

Abstract
The anthraquinones emodin and aloe-emodin are abundant in rhubarb. Several lines of evidence indicate that emodin and aloe-emodin have estrogenic activity as phytoestrogens. However, their effects on estrogen receptor α (ERα) activation and breast cancer cell growth remain controversial. The goal of this study is to investigate the effects and molecular mechanisms of emodin and aloe-emodin on breast cancer cell proliferation. Our results indicate that both emodin and aloe-emodin are capable of inhibiting breast cancer cell proliferation by downregulating ERα protein levels, thereby suppressing ERα transcriptional activation. Furthermore, aloe-emodin treatment led to the dissociation of heat shock protein 90 (HSP90) and ERα and increased ERα ubiquitination. Although emodin had similar effects to aloe-emodin, it was not capable of promoting HSP90/ERα dissociation and ERα ubiquitination. Protein fractionation results suggest that aloe-emodin tended to induce cytosolic ERα degradation. Although emodin might induce cytosolic ERα degradation, it primarily affected nuclear ERα distribution similar to the action of estrogen when protein degradation was blocked. In conclusion, our data demonstrate that emodin and aloe-emodin specifically suppress breast cancer cell proliferation by targeting ERα protein stability through distinct mechanisms. These findings suggest a possible application of anthraquinones in preventing or treating breast cancer in the future.
1. Introduction
Many phytochemicals derived from plants, including anthraquinone, have been reported to have anticancer potential. The anthraquinone derivatives emodin (1,3,8-trihydroxy-6-methylanthraquinone) and aloe-emodin (1,8-dihydroxy-3-hydroxyl-methylanthraquinone) are the main bioactive components of rhubarb (Rheum palmatum), which has been used in traditional Chinese medicine [1]. Aloe-emodin is also abundant in the leaves of the common plant Aloe vera [2]. Emodin has been widely investigated for its antibacterial [3], anti-inflammatory [4], and antiproliferative effects in several types of cancer [1]. Notably, emodin may downregulate androgen receptor (AR) and lead to the inhibition of prostate cancer cell growth [5], suggesting that anthraquinone derivatives might modulate steroid receptor activity. Several lines of evidence indicate that emodin and aloe-emodin have estrogenic activity and modulate breast cancer cell proliferation as phytoestrogen compounds [6, 7]. However, the pharmacological effects and molecular mechanisms of emodin and aloe-emodin in estrogen receptor α (ERα) modulation and breast cancer cell growth remain elusive.
Breast cancer is a common malignancy with high lethality in women. Because ERα activation plays an important role in the initiation, development, and progression of breast cancer, estrogen replacement therapy is the most common strategy to suppress breast cancer progression [8]. By mimicking the structure of estrogen, synthetic estrogen-like compounds are used to compete for the binding of endogenous estrogen with ERα and therefore inhibit ERα-dependent growth of breast cancer cells [9, 10]. However, synthetic estrogens have side effects that increase the risk of cancer development due to unselective estrogenic action [11]. Although the potency of natural phytoestrogens is generally lower than that of synthetic estrogens in terms of estrogenic action, natural phytoestrogens are relatively safer with fewer side effects [12]. Therefore, studies investigating the effects and molecular mechanisms of natural herbal medicines that contain phytoestrogens as potential treatments to breast cancer are of interest.
We found that the inhibition of proliferation by emodin and aloe-emodin was ERα-dependent in breast cancer cell lines. Importantly, aloe-emodin treatment promotes ERα protein degradation by repressing the association of ERα and heat shock protein 90 (HSP90). Moreover, the dissociated ERα is ubiquitinated and targeted for proteasome-dependent degradation in the cytosol. The findings for aloe-emodin are distinct from those for emodin. Based on the above observations, these two anthraquinones could potentially be used as specific phytoestrogens to treat breast cancer.
2. Materials and Methods
2.1. Cell Lines and Cell Culture
The human breast cancer cell lines MCF-7 and MDA-MB-453 were obtained from the Bioresource Collection and Research Center (BCRC), Food Industry Research and Development Institute, Taiwan. MCF-7 cells were grown in minimum essential medium (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (Gibco), 1.5 g/L NaHCO3, 0.1 mM nonessential amino acids (Gibco), 1 mM sodium pyruvate (Gibco), and 1% penicillin-streptomycin (Sigma, St. Louis, MO, USA). MDA-MB-453 cells were maintained in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum, 1.5 g/L NaHCO3, and 1% penicillin-streptomycin. All cells were incubated at 37°C in a humidified atmosphere with 5% CO2.
2.2. Cell Viability Assay
Cells were incubated for 24 hours after attachment. Cell numbers were calculated by direct counting of cells, excluding cells that stained positive for 0.2% trypan blue stain (Sigma, St. Louis, MO, USA) [13]. Cells were treated with different concentrations of aloe-emodin or emodin (ChromaDex, Irvine, CA, USA) for the indicated number of days, and then, the 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT, Sigma, St. Louis, MO, USA) assay was used to quantify cell proliferation. The MTT stock solution (5 mg/mL) was diluted to 0.5 mg/mL with complete culture medium, and 0.1 mL was added to each well. The yellow MTT was converted to blue formazan by living cells, a reaction that is dependent on mitochondrial enzyme activity. After using DMSO to dissolve the blue formazan, the absorbance of converted MTT could be measured at 570 nm λ [14].
2.3. Cell Fractionation and Western Blot Analysis
Cells were collected using a rubber scraper and homogenized with Na3VO4 diluted in PBS (1 : 100). After centrifugation, cells were resuspended with lysis buffer as previously described [15–19], and protease inhibitor cocktail (Roche Applied Science, Mannheim, Germany) was added, followed by incubation on ice for 45 minutes. The cell lysate was centrifuged, and the supernatant was collected as total protein extract. The protein extract was mixed with sample buffer and boiled for 10 minutes. Then, western blotting was performed as previously described [19, 20]. Briefly, SDS-polyacrylamide gel electrophoresis (SDS-PAGE) was performed, and proteins were transferred from the SDS-PAGE gel onto a polyvinylidene fluoride (PVDF) membrane. Primary antibodies were incubated with the membrane overnight, and horseradish peroxidase- (HRP-) conjugated secondary antibodies (Jackson ImmunoResearch Laboratory, West Grove, PA, USA) were applied. The ECL (western lighting chemiluminescence reagent plus, PerkinElmer Life Sciences, Shelton, CT, USA) reaction was performed, and the membranes were exposed to X-ray films to visualize protein staining (Fujifilm, Tokyo, Japan). Antibodies directed against the following proteins were used in this study: poly(ADP-ribose) polymerase (PARP, 06-557, Upstate Biotechnology, Lake Placid, NY, USA), α-tubulin (05-829, Upstate Biotechnology), ERα (sc-543 and sc-8005, Santa Cruz Biotechnology, Santa Cruz, CA, USA), cyclin D1 (sc-20044, Santa Cruz Biotechnology), HSP 90 α/β (sc-59577, Santa Cruz Biotechnology), ubiquitin (sc-8017, Santa Cruz Biotechnology), and β-actin (MAB1501, Millipore, Temecula, CA, USA). The quantification software used was MCID Image Analysis Evaluation.
2.4. Immunoprecipitation
Cell lysate was extracted in lysis buffer, and immunoprecipitation was performed as previously described [19]. Briefly, the beads/antibody precipitated complex was prepared by mixing Protein G Mag Sepharose Xtra beads (GE Healthcare, Waukesha, WI, USA) and specific antibody at 10 μg : 1 μg for 2 h at room temperature. Cell lysates were mixed with beads/antibody for 12 h at 4°C, and protein was isolated by precipitation under magnetic attraction with three rounds of PBST washes. The precipitated proteins were analyzed by western blotting after denaturation following dilution in sample buffer and boiling for 10 minutes.
2.5. Quantitative Real-Time PCR
Total RNA was extracted from cells using a Miniprep Purification Kit (Genemark, Taipei, Taiwan), and reverse transcription-PCR was performed using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) following the standard procedures recommended by the manufacturer. For reverse transcription, 2 μg of total RNA was used as the first-strand cDNA template for the subsequent amplification procedure. The following primers were used to amplify the cDNAs: ERα (5′-TGGAGATCTTCGACATGCTG-3′ and 5′-TCCAGAGACTTCAGGGTGCT-3′) [21] and β-actin (5′-TTGCCGACAGGATGCAGAA-3′ and 5′-GCCGATCCACACGGAGTACT-3′). cDNA and primers were mixed within FastStart Universal SYBR Green Master (Roche Applied Science) and measured using a real-time PCR instrument (Applied Biosystems). Data presented by Ct values were analyzed and adjusted relative to levels of the β-actin house-keeping gene.
2.6. Transfection and Reporter Assays
Cells were plated for at least 24 h and had reached 80% confluency prior to transfection. Expression plasmid was premixed within Lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. The liposome/plasmid complex was transfected into MCF-7 cells incubated with Opti-MEM (Gibco) for 6 h, and then culture medium was added for exogenous protein expression. The pCMV5-ERα expression plasmid was kindly provided by Professor Chih-Yang Huang, Graduate Institute of Basic Medical Science, China Medical University, Taichung, Taiwan. A reporter assay was performed to detected ERα transcriptional activity after cells were transfected with 3 × ERE-containing-promoter luciferase reporter gene (pGL2-TK-3 × ERE, gift from Dr. Chih-Yang Huang, China Medical University, Taichung, Taiwan) and internal control pSV-β-galactosidase expression plasmid (gift from Dr. Jeremy J. W. Chen, National Chuang Hsing University, Taichung, Taiwan). Reporter gene luciferase production was performed using the Dual-Light System (Applied Biosystems), and measurements were performed using a 1420 Multilabel Counter VICTOR3 instrument (PerkinElmer Life Sciences). The raw data were normalized to β-galactosidase activity to control for varying transfection efficiencies [19].
2.7. Statistics
All values are presented as the mean ± standard error of the mean (SEM). In all cases, Student's t-test was used to assess the results of cell proliferation and reporter assays. A difference between two means was considered statistically significant when P < 0.05.
3. Results
3.1. ERα Is Important for the Growth Inhibition Induced by Emodin and Aloe-Emodin
The effects of different concentrations (0, 6, 12.5, 25, 50, or 100 μM) of emodin and aloe-emodin on the growth of the ERα-positive breast cancer cell line MCF-7 were determined by cell number counting (0–6 days) and MTT assays (4 days). Emodin and aloe-emodin treatment led to dose-dependent suppression of MCF-7 growth (Figures 1(a) and 1(b)). Notably, 12.5 μM or higher concentrations of aloe-emodin had stronger effects on MCF-7 growth compared to the same dosage of emodin. MTT assays showed significant inhibition of MCF-7 proliferation in a dose-dependent manner following emodin treatment at concentrations of 25 to 100 μM (Figure 1(c)) and of aloe-emodin treatment at concentrations of 6 to 100 μM (Figure 1(d)). Whereas emodin was more effective against the ERα-negative breast cancer cell line MDA-MB-453 than against MCF-7 (Figure 1(e)), the inhibitory effects of aloe-emodin on the growth of MDA-MB-453 was moderate compared to the effects on MCF-7 cells (Figure 1(f)). In which, 25 μM of aloe-emodin was not able to affect MDA-MB-453 cell proliferation (Figure 1(f)), while 25 μM of emodin significantly reduced that cell proliferation (Figure 1(e)) implies that the effects of aloe-emodin might be associated with ERα and distinct to emodin. To further investigate whether ERα was involved in the inhibitory effects of the emodin and aloe-emodin treatments, the effects of emodin and aloe-emodin on the growth of MCF-7 with or without ERα overexpression were investigated. Following a 25 μM treatment with emodin (Figure 1(g)) or aloe-emodin (Figure 1(h)), the ERα-overexpressing cells were more sensitive to drug treatments compared to control cells. Similar results were also observed in another ERα-positive cell line, T47D (data not shown). These data indicate that ERα protein plays an important role in the emodin- and aloe-emodin-induced suppression of breast cancer cell proliferation, although the potency of these two compounds are slightly different.
Figure 1.
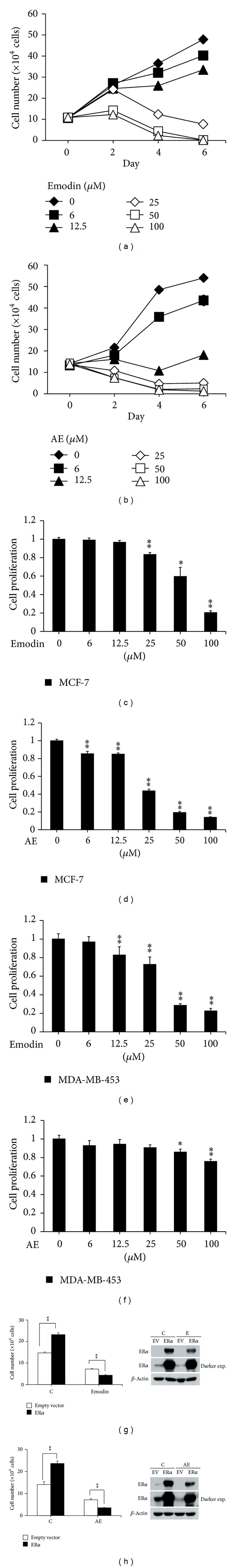
Emodin and aloe-emodin inhibit breast cancer cell growth. MCF-7 cells were treated with various concentrations of emodin (a) or aloe-emodin (b) for 0 to 6 days. Cell numbers were calculated by trypan blue staining. (c–f) MCF-7 and MDA-MB-453 cells were treated with various concentrations of emodin or aloe-emodin for 4 days. Cell proliferation was determined by an MTT assay. The control cell proliferation rate was set at 1. MCF-7 cells were transiently transfected with empty vector or pCMV5-ERα and treated with 25 μM emodin (g) or aloe-emodin (h) for 4 days. The transfected efficiencies of ERα overexpression and cell numbers were detected by western blotting and trypan blue staining, respectively. The results were presented as the mean ± SEM. * and ** correspond to P < 0.05 and P < 0.01, respectively, versus the control group or empty vector group.
3.2. Emodin and Aloe-Emodin Decrease ERα Protein Levels in a Time- and Dose-Dependent Manner
ERα activation is triggered by estrogen and promotes the initiation and progression of breast cancer [22, 23]. ERα protein levels were detected following treatment with various dosages of emodin or aloe-emodin (0–100 μM, Figures 2(a) and 2(c)) for different time intervals (0–48 h, Figures 2(b) and 2(d)). The quantitative results were provided in the lower panels of the accompanying figures. The data suggest that emodin and aloe-emodin triggered a decrease in ERα protein level in a time- and dose-dependent manner.
Figure 2.
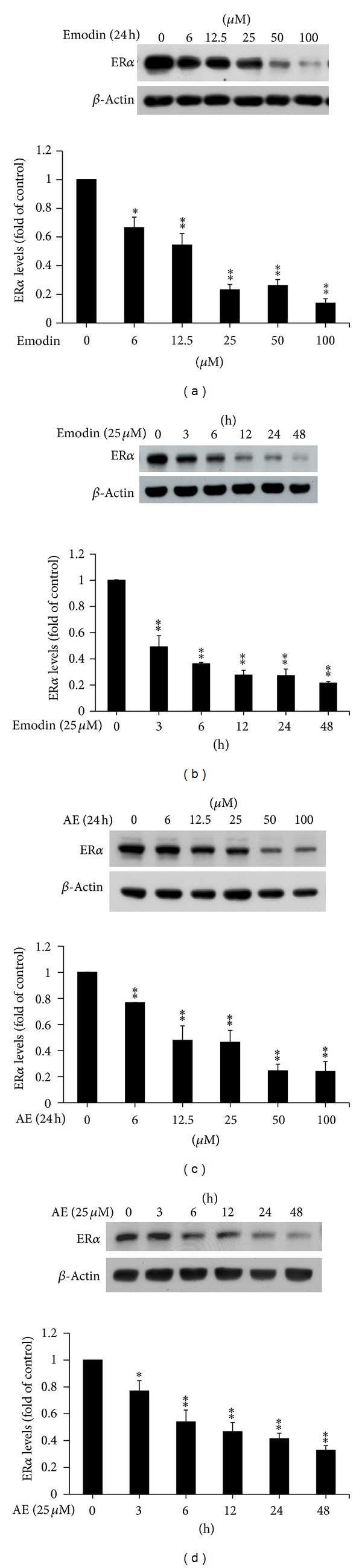
Emodin and aloe-emodin reduce ERα protein levels. MCF-7 cells were treated with emodin or aloe-emodin in a dose-dependent manner for 24 h (a, c) and via a time course using 25 μM of the drug (b, d). ERα protein levels were detected by western blotting. β-Actin was used as an internal control. The quantification of ERα levels is presented as fold increase or decrease compared to control levels and was evaluated in three independent experiments. The results were presented as the mean ± SEM. * and ** correspond to P < 0.05, and P < 0.01, respectively, versus the control group.
3.3. Emodin and Aloe-Emodin Decrease Both Nuclear and Cytosolic ERα and Inhibit ERα Activation
Because ERα protein levels were affected by emodin and aloe-emodin, the activation status of ERα was investigated. Fractionation of cellular protein was performed and indicated that emodin and aloe-emodin repressed both the nuclear and cytosolic distribution of ERα protein (upper panels in Figures 3(a) and 3(b)). The quantitative results are presented in the lower panels of Figures 3(a) and 3(b). Additionally, cytosolic ERα was more sensitive to treatment than nuclear ERα. An ERα reporter assay was performed in MCF-7 cells. The data showed that aloe-emodin significantly inhibited ERα-targeted promoter activity in a dose-dependent manner (Figure 3(d)). Compared to aloe-emodin, emodin in high dosages (25–100 μM) moderately inhibited ERα activation, whereas low dosages of emodin (6 and 12.5 μM) tended to increase ERα activation. Furthermore, the expressions of ERα downstream genes might be affected by those two compounds. Figures 3(e) and 3(f) were the evidence indicating that the protein expression of cyclin D1 which is one of ERα-regulated protein was indeed decreased by treatments of 25 μM emodin or 25 μM aloe-emodin for 48 h.
Figure 3.
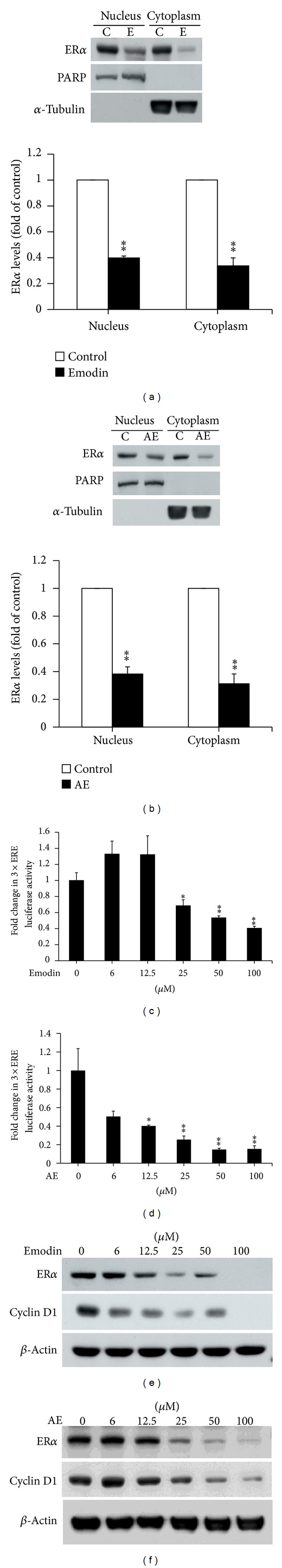
Emodin and aloe-emodin diminish both nuclear and cytoplasmic ERα levels and transcriptional activity. MCF-7 cells were treated with 25 μM emodin (a) or aloe-emodin (b) for 24 h prior to cell lysis and protein fractionation as described in Section 2. ERα protein was detected by western blotting, and PARP and α-tubulin served as markers for the nuclear and cytoplasmic fractions, respectively. The quantification of ERα levels was presented as fold increase or decrease relative to controls and was evaluated in three independent experiments. The effects of emodin (c) and aloe-emodin (d) on ERα activation were evaluated using a 3 × ERE- (estrogen-responsive element-) containing luciferase reporter assay in MCF-7 cells for 48 h. β-galactosidase expression served as the internal control. The data were presented as the fold change compared to control levels (0 μM). The results were presented as the mean ± SEM. * and ** correspond to P < 0.05 and P < 0.01, respectively, versus control group. MCF-7 cells were treated with emodin (e) or aloe-emodin (f) in a dose-dependent manner for 48 h, and the expressions of ERα-regulated protein cyclin D1 were detected by western blotting.
3.4. Emodin and Aloe-Emodin Treatment Leads to Decreased ERα Protein through Proteasomal Degradation
Because ERα protein levels were affected by both emodin and aloe-emodin in the previous experiments, the gene expression and protein stability of ERα were investigated. First, ERα messenger RNA expression was detected by quantitative real-time PCR following treatment. We found that treatments of 25 μM emodin and 25 μM aloe-emodin for 24 h did not have a significant effect compared to the control group (Figure 4(a)). Second, because ubiquitin-proteasome-dependent degradation is the main process involved in ERα proteolysis [23], the proteasome inhibitor MG132 was used to prevent ERα protein degradation, and drug effects can be observed. The results of this experiment indicated that MG132 could rescue ERα degradation after emodin or aloe-emodin treatment (Figures 4(b) and 4(c)). These findings suggest that the decreased ERα protein levels observed following emodin or aloe-emodin treatment resulted from proteasome degradation.
Figure 4.
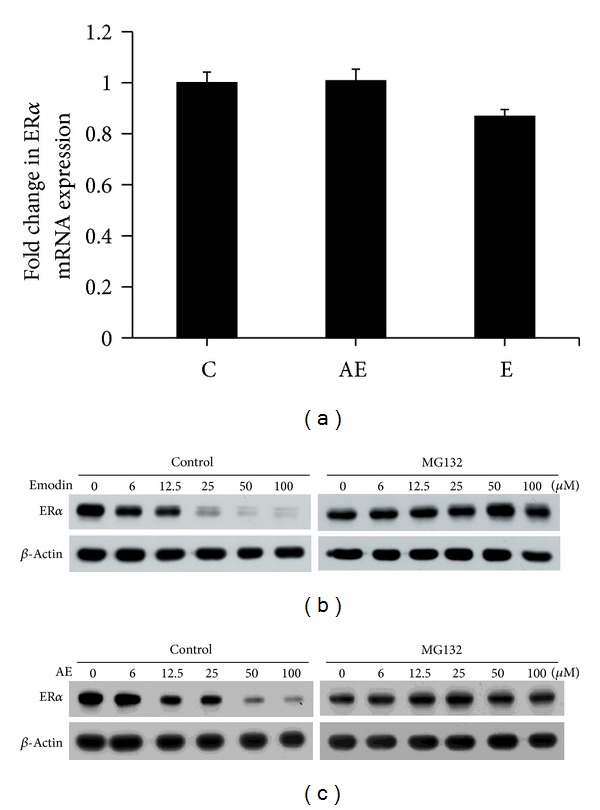
Emodin and aloe-emodin decrease ERα protein levels by promoting protein degradation. (a) MCF-7 cells were treated with 25 μM of emodin or aloe-emodin for 24 h prior to quantitative real-time PCR to assess ERα mRNA expression. Data were presented as the fold change compared to control levels. The experiments were performed in triplicate, and each condition was replicated four times within each experiment. The results are presented as the mean ± SEM. (b, c) MCF-7 cells were treated with various concentrations of emodin or aloe-emodin in the presence of MG132 (proteasome inhibitor, 5 μM) for 24 h. ERα protein levels were detected by western blotting. β-Actin was used as an internal control.
3.5. Aloe-Emodin Promotes the Dissociation of ER and Heat Shock Protein 90 and Causes ERα Ubiquitination
ERα is processed by ubiquitination after disassociating from heat shock protein 90 (HSP90) [24]. Using immunoprecipitation in the presence of MG132 to prevent protein degradation, we found that aloe-emodin apparently blocked the protein interaction between ERα and HSP90 (Figure 5(a)); however, the effect induced by emodin was not as significant (Figure 5(b)). The quantitative graphs were provided in the lower panels of accompanying figures. Subsequently, ERα ubiquitination following drug treatment was investigated by detecting the levels of ubiquitinated ERα in drug-treated cell extracts following MG132 administration. The data indicated that aloe-emodin treatment enhanced ubiquitin-conjugated ERα levels, and protein degradation was prevented by MG132 treatment regardless of whether ERα or ubiquitin was immunoprecipitated first (Figure 5(c)). Although emodin slightly promoted ERα/HSP90 dissociation (Figure 5(b)), no increase in ERα ubiquitination was observed following emodin treatment (Figure 5(d)). The related quantitative results were shown in the lower panels. It suggests that ubiquitination was not required for emodin-induced ERα degradation by proteasome. These results indicate that aloe-emodin specifically reduces ERα protein levels by promoting ERα ubiquitination, which results in proteasome-dependent degradation.
Figure 5.
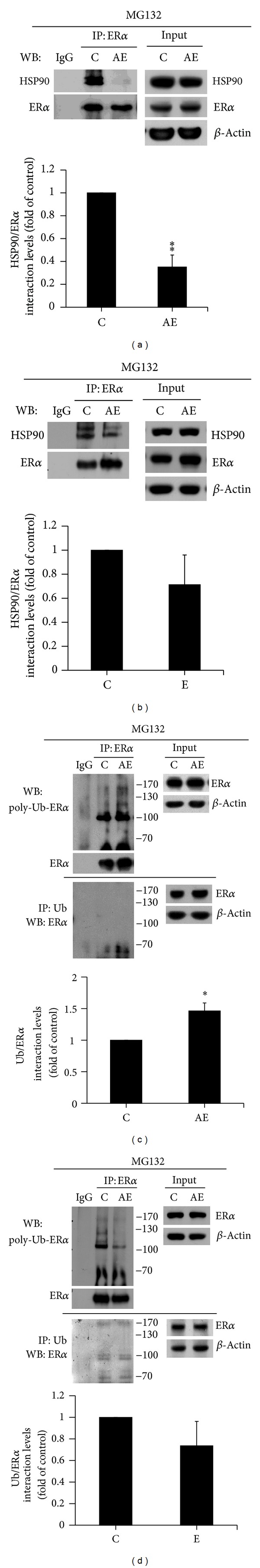
Only aloe-emodin promotes the dissociation of HSP90/ERα and subsequent ERα ubiquitination. The interaction between ERα and HSP90 in MCF-7 cells following 25 μM treatment of aloe-emodin (a) or emodin (b) in the presence of MG132 (5 μM) for 24 h was evaluated by ERα immunoprecipitation followed by western blotting for HSP90 with specific antibodies. The ubiquitin-conjugated ERα levels in MCF-7 cells following 25 μM treatment of aloe-emodin (c) and emodin (d) in the presence of MG132 (5 μM) were examined by immunoprecipitation with anti-ERα or antiubiquitin antibodies followed by western blotting. The lysates were used in the western blotting experiments to indicate protein levels. Immunoprecipitation by IgG served as a negative control. β-Actin served as an internal control for western blotting. The quantitative data were obtained from the results of three times immunoprecipitation experiments and presented as fold change of control. The quantitative graphs were presented as the mean ± SEM. * and ** correspond to P < 0.05 and P < 0.01, respectively, versus the control group.
3.6. Comparison of Emodin/Aloe-Emodin and Estrogen on ERα Behaviors
Ligand binding by estrogen or estrogen-like molecules promotes ERα transactivation, during which ERα first dissociates from HSP90 in the cytoplasm and then translocates into the nucleus [8]. Because the effects of aloe-emodin and emodin on the ERα ubiquitination process are distinct (Figure 5), it is of interest to understand how the effects of these two compounds compare to those of estrogen to mediate ERα behavior. Immunoprecipitation experiment results indicate that aloe-emodin had similar effects to synthetic estrogen (estradiol benzoate (EB)) on ERα/HSP90 dissociation (Figure 6(a)), whereas emodin did not (Figure 5(b)). However, in contrast to EB treatment, aloe-emodin treatment led to a decrease in the nuclear and cytoplasmic levels of ERα protein. Furthermore, the decrease in ERα protein levels following aloe-emodin treatment was blocked by MG132, suggesting that aloe-emodin promotes ERα degradation (Figure 6(b)). Comparing the data in Figure 3(b), we suggest that aloe-emodin preferentially induces ERα degradation in the cytoplasm of breast cancer cells. Interestingly, nuclear ERα was increased by emodin treatment, and protein degradation was prevented by MG132 (Figure 6(c)), suggesting that although emodin induces ERα degradation in both the nucleus and cytoplasm (compared to Figure 3(a)), it seems that emodin is able to promote ERα translocation into the nucleus, which is in contrast to the actions of aloe-emodin. This might explain why low concentrations of emodin showed slight stimulation in the ERα reporter assay (Figure 3(c)). Taken together, these results demonstrate that aloe-emodin induces the dissociation of ERα/HSP90 and subsequently promotes ERα ubiquitin-proteasome-dependent degradation in the cytoplasm, thereby inhibiting ERα translocation into nucleus, where ERα would otherwise be activated as a positive modulator.
Figure 6.
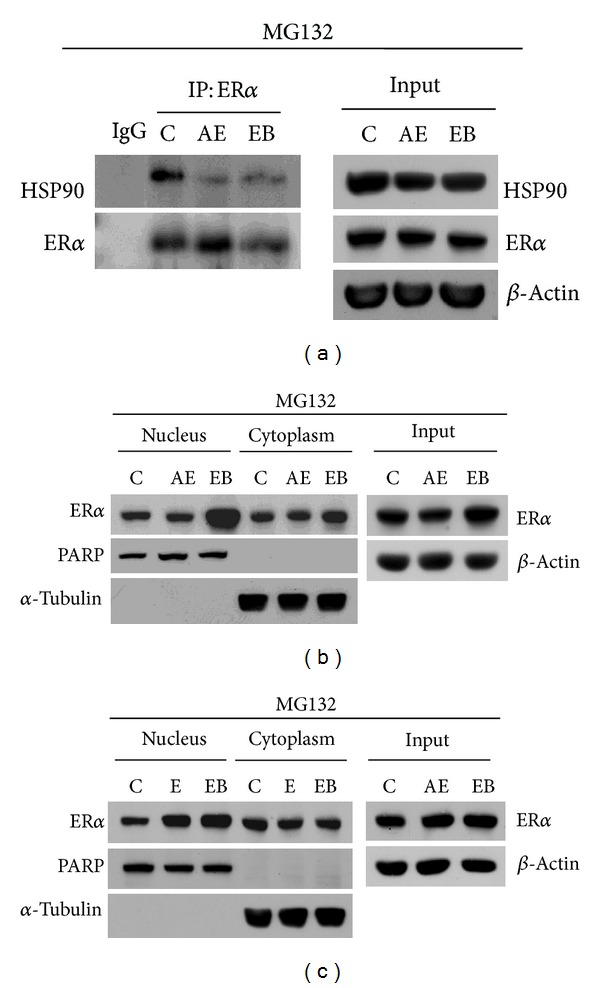
Effects of aloe-emodin and emodin on ERα subcellular distribution in comparison to EB. (a, b) MCF-7 cells were treated with aloe-emodin (25 μM) or estradiol benzoate ((EB) 10 nM) in the presence of MG132 (5 μM) for 24 h. The interaction between HSP90 and ERα was evaluated by ERα immunoprecipitation followed by western blotting of HSP90 with specific antibodies. Immunoprecipitation by IgG served as a negative control. The fractionation of cellular proteins was performed, and ERα protein was detected by western blotting. (c) MCF-7 cells were treated with emodin (25 μM) or EB (10 nM) in the presence of MG132 (5 μM) for 24 h. The fractionation of cellular proteins was performed, and ERα protein was detected by western blotting. PARP and α-tubulin served as markers for the nuclear and cytoplasmic fractions, respectively. The lysates were used in the western blotting experiments to indicate protein levels. β-Actin served as an internal control for western blotting.
4. Discussion
Breast cancer is a common malignancy in women, and estrogen plays an important role in early cancer development [25]. Estrogen, which usually denotes 17β-estradiol (E2), binds to its primary receptor ERα and stimulates ERα transcriptional activity to regulate downstream gene expression and cell growth [26, 27]. Tamoxifen is designed to interfere with E2 binding and thus block ERα transcriptional activity to treat ERα-dependent diseases such as breast cancer [28]. However, alternative compounds that are safer and associated with fewer side effects than Tamoxifen are desired. Numerous studies suggest that phytoestrogens possess organ-specific estrogenic and antiestrogenic effects [29]. Phytoestrogens mainly consist of isoflavones, such as genistein and daidzein, and are used in the treatment of menopausal symptoms as well as breast cancer [30, 31]. In this study, we provide evidence indicating that two phytoestrogens, emodin and aloe-emodin, might significantly inhibit the proliferation of ERα-positive breast cancer cells through ERα degradation. Although both drugs had dose-responsive effects on inhibiting proliferation of breast cancer cells, low doses (25 μM or less) of aloe-emodin could not affect proliferation of ERα-negative cells (Figure 1(f)). Therefore, 25 μM of drugs were utilized in other experiments (Figure 3 to Figure 6). Interestingly, both compounds had inhibitory effects on ERα, but they utilized different inhibitory mechanisms. Aloe-emodin inhibited ERα activation through HSP90/ERα dissociation and ubiquitin-dependent degradation, whereas emodin did not share the same molecular pathway. Therefore, these findings illustrate that emodin and aloe-emodin might serve as estrogen receptor modulators with different molecular mechanisms. The therapeutic application of these two compounds should be investigated further in the future.
Anthraquinones are phytoestrogens that have been demonstrated to possess anticancer properties through the inhibition of cell proliferation, the induction of apoptosis, and the prevention of metastasis [1]. The effects of the anthraquinone derivatives emodin and aloe-emodin have been extensively investigated in several cancer types through studies on their signaling targets. Emodin has been reported to sensitize Her2/neu-overexpressing lung cancer cells to chemotherapeutic treatments and to suppress Her2/neu-overexpressing breast cancer growth by inhibiting tyrosine kinase activity [32–34]. Notably, emodin has been shown to downregulate androgen receptor (AR) and inhibit prostate cancer growth [5], suggesting that there is a connection between anthraquinone derivatives and steroid receptor in hormone-related cancers. Although the estrogenic ability of emodin is higher than that of aloe-emodin, as determined by ERα binding studies, the antiproliferation effect of aloe-emodin is more efficient than that of emodin in breast cancer cells [7]. Kang et al. also showed that the cytotoxicity of aloe-emodin is higher in ERα-positive cells than in ERα-negative cells [7]. These observations suggest that aloe-emodin might be a stronger inhibitor of ERα-positive cancer cell growth than emodin.
The goal of this study was to investigate the differences between emodin and aloe-emodin in their efficiency and mechanism of blocking breast cancer cell growth. Our data showed that aloe-emodin is a more potent growth inhibitor than emodin and has a unique mechanism for the growth inhibition of breast cancer cells. MCF-7 cell growth was abolished following treatment with 12.5 μM aloe-emodin for 6 days, and only 50% of cells survived at 25 μM aloe-emodin treatment for 4 days (Figure 1(b)). Although a previous study suggested that the IC50 of aloe-emodin is 12.6 ± 0.83 μg/mL (approximately 46 μM) on MCF-7 cells [7], the IC50 of aloe-emodin in our system was only approximately 25 μM (Figure 1(d)). With regard to side effects on normal cells, the previous report indicated that 25 μM aloe-emodin is a more significant proliferation inhibitor in human skin epidermoid carcinoma cells than in noncancerous cells [35]. However, aloe-emodin did not significantly affect the proliferation of MDA-MB-453 ERα-negative cells (Figure 1(f)). The overexpression of ERα in MCF-7 cells increased the sensitivity to aloe-emodin treatment (Figure 1(h)). These results are consistent with previous findings that aloe-emodin has a higher cytotoxic potential to MCF-7 (ERα-positive) cells than to MDA-MB-231 (ERα-negative) cells [7]. A low dosage (10 μM) of aloe-emodin [6] showed that both cell growth (Figures 1(b) and 1(d)) and ERα activation (Figure 3(d)) were significantly repressed by aloe-emodin in a dose-dependent manner, even at low dosages (6 μM). Additionally, aloe-emodin treatment reduced ERα protein levels not only in total lysates (Figures 2(c) and 2(d)) but also in nuclear and cytoplasmic fractions (Figure 3(b)). In the nuclear and cytoplasmic fractions, the impact of aloe-emodin treatment on cytoplasmic ERα levels seems stronger than that on nuclear ERα levels (Figure 3(b)). As shown in Figures 2 and 4, we conclude that the decrease in ERα levels induced by aloe-emodin was due to protein degradation. This correlates with ERα proteasome-dependent degradation because of the observed elevation in ubiquitin-conjugated levels (Figure 5(c)). Interestingly, the dissociation of ERα and HSP90 was increased by aloe-emodin treatment (Figure 5(a)), and ERα released from HSP90 protection was subjected to ubiquitination for degradation rather than translocation to the nucleus for activation (Figure 6(b)). These phenomena are similar to the actions of antiestrogens such as ICI 164384, ICI 182780, and RU 58668, which may induce ERα degradation and result in rapid turnover [24]. As a ligand with an inhibitory effect on ERα, we suggest that aloe-emodin might qualify as an estrogen receptor modulator to negatively regulate ERα activity and inhibit breast cancer cell growth.
The effect of emodin was also investigated in this study. Although the inhibitory effects of emodin on breast cancer growth and ERα activation were similar to those of aloe-emodin, the molecular mechanism of emodin inhibition was unexpectedly distinct. In Figures 1(a) and 1(c), the inhibitory potency of emodin on MCF-7 cell growth was lower than that of aloe-emodin (Figures 1(b) and 1(d)). Interestingly, emodin significantly suppressed cell proliferation of the ERα-negative cell line MDA-MB-453 (Figure 1(e)). Because emodin may act as a tyrosine kinase inhibitor by inhibiting the binding of Her2/neu and HSP90, thereby depleting Her2/neu via proteasomal degradation [36], it is possible that emodin might differ in its effect on the growth of MDA-MB-453 cells (which are characterized by Her2 overexpression) in ERα-independent regulation in comparison to aloe-emodin. Like aloe-emodin, emodin targeted ERα by proteasomal degradation (Figures 2 and 4); however, the degradation pathway was independent of HSP90/ERα dissociation and ubiquitination (Figures 5(b) and 5(d)). Although the ubiquitin-proteasome degradation is an important process for modulation of many proteins, recent studies demonstrate that the proteasome-dependent protein degradation possibly goes through ubiquitination-independent pathway [37, 38]. Previous reports indicate that the estrogenic activity of emodin is higher than that of aloe-emodin [6, 7]. Our ERE reporter assay results showed that emodin induced the inhibition of ERα activation only at high dosages (over 25 μM, Figure 3(c)), whereas the aloe-emodin data showed inhibitory effects starting at low dosages (6 μM, Figure 3(d)). Notably, emodin treatment led to a slight increase in ERα activation at 6 and 12.5 μM, which differed from the aloe-emodin treatment results (Figure 3(c)). Unlike aloe-emodin, emodin treatment elevated nuclear ERα protein levels in the presence of MG132, similar to the effects of estradiol benzoate (EB) treatment (Figure 6(c)). Taken together with the data in Figure 3(a), we suggest that emodin, using a distinct mechanism from aloe-emodin, might cause ERα shuttling into the nucleus and subsequently promote nuclear ERα degradation through ubiquitination-independent pathway, whereas cytoplasmic ERα is less affected. This hypothesis is similar to previous studies indicating that the chemical structure of the ligand directly affects the nuclear fate and protein turnover rate of ERα independently of transcriptional regulation [39]. These observations suggest that the essential difference between emodin and aloe-emodin lies in the mechanism of ERα regulation and Her2 inhibition.
In conclusion, we provide evidence showing that the anticancer mechanisms of the anthraquinone derivatives emodin and aloe-emodin are mediated via an ERα-dependent pathway in breast cancer cells. Although emodin and aloe-emodin display distinct differences in efficiency and ERα activation mechanism on breast cancer cell growth, both compounds are potential selectively therapeutic treatments for breast cancer.
Conflict of Interests
The authors declare that they have no conflict of interests.
Acknowledgments
The authors thank Dr. H. C. Chen, Dr. T. H. Lee, Dr. Y. M. Liou, Dr. J. W. Chen, and Dr. H. C. Cheng (National Chung Hsing University, Taiwan) for technical support. This work was supported by the following Grants: NSC100-2320-B-005-002 and NSC101-2320-B-005-004-MY3 from the Taiwan National Science Council (to H. Lin); Taichung Veterans General Hospital/National Chung Hsing University Joint Research Program (TCVGH-NCHU1017607 and TCVGH-NCHU1027606); Chang Bing Show Chwan Memorial Hospital Research Grant (CBBC001, to Y.-T. Lee); and the Taiwan Ministry of Education under the Aiming for the Top University plan.
Abbreviations
- ERα:
Estrogen receptor α
- HSP90:
Heat shock protein 90
- AE:
Aloe-emodin
- E:
Emodin.
References
- 1.Huang Q, Lu G, Shen H-M, Chung MCM, Choon NO. Anti-cancer properties of anthraquinones from rhubarb. Medicinal Research Reviews. 2007;27(5):609–630. doi: 10.1002/med.20094. [DOI] [PubMed] [Google Scholar]
- 2.Dutta A, Bandyopadhyay S, Mandal C, Chatterjee M. Aloe vera leaf exudate induces a caspase-independent cell death in Leishmania donovani promastigotes. Journal of Medical Microbiology. 2007;56(5):629–636. doi: 10.1099/jmm.0.47039-0. [DOI] [PubMed] [Google Scholar]
- 3.Wang H-H, Chung J-G. Emodin-induced inhibition of growth and DNA damage in the Helicobacter pylori . Current Microbiology. 1997;35(5):262–266. doi: 10.1007/s002849900250. [DOI] [PubMed] [Google Scholar]
- 4.Chang C-H, Lin C-C, Yang J-J, Namba T, Hattori M. Anti-inflammatory effects of emodin from Ventilago leiocarpa . American Journal of Chinese Medicine. 1996;24(2):139–142. doi: 10.1142/S0192415X96000189. [DOI] [PubMed] [Google Scholar]
- 5.Cha T-L, Qiu L, Chen C-T, Wen Y, Hung M-C. Emodin down-regulates androgen receptor and inhibits prostate cancer cell growth. Cancer Research. 2005;65(6):2287–2295. doi: 10.1158/0008-5472.CAN-04-3250. [DOI] [PubMed] [Google Scholar]
- 6.Matsuda H, Shimoda H, Morikawa T, Yoshikawa M. Phytoestrogens from the roots of Polygonum cuspidatum (Polygonaceae): structure-requirement of hydroxyanthraquinones for estrogenic activity. Bioorganic and Medicinal Chemistry Letters. 2001;11(14):1839–1842. doi: 10.1016/s0960-894x(01)00318-3. [DOI] [PubMed] [Google Scholar]
- 7.Kang SC, Lee CM, Choung ES, et al. Anti-proliferative effects of estrogen receptor-modulating compounds isolated from Rheum palmatum . Archives of Pharmacal Research. 2008;31(6):722–726. doi: 10.1007/s12272-001-1218-1. [DOI] [PubMed] [Google Scholar]
- 8.Acconcia F, Kumar R. Signaling regulation of genomic and nongenomic functions of estrogen receptors. Cancer Letters. 2006;238(1):1–14. doi: 10.1016/j.canlet.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 9.Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nature Reviews Cancer. 2002;2(2):101–112. doi: 10.1038/nrc721. [DOI] [PubMed] [Google Scholar]
- 10.Osborne CK. Tamoxifen in the treatment of breast cancer. The New England Journal of Medicine. 1998;339(22):1609–1618. doi: 10.1056/NEJM199811263392207. [DOI] [PubMed] [Google Scholar]
- 11.Colditz GA, Hankinson SE, Hunter DJ, et al. The use of estrogens and progestins and the risk of breast cancer in postmenopausal women. The New England Journal of Medicine. 1995;332(24):1589–1593. doi: 10.1056/NEJM199506153322401. [DOI] [PubMed] [Google Scholar]
- 12.Adlercreutz H, Mazur W. Phyto-oestrogens and Western diseases. Annals of Medicine. 1997;29(2):95–120. doi: 10.3109/07853899709113696. [DOI] [PubMed] [Google Scholar]
- 13.Chen MC, Huang CY, Hsu SL, et al. Retinoic acid Induces apoptosis of prostate cancer DU145 cells through Cdk5 overactivation. Evidence-Based Complementary and Alternative Medicine. 2012;2012:11 pages. doi: 10.1155/2012/580736.580736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin H, Wang PS. Inhibitory effects of digoxin on testosterone production in rat Leydig cells. Adaptive Medicine. 2012;4:165–172. [Google Scholar]
- 15.Lin H, Lin T-Y, Juang J-L. Abl deregulates Cdk5 kinase activity and subcellular localization in Drosophila neurodegeneration. Cell Death and Differentiation. 2007;14(3):607–615. doi: 10.1038/sj.cdd.4402033. [DOI] [PubMed] [Google Scholar]
- 16.Lin H, Chen M-C, Chiu C-Y, Song Y-M, Lin S-Y. Cdk5 regulates STAT3 activation and cell proliferation in medullary thyroid carcinoma cells. Journal of Biological Chemistry. 2007;282(5):2776–2784. doi: 10.1074/jbc.M607234200. [DOI] [PubMed] [Google Scholar]
- 17.Lin H, Chen M-C, Ku C-T. Cyclin-dependent kinase 5 regulates steroidogenic acute regulatory protein and androgen production in mouse Leydig cells. Endocrinology. 2009;150(1):396–403. doi: 10.1210/en.2008-0496. [DOI] [PubMed] [Google Scholar]
- 18.Lin H, Juang J-L, Wang PS. Involvement of Cdk5/p25 in digoxin-triggered prostate-cancer cell apoptosis. Journal of Biological Chemistry. 2004;279(28):29302–29307. doi: 10.1074/jbc.M403664200. [DOI] [PubMed] [Google Scholar]
- 19.Hsu F-N, Chen M-C, Chiang M-C, et al. Regulation of androgen receptor and prostate cancer growth by cyclin-dependent kinase 5. Journal of Biological Chemistry. 2011;286(38):33141–33149. doi: 10.1074/jbc.M111.252080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen M-C, Wang S-W, Kan S-F, Tsai S-C, Wu Y-C, Wang PS. Stimulatory effects of propylthiouracil on pregnenolone production through upregulation of steroidogenic acute regulatory protein expression in rat granulosa cells. Toxicological Sciences. 2010;118(2):667–674. doi: 10.1093/toxsci/kfq302.kfq302 [DOI] [PubMed] [Google Scholar]
- 21.Dalvai M, Bystricky K. Cell cycle and anti-estrogen effects synergize to regulate cell proliferation and er target gene expression. PLoS One. 2010;5(6) doi: 10.1371/journal.pone.0011011.e11011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heldring N, Pike A, Andersson S, et al. Estrogen receptors: how do they signal and what are their targets. Physiological Reviews. 2007;87(3):905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 23.Miyoshi Y, Murase K, Saito M, Imamura M, Oh K. Mechanisms of estrogen receptor-α upregulation in breast cancers. Medical Molecular Morphology. 2010;43(4):193–196. doi: 10.1007/s00795-010-0514-3. [DOI] [PubMed] [Google Scholar]
- 24.Fan M, Park A, Nephew KP. CHIP (carboxyl terminus of Hsc70-interacting protein) promotes basal and geldanamycin-induced degradation of estrogen receptor-α . Molecular Endocrinology. 2005;19(12):2901–2914. doi: 10.1210/me.2005-0111. [DOI] [PubMed] [Google Scholar]
- 25.Walker RA. Oestrogen receptor and its potential role in breast cancer development. The Journal of Pathology. 1999;188(3):229–230. doi: 10.1002/(SICI)1096-9896(199907)188:3<229::AID-PATH341>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 26.Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor super-family: the second decade. Cell. 1995;83(6):835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. Journal of Biological Chemistry. 2001;276(40):36869–36872. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- 28.Jordan VC. Tamoxifen (ICI46,474) as a targeted therapy to treat and prevent breast cancer. British Journal of Pharmacology. 2006;147(1):S269–S276. doi: 10.1038/sj.bjp.0706399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murkies AL, Wilcox G, Davis SR. Phytoestrogens. Journal of Clinical Endocrinology and Metabolism. 1998;83(2):297–303. doi: 10.1210/jcem.83.2.4577. [DOI] [PubMed] [Google Scholar]
- 30.Nikander E, Kilkkinen A, Metsä-Heikkilä M, et al. A randomized placebo-controlled crossover trial with phytoestrogens in treatment of menopause in breast cancer patients. Obstetrics and Gynecology. 2003;101(6):1213–1220. doi: 10.1016/s0029-7844(03)00232-1. [DOI] [PubMed] [Google Scholar]
- 31.Imai Y, Tsukahara S, Asada S, Sugimoto Y. Phytoestrogens/flavonoids reverse breast cancer resistance protein/ABCG2-mediated multidrug resistance. Cancer Research. 2004;64(12):4346–4352. doi: 10.1158/0008-5472.CAN-04-0078. [DOI] [PubMed] [Google Scholar]
- 32.Jayasuriya H, Koonchanok NM, Geahlen RL, McLaughlin JL, Chang C-J. Emodin, a protein tyrosine kinase inhibitor from Polygonum cuspidatum . Journal of Natural Products. 1992;55(5):696–698. doi: 10.1021/np50083a026. [DOI] [PubMed] [Google Scholar]
- 33.Zhang L, Chang C-J, Bacus SS, Hung M-C. Suppressed transformation and induced differentiation of HER-2/neu-overexpressing breast cancer cells by emodin. Cancer Research. 1995;55(17):3890–3896. [PubMed] [Google Scholar]
- 34.Zhang L, Hung M-C. Sensitization of HER-2/neu-overexpressing non-small cell lung cancer cells to chemotherapeutic drugs by tyrosine kinase inhibitor emodin. Oncogene. 1996;12(3):571–576. [PubMed] [Google Scholar]
- 35.Chou T-H, Liang C-H. The molecular effects of Aloe-Emodin (AE)/liposome-AE on human nonmelanoma skin cancer cells and skin permeation. Chemical Research in Toxicology. 2009;22(12):2017–2028. doi: 10.1021/tx900318a. [DOI] [PubMed] [Google Scholar]
- 36.Yan Y-Y, Zheng L-S, Zhang X, et al. Blockade of Her2/neu binding to Hsp90 by emodin azide methyl anthraquinone derivative induces proteasomal degradation of Her2/neu. Molecular Pharmaceutics. 2011;8(5):1687–1697. doi: 10.1021/mp2000499. [DOI] [PubMed] [Google Scholar]
- 37.Murakami Y, Matsufuji S, Kameji T, et al. Ornithine decarboxylase is degraded by the 26S proteasome without ubiquitination. Nature. 1992;360(6404):597–599. doi: 10.1038/360597a0. [DOI] [PubMed] [Google Scholar]
- 38.Asher G, Shaul Y. p53 proteasomal degradation: poly-ubiquitination is not the whole story. Cell Cycle. 2005;4(8):1015–1018. doi: 10.4161/cc.4.8.1900. [DOI] [PubMed] [Google Scholar]
- 39.Kocanova S, Mazaheri M, Caze-Subra S, Bystricky K. Ligands specify estrogen receptor alpha nuclear localization and degradation. BMC Cell Biology. 2010;11(article 98) doi: 10.1186/1471-2121-11-98. [DOI] [PMC free article] [PubMed] [Google Scholar]