLS force fields22 for treating molecules containing such substructures. The parameterization for the structures and torsional energetics has been based on results of ab initio calculations for more than 30 biaryl systems. The results are also of value as a quick reference for molecular designers and for the improvement of other force fields.
2. COMPUTATIONAL DETAILS
Torsional energy profiles were calculated using Gaussian09.23 Geometries of the compounds were optimized at different fixed aryl-aryl dihedral angles, using dihedral scans at the MP2(full)/6-311G(d,p) level. Single point energies were subsequently calculated for all geometries at the MP2(full)/aug-cc-pVTZ level. For the negatively charged tetrazole, the dihedral scan was performed at the MP2(full)/6-311G++(d,p) level. Calculations at similar levels have been found to yield highly accurate results for conformational energetics.24
Force fields use atom types to designate parameters. For OPLS, two letter codes are employed such as CT, CA, and HA for a saturated carbon, aromatic carbon in a six-membered ring, and a hydrogen attached to an aromatic carbon. Then, bond stretching, angle bending, and torsion parameters are represented by pairs, triples, and quadruples of atom types such as CA-CA, CA-CA-HA, and CA-CA-CT-CT. The relevant OPLS atom types used here are illustrated by the ring templates in Figure 1; the atom types are assigned automatically by the BOSS program,25 which was used for all force field calculations. CM and C= are the atom types for alkene carbons and C2 and C3 in 1,3-dienes, while C! designates the ipso carbon for biphenyl-like junctions. New atom types are only introduced when necessary to avoid errors in structures or conformational energetics. For example, if CA were used for the ipso carbons in biphenyl, the inter-ring bond length (CA-CA) would be the same as for a C-C bond in benzene; there also has to be a significant difference between the CA-CA-CA-CA and CA-C!-C!-CA torsional parameters.
Figure 1.

OPLS atom types for prototypical aryl rings.
The OPLS-AA atom types and parameters for azines, azoles, oxazoles, and furan were previously reported.26 The possible carbon symbols are CW, CS, CR, CU, and CV (Figure 1). When a 6-membered arene is attached to a 5-membered heterocycle, a special atom type like C! is not needed at the junction for the 5-membered ring, e.g., occurrence of a C!-CW or C!-CS is unambiguous in referring to the inter-ring connection. Only two modifications to the former atom types were introduced here. An unsubstituted pyrrole nitrogen remains an NA, while one with an aryl or saturated substituent is now NX or NS, and C2 in thiophenes is now a CP instead of the CW of a furan or pyrrole. Satisfactory structures and torsional profiles could not be obtained without these additions. The new bond stretching and angle bending constants typically only required small adjustments to previously reported values to yield improved agreement with the MP2 results. The default Lennard-Jones parameters for NX and NS (3.25Å, 0.17 kcal/mol) and CP (3.55, 0.07) are unchanged from those for NA and an aromatic carbon, e.g., CA, CS, CV, or CW. The focus was primarily on deriving torsion parameters for the biaryl junctions. For the intra-ring torsional energetics, standard OPLS-AA dihedral parameters are used to promote planarity.22,26
There are two OPLS all-atom force fields, OPLS-AA and OPLS/CM1A.22b The latter is the general force field for arbitrary organic molecules in which the partial atomic charges are taken from CM1A calculations.27 The CM1A charges are scaled by a factor of 1.14 for neutral systems in order to maximize the agreement between computed and experimental free energies of hydration for small molecules.22b All other parameters (bonded and non-bonded) are taken to be the same for OPLS-AA and OPLS/CM1A. Scaled CM1A charges were used for the present molecules to be consistent with the normal practice for representation of designed molecules in ligand optimization projects.20 The partial charges are computed with a BOSS script, which executes several cycles of energy minimization and CM1A calculations until the charges converge for the lowest-energy conformer. OPLS/CM1A torsional energy profiles are computed with the ‘dihedral angle driving’ feature in BOSS. The torsional energy consists of the Fourier series sum over all dihedral angles in eq 1. The present fitting focused on the Fourier coefficients for the inter-ring dihedral angles. The target for them is to make up the difference between the computed MP2 energy profile and the OPLS/CM1A one with these Fourier coefficients set to zero. The optimal choices were determined by ordinary least-squares
| (1) |
regression analyses. Resulting Fourier coefficients below ca. 0.05 kcal/mol in magnitude were set to zero.
For symmetrical cases like biphenyl, only one dihedral parameter had to be fitted, e.g., CA-C!-C!-CA. For unsymmetrical case like 3-phenylfuran, the two dihedrals CA-C!-CS-CS and CA-C!-CS-CW were treated as equal and the same parameters were assigned for both. The analogous CA-C!-CS-CP of thiophene was also assigned to be the same as CA-C!-CS-CS and CA-C!-CS-CW. For 2-phenyl substituted furans, thiophenes, and pyrroles the CA-C!-CW-CS parameters were also taken to be the same as CA-C!-CS-CS. In this way, only CA-C!-CW-OS, CA-C!-CW-S, and CA-C!-CW-NA had to be optimized. When such equivalences were invoked, a compromise was sought that would yield a good fit for all affected molecules. The biaryl torsions involving the imidazole C2-NH atoms (CR-NA) in aryl-imidazoles were assigned the CW-NA parameter from the corresponding aryl-pyrrole. For example, the parameters for the CA-C!-CW-NA were taken to be the same as for CA-C!-CR-NA. Thus, only the torsions involving CR-NB had to be parameterized for aryl-imidazoles. For pyridinone connected at C! to the 3-position of furan, individual parameters had to be assigned to the NA-C!-CS-CS and NA-C!-CS-CW dihedrals, while it was possible to use the NA-C!-CS-CS parameter for the NA-C!-CW-CS torsion. Parameters for the dihedrals involving the C= and C! atoms of pyridinone were assigned the same values as for the corresponding phenyl fragments, i.e. the C=-C! string is treated to be equivalent to CA-C! for dihedral parameters. In this way, parameters for a maximum of two dihedral angles had to be fitted for the substituted pyridinones.
3. RESULTS AND DISCUSSION
To assess the diversity of biaryl structures in pharmacologically relevant molecules, we analyzed a database of oral drugs. Specifically, the database was assembled by adding the ca. 200 FDA-approved entries for 1998–2012 to the compilation of ca. 1800 oral drugs for 1937–1997 from Proudfoot.28 The new entries came mostly from Annual Reports of Medicinal Chemistry, which includes specifications on administration.29 Visual inspection of the database and substructure search identified 100 drugs (5%) containing at least one biaryl fragment. As summarized in Figure 2, the four most frequent biaryl fragments are biphenyl, 5-phenyl-1H-tetrazole, 1-phenyl-1H-pyrazole, and 3-phenylisoxazole. In all, 64 unique biaryl fragments were found in the database. Biaryls consisting of two six-membered rings (6:6) or one six-membered ring and one five-membered ring (6:5) were the most frequent. 26 biaryls included fused bicyclic ring systems, though no specific one occurred more than twice. Curiously, no 5:5 biaryls were found among the oral drugs, which may reflect historical synthetic limitations. Notable exceptions are arotinolol, which is not FDA-approved, and bleomycins, which are not oral drugs. They contain 4-(thiopen-2-yl)thiazole and 2,4′-bithiazole substructures.
Figure 2.

The 64 biaryl fragments found in the database of oral drugs. The number of occurrences of each fragment is given below the structure. Each fragment got a maximum of one count per drug.
Based on these observations, 20 well represented 6:6 and 6:5 biaryls were selected for parameterization (1–20 in Figure 3). In addition, as part of an ongoing effort to develop improved inhibitors of the von Hippel-Lindau E3 ubiquitin ligase/HIF-1α interaction, we have been exploring 6-arylpyridinones as alternatives to previous biaryl cores.3 Arylpyrimidinone substructures have also been featured in some of our FGFR1 kinase inhibitors.2 Furthermore, 6-aryl-pyridinones have been reported as orally active inhibitors of the EP3 receptor,30 and as core structures of different non-peptidic inhibitors of human leukocyte elastase31 and interleukin-1β converting enzyme.32 Thus, ten 6-aryl-pyridinones were also selected for parameterization (21–30 in Figure 3).
Figure 3.


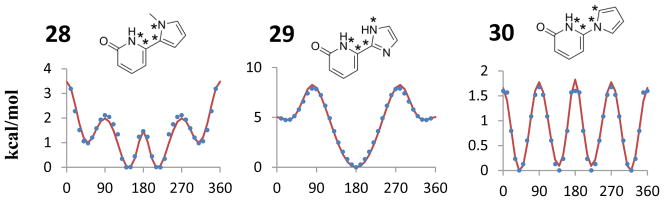
Structures and torsional energy profiles for the indicated dihedral angles of biaryls 1–30. Blue dots represent the MP2 single point energies and the red solid lines are the OPLS/CM1A energy profiles obtained using the new parameters. The four starred atoms indicate the dihedral angle for which the relative energies are plotted.
As noted above, three new atom types were added for biaryls (Figure 1). Atom type NX was specifically needed to distinguish between an internal pyridinone C!-NA bond (1.38 Å) and an inter-ring bond between a pyridinone C! atom and an NA atom of an N-substituted heterocycle (1.44 Å, Figure 4). Nitrogen atoms in rings with substituents other than a hydrogen atom or an aromatic ring are assigned atom type NS. This provided beneficial flexibility for torsional parameterization of the biaryls containing N-methylpyrrole or 1H-pyrrole. Finally, atom type CP was needed to distinguish C2 and C5 of thiophenes from the corresponding atoms in pyrroles and furans. This enabled the addition of new bond stretching and angle bending parameters, specific for thiophenes, which significantly reduced former high internal strain energies. Overall, the new bond stretching (Table S1) and angle bending parameters (Table S2) were set to reproduce average values from the MP2 optimized geometries of the relevant biaryl fragments (see Supporting Information). A few existing parameters were also modified to reduce angle-bending strain, such as the NA-CR-NB angle of imidazole and histidines. The effect on geometries is not large, e. g., the N-C-N angle in imidazole is reduced from the former 113° to 110°.
Figure 4.
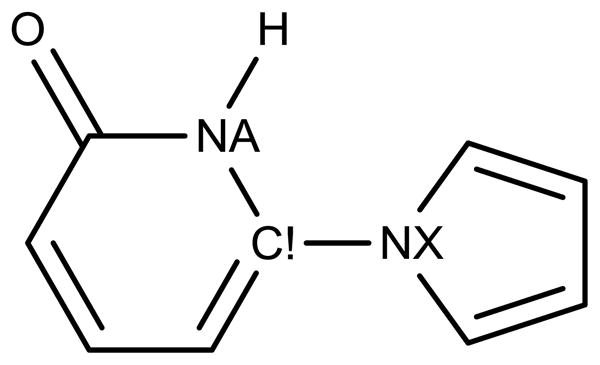
Atom type NX was added to allow a longer bond between pyridinones and aryls (1.44 Å), while keeping a shorter bond for the internal pyridinone C!-NA bond (1.38 Å).
Torsional Energetics
The results for biphenyl (1) are given in Figure 3 and Table 1. The optimal dihedral angle from the MP2 calculations is 43.4°, which agrees well with the experimental value and results from other high-level calculations.8 The present results yield a slightly higher energy barrier at 90° (2.2 kcal/mol) than at 0° (2.1 kcal/mol) in accord with the observed pattern. There has been some controversy on this point, though the highest level results indicate barriers at 0 K of 1.9 and 2.0 kcal/mol, respectively.9 Regression fitting of the Fourier coefficients in eq 1 gave V2 = 1.97 for CA-C!-C!-CA (Table S3) and an essentially perfect fit between the MP2 and OPLS/CM1A energy profiles (Figure 3). Zero-point energy corrections have not been made for the present energy profiles. For biphenyl, the corrections are in the range of ±0.1 kcal/mol.9
Table 1.
Experimental and Calculated Optimal Dihedral Angle and Rotational Barriers (kcal/mol) for Biphenyl.
Ultimately, 30 new OPLS dihedral parameters were developed for the 30 biaryls (Table S3) yielding the energy curves illustrated in red for 1–30 in Figure 3. The occurrence of small coefficients was minimized, especially for V4 terms. Such coefficients were nullified if there was little or no effect on the torsional profiles. For instance, a perfect fit between the OPLS/CM1A relative energies and the MP2 results could be obtained for 25 with a small V4 term for NA-C!-CW-OS; however, the fit in Figure 3 with no V4 component was judged to be more than adequate. For most of the cases, it was only necessary to introduce V2 terms. A few of the biaryls with more complicated torsional energy surfaces and non-planar minima, such as for 8 and 12, required a V4 term. However, many of the aryl-pyridinones (21–30) are comparatively asymmetric and yielded complex torsional energy profiles that often required V1, V2, and V3 terms, e.g., for 24, 26, and 28. The complexity is such that it is clear that force fields are not going to represent such systems well in the absence of detailed parameterization.
A literature search was also done for single molecule X-ray data of molecules containing any of the fragments 1–30 that were unsubstituted in the ortho positions. The observed biaryl torsion angles are compared with the results of energy minimizations using the force field parameters in Tables S1–S4. There is good agreement between the computed and crystallographic results, especially since the gas and solid phases are being compared. Three systems (2, 9, 11) prefer to be nearly planar, while the other three (10, 12, 21) are twisted by about 30°. Though patterns are discussed more below, it can be noted that the twisted ones have either the full complement of four ortho hydrogens or, for 12, clear electrostatic repulsion in the planar form.
Testing for Additional Cases
There are many biaryl fragments in the oral drugs database that were not explicitly parameterized. The transferability of some of the parameters was tested by calculating OPLS/CM1A torsional profiles for three additional biaryls, 31–33, in Figure 5. 5-Phenyl-1H-tetrazole (33) is the second most frequent biaryl fragment with seven occurrences in the database of oral drugs (Figure 2). 2-Phenylpyridine (31) was found twice and there was one appearance of 6-phenyl-1,2,4-triazine (32). The requisite torsions for 2-phenylpyridine (31) and 6-phenyl-1,2,4-triazine (32) have the CA-C!-C!-CA and CA-C!-C!-NC patterns derived for biaryls 1 and 2, while 5-phenyltetrazole (33) uses the CA-C!-CR-NB parameters derived from biaryl 9. 5-Phenyl-1H-tetrazole has a pKa of about 4.5 and can be expected to be deprotonated under physiological conditions,41 so the deprotonated tetrazole 33 was of prime interest. MP2 torsional energy profiles were computed in the same manner as above for 31–33. As shown in Figure 5, there is good accord between the MP2 and OPLS/CM1A results for the positions of the energy minima and barriers and for the shapes of the barriers. The barrier heights are underestimated with OPLS/CM1A by ca. 0.5 – 0.8 kcal/mol. Thus, no qualitative errors are apparent, though the transitions between conformers would be somewhat accelerated in these cases.
Figure 5.
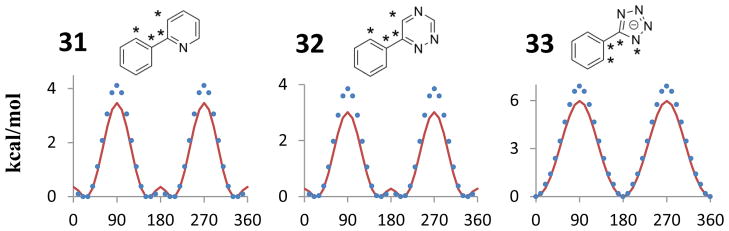
Transferability of some of the dihedral parameters was evaluated by comparing computed OPLS/CM1A energies (red line) with MP2 results (blue circles) for three additional biaryl fragments found in the oral drugs database.
In developing the OPLS force fields, testing has included performance in simulations of pure organic liquids including benzene and monocyclic heterocycles.22,26 The present biaryl molecules are all solids at room temperature, and there is a dearth of experimental data on their liquids. For biphenyl, the melting point is 71 C, the boiling point is 255 C, and the heat of vaporization at the boiling point was reported in 1929 as 11.47 ± 0.05 kcal/mol.42 We did run Monte Carlo simulations of liquid biphenyl at 255 C and 1 atm using standard protocols,22 including a periodic cube with 267 monomers and 12-Å non-bonded cutoffs. The resultant heat of vaporization with the OPLS-AA force field is 11.53 ± 0.07 kcal/mol and the computed density is 0.8081 ± 0.0022 g cm−3. With OPLS/CM1A the corresponding values are 12.91 ± 0.09 kcal/mol and 0.8389 ± 0.0022 g cm−3. The only difference between OPLS-AA and OPLS/CM1A is the partial atomic charges. For the CH units in biphenyl the charges are ±0.115 e with OPLS-AA and ca. ±0.15 e with OPLS/CM1A, which is consistent with the pattern in the computed heats of vaporization and densities.
The importance of twist
Detailed knowledge of molecular geometry is essential for medicinal chemists. During the course of a lead identification or lead optimization campaign a chemist might be considering the effect of replacing one aryl ring in a biaryl fragment with another. In addition to changes in molecular properties, change in the biaryl core can affect the molecular geometry with concomitant effects on intermolecular interactions. For instance, an N-phenylpyrrole core (10), which has an optimal dihedral angle of 40°, can convert to favor planar structures upon replacement of the pyrrole with an imidazole (9) or pyrazole (11) ring. For less symmetric cases such as pyridinones substituted with an asymmetric aryl such as 2-furanyl, it is also important to have knowledge of the pseudo E/Z preference. This is apparent for 25 in Figure 3, where the preference is seen to be reversed for the 2-pyrrolyl analog 27. Below, biaryl fragments are classified into planar or non-planar categories and the preferences are rationalized. Key factors determining the preference for planarity or non-planarity of biaryls are steric clashes between substituents attached to ortho atoms and electrostatic interactions between the ortho groups.
Non-planar biaryls
All biaryls investigated (1 – 33) with two hydrogen or other atoms attached to the ortho positions on both rings are non-planar. This can be attributed to steric clashes between the ortho groups that are relieved upon rotation to non-planar geometries. The preference is stronger for 6:6 systems (1, 21) than for 6:5 ones owing to the increased bond angles to the 5-membered ring. The barrier at 0° is very small for most 6:5 cases with four ortho hydrogen atoms, i.e., 3, 4, 7, 23, 24, 27. Of course, the energy barrier at 0° can be much increased by adding bulkier substituents to the ortho positions, as in 8 vs. 7 and 28 vs. 27.
Non-planarity is also induced when there is significant electrostatic repulsion between heteroatoms in the 2- and 2′-positions. 2,2′-bipyrimidine (12) is an example that lacks ortho steric clashes, but prefers to be twisted to diminish the N-N electrostatic repulsions. Related 6:5 systems with the 2-2′ electrostatic interactions all repulsive are also expected to favor non-planar geometries, e.g., 2-(pyrimidin-2-yl)oxazole. Electrostatic effects are also overlaid on the steric preference for a non-planar geometry for 27; the pseudo-E conformation with the two NH groups anti is preferred by ca. 1.5 kcal/mol over pseudo-Z.
Planar biaryls
Biaryls favor planar geometries when there is one or more unsubstituted heteroatom in the 2 or 2′ positions and there is at least one favorable electrostatic interaction. For the 6:6 systems, 2-phenylpyrimidine (2) is strongly planar, while any barrier at 0° for 2-phenylpyridine (31) or the triazene 32 is miniscule. 6:5 biaryls that lack one or more hydrogens at ortho positions are normally planar and those with four ortho hydrogens have very small barriers to planarity. Thus, 2-phenylfuran (5), 2-phenylimidazole (9), 1-phenylpyrazole (11), 13 – 17, 19, 20, 25, 29, and 33 are all favor planar geometries. The non-planarity of 26 is a bit surprising; it appears that sulfur is more dominated by steric effects than possible electrostatic ones. For 29, the planar conformer with the NH groups anti is also expected to be strongly favored to optimize the 2-2′ electrostatic interactions. The greatest preferences for planarity and largest rotational barriers, ca. 10 kcal/mol, occur when there is a lack of ortho hydrogen clashes and strong electrostatic reinforcement for planar structures, e.g., for 17, 20, and 22.
4. CONCLUSIONS
Biaryl substructures are found in many drugs and drug-like compounds. In our analysis, 100 oral drugs out of ca. 2000 were found to contain at least one of 64 unique biaryl fragments. The torsional energetics of 20 prototypical biaryls and 10 additional arylpyridinones were designated for investigation with MP2 calculations to provide a basis for parameterization of the OPLS all-atom force fields. The torsional parameters developed from this set are representative of the majority of the biaryls in the oral drugs database. In addition to the optimization of the torsional parameters, new bond stretching and angle bending parameters were also created to reproduce the MP2-optimized geometries. Using the new parameters, the torsional energy profiles from OPLS/CM1A force field calculations are in close agreement with the MP2 profiles (Figure 3). It was also shown that the new parameters were capable of reproducing well the MP2 torsional energy profiles for three additional biaryls that were not in the original set of 30. General observations on the structural features that lead to preferences for planar and non-planar geometries of biaryls were also identified. This information should be of value to the molecular design community.
Supplementary Material
Table 2.
X-Ray Data for Biaryl Dihedral Angles in Small Molecules Containing any of the Fragments 1–30a
Acknowledgments
Gratitude is expressed to the National Institutes of Health (GM32136) for support of this work and to IF’s Foundation for Pharmaceutical Research for a postdoctoral fellowship (MKD). Dr. John R. Proudfoot kindly provided a listing of his oral drugs database.28
Footnotes
Supporting Information. Tables of all force field parameters used and developed in this work, and structure files as Z-matrices that include the non-bonded parameters (atomic charges and Lennard-Jones σs and εs) for the 33 biaryls. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Brameld KA, Kuhn B, Reuter DC, Stahl M. Small molecule conformational preferences derived from crystal structure data. A medicinal chemistry focused analysis. J Chem Inf Model. 2008;48:1–24. doi: 10.1021/ci7002494. [DOI] [PubMed] [Google Scholar]
- 2.Ekkati AR, Mandiyan V, Ravindranathan KP, Bae JH, Schlessinger J, Jorgensen WL. Aryl extensions of thienopyrimidinones as fibroblast growth factor receptor 1 kinase inhibitors. Tetrahedron Lett. 2011;52:2228–2231. doi: 10.1016/j.tetlet.2010.12.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buckley DL, Molle IV, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A, Crews CM. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J Am Chem Soc. 2012;134:4465–4468. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dahlgren MK, Garcia AB, Hare AA, Tirado-Rives J, Leng L, Bucala R, Jorgensen WL. Virtual screening and optimization yield low-nanomolar inhibitors of the tautomerase activity of Plasmodium falciparum macrophage inhibitory factor. J Med Chem. 2012;55:10148–10159. doi: 10.1021/jm301269s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jorgensen WL, Gandavadi S, Du X, Hare AA, Trofimov A, Leng L, Bucala R. Receptor agonists of macrophage migration inhibitory factor. Bioorg Med Chem Lett. 2010;20:7033–7036. doi: 10.1016/j.bmcl.2010.09.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miyaura N, Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem Rev. 1995;95:2457–2483. [Google Scholar]
- 7.Fanta PE. The Ullman synthesis of biaryls. Synthesis. 1974;1:9–21. [Google Scholar]
- 8.Sancho-García JC, Cornil J. Anchoring the torsional potential of biphenyl at the ab initio level: the role of basis set versus correlation effects. J Chem Theory Comput. 2005;1:581–589. doi: 10.1021/ct0500242. [DOI] [PubMed] [Google Scholar]
- 9.Johansson MP, Olsen J. Torsional barriers equilibrium angle of biphenyl: reconciling theory with experiment. J Chem Theory Comput. 2008;4:1460–1471. doi: 10.1021/ct800182e. [DOI] [PubMed] [Google Scholar]
- 10.Almenningen A, Bastiansen O, Fernholt L, Cyvin BN, Cyvin SJ, Samdal S. Structure and barrier of internal rotation of biphenyl derivatives in the gaseous state. Part 1. The molecular structure and normal coordinate analysis of normal biphenyl and perdeuterated biphenyl. J Mol Struct. 1985;128:59–76. [Google Scholar]
- 11.Almenningen A, Bastiansen O, Fernholt L, Gundersen S, Kloster-Jensen E, Cyvin BN, Cyvin SJ, Samdal S, Skancke A. Structure and barrier to internal rotation of biphenyl derivatives in the gaseous state. Part 2. Structure of 3,3′-dibromo-,3,5,4′-tribromo- and 3,5,3′,5′-tetrabromobiphenyl. J Mol Struct. 1985;128:77–93. [Google Scholar]
- 12.Almenningen A, Bastlansen O, Gundersen S, Samdal S, Skancke A. Structure and barrier of internal rotation of biphenyl derivatives in the gaseous state. Part 3. Structure of 4-fluoro-, 4,4′-difluoro-, 4-chloro- and 4,4′-dichlorobiphenyl. J Mol Struct. 1985;128:95–114. [Google Scholar]
- 13.Bastiansen O, Samdal S. Structure and barrier of internal rotation of biphenyl derivatives in the gaseous state. Part 4. Barrier of internal rotation in biphenyl, perdeuterated biphenyl and seven non-ortho-substituted halogen derivatives. J Mol Struct. 1985;128:115–125. [Google Scholar]
- 14.Bastiansen O, Gundersen S, Samdal S. Structure and barrier to internal rotation of biphenyl derivatives in the gaseous state. Part 5. A reinvestigation of the molecular structure and internal rotation of perfluorobiphenyl. Acta Chem Scand. 1989;43:6–10. [Google Scholar]
- 15.Casarini D, Lunazzi L, Mazzanti A. Recent advances in stereodynamics and conformational analysis by dynamic NMR and theoretical calculations. Eur J Org Chem. 2010:2035–2056. [Google Scholar]
- 16.Grein F. Twist angles and rotational energy barriers of biphenyl and substituted biphenyls. J Phys Chem A. 2002;106:3823–3827. [Google Scholar]
- 17.Göller A, Grummt U-W. Torsional barriers in biphenyl, 2,2′-bipyridine and 2-phenylpyridine. Chem Phys Lett. 2000;321:399–405. [Google Scholar]
- 18.Pérez-Jiménez ÁJ, Sancho-García JC, Pérez-Jordá JM. Torsional potential of 4, 4′-bipyridine: Ab initio analysis of dispersion and vibrational effects. J Chem Phys. 2005;123:134309. doi: 10.1063/1.2043107. [DOI] [PubMed] [Google Scholar]
- 19.Brock CP, Minton RP. Systematic effects of crystal-packing forces: biphenyl fragments with H atoms in all four ortho positions. J Am Chem Soc. 1989;111:4586–4593. [Google Scholar]
- 20.(a) Jorgensen WL. The many roles of computation in drug discovery. Science. 2004;303:1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]; (b) Jorgensen WL. Acc Chem Res. 2009;42:724–733. doi: 10.1021/ar800236t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.For a review see: Ponder JW, Case DA. Force Fields for Protein Simulations. Adv Prot Chem. 2003;66:27–85. doi: 10.1016/s0065-3233(03)66002-x.
- 22.(a) Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]; (b) Jorgensen WL, Tirado-Rives J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc Nat Acad Sci USA. 2005;102:6665–6670. doi: 10.1073/pnas.0408037102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision A02. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 24.Murphy RB, Pollard WT, Friesner RA. Pseudospectral localized generalized Møller-Plesset methods with a generalized valence bond reference wave function: Theory and calculation of conformational energies. J Chem Phys. 1997;106:5073–5084. [Google Scholar]
- 25.Jorgensen WL, Tirado-Rives J. Molecular modeling of organic and biomolecular systems using BOSS and MCPRO. J Comput Chem. 2005;26:1689–1700. doi: 10.1002/jcc.20297. [DOI] [PubMed] [Google Scholar]
- 26.Jorgensen WL, McDonald NA. Development of an all-atom force field for heterocycles. Properties of liquid pyridine and diazenes. J. Mol. Struct. (Theochem) 1998; 424: 145–155. (b) McDonald, N. A., Jorgensen, W. L. Development of an all-atom force field for heterocycles. Properties of liquid pyrrole, furan, diazoles, and oxazoles. J Phys Chem B. 1998;102:8049–8059. [Google Scholar]
- 27.Storer JW, Giesen DJ, Cramer CJ, Truhlar DG. Class IV charge models: A new semiempirical approach in quantum chemistry. J Comput Aid Mol Des. 1995;9:87–110. doi: 10.1007/BF00117280. [DOI] [PubMed] [Google Scholar]
- 28.Proudfoot JR. The evolution of synthetic oral drug properties. Bioorg Med Chem Lett. 2005;15:1087–1090. doi: 10.1016/j.bmcl.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 29.Annual Reports in Medicinal Chemistry. 34–47. American Chemical Society; Washington, D. C: 1999–2012. [Google Scholar]
- 30.Jin J, Morales-Ramos A, Eidam P, Mecom J, Li Y, Brooks C, Hilfiker M, Zhang D, Wang N, Shi D, Tseng P, Wheless K, Budzik B, Evans K, Jaworksi J, Jugus J, Leon L, Wu C, Pullen M, Karamshi B, Rao P, Ward E, Laping N, Evans C, Leach C, Holt D, Su X, Morrow D, Fries H, Thorneloe K, Edwards R. Novel 3-oxazolidinedione-6-aryl-pyridinones as potent, selective, and orally active EP3 receptor antagonists. ACS Med Chem Lett. 2010;1:316–320. doi: 10.1021/ml100077x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Damewood JR, Edwards PD, Feeney S, Gomes BC, Steelman GB, Tuthill PA, Williams JC, Warner P, Woolson SA, Wolanin DJ, Veale CA. Nonpeptidic inhibitors of human Leukocyte elastase. 2. Design, synthesis, and in vitro activity of a series of 3-amino-6-arylopyridin-2-one trifluoromethyl ketones. J Med Chem. 1994;37:3303–3312. doi: 10.1021/jm00046a015. [DOI] [PubMed] [Google Scholar]
- 32.Golec JMC, Mullican MA, Wilson KP, Kay DP, Jones SD, Murdoch R, Bemis GW, Raybuck SA, Luong Y, Livingston DJ. Structure-based design of non-peptidic pyridone aldehydes as inhibitors of Interleukin-1β converting enzyme. Bioorg Med Chem Lett. 1997;7:2181–2186. [Google Scholar]
- 33.Cieplik J, Pluta J, Bryndal I, Lis T. N-(2-fluorophenyl)-5-[(4-methoxy-phenyl)aminomethyl]-6-methyl-2-phenyl-pyrimidin-4-amine. Acta Crystallogr, Sect E: Struct Rep Online. 2011;E67:o3162. doi: 10.1107/S1600536811045028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barforoush MM, Naderi S, Ghanbarpour AR, Tehrani AA, Khavasi HR. 2-Phenyl-1H-imidazole. Acta Crystallogr Sect E: Struct Rep Online. 2011;E67:o3248. doi: 10.1107/S160053681104699X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thallapally PK, Chakraborty K, Katz AK, Carrell HL, Kotha S, Desiraju GR. Matching of molecular and supramolecular symmetry. An exercise in crystal engineering. Cryst Eng Comm. 2001;3:134–136. [Google Scholar]
- 36.Isloor AM, Malladi S, Gerber T, Brecht B, Betz R. (2E)-1-(2, 4-dichlorophenyl)-3-[3-(4-nitrophenyl)-1-phenyl-1H-pyrazol-4-yl]-prop-2-en-1-one. Acta Crystallogr Sect E: Struct Rep Online. 2012;E68:o616–o617. doi: 10.1107/S1600536812003960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asiri AM, Faidallah HM, Sobahi TR, Ng SW, Tiekink ERT. 1-Phenyl-1H-pyrazole-4-carbaldehyde. Acta Crystallogr Sect E: Struct Rep Online. 2012;E68:o1088. doi: 10.1107/S1600536812010896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamatchi P, Jagadeesan G, Pramesh M, Perumal PT, Aravindhan S. Ethyl 2-benzyl-3-[3-(4-chlorophenyl)-1-phenyl-1H-pyrazol-4-yl]-4,6-dioxo-5-phenyloctahydropyrrolo[3,4-c]pyrrole-1-carboxylate. Acta Crystallogr Sect E: Struct Rep Online. 2012;E68:o552. doi: 10.1107/S1600536812002450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ren T, Zhang Z, Zhong C, Yang Z, Shi Z. 4, 6-Dichloro-5-(2-methoxyphenoxy)-2, 2′-bipyrimidine. Acta Crystallogr Sect E: Struct Rep Online. 2011;E67:o1334. doi: 10.1107/S160053681101419X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chopra D, Mohan TP, Vishalakshi B, Row TNG. 4-(4-Fluoro-3-phenoxyphenyl)- 6-(4-fluorophenyl)-2-oxo-1,2-dihydro-pyridine-3-carbonitrile and the 6-(4-methylphenyl)- analogue. Acta Crystallogr, Sect C: Cryst Struct Commun. 2006;C62:o540–o543. doi: 10.1107/S010827010602378X. [DOI] [PubMed] [Google Scholar]
- 41.Herr RJ. 5-Substituted-1H-tetrazoles as carboxylic acid isosteres: Medicinal chemistry and synthetic methods. Bioorg Med Chem. 2002;10:3379–3393. doi: 10.1016/s0968-0896(02)00239-0. [DOI] [PubMed] [Google Scholar]
- 42.Chipman J, Peltier SB. Vapor Pressure and Heat of Vaporization of Diphenyl. Indust Engin Chem. 1929;21:1106–1108. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.