

Abstract
Live attenuated measles virus (MV) vaccines have an impressive record of safety, efficacy and ability to induce life-long immunity against measles infection. Using reverse genetics technology, such negative-strand RNA viruses can now be rescued from cloned DNA. This technology allows the insertion of exogenous genes encoding foreign antigens into the MV genome in such a way that they can be expressed by the MV vaccine strain, without affecting virus structure, propagation and cell targeting. Recombinant viruses rescued from cloned cDNA induce immune responses against both measles virus and the cloned antigens. The tolerability of MV to gene(s) insertion makes it an attractive flexible vector system, especially if broad immune responses are required. The fact that measles replication strictly occurs in the cytoplasm of infected cells without DNA intermediate has important biosafety implications and adds to the attractiveness of MV as a vector. In this article we report the characteristics of reporter gene expression (GFP, LacZ and CAT) and the biochemical, biophysical and immunological properties of recombinant MV expressing heterologous antigens of simian immunogeficiency virus (SIV).
Keywords: Recombinant vaccines, Viral vectors, Protein expression
1. Introduction
Modern approaches in vaccinology and immunology have lead to the discovery of a number of antigens potentially allowing to address complex diseases such as AIDS, cancer and malaria, if administered in an appropriate way. Therefore, the establishment of potent antigen delivery systems became essential for the progress in this field. The use of live attenuated viral vectors appears promising especially since multiple antigens can efficiently be presented to the immune system. Attenuated measles virus (MV) vaccine represents a number of essential features for the development of candidate prophylactic and therapeutic vaccines. Among these features are safety, immunogenicity and cost effectiveness of the recombinant vaccines.
MV belongs to the genus Morbillivirus in the family Paramyxoviridae. It is an enveloped virus with a long non-segmented RNA genome of negative polarity [12]. In 1954, Enders and Peebles inoculated primary human kidney cells with the blood of David Edmonston, a child with measles [7], and the resulting Edmonston strain of MV was subsequently adapted to growth in a variety of cell lines. Adaptation to chicken embryos, chick embryo fibroblasts (CEF), and/or dog kidney cells and human diploid cells produced the attenuated Edmonston A and B, Zagreb (EZ) and AIK-C seeds [12], [13], [25]. Edmonston-B was licensed in 1963 as the first MV vaccine. However, being reactogenic it was abandoned in 1975. Further passages of Edmonston A and B on CEF produced the more attenuated Moraten vaccines [26].
Today, the Schwarz/Moraten, AIK-C and EZ vaccines are commonly used. MV vaccines induces a life-long immunity after a single or two low-dose injections [12], [13]. Persistence of antibodies and CD8 cells has been shown for as long as 25 years after vaccination [22]. MV vaccines are easily produced on a large scale in most countries and can be distributed at low cost. Because the attenuation of MV genome results from an advantageous combination of numerous mutations, the vaccine is very stable and reversion to pathogenicity has never been observed [13]. Regarding safety, MV replicates exclusively in the cytoplasm, ruling out the possibility of integration into host DNA. These characteristics make live attenuated MV vaccine an attractive candidate for use as a multivalent vaccination vector.
A reverse genetics system for MV was established allowing the rescue of infectious MV from cloned cDNA [24]. Additional MV-specifc transcription units containing multiple cloning sites were introduced into the MV antigenome (i.e. cDNA sequence) allowing the addition and simultaneous expression of several exogenous genes. Genes from Hepatitis B virus (HBV), Simian or Human Immunodeficiency viruses (SIV, HIV), Mumps virus (MV), West Nile virus (WNV) and human IL12 were inserted and expressed from different loci of the MV genome [3], [5], [6], [16], [21], [23], [27], [28], [29], [31].
Here we show that multiple marker genes coding for chloramphenicol acetyltransferase (CAT), green fluorescent protein (GFP) and beta-galactosidase (B-gal) can be expressed simultaneously by the same progeny. In addition, recombinant MVs expressing single or multiple antigenes of SIV induced strong and enduring humoral immune responses in experimental animals.
2. Materials and methods
2.1. Cells
Cells were maintained as monolayers in Dulbecco's modified Eagle's medium (DMEM), supplemented with 5% fetal calf serum (FCS) for Vero cells (African green monkey kidney), and with 10% FCS and 1.2 mg of G418 per millilitre for stably transfected human embryonic kidney cells 293 (293-3-46) cells.
2.2. Plasmids and viruses
All constructs described in this report are derivatives of plasmid p(+)MV [24]. From this plasmid several vector plasmids were generated bearing additional transcription units that contain suitable restriction sites for easy exchange of ORFs. These sites include BssHII, SnaBI, AatII, MluI and NruI that flank a GFP ORF. The GFP ORF can, thus be swapped with transgene inserts derived from pathogens by the specific unique restriction sites that are also unique at the extremities of the transgenes. Care has to be taken that, in the final constructs, the total number of nucleotides in the recombinant antigenomic MV sequence has to be a multiple of six [2].
A full-length MV plasmid containing three exogenous marker genes was generated by insertion of green fluorescence protein (GFP) upstream of the N gene (position 1), Beta-galactosidase (LacZ) between P and M genes (position 2) and chloramphenicol acetyltransferase (CAT) between H and L genes (position 3) (Fig. 1 ). The generation of multi-reporter plasmid is basically originated from three independent plasmids expressing eGFP [5], LacZ [21] and CAT [15]. The SacII–PacI segment containing the LacZ of the plasmid p(+)MPLacZV expressing LacZ from position 2, was exchanged with the SacII–PacI segment of p(+)MeGFPNV plasmid giving rise to an intermediate p(+)MeGFPNLacZPV. The final triporter plasmid was generated by replacement of the PacI–NotI fragment containing the CAT gene of the plasmid p(+)MHCATV expressing CAT from position 3 with the PacI–NotI fragment of the p(+)MeGFPNLacZPV. The cloning procedures of plasmids carrying the SIV genes were carried out basically as described by Wang et al. [31], PCR amplifications were performed using the proof reading pfu DNA polymerase (Stratagene).
Fig. 1.
Schematic representation of the modifications introduced into the antigenomic p(+)MV of measles virus. The GFP, LacZ and CAT were inserted simultaneously into the antigenomic p(+)MV plasmid (see Section 2). SIV genes, env, pol and gag were inserted as reported previously [31]. Plasmids containing env gene in position 2 and gag gene in position 3, p(+)MV2env-SIV and p(+)MV3gag-SIV, were digested with SacII–PacI. The SacII–PacI insert of p(+)MV2env-SIV replaced that of p(+)MV3gag-SIV, to generate p(+)MV2,3env,gag-SIV. Arrows indicate the insertion positions of additional transcription units. T7 is the T7 RNA polymerase promotor. T7t is the T7-terminator. δ is the hepatitis delta ribozyme.
A PCR amplification of the SIV-gag and env genes was done by specific primers flanked by SnaBI and BssHII and inserted in positions 2 or 3, respectively (Fig. 1) generating two different full-length plasmids [31]. Both plasmids p(+)MV2env-SIV and p(+)MV3gag-SIV (Fig. 1) were digested with SacII and PacI. The fragment SacII–PacI from p(+)MV2env-SIV was swapped by that of the p(+)MV3gag-SIV, resulting with the plasmid p(+)MV2,3env,gag-SIV. All recombinant viruses were rescued according to Radecke et al [24], by a slightly modified protocol [31].
2.3. Assays for stability of protein expression
Rescued viruses were serially passaged 10 times on Vero cells seeded into 35-mm-diameter plates that were infected with the standard and recombinant MVs at an MOI of 0.01 PFU/cell. After monolayer was fully infected (100%), 1% of the supernatant of each culture was used to infect the subsequent Vero cells monolayer. To test transgene expression and stability, viruses from passage 1 and 10 were used for further characterization of expression by immunofluorescence and Western blots essentially as described before [29].
2.4. Immunofluorescence
To visualize expression of proteins, infected cells on coverslips were fixed with 3% paraformaldehyde in PBS, and permeabilized with 0.1%TX100 in PBS, washed with blocking solution (PBS containing 1% BSA) for 1 h, and stained with the specific antibody [19], [20]. Monoclonal anti-SIV gp120 and a polyclonal anti-SIV-gag antibodies were used in a dilution of 1:300 v/v followed by FITC or Cy3 conjugated goat anti-mouse or anti rabbit serum, respectively. Cells were analyzed on a confocal microscope.
2.5. Confocal microscopy
The imaging system consisted of a Leica inverted microscope DM IRB/E, a Leica true confocal scanner TCS NT and a Silicon Graphics workstation. The images were recorded using a Leica PL APO 63× oil or a PL APO 40× oil immersion objective. The system was equipped with an argon/krypton mixed gas laser. Image processing was done on a Silicon Graphics workstation using “Imaris” (Bitplane AG, Zurich, Switzerland), a 3D multi-channel image processing software specialised for confocal microscopy images.
2.6. Sucrose gradient purification of MV and recombinant MVs
Purification of MV and recombinant MVs is performed essentially according to Spielhofer et al. [29]. Supernatants from approximately 6 × 106 PFUs of MV and recombinant MVs from infected cells were first clarified by centrifugation, at 7000 × g for 20 min, and layered on a cushion of 1 ml each of 20% and 60% sucrose in TNE (1 mM Tris pH 7.8, 100 mM NaCl, 10 mM EDTA). After centrifugation at 28,000 rpm for 90 min in a SW41 rotor, the sucrose interphase (1 ml) was taken, diluted to 20% sucrose and applied on the top of a 20%, 30%, 40%, 50% and 60% (2 ml each) sucrose step gradient, followed by centrifugation in an SW41 rotor at 29,000 rpm for 16 h (to equilibrium). Gradients were prepared using Auto Densi-flow (Labconco, USA). A control gradient (containing no virus) was run in parallel to measure fluctuation of the density. Fractions, approximately 500 μl, were collected and density was measured by weighing twice 100 μl from each fraction. Similarly, the density of the control fractions was determined. Less than 6% differences in density among corresponding fractions of all the gradients were detected. Protein contents in each fraction were analyzed by SDS-PAGE followed by immunoblotting.
2.7. Immunoelectron microscopy
Sucrose gradient purified MV and rMV-SIV viruses were pelleted by ultracentrifugation (30,000 rpm/min for 2 h) and re-suspended in 30 μl PBS pH 7.6, 1 μl was placed on grids for negative stain or immunogold staining. Polymorphic MV virus virions with a size of 300–400 nm containing ribonucleoproteins (RNP) were visualized. For immunogold staining, grids treated with anti-MV-H or anti-SIV-gp120 monoclonal antibodies were washed in PBS and further incubated for 60 min at room temperature with a dilution of 1:100 of 6 nm colloidal gold particles coated with protein A (Aurion, the Netherlands). Free protein A-gold complexes were washed away according to standard protocols.
2.8. Animals and immunization
Genetically modified mice IFNARtm-hCD46Ge [18] susceptible to MV infections were used for immunization experiments. These mice express the human CD46 gene with human-like tissue specificity, with in addition, a target mutation inactivating the interferon receptor type I. The animals were kept under optimal hygienic conditions and were immunized at 6–8 weeks of age. Immunization was performed intraperitoneally (i.p.) with 2 × 104 PFU of each recombinant MV in two injections, at 0 and 4 weeks. Non-infected mice served as control. UV inactivated MV was used as a control to determine the effect of virus replication on activation of immune responses.
2.9. MV RNA quantification by real-time PCR
Total RNA from mouse spleens was extracted as previously described [18]. For reverse transcription, the minus-strand primer 5′-TTATAACAATGATGGAGGGTAGGC, hybridizing to the last 24 nucleotides of the N mRNA, was used. Real-time quantitative TaqMan PCR based on the primer pair 5′-GGGTACCATCCTAGCCCAAATT and 5′-CGAATCAGCTGCCGTGTCT, amplifying 73 bases of the N mRNA, and the molecular beacon 5′-FAM-CGCAAAGGCGGTTACGGCCC-DABCYL, where 6-carboxyfluorescein (FAM) serves as the reporter fluorochrome and 4-dimethylaminophenylazobenzoic acid (DABCYL) serves as the quencher, was performed according to the protocol of the supplier (Perkin-Elmer, Applied Biosystems). Briefly, each 25 μl reaction mixture contained 2 μl of cDNA from the RT reaction, 12.5 μl of TaqMan PCR Master Mix (Perkin-Elmer), a 240 nM concentration of each primer, and 160 nM molecular beacon. One cycle of denaturation (95 °C for 10 min) was applied, followed by 45 cycles of amplification (95 °C for 15 s and 60 °C for 1 min). PCR was carried out in a spectrophotometric thermal cycler (ABI PRISM 7700 Sequence Detection System; Perkin-Elmer) that monitors changes in the fluorescence spectrum of molecular beacon FAM in each reaction tube during the course of the reaction, resulting in a real-time analysis.
For real-time PCR quantification, a standard curve was generated from triplicate samples of purified MV N RNA transcribed in vitro. Briefly, an MV N gene-containing plasmid, p(+)MNPCAT, was linearized with StuI and transcribed in vitro by using T7 RNA polymerase. After digestion of the DNA template with RNase-free DNase I (Roche, Basel, Switzerland), the generated MV N RNA transcript was purified and analyzed on an agarose gel, followed by determination of the concentration by spectrophotometry. The linear copy number range from 4 × 1010 to 4 × 103 copy equivalents per reaction (10-fold dilutions) was taken for RT-PCR amplification and detection of corresponding threshold cycles, which ranged from 14 to 31, corresponding to 4 × 1010 to 4 × 103 MV N RNA copy equivalents per RT-PCR, respectively. The determined viral RNA load was expressed as MV N RNA copy number per 1 μg of total RNA, and the calculation of MV N RNA copies per average cell was done by considering that about 2 × 105 splenic cells contain approximately 1 μg total RNA.
2.10. ELISA assays
2.10.1. MV ELISA
The presence of MV-specific antibodies in the sera from the immunized IFNARtm-hCD46Ge mice (10 per group) was determined by ELISA using microwell plates coated with measles virus EIA bulk (ATCC VR-24) for IgG antibody detection (Virion, Zurich). Protein was diluted to 0.6 μg/ml with 0.05 M carbonate buffer (pH 9.4), and 100 μl/well was added to 96-well microtiter plates. The plates were incubated overnight at 4 °C, washed with PBS-0.05% Tween 20 (PT), incubated with PT (0.1 ml/well)-10% bovine serum albumin for 60 min at 37 °C, and washed again with PT. Serial two-fold dilutions of the tested sera were added (100 μl/well), and the plates were incubated for 60 min at 37 °C.
The plates were washed with PT and were incubated with 100 μl per well of goat anti-mouse IgG (Gamma) HRP (074–1802 KLP) diluted 1:5000 in PBS-0.05% Tween 20 for 30 min at 37 °C. The plates were washed with PT and incubated with 100 μl OPD (o-Phenylendiamin, Fluka, 78411). The reaction was stopped after 3–4 min. Plates were read on a MicroElisa reader at a wavelength of 492 nm. Readings higher than three-fold above negative controls were scored as positive reactions.
2.10.2. SIV env ELISA
Anti-SIV env IgG antibody titers in the sera of immunized mice were determined by ELISA assay. Briefly, 96-well plates were coated with SIV mac239 gp130 antigen kindely obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (SIV mac239 gp130, Catalog Number: 2322) diluted with carbonate buffer pH 9.4 at a concentration of 50 ng/well. The plates were incubated overnight at 4 °C and washed with PT. Subsequently unspeciic interaction were blocked with 10% defatted milk dissolved in PT for an hour at 37 °C and wells were washed again with PT. The plates were consecutively incubated with various dilutions of mouse sera (starting at 1:100, followed by serial two-fold dilutions), peroxidase-conjugated goat anti-mouse IgG (074–1802 KLP) and with OPD substrate (o-Phenylendiamin, Fluka). Optical density values were measured at 492 nm. Values above the cut-off background level (mean value of sera from MV immunized mice multiplied by a factor of 2.1) were considered positive. Titers were depicted as reciprocal end-dilutions.
3. Results
3.1. Protein expression by recombinant MV vectors
We have shown earlier that large transgenes (2–3.2 kb) [21], [31] are stably expressed by MV vector. Multiple insertions, and simultaneous expression of multiple genes appeared challenging. Fig. 2A shows that three reporter genes of GFP, LacZ and CAT are actively expressed by a single MV construct. The total size of inserted genetic material, around 30% of MV-genome, did not restrict virus assembly or gene expression. Since reporter genes are cytosolically expressed, it is possible that their expression may not be hampered by virus assembly and growth that usually takes place at the cell surface. We therefore, determined the efficacy of expression of heterologous antigens of SIV env (a transmemberane protein) and gag (a cytosolic protein). Fig. 2B indicates the expression of both env (green) and gag (red) in the same syncytia. The stability of expression was determined after 10 passages on Vero cells demonstrating the stability of protein expression of single transgene (pol), previously reported and used here as a control, or double transgenes (env and gag) (Fig. 2C). These results support the use of MV as a vector for expression of single or multiple genes.
Fig. 2.
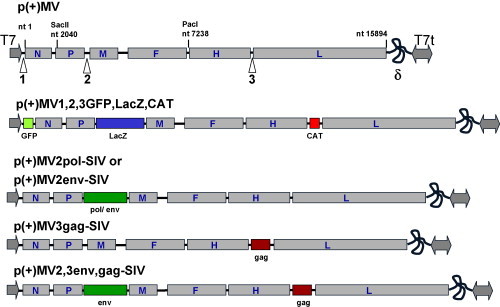
Expression and stability of marker genes (GFP, LacZ and CAT) and heterologous antigens of SIV (env, gag and pol). (A) Expression of GFP, LacZ and CAT by a single recombinant virus. Vero cells were infected with recombinant MV and syncytia were visualized by UV for GFP, and light microscopy for LacZ and CAT after beta-gal and CAT assays. (B) Co-expression of SIV env and gag simultaneously by recombinant MV. Indirect immunofluorescence assays performed on single syncytia of cells infected recombinant MV expressing SIV env and gag. Monoclonal anti-SIV gp120 coupled to FITC and polyclonal anti-gag antibody coupled to Cy3 visualized the localization of env and gag by confocal microscopy. (C) Stability of transgene (s) expression by recombinant MV. Vero cells were infected with recombinant MVs expressing either pol or both env and gag genes. Shed virions were passaged 10 time (see Section 2). Virions of passages 1 and 10 were used to infect equal Vero cells cultures at an MOI of 0.1. Appearance of syncytia was monitored by microscopy. After 36–48 h p.i. cells were lysed and equal amounts of lysates were analyzed by SAS-PAGE and immunoblots using polyclonal anti-SIV antibody. Standard MV was used as a negative control.
3.2. Density and composition of MV and infectious recombinant MV particles
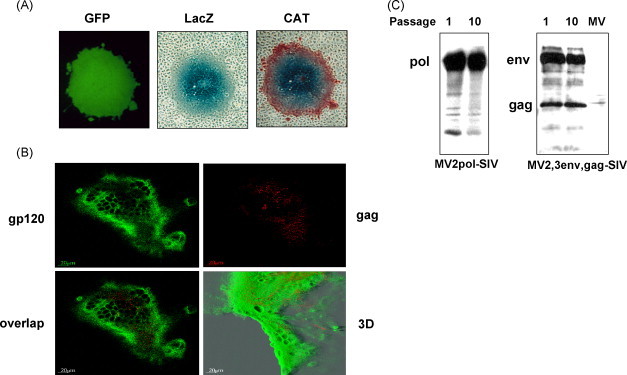
We were concerned that envelope proteins (with transmembrane domains) of foreign viruses (in this case the SIV-env) might be integrated into the lipid envelope of matured recombinant virus, thus affecting the tropism of the virus. Therefore, cell-free virus particles (recombinant MV-SIV expressing either env alone or both env and gag) from infected Vero cell cultures were sedimented to equilibrium on a 20–60% sucrose gradient by ultracentrifugation. To determine the protein contents of each fraction, gradient fractions were analyzed by Western blots and probed with monoclonal anti-gp120 (for SIV-env) and a mixture of monoclonal antibodies to MV P, and M and, in a separate experiment, using a monoclonal anti-MV-H antibody (for MV). In addition, infectivity of the gradient fractions and density were determined (Fig. 3 ). The MV infectious particles were detectable in fractions 10–15 of both gradients. Subsequently, MV P, M, components of the infectious particle were also detected in these fractions (10–15) (Fig. 3A). Some SIV-env protein was detectable in the top fractions of the gradient (Fig. 3A, fractions 2–7), likely due to membrane associated env that was not clarified by centrifugation. Upon expression of SIV-env and gag together, a slight shift of SIV-env into heavier fractions was observed (Fig. 3B). However, the env-gag complex did not co-localize with MV particles. The fact that only in the presence of gag and env, the desity was shifted heavier, only if env and gag are co-expressed indicating that these antigens may have formed a complex or virus-like particles (VLP) of higher density than the env alone (Fig. 3B). Determination of the buoyant densities of all particles did not show differences between recombinant MVs and standard MV (1.16 g/ml–1.20 g/ml). In summary, we demonstrate that SIV-env protein does not incorporate into the MV lipidic envelope and that the only structural modification made to virions was the addition of the env, gag or env-gag genes into the viral genomic RNA.
Fig. 3.
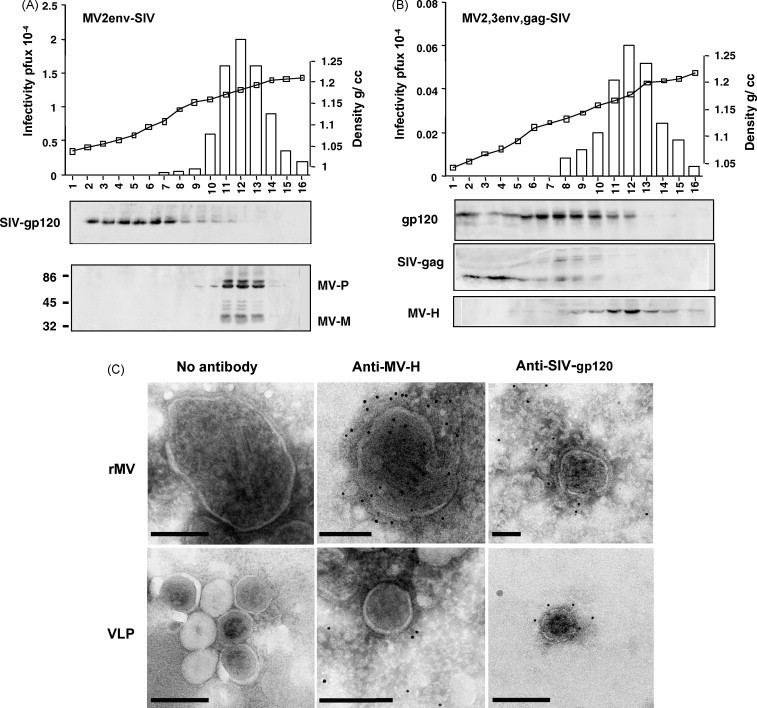
SIV env protein containing the transmembrane domain does not incorporate into mature recombinant measles viruses. Cell-free viruses were analysed by ultracentrifugation on a 20–60% sucrose gradient. (A, B) The protein-content, infectivity and density of each fraction was analyzed by immunoblots, plaque assays and precision wt determination, respectively. The SIV env protein was detected using monoclonal anti-SIV-gp120 antibodies. MV proteins were identified using monoclonal anti N and M antibodies (A) and anti-H (B). (A) The localization of SIV env and MV-structural proteins, respectively, is shown for the same gradient fraction. (B) The localization of SIV env and gag after co-expression by a single recombinant was compared to the localization of MV H. A shift in the density of the SIV env upon concommitant expression of gag from the double recombinant MV. (C) Immunoelectron microscopic visualization of the double recombinant MV (expressing env and gag simultaneously). Cell free viruses were pelletted over a 20% sucrose cushion and fixed and stained on EM grids and further treated with specific antibodies. Monoclonal anti-MV-H antibody coupled to 10 nm gold particles, or monoclonal anti-SIV-gp120 coupled to 6 nm gold particles were used. Bars = 200 nm.
3.3. Immunoelectron microscopy of MV and recombinant MV
In order to visualize envelope protein localization and distribution as well as potential incorporation of SIV-env into the MV envelope and to corroborate the above biochemical data, cell-free MV and recombinant MV-SIVs were examined by immunoelectron microscopy. Viruses were immunolabeled with an indirect reaction employing anti-MV-H monoclonal antibody cl5, or monoclonal anti-SIV-env antibody (gp120) followed by protein A coated with 6 nm or 10 nm colloidal gold. The anti-H antibody was reactive to the envelope of the large pleomorphic MV virions (Fig. 3C) which show typical electron-dense RNP structures accumulated inside the H-containing envelope. In contrast, anti-SIV antibody did not localize to on the surface of recombinant MV particles. Few gold particals were sporadically observed away of the virus (Fig. 3C). Interestingly, VLPs were observed only in the preparation of recombinant MV expressing both SIV-env and gag (MV2,3env,gag-SIV) (Fig. 3C) while no VLPs were observed in recombinants expressing SIV-env alone (not shown). The diameter of these round-shape structures varried from 50–100 nm were lightly coated with anti-SIVgp120-colloidal gold particles suggesting that SIV env is likely present on these particles. Since this phenomenon is only occuring upon coexpression of SIV-env and gag, we conclude that the presence of VLPs in this preparation is due to env-gag association. These results are in accord with previous findings from a different lab [32].
3.4. In vivo replication of MV and recombinant MVs
TaqMan technology was used to quantify the in vivo replication of the recombinant viruses. Ten mice susceptible to measles infection were immunized i.p. by 104 PFU MV, and spleens were collected at an interval of 2 days during 10 days post immunization (Fig. 4A). Isolated total RNA was reverse transcribed, using a minus-strand primer that hybridizes to the last 24 nucleotides of the N mRNA, and a segment of 73 bases of the cDNA was amplified by PCR (see Section 2). As a standard for quantification, in vitro transcribed MV N RNA was used. The results of triplicate experiments show that the MV replication was highest at day 2 post immunization (108 copies per μg total RNA) and then gradually decreased reaching a three logs reduction at day 10 (Fig. 4A).
Fig. 4.
Immunization of transgenic mice with MV and recombinant MVs. (A, B) Kinetics of the in vivo replication of MV (A) and the magnitude of replication of recombinant MVs was compared to that of the standard MV (B). The kinetics of replication was determined by TaqMan (see Section 2). (A) Mice were first immunized with 104 PFU of standard MV. Every 2 days, the number of MV-N copies were determined from spleens of two mice. (B) Mice were immunized i.p. with equal MOI of standard MV or recombinant MVs. Spleens were collected 5 days post immunization and total RNA was purified and the number of MV-N copies per μg RNA was determined (see Section 2). (C, D) Induction of humoral immune responses by MV and recombinant MVs against MV and SIV env. The induction of anti-MV antibodies in CD46 transgenic mice persisted up to 50 weeks post immunization. As shown in the different panels, MV and recombinant MV-SIVs induced comparable anti-MV IgG profiles, irrespective of the size of the gene insert. (C) Anti-MV antibody was measured by standard ELISA assays, and anti-SIV env antibody was measured by end-dilution (see Section 2). (D) For SIV env ELISA, sera from five mice were pooled, and coated plates were treated as described in Section 2. Values above the cut-off background level, mean value of sera from MV immunized mice (negative controls) multiplied by a factor of 2.1, were considered positive. Titers were depicted as reciprocal end-dilutions.
To determine whether rMV-SIV viruses replicate in the same fashion, similar experiments were done on spleen samples withdrawn 5 days post immunization. Mice immunized with MV and rMV-SIVs (MV2env-SIV, MV3env-SIV, and MV2,3env,gag-SIV) showed replication of these viruses at a comparable level (Fig. 4B). These results indicate that in vivo, MV and recombinant MVs replicate with similar kinetics. Although the double recombinant MV2,3env,gag-SIV showed a slightly lower copy number of N mRNA, this did not significantly affect the induction of immune responses (see below).
3.5. MV vectors induce persistening humoral immune responses against MV and SIV antigens
The expression of SIV-env, and gag was documented earlier. The recombinant viruses expressing env in postion 2, or 3, and a recombinant virus expressing simultaneously env in position 2 and gag in position 3 were also used to immunize transgenic mice. Immune responses towards MV, and SIV-env were determined by ELISA. MV specific antibody responses induced by the standard MV vaccine or rMV-SIV were comparable up to 50 weeks post immunization (Fig. 4C). UV inactivated MV induced only marginal levels of antibody titers fading away very quickly, indicating that replication of MV or rMV is necessary to induce high and persisting immune reponses. For the three rMV-SIV vectors the initial induction of IgGs against SIV-env protein appeared very different at 8–20 weeks post immunization, but the antibody levels reached rather similar end points at 50-week post immunization (Fig. 4D). While very high early (8 weeks) anti-env antibody titers were induced by the double recombinant MV2,3env,gag-SIV, recombinant MV expressing env from position 3, MV3env-SIV, appeared to elicit the lowest initial anti-SIV-env antibody titer at week 8 post immunization. This is remarkable in view of the fact that the double recombinant grew to slightly lower titers than the other two recombinants (Fig. 4B). We observed that anti-SIV-env antibody titers remained rather high, declining only slowly after 20 weeks, similar to anti-MV antibodies. Therefore, the longevity of humoral immune response against foreign antigens appear to follow kinetics similar to those observed for the MV specific antibody response (Fig. 4D).
4. Discussion
The results described in this report demonstrate that the non-segmented, negative strand RNA measles virus used as a vector (i) can stably express heterologous genes singly or in combination, (ii) excludes foreign antigens, shown for SIV-env, from incorporation into the envelope of viral progeny, and (iii) single or multiple genes expression induce long-lived humoral immune responses against the vector (MV) and the inserted gene-products (SIV).
Due to the helical structure of the MV nucleocapsid, it appears that there is no definite size limitation for the insertion of foreign genetic material (size of inserts can exceed 5 kb). Whereas this value was established for reporter genes, insertions of similar size encoding SIV antigens was also successfully achieved. Although the genetically heavily loaded viruses propagated slightly slower than standard MV, the resulting end viral titers were similar. Even more gratifying was the observation that after 10 serial passages, with a total amplification of around 1020-fold, the high genetic stability of rMVs ensured expression of different genes, including pathogens derived heterologous genes [30]. While large inserts may be tolerated mainly in view of the little constraint on genome size for such a pleomorphic virus, with a helical nucleocapsid, we believe that the stability of gene expression is likely due to other factors. The tight RNP structure, the adherence of MV-genome to the rule of six defined by Calain and Roux [2] and the very slow elongation rate of RNA during replication may allow internal proof-reading control by the viral polymerase.
The fact that MV is a live attenuated vaccine strain, genetically engineered live recombinant MVs should be carefully analyzed, e.g., confirmation of sequence identity of vector moity to corresponding sequence of the original vaccine strain. Indeed, regulatory hurdle may arise if essential biological properties of genetically modified organisms (GMOs) are modified or completely changed. This also applies to recombinant MV. Importantly, the maintained fuctionality and targeting of the recombinant vaccine virus, i.e. its in vivo tropism, should be maintained [19]. We analyzed the incorporation of foreign transmembrane protein (SIV-env) into the cell-free recombinant MV, biochemically and by immunoelectron microscopy and could demonstrated that SIV env was excluded from the envelope of MV. Therefore, the induction of immune responses towards SIV-env is due to the in vivo expression of the transgene during replication of recombinant MV in target cells of the experimental animals (reported here). In fact, the results presented here demonstrate for the first time the replication of recombinant MVs in transgenic mice.
The MV vectors carrying either single or multiple genes at different genomic locations find various direct medical applications, specifically for the development of prophylactic multivalent vaccines. Interestingly, since MV efficiently infects professional antigen presenting cells, macrophages and particularly dendritic cells [8], MV-based vectors deliver their foreign cargo directly to the most efficient antigen presenting cells. Therefore, the wide experience with MV as a measles vaccine favours the use of MV vector as a model for the development of novel multivalent vaccines, including HIV. MV and HIV have several properties in common, since they both infect monocytes, macrophages and dendritic cells [8], [14]. In this respect, an MV vector that targets the HIV proteins in the same compartment as HIV itself may provide advantages over alternative vector systems, particularly in the induction of “danger signals” [17]. In addition, replicating live attenuated MV has the advantageous capacity to induce long-lasting immunity [13]. We also demonstrated here that recombinant MVs, expressing SIV antigens, induced significantly high and long-lived antibody responses against both MV and SIV-env antigen. Therefore, with its capacity to induce a strong and long lasting protective immunity, the live-attenuated MV expressing the appropriate HIV genes could potentially serve as a pediatric vaccine protecting simultaneously against measles and AIDS. The immunogenicity induced by our system is likely due to (i) quantitative levels of expression and presentation of the antigens in cells infected by live recombinant MV and, (ii) the proper and efficient presentation to the immune system.
The immunogenicity of recombinant MV expressing heterologous antigens, beta-Gal [21], HBV [28], HIV [16] or WNV [3], was tested in transgenic mice [18]. The recombinant MV vector expressing Beta-Gal significantly activated cellular immune responses [21]. Immune responses to HBV surface and core antigens were tested both after intranasal and intraperitoneal inoculation, revealing high levels of anti-MV antibodies and reasonable amounts of antibodies to HBV [28]. rMV vectors expressing the secreted form of the E-glycoprotein of WNV strain IS-98-ST1, and the surface S antigen of SARS-CoV induced neutralizing antibodies to both WNV [3] and SARS-CoV (Liniger et al., submitted). Furthermore, immunization with chimeric MV-VSV, envelope swaps between MV (F and H) and vesicular stomatitis virus G (VSV-G) protected immunocompromized mice from lethal doses of VSV [9], [29]. Most importantly, it was demonstrated that pre-existing antibodies MV did not block the immunogenic potential of recombinant virus that was able to induce anti-HIV antibodies in pre-vaccinated mice and macaques, provided that two injections were administered [16].
The last feature is highly relevant to the potential widspread use of MV as a vaccine vector. Indeed, the presence of anti-MV immunity in nearly the entire adult human population would seem to restrict the use of MV recombinants to infants, an already worthy goal in any event. However, large vaccination studies showed that re-vaccinating already immunized individuals results in a boost of anti-MV antibodies, suggesting that the attenuated live vaccine replicated and expressed its proteins in spite of pre-existing immunity [4]. Studies of cell-mediated immunity in infants given measles vaccine during the first year of life show that the presence of maternal anti-measles antibodies does not preclude the induction of anti-measles cellular responses and that responses can be very efficiently boosted [10], [11]. Under such circumstances one might hope to be able to vaccinate adults against a foreign antigen with rMV. Indeed, this has been demonstrated earlier, in preclinical models, by Lorin et al. [16], and results from our laboratory (Liniger et al., not published). However, it remains to be seen whether MV vectors can induce immunity to carrier proteins in already immunized individuals and to evaluate the safety of MV vectors in phase I and II clinical studies. Such trials will aim at: (i) defining the appropriate dose of rMV and (ii) demonstrating an acceptable reactogenicity profile of recombinant viruses and at confirming the absence of in vivo virus shedding from vaccinees.
Finally, recombinant MV vaccine intended to immunize humans simultaneously against measles and against additional infectious disease must be derived from an approved strain of MV vaccine. For this purpose, a clinically approved MV vaccine Edmonston Zagreb (EZ) vaccine strain (Berna Biotech LTD), that is widely used in Europe, Asia and Africa and whose safety was also demonstrated as an aerosolized vaccine in South Africa and Mexico [1] will be used.
Acknowledgments
This work was supported by the National Institute of Health (NIH AI46007). Thanks to Drs. M. Feinberg, R. Andino, F. Tangy and colleagues for the challenging collaboration in frame of the HIVRAD grant. We thank Dr. E. Ehler, department of Cell biology, ETH and Elisabeth Schraner, Institute of Vet. Virology, for support with microscopy. Thanks to Dr F. Wild for supplying the monoclonal anti-MV antibodies, and the NIH AIDS research and reference reagent program for supplying different antibodies to SIV.
References
- 1.Bennett J.V., Fernandez D.C., Valdespino-Gomez J.L., Garcia-Garcia M.L., Islas-Romero R., Echaniz-Aviles G. Aerosolized measles and measles-rubella vaccines induce better measles antibody booster responses than injected vaccines: randomized trials in Mexican schoolchildren. Bull World Health Organ. 2002;80:806–812. [PMC free article] [PubMed] [Google Scholar]
- 2.Calain P., Roux L. The rule of six, a basic feature for efficient replication of Sendai virus defective interfering RNA. J Virol. 1993;67:4822–4830. doi: 10.1128/jvi.67.8.4822-4830.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Despres P., Combredet C., Frenkiel M.P., Lorin C., Brahic M., Tangy F. Live measles vaccine expressing the secreted form of the West Nile virus envelope glycoprotein protects against West Nile virus encephalitis. J Infect Dis. 2005;191:207–214. doi: 10.1086/426824. [DOI] [PubMed] [Google Scholar]
- 4.Dilraj A., Cutts F.T., de Castro J.F., Wheeler J.G., Brown D., Roth C. Response to different measles vaccine strains given by aerosol and subcutaneous routes to schoolchildren: a randomised trial. Lancet. 2000;355:798–803. doi: 10.1016/s0140-6736(99)95140-1. [DOI] [PubMed] [Google Scholar]
- 5.Duprex W., McQuaid H., Hangartner L., Billeter M., Rima B. Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescenct protein expressing recombinant virus. J Virol. 1999;73:9568–9575. doi: 10.1128/jvi.73.11.9568-9575.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ehrengruber M.U., Ehler E., Billeter M.A., Naim H.Y. Measles virus spreads in rat hippocampal neurons by cell-to-cell contact and in a polarized fashion. J Virol. 2002;76:5720–5728. doi: 10.1128/JVI.76.11.5720-5728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Enders J.F., Peebles T.C. Propagation in tissue cultures od cytopathogenic agents from patients with measles. Proc Soc Exp Biol Med. 1954;86:277–286. doi: 10.3181/00379727-86-21073. [DOI] [PubMed] [Google Scholar]
- 8.Esolen L.M., Ward B.J., Moench T.R., Griffin D.E. Infection of monocytes during measles. J Infect Dis. 1993;168:47–52. doi: 10.1093/infdis/168.1.47. [DOI] [PubMed] [Google Scholar]
- 9.Fehr T., Naim H., Bachmann M., Ochsenbein A., Spielhofer P., Bucher E. T-cell independent IgM and enduring protective IgG antibodies induced by chimeric measles viruses. Nat Med. 1998;4:945–949. doi: 10.1038/nm0898-945. [DOI] [PubMed] [Google Scholar]
- 10.Gans H., Yasukawa L., Rinki M., DeHovitz R., Forghani B., Beeler J. Immune responses to measles and mumps vaccination of infants at 6, 9, and 12 months. J Infect Dis. 2001;184:817–826. doi: 10.1086/323346. [DOI] [PubMed] [Google Scholar]
- 11.Gans H., DeHovitz R., Forghani B., Beeler J., Maldonado Y., Arvin A.M. Measles and mumps vaccination as a model to investigate the developing immune system: passive and active immunity during the first year of life. Vaccine. 2003;21:3398–3405. doi: 10.1016/s0264-410x(03)00341-4. [DOI] [PubMed] [Google Scholar]
- 12.Griffin D. 4th ed. Lippincott Williams & Wilkins; Philadelphia: 2001. Measles virus. [Google Scholar]
- 13.Hilleman M.R. Current overview of the pathogenesis and prophylaxis of measles with focus on practical implications. Vaccine. 2002;20:651–665. doi: 10.1016/s0264-410x(01)00384-x. [DOI] [PubMed] [Google Scholar]
- 14.Hilleman M.R. Vaccinology, immunology, and comparative pathogenesis of measles in the quest for a preventive against AIDS. AIDS Res Hum Retroviruses. 1994;10:3–12. doi: 10.1089/aid.1994.10.3. [DOI] [PubMed] [Google Scholar]
- 15.Howley P.M., Lafont B., Spehner D., Kaelin K., Billeter M.A., Drillien R. A functional measles virus replication and transcription machinary encoded by the vaccinia virus. J Virol Methods. 1999;79:65–74. doi: 10.1016/s0166-0934(99)00012-9. [DOI] [PubMed] [Google Scholar]
- 16.Lorin C., Mollet L., Delebecque F., Combredet C., Hurtrel B., Charneau P. A single injection of recombinant measles virus vaccines expressing human immunodeficiency virus (HIV) type 1 clade B envelope glycoproteins induces neutralizing antibodies and cellular immune responses to HIV. J Virol. 2004;78:146–157. doi: 10.1128/JVI.78.1.146-157.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 18.Mrkic B., Pavlovic J., Rulicke T., Volpe P., Buchholz C.J., Hourcade D. Measles virus spread and pathogenesis in genetically modified mice. J Virol. 1998;72:420–7427. doi: 10.1128/jvi.72.9.7420-7427.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naim H.Y., Ehler E., Billeter M.A. Measles virus matrix protein specifies apical virus release and glycoprotein sorting in epithelial cells. EMBO J. 2000:3576–3585. doi: 10.1093/emboj/19.14.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Naim H.Y., Roth M.G. Basis for selective incorporation of glycoproteins into the influenza virus envelope. J Virol. 1993;67:4831–4841. doi: 10.1128/jvi.67.8.4831-4841.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neumeister C., Nanan R., Cornu T., Luder C., ter Meulen V., Naim H. Measles virus and canine distemper virus target proteins into a TAP-independent MHC class I-restricted antigen-processing pathway. J Gen Virol. 2001;82:441–447. doi: 10.1099/0022-1317-82-2-441. [DOI] [PubMed] [Google Scholar]
- 22.Ovsyannikova I.G., Dhiman N., Jacobson R.M., Vierkant R.A., Poland G.A. Frequency of measles virus-specific CD4+ and CD8+ T cells in subjects seronegative or highly seropositive for measles vaccine. Clin Diagn Lab Immunol. 2003;10:411–416. doi: 10.1128/CDLI.10.3.411-416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radecke F., Billeter M.A. Reverse genetics meets the nonsegmented negative-strand RNA viruses. Rev Med Virol. 1997;7:49–63. doi: 10.1002/(sici)1099-1654(199704)7:1<49::aid-rmv181>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 24.Radecke F., Spielhofer P., Schneider H., Kaelin K., Huber M., Dotsch C. Rescue of measles viruses from cloned DNA. EMBO J. 1995;14:5773–5784. doi: 10.1002/j.1460-2075.1995.tb00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rota J., Wang Z., Rota A.P., Bellini W. Comparison of sequences of the H, F and N coding genes of measles virus vaccine strains. Virus Res. 1994;31:317–330. doi: 10.1016/0168-1702(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 26.Schwarz A. Preliminary tests of a highly attenuated measles vaccine. Am J Dis Child. 1962;103:216–219. doi: 10.1001/archpedi.1962.02080020398042. [DOI] [PubMed] [Google Scholar]
- 27.Singh M., Billeter M.A. A recombinant measles virus expressing biologically active human interleukin-12. J Gen Virol. 1999;80:101–106. doi: 10.1099/0022-1317-80-1-101. [DOI] [PubMed] [Google Scholar]
- 28.Singh M., Cattaneo R., Billeter M.A. A recombinant measles virus expressing hepatitis B virus surface antigen induces humoral immune responses in genetically modified mice. J Virol. 1999;73:4823–4828. doi: 10.1128/jvi.73.6.4823-4828.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spielhofer P., Bachi T., Fehr T., Christiansen G., Cattaneo R., Kaelin K. Chimeric measles viruses with a foreign envelope. J Virol. 1998;72:2150–2159. doi: 10.1128/jvi.72.3.2150-2159.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tangy F., Naim H.Y. Live attenuated measles vaccine as a potential multivalent pediatric vaccination vector. Viral Immunol. 2005;18:317–326. doi: 10.1089/vim.2005.18.317. [DOI] [PubMed] [Google Scholar]
- 31.Wang Z., Hangartner L., Cornu T.I., Martin L.R., Zuniga A., Billeter M.A. Recombinant measles viruses expressing heterologous antigens of mumps and simian immunodeficiency viruses. Vaccine. 2001:19. doi: 10.1016/s0264-410x(00)00523-5. [DOI] [PubMed] [Google Scholar]
- 32.Yao Q., Bu Z., Vzorov A., Yang C., Compans R.W. Virus-like particle and DNA-based candidate AIDS vaccines. Vaccine. 2003;21:638–643. doi: 10.1016/s0264-410x(02)00572-8. [DOI] [PubMed] [Google Scholar]