

Abstract
Methylation is a ubiquitous covalent modification used to control the function of diverse biomolecules including hormones, neurotransmitters, xenobiotics, proteins, nucleic acids and lipids. Histone methyltransferases (HMTs) are currently of high interest as drug targets because of their role in epigenetic regulation, however most HMT assay methods are either not amenable to an HTS environment or are applicable to a limited number of enzymes. We developed a generic methyltransferase assay method using fluorescent immunodetection of AMP, which is formed from the MT reaction product S-adenosylhomocysteine in a dual enzyme coupling step. The detection range of the assay, its suitability for HTS, including stability of reagents following dispensing and after addition to reactions as well as the potential for interference from drug like molecules was investigated. In addition, the use of the assay for measuring inhibitor potencies with peptide or intact protein substrates was examined through pilot screening with selected reference enzymes including HMT G9a. By combining a novel enzymatic coupling step with the well characterized Transcreener® AMP/GMP assay, we have developed a robust HTS assay for HMTs which should be broadly applicable to other types of methyltransferases as well.
Keywords: epigenetics, histone methyltransferase, DNA methyltransferase, methyltransferase assay, high throughput screening
Introduction
Methyltransferases are a diverse family of enzymes that catalyze the transfer of a methyl group from S-adenosylmethionine (SAM) to amino, thiol, or hydroxyl groups of acceptor molecules, generating S-adenosylhomocysteine (SAH) as a byproduct. Acceptor substrates include endogenous and xenobiotic small molecules 11; 29, proteins 3, DNA 8 and RNA 14, and lipids 20. Their role in epigenetic regulation includes methylation of histones at lysine and arginine residues 16 and methylation of DNA at cytosines in hemi-methylated CpG sites 6. There are over 50 protein lysine methyltransferases (PKMTs) in humans, at least 10 protein arginine methyltransferases (PRMTs), and two DNA methyltransferases (DNMTs) 13. Together these enzymes play a critical role in the dynamic modification of chromatin, and they are increasingly being targeted for cancer and other diseases with an epigenetic component 3; 8; 23.
Assay methods for methyltransferases rely on either detection of the methylated product or detection of SAH 13. Filter- or flash plate-based radioassays using 3H-SAM to generate 3H-methylated products 4; 22 are the most quantitative and reliable method, however the associated regulatory and disposal costs are a liability for HTS. Immunoassays for methylated lysine, arginine and cytosine have been used for both PKMT and DNMT enzyme assays, either in an ELISA format or in a homogenous format such as TR-FRET 1; 18. Antibody selectivity is critical for this approach and can limit the utility of the assay. For instance some histone methyltransferases can generate both mono- and di-methylated lysine products, and the known antibodies do not recognize both forms 13. Assays based on enzymatic cleavage (or protection) of products have been applied to both PKMTs 26 and DNMTs 27 utilizing methylation state–dependent restriction enzymes or endoproteinase Lys C, which is unable to cleave at methylated lysine residues. Though application of this approach has generally relied on a solid phase method such as ELISA, or a separation step, a more HTS-friendly fluorescence de-quenching configuration was recently developed for DNMT1 27.
Detection of SAH formation has the advantage of providing universal detection of MT enzymes regardless of the acceptor substrate or the mix of methylated reaction products. Several coupled enzyme assays have been deployed for SAH detection. For instance, SAH can be converted to homocysteine and adenosine using SAH hydrolase, and homocysteine is then detected using covalent reaction with a thiol-sensitive fluor 2; 10. Alternatively, the adenine portion of SAH can be converted to urate by the sequential action of three coupling enzymes, with co-production of hydrogen peroxide in the final step 5. Hydrogen peroxide can be detected colorimetrically or fluorescently, via formation of resorufin. Recently, a luciferase-based coupled assay was developed that relies on the sequential conversion of SAH to adenine, AMP, and ATP, with the final step catalyzed by pyruvate phosphate dikinase 12.
BellBrook has developed a line of HTS assays, called Transcreener, that use highly specific immunodetection of nucleotide reaction products with homogenous fluorescent readouts including FP, TRF and FI 15; 19; 24. Direct immunodetection of SAH would be advantageous as it would eliminate the potential for compound interference with coupling enzymes, however it requires an antibody that specifically binds SAH in the presence of excess SAM; ie, that differentiates on the basis of a single methyl group. Though we have produced polyclonal antibodies in rabbits with more than 100-fold selectivity for SAH vs. SAM, (data not shown), we have been unable to produce sufficient quantities of high titer antibodies to support commercialization of an HTS assay kit. There is one literature report of an FP-based methyltransferase assay using an anti-SAH antibody from a diagnostic assay kit for homocysteine 7. However we and others have been unable to obtain the antibody from the commercial suppliers.
To overcome this hurdle, we used two coupling enzymes, SAH hydrolase and adenosine kinase, to convert the SAH consecutively to adenosine and AMP. The AMP can be measured with nanomolar sensitivity using an existing Transcreener monoclonal antibody and tracer with negligible cross-reactivity with the other nucleotides present, SAM 24 and dGTP (Figure 1). We optimized the assay components for measuring methyltransferase initial velocity using initial SAM concentrations ranging from 0.20 μM to 50 μM and tested reagent and signal stability and interference from drug molecules. We next showed that the assay could be used to detect histone methyltransferases and DNA methyltransferases and examined the response to known inhibitors. Last, we performed a pilot screen of 8,800 small molecules with HMT G9a in 1,536 well format. The results show that combining coupling enzymes for conversion of SAH with the Transcreener AMP/GMP Assay provides a robust, HTS-compatible approach for screening methyltransferases.
Figure 1. Transcreener Epigen methyltransferase assay principle.

SAH produced in a methyltransferase reaction is converted to AMP in two sequential enzymatic steps. AMP is detected using a competitive fluorescence polarization immunoassay.
Materials and Methods
Materials
The monoclonal antibody and Alexa Fluor®633 tracer for immunodetection of AMP were developed at BellBrook Labs (Madison, WI). Alexa Fluor®633 succinimidyl ester used was purchased from Invitrogen (Carlsbad, CA) and conjugated to AMP. The Tocriscreen Mini compound library was from Tocris Bioscience (Ellisville, MO). SAM was obtained from AK Scientific (Mountain View, CA). SAH, AMP and other nucleotides were from Sigma (St. Louis, MO). Buffer components were purchased from Sigma (St. Louis, MO) or Fisher Scientific (Hampton, NH). All methyltransferases were recombinant human proteins with an N-terminal GST tag. G9a HMT, SUV39H1 HMT, DNMT1 MT, and DNMT3a MT purchased from BPS Biosciences (San Diego, CA) and Set7/Set9 HMT was from Prospec Protein Specialists (Ness-Ziona, Israel). G9a (amino-acids 785–1210) was expressed using E. coli (specific activity = 146 pmol/min/mg) and baculovirus infected Sf9 insect cells (specific activity = 10 pmol/min/mg). SUV39H1 (full length, specific activity 400 pmol/min/mg) was expressed in Sf9 cells. Set7/Set9 was expressed in E. coli (366 amino-acids, specific activity unknown). DNMT1, (amino-acids 2–1632, specific activity = 0.14 pmol/min/mg) and DNMT3a (full length, specific activity unknown) were expressed in Sf9 cells.
Instrumentation and analysis
Assays were performed in black Corning® 384 Well Flat Bottom Microplates (Part #3654) (Corning, NY) or black Corning 384 Well Round Bottom Low Volume Polystyrene Non-Binding Surface Microplates (Part # 3676). Mixing after additions was performed by orbital shaking for one minute. Unless otherwise noted, 20 μL assays were equilibrated for one hour at room temperature before reading the plate on the instrument.
Fluorescence polarization measurements utilizing the AMP-Alexa Fluor®633 tracer were performed on a Tecan Safire2™ (Tecan, Durham, NC), Perkin Elmer Envision (Perkin-Elmer, Waltham MA) or BMG PheraStar Plus (BMG Labtech, Cary, NC) plate reader using 635 nm excitation (LED) and 670 emission (20 nm bandwidth) settings. The free tracer reference was set to 20 mP, and the buffer (with or without antibody) was used as the buffer blank for both the sample and free tracer reference wells.
Equilibrium binding data, standard curves, and enzyme velocity data was analyzed and graphed using GraphPad Prism (GraphPad Software, San Diego, CA).
Assay Development
Antibody specificity
The AMP detection component for the assay is a competitive immunoassay that relies on displacement of a fluorescent tracer from antibody by analytes 24. The affinity of AMP and cross-reacting molecules was measured using competition binding experiments and quantified by IC50 determinations. Competition binding experiments were carried out by titrating AMP, SAM, SAH, adenosine, and dGTP independenty into wells containing AMP antibody and Alexa Fluor®633 AMP tracer. Final conditions were 20 mM HEPES (pH 7.5), 100 mM KPO4 (pH 7.5), 20 mM KCl, 40 mM MgCl2, 4 nM tracer; the AMP antibody was present at 3.0 μg/ml, which was 85% of saturation. EC85 was calculated by using the EC50 and hillslope values, calculated from fitting the equilibrium binding data to a variable slope sigmoidal dose-response curve using the equation below.
Assay development was carried out using mock reactions containing SAM and SAH at ratios representing quenched MT enzyme reactions. Mock reactions were run similarly to endpoint enzymatic reactions; i.e., with the inclusion of a quench step prior to addition of detection reagents. SAM/SAH mixtures were added to wells at the indicated concentrations in 10 μl of HMT Buffer (50 mM Tris, 5 mM MgCl2, 4 mM DTT, pH 8.5), followed by addition of 5μl of HMT Stop Buffer (400 mM MES, 1M NaCl, and 4 mM MgCl2, pH of 6.0) and by 5 μl of Detection Mix (40 mM HEPES, 0.04% Brij-35, 16 nM Alexa Fluor®633 AMP tracer and AMP antibody as indicated, pH 7.25). Unless otherwise indicated, final read conditions were 10 mM HEPES (pH 7.2), 12.5 mM Tris (pH 8.5), 100 mM MES (pH 6.0), 250 mM NaCl, MgCl2, 2 mM DTT, 250 μM Cofactor, 0.01% Brij-35, 4 nM tracer, 2 ng/μL SAH hydrolase, 5 ng/μL adenosine kinase and antibody.
Coupling Enzymes
The two coupling enzymes, SAH hydrolase and adenosine kinase were first tested separately for each of the two steps in the conversion of SAH to AMP, and then tested together in a matrix fashion to determine the relative amounts of each to use in the assay. Enzymes from different commercial sources as well as enzyme produced in house were tested. An FP-based immunoassay for the disappearance of SAH was used to monitor SAH hydrolase (BellBrook Labs, unpublished data), and AMP immunodetection was used to monitor the adenosine kinase. A recombinant human SAH hydrolase and a recombinant human adenosine kinase were selected for use in the assay; both are available from commercial sources. The final SAH hydrolase and adenosine kinase concentrations used for the assay were 2.5-fold and 100-fold in excess of what was required for a maximal signal in mock reactions representing initial velocity MT reactions with initial SAM concentrations ranging from 0.5 μM to 100 μM.
Antibody Optimization
In a competitive immunoassay with antibody in excess, the antibody concentration determines the dynamic range of the assay. The optimal amount of AMP antibody was determined by titrating it in two-fold dilutions into mock reactions representing 20% conversion of initial SAM concentrations of 0.5, 1.0, 5.0, 10, 50, and 100 μM (e.g., 0.2 μM SAH/0.8 μM SAM for the 1.0 μM initial SAM reaction) and polarization values were determined relative to control reactions with only SAM present (the equivalent of no-enzyme controls). Antibody concentrations of 1.0 μg/ml, 2.5 μg/ml, 5.0 μg/ml, 7.5 μg/ml and 10 μg/ml, respectively were selected as optimal for initial velocity reactions with SAM concentrations of <1, 1–4 μM, 5–9 μM, 10–24 μM and 25–50 μM.
SAM/SAH Standard Curves
Mock reactions mimicking enzymatic conversion (0, 1.0, 2.5, 5..0, 7.5, etc.……100%) of SAM to SAH were dispensed into wells in 10 μl of HMT Buffer, followed by the addition of 5 μl of Stop Buffer and 5 μl of Detection Mix as described above. For each standard curve, total [SAM + SAH] remained constant at 0.20, 0.5, 1.0, 5.0, 10, or 50 μM, representing the initial SAM concentrations in MT enzyme reactions and antibody was present at the optimal concentrations for each SAM concentration as determined above. Polarization was measured after 1 hour at ambient temperature. The data were plotted as ΔmP vs % SAM conversion using four-parameter nonlinear regression curve fitting. ΔmP = mPinitial [SAM] − mPsample. Z′ values were calculated using replicates of either 12 or 24 for each standard point. 28
Reagent and Signal Stability
To determine the equilibration time following addition of detection reagents and the stability of the signal, a 10 μM SAM/SAH standard curve (n = 12) was prepared as described above and the FP signal was read periodically. To assess the stability of the SAH Detection Mixture prior to dispensing, 10 μM SAM/SAH standard curves were prepared periodically using Detection Mix that had been stored at room temperature for up to 48 hours and freshly prepared SAM/SAH mock reactions. Reactions were allowed to equilibrate for 90 min prior to reading.
Compound Interference Screen
The potential for interference of drug molecules was assessed by screening the Tocriscreen Mini compound library (1120 compounds) in triplicate mock reactions representing 4% conversion of 10 μM SAM (9.6 μM SAM/0.4 μM SAH). Mock reactions were added to compound wells for a final concentration of 10 μM compound, 1% DMSO in 10 μl HMT Buffer, followed by addition of 5 μl of Stop Buffer and 5 μl of Detection Mix. Reactions were allowed to equilibrate for 90 min prior to reading. Control reactions representing 0%, 4% and 100% SAM conversion with no compound were included on each plate (n = 16). Z factor was determined using 0% control and compound wells and Z′ factor was determined using 0% control and 4% control wells 28.
Enzyme and Inhibitor Studies
The protocol used for enzyme assays was identical to that used for mock reactions. MT enzyme reactions (10 μl) were incubated at RT for the indicated time followed by addition of 5 μl Stop Buffer and 5 μl of Detection Mix, and plates were read following a 90 min equilibration period. The period between addition of Stop Buffer and Detection Mix was not carefully controlled, but generally was within one hour. HMT Buffer (50 mM Tris, 5 mM MgCl2, 4 mM DTT, pH 8.5) was used for HMTs G9a, Set7/Set9 and SUV39H1 and DNMT Buffer (50 mM Tris, 4 mM MgCl2, 2 mM EDTA, pH 8.0) was used for DNMT 1 and DNMT 3A. The stop buffer used for HMTs was 400 mM MES, 1M NaCl, and 4 mM MgCl2, pH of 6.0, and the same buffer with 14 mM MgCl2 was used to stop DNMTs. All enzymes were highly purified recombinant preparations from commercial sources (see Materials). A peptide representing amino acids 1–25 of Histone H3, generally at ≥ 2x Km, was used for HMT reactions except in indicated cases when full length Histone H3 was used at 1 μM. Poly d(I-C) at 5 μM was used as substrate for DNMT reactions. SAM was generally used at a concentration at or near its reported Km for each enzyme. Reactions were initiated by the addition of SAM. Enzyme assay signal, the decrease in polarization relative to control reactions lacking enzyme, was reported as a positive value, ΔmP. For some reactions, including inhibitor studies, SAM/SAH standard curves were used to convert the polarization values to amount of SAH formed so that accurate enzyme velocity and IC50 values could be determined.
Assay miniaturization and pilot screen
Using the chemical and biological reagents provided by Bellbrook as described above, the assay was run in black Greiner 1536-well Microplates (Part #789176-A, Greiner, Germany). The reagents were dispensed by the Synquad (Digilab Inc., MA) and the Multidrop Combi (Thermo Scientific, IL). Library compounds in 90% DMSO (50 nl of 2 mM stock for primary screening, 100 nl of serial dilutions for dose-response experiments and counterscreening) were transferred with a Hummingbird (Digilab Inc., MA) directly into the destination plate and incubated for 15 minutes at room temperature together with 2 μl of 2x concentrated enzyme (2 nM final concentration), 2x concentrated SAH (0.7 uM final concentration) for the counterscreen or buffer for controls. After addition of 2 μl of a 2x concentrated substrate mix (4 μM SAM and 4 μM H3K9 final concentration), incubation at room temperature was continued for a further 30 minutes at room temperature. The reaction was stopped by 2 μl of the Stop Buffer and the AMP production was initiated with 2 μl of SAH Detection Mixture for 90 minutes at room temperature and in the darkness before reading fluorescence polarization of the AMP-Alexa Fluor®633 tracer on an Analyst GT plate reader (Molecular Devices, provided through Bucher biotec, Switzerland) using 620 nm (40 nm bandwidth) excitation and 688 nm (45 nm bandwidth) emission settings. Data was analysed using the proprietary Helios software package (Gubler et al. 2006) as well as GraphPad Prism (GraphPad Software, San Diego, CA).
Results
Assay development and optimization
Antibody specificity
The key detection reagents are a highly specific antibody for AMP 24, two coupling enzymes, and a nucleotide cofactor, dGTP, (Figure 1). Minimal antibody cross-reactivity with SAM and dGTP is critical for assay feasibility. The competition binding curves in Figure 2 and the IC50 values (see legend to Figure 2) show that none of these molecules cross-react significantly with the AMP antibody, and thus will not contribute to background signal in the assay. Cross-reactivity with adenosine, the intermediate in the coupling reactions, was detectable but low. dGDP, the byproduct of the adenosine kinase reaction was not tested, however both ADP and GDP were previously shown to have negligible cross-reactivity (ref 23). Moreover, it is produced in stoichiometric amounts with SAH, so cross-reactivity with the AMP antibody would result in enhancement of the signal, not background.
Figure 2. Competition binding assays with nucleotides used or produced in the assay.
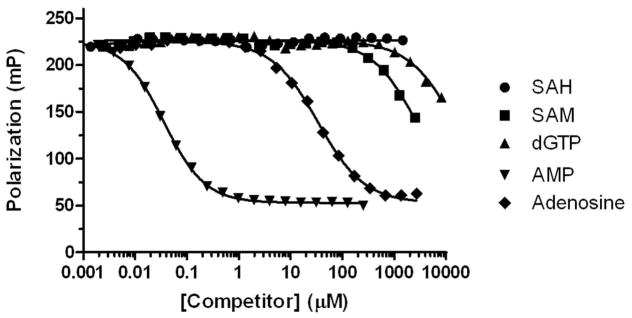
Serial dilutions of nucleotides were added independently to 3 μg/mL AMP antibody and 4 nM tracer, allowed to come to equilibrium and polarization was read. IC50 values for AMP, SAM, SAH, adenosine, and dGTP were 0.035, 2,624, >20,000, 35.2, and 14,491 μM, respectively.
Coupling enzymes and cofactor
To show proof of concept and optimize the key detection components, we used mock reactions lacking MT enzyme and containing various ratios of SAM and SAH to mimic a completed enzyme reaction. For instance, a mock reaction representing 10% conversion of 1 μM SAM contained 0.9 μM SAM, 0.1 μM SAH and all of the detection reagents. In general, the desired endpoint was a maximal polarization change in mock reactions representing low fractional conversion of SAM to SAH (5–20%).
AMP is produced in two enzymatic steps catalyzed by SAH hydrolase and adenosine kinase (Figure 1), the second of which requires a phosphate donor. Following evaluation of several enzyme preparations, including native and recombinant, some that we cloned and expressed, we selected two for further assay development. To minimize the potential for assay interference caused by inhibition of the coupling enzymes, we chose concentrations of each of the two coupling enzymes that were well in excess of what was required for a maximal signal. Titrating adenosine kinase in the presence of all other assay components resulted in a signal plateau at 0.01 ng/μl; we chose 5 ng/μl as the final assay concentration. SAH hydrolase exhibited more of a bell-shaped response, with signal starting to level off at 0.25 ng/μl; we selected 2 ng/μl as a final concentration.
We tested several nucleotide triphosphates reported to serve as phosphate donors for adenosine kinase (Long, Escuyer, and Parker, J Bacteriology, Vol 185, No 22, 2003,p 6548–6555), including ATP, GTP, dGTP and UTP for cross reactivity with the AMP antibody and concentration dependence in the adenosine kinase reaction. We selected dGTP based on its relatively low Km and minimal cross-reactivity with the AMP antibody (Figure 2). We then tested a range of concentrations and selected one that is approximately 100-fold over its reported Km concentration, to drive the equilibrium toward product (AMP) formation (data not shown).
Antibody optimization
In a competitive binding binding assay, the concentration of antibody defines the dynamic range of the assay, which is generally about two logs. Therefore, it is necessary to determine the optimal antibody concentration for detecting different ranges of analyte. We examined dependence of assay signal on antibody concentration in mock reactions representing 20% SAM conversion with a range of initial SAM concentrations. As shown in Figure 3, increasing the total SAM/SAH concentration requires more antibody for a maximal signal. Note that the maximal assay response increases as total SAM/SAH increases up to 50 μM, and then falls off. Investigation of this effect has suggested that the decrease at higher SAM concentrations is due to production of AMP from SAM, either from SAH contamination in the SAM preparation or from direct action of SAH hydrolase on SAM (data not shown). The former is more likely the predominant cause, since the assay signal is quite stable once it has equilibrated (see Figure 4B, below). Based on these results, we selected five antibody concentrations for use over initial SAH concentrations ranging from 0.25 to 50 μM. Though the assay can be used at higher or lower SAM concentrations, the assay window may not be sufficient for HTS applications.
Figure 3. Effect of AMP antibody concentration on assay signal in mock reactions representing 20% conversion of SAM.
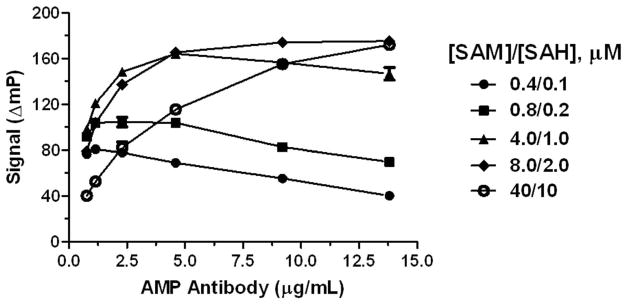
Mock reactions (10 μl) contained total [SAM + SAH] concentrations of 0.5, 1.0, 5.0, 10, 25, 50, and 100 μM at a 4:1 SAM:SAH ratio. Control reactions contained only SAM. Stop Buffer and Detection Mix were added in sequential 5 μl aliquots just as in an enzymatic assay, and plates were read following a 90 min equilibration period. The assay signal is shown as the magnitude of the decrease in polarization of mock reactions relative to controls: ΔmP = mPcontrol − mP20%.
Figure 4. Standard curves mimicking enzymatic conversion of SAM to SAH.

A) SAM/SAH standard curves (n = 16) for initial SAM concentrations of 0.2, 0.5, 1.0, 5.0, 10, and 50 μM. Stop Buffer and Detection Mix were added in sequential 5 μl aliquots to mock reactions with various SAM/SAH concentrations and plates were read after 90 min. B) Signal equilibration and stability measured using 10 μM SAM/SAH standard curve (n = 12). Following addition of Stop Buffer and Detection Mix, plates were kept at room temperature and read periodically. C) Reagent stability measured using 10 μM SAM/SAH standard curves (n = 12). Standard curves were prepared from Detection Mix that had been stored at room temperature for the indicated time periods.
Standard curves
Standard curves mimicking enzyme reactions were used to test the optimized reagent concentrations over a range of initial SAM concentrations. As a rule of thumb, we consider a polarization shift of 60–100 mP and a Z′ value of 0.5 to be indicative of robust assay performance for HTS applications. For initial SAM concentrations of 5 μM, 10 μM and 50 μM, these criteria were achieved when less than 5% of the SAM was converted to SAH, whereas approximately 10% conversion was required at 1 μM initial SAM and 25% conversion for 0.5 μM SAM (Figure 4A). For instance a Z′ value of 0.59 and a polarization shift of 74 mP was reached at 3% conversion of 5 μM SAM. At 0.20 SAM, 40% conversion - which is beyond the initial velocity region - was required to generate an assay window suitable for HTS (Figure 4A), reflecting the limits of sensitivity for the AMP immunodetection component of the assay. Efforts are currently underway to increase the sensitivity of the assay by optimizing conditions for antibody binding (i.e., decreased Kd for tracer and antigen), and we have observed a 2–4-fold increase by limiting the dilution resulting from addition of quenching and detection reagents and decreasing the ionic strength (BellBrook Labs, unpublished data).
Deck and Signal Stability
Implementation of an assay in HTS typically requires stable reagents during dispensing and sustained signal stability after addition of detection reagents. To assess these parameters we monitored the assay signal using standard curves mimicking MT enzyme reactions initially containing 10 μM SAM. The maximal FP signal was reached by 60 minutes, reflecting complete equilibration of all the detection reagents (Fig. 4B). After that, the curve remained unchanged for at least 8 hours, with only a very slight downward shift after 48 hours, reflecting excellent signal stability. To assess the “deck stability” of the assay, pre-combined detection reagents were used to develop standard curves following storage at room temperature. There was no detectable decay in the standard curve at 18 hours (Fig 4C), indicating excellent stability for the pre-combined detection mixture.
Effect of drug compounds on detection reagents
Inhibition or activation of coupling enzymes by screening compounds is a common source of assay interference for coupled assays. Other potential sources of interference would be disruption of antibody-tracer binding or extraneous fluorescence. To investigate the potential for compound interference, we screened a collection of 1,120 bioactive compounds (Tocriscreen Mini) using mock reactions representing 4% conversion of 10 μM SAM. As shown in Figure 5, the vast majority of the compounds clustered within the data variability bands drawn at 3 standard deviations from the mean. The Z factor for the screen, which includes all of the sample wells was 0.59 and the Z′, which is determined from control wells lacking sample, was 0.77 28 (using 0 and 4% conversion). Nine compounds fell outside of the signal variability bands, reflecting a 0.8% interference rate. Further analysis showed that 5 of the interfering compounds interfered with the AMP antibody and tracer alone, so it can be surmised that the remaining 4 inhibited the coupling enzymes or both.
Figure 5. Assay interference screen.

An 1120 compound library of bioactives (Tocriscreen Mini) was screened at 10 μM against mock reactions representing 4% conversion of 10 μM SAM (9.6 μM SAM/0.4 μM SAH). Upper solid line is drawn at the average polarization of 0% conversion, no compound controls; lower solid line is drawn at the average polarization of 100% conversion, no compound controls. The 4% conversion, no compound controls are clustered with the majority of the compound wells at 220 mP. The dashed lines are drawn at +/− 3 standard deviations from the mean of the compound wells.
Detection of Enzymes and Inhibitors
We tested the ability to detect enzyme activity with the Transcreener Epigen MT Assay using three HMTs, and two DNMTs. We used an endpoint assay format incorporating an enzyme quench step followed by addition of a single detection mix containing the coupling enzymes and AMP immunodetection reagents. HMTs were quenched using a low pH buffer and DNMTs were quenched by MgCl2; note that we have not tested these quenching reagents for other methyltransferases.
A peptide representing the first 25 amino acids of Histone H3 was used as acceptor substrates for HMTs G9a, Set7/Set9 and SUV39H1, full length Histone H3 was also used for HMT G9a, and poly d(I-C) was used for DNMT 1 and DNMT 3a. SAM was used near its reported Km concentration, except where indicated, and the peptide and DNA substrates were present in excess. Histone H3 was used at a limiting concentration for reasons explained below. The signal increased in a dose dependent manner as increasing amounts of enzyme were added to reactions (Figures 6A and B), and in all cases except DNMT 3a, a total polarization change of at least 100mP was achieved, indicating a good assay window. Where activity data was provided by the vendor, our results were in agreement with the expected relative activity levels for the different enzymes tested. These results suggest that the assay will have broad applicability across the MT enzyme family.
Figure 6. Detection of SAM produced in HMT and DNMT enzyme reactions.

MT enzyme reactions, initiated by the addition of SAM were incubated, quenched with 5 μl Stop Buffer, 5 μL of Detection Mix was added and plates were read after 90 min. Control reactions lacked enzyme; ΔmP = mPcontrol − mPenzyme. A) Titrations of HMTs Set7/Set9 and SUV39H1 and DNMTs 1 and 3a. Enzyme incubations were 120 min. Set7/Set9 and SUV39H1 reactions contained 50 μM peptide substrates and 6 and 12 μM SAM, respectively. DNMT reactions contained 5 μM poly d(I-C) and 3 μM SAM. B) Titration of HMT G9a from two different sources with peptide and full length histone substrates. Enzyme incubations were 120 min. Peptide substrate was present at 10 μM and Histone H3 was used at 1 μM. C) Rates of SAH formation in HMT G9a reactions. SAM/SAH standard curves were used to convert polarization data to SAH formation. D) Inhibition of HMT G9a by sinefungin. HMT G9A and detection reactions were performed as described in Materials and Methods. Enzyme incubations were 60 min. Peptide substrate was present at 20 μM and SAM was at 2 μM.
We carried out more detailed studies with HMT G9a 21, because it was to be the target for a pilot screen (Fig. 8). In a comparison of two commercially available recombinant enzymes, the preparation from BaV-infected Sf9 insect cells was significantly more active than the enzyme expressed in E. coli (Fig. 6B), therefore the Sf9 material was used for further studies. HMT G9a had much higher activity with the peptide substrate than full length histone (Fig 6B), and use of higher histone concentrations resulted in even lower activity (data not shown). In investigating this, we determined that the full length histone interferes with the immunodetection component of the assay: it caused polarization values to decrease when incubated with the AMP antibody and tracer in the absence of other assay components. To minimize this effect, we used a limiting amount of histone H3 (1μM).
Figure 8. Hit validation in 1536-well format for different types of pilot screen hits.

Shown are results in the G9a assay (top) or the SAH counterassay (bottom) for one of the 4 primary hits specifically inhibiting G9a (left), for one of the 21 primary hits affecting both the G9a-dependent and the counterassay (middle) and for one of the 17 primary hits that were inactive in a concentration-response experiment. Note that the specific inhibition curve (left) has the inflection point around −50% inhibition and a plateau at −100% inhibition, whereas the interfering compounds (middle) reach inhibition values that exceed the no-enzyme control.
The Transcreener AMP assay relies on a competitive binding reaction, therefore it generates a non-linear signal in response to enzyme. To demonstrate linearity of the assay with respect to SAH formation, we performed HMT G9a reactions using 2 μM SAM and 50 μM peptide substrate and used a standard curve to convert polarization values to SAH formation. The resulting progress curves were linear over time, and the slopes increased with the HMT concentration (Fig. 6C). Moreover, the polarization shifts at the 60 min time points (data not shown) confirmed that HMT linear velocity can be detected with an assay window suitable for HTS; e.g. 60–100 mP.
We tested the assay for measuring inhibitor potency using sinefungin, a SAM-competitive inhibitor of HMT G9a, and it displayed a typical dose response curve (Fig. 6D); SAM was present at its Km concentration of 2 μM 21. The observed IC50 of 9 μM for sinefungin agrees well with previously reported value of 18 μM determined using an assay based on detection of methylated peptides 26.
Pilot Screening
A miniaturized (1,536 well) assay protocol was used to screen a proprietary 8800 compound pilot library at a concentration of 25 μM for inhibitors of HMT G9a. Data were normalized based on controls that either contained or lacked G9a. The assay window was stable during screening of the 7 plates and fluctuated only slightly between 78 and 84 mP. The overall quality of the screen was acceptable as judged from “robust Z′ values” (an equivalent of the well-known Z′ value based on robust statistics instead of standard deviation and thus is less affected by outliers 9) of around 0.6. The more widely-used Z′ values 28 were around 0.5. At a compound concentration of 16.5 μM during the reaction, just 0.41% of the compounds showed an effect of more than −30%.
Primary hits were cherry-picked and tested in 1536-well format at concentrations ranging from 33 μM down to 10 nM, allowing determination of IC50 values by non-linear least square fitting of a sigmoidal concentration-response curve. In parallel, the same compounds were tested at the same concentration in an assay set-up in which a mixture of SAM and SAH simulated the roughly 20% conversion at the end of the standard reaction time. Of the 41 hits compounds tested in this way, 17 were inactive in either set-up, 20 showed an effect in both set-ups (and thus appear to interfere with the detection system) and 4 showed a specific effect in the G9a-dependent assay format only. Two previously known reference compounds were also active in just the G9a dependent assay variant.
Discussion
The Transcreener Epigen MT assay was designed, developed and validated specifically for HTS, with the key parameters being facile incorporation into an automated environment and robust performance under relevant enzyme conditions. As BellBrook is a drug discovery solutions provider, we cannot provide an unbiased comparison to alternative MT assays, many of which are also available commercially. However, we can provide a summary of the key characteristics of the assay protocol, reagents and performance that make it well suited for HTS.
The assay protocol is a simple, mix and read format, with homogenous detection, and it is miniaturizable to 384- and 1536-well densities, properties that make incorporation into a typical automated HTS environment very straightforward. Endpoint assays are generally easier to incorporate into an automated protocol, and use of the assay in this mode allows determination of initial rates using a standard curve, if desired (Fig. 6C). Due to the difference in pH optimum between the G9a methyltransferase and the coupling enzymes, a two-step protocol with an enzyme quenching step prior to detection (Fig. 6) was used for enzyme studies and the pilot screen (Figures 7, 8). However, since quenching conditions that would also be compatible with the detection reagents have not been identified for all methyltransferases, the ability to use continuous detection may be advantageous in some situations, and we have preliminary evidence that the assay can also be used in this fashion (data not shown / to be published later). Note that use of the assay in continuous mode yields an approximation of initial rates since the detection reactions require more than 30 minutes to reach equilibrium (Fig. 4A).
Figure 7. Histogram of HMT G9a primary screening results.

Shown are a typical distribution of compounds, hits, artifacts, and outliers of the 8800 compound library screen of G9a HMT.
The sensitivity, dynamic range and data quality of the assay combine to yield a very good signal window for initial velocity measurements within the physiological range of SAM concentrations (0.20–50 μM). Assay performance at the lower end of this range is important, because screening is often done using sub-saturating substrate to avoid masking competitive inhibitors and most HMTs have SAM Km’s of less than 5 μM. The main determinants of assay performance are the sensitivity and selectivity of the AMP antibody/tracer pair and the efficiency of SAH conversion to AMP. The IC50 for AMP for binding to the AMP antibody in competition with tracer was 35 nM, and selectivity versus SAH and SAM was greater than 30,000 (Fig. 2). Thus if conversion of SAH by coupling enzymes is nearly complete, the assay should yield a good signal with SAH concentrations of 70–100nM, even in the presence of excess SAM. In practice, we found that this was the case, with acceptable HTS performance (Z′ > 0.5, ΔmP = 60–100) occurring right around 100 nM SAH for the lower SAM concentrations tested; e.g., 10%, 25%, and 40% conversion of 1 μM, 0.5 μM and 0.25 μM SAM, respectively (Fig. 4A). Preliminary studies have indicated that the sensitivity of the assay can be increased by decreasing the volume and ionic strength of the detection reactions, thus increasing the assay window at the lower SAM concentrations (BellBrook Labs, unpublished data).
Even in the miniaturized 1536-well format with 4 μl reaction volume, the assay yielded an acceptable assay quality when SAM was employed at a concentration of roughly 2x its KM value of 1.8 μM 21. The assay window could have undoubtedly been increased further by using higher concentrations of SAM, which would have improved assay statistics but also might have jeopardized assay sensitivity.
The histogram of the primary screening results (Fig. 7) shows a normal distribution centered around 0% inhibition for the vast majority of the library compounds tested. Some compounds, among them the true inhibitors such as the one in Fig. 8 (left), are observed between the threshold of −50% and full inhibition (which can be assumed at −100 ± 30% inhibition), but not on the other side of the normal distribution. Only a handful of compounds - presumably readout or other artifacts and outliers - show results significantly below −100% or significantly above 0% that cannot be explained by stoichiometric inhibition. Overall, this distribution profile is very typical of the observations we make both in small-scale pilot screens such as the one described here and large-scale High-Throughput Screens of up to 1.5 million compounds for multiple targets (both methyltransferases and others) with a variety of readouts.
The significant fraction of primary hits that show activity in the counterscreen reflects the nature of the coupled assay system as well as the fact that typical screening libraries inevitably contain some nonstoichiometric and hence non-specific compounds. As long as there is an efficient way to rapidly distinguish them from the desired stoichiometric compounds, which we demonstrate in Fig. 8, this is no obstacle to finding leads 17
The hit rate of 0.41% against our pilot library is in the range that we would expect for a robust screening assay which is not overly affected by readout artifacts. Nevertheless, inhibitors of the auxiliary enzymes as well as other compounds interfering with the readout undoubtedly can show up in the assay, but are efficiently eliminated by appropriate counterscreening (Fig. 8). Although we demonstrate this by means of concentration-response experiments, the principle can be just as well applied at a single concentration, which would allow efficient work-up of larger hit-lists. Our confidence in this approach is corroborated by the results for the reference compounds as well as the fact that three of the four G9a-dependent hits had been observed previously in an orthogonal TR-FRET-based assay (data not shown) similar to the DELFIA-assay described by Kubicek et al 18.
As noted in the Introduction, with the exception of direct immunodetection of SAH - an approach which has proven difficult to commercialize - all of the SAH-based MT assay methods rely on coupling enzymes 2; 5; 10; 12, with the associated risk of compound interference. To mitigate this risk, we used coupling enzymes in significant excess of what was required for a maximal signal. Thus, nearly complete inhibition would be required to affect the assay signal. Interference with the AMP detection component of the assay is likely to be low, because of the very high selectivity of the antibody 24 (Fig. 2) and because the tracer emits in the far red, which minimizes interference from fluorescent compounds and light scattering 25. Though obviously not a comprehensive study, the low level of interference that we observed in the compound interference screen (Fig. 5), and the reasonable hit rate in the pilot screen (Fig. 7) are both encouraging. A more complete picture of interference levels will emerge from screens by end users using larger, more diverse libraries.
Reagent stability and cost are sometimes overlooked in assay development, but these factors frequently prohibit the practical use of an assay for high volume screening. All of the reagents that were used for the Transcreener Epigen MT Assay are available with continuous supply and at a scale and cost that will support many millions of wells of screening annually. Moreover the reagents are stable to multiple freeze thaws (data not shown) and for extended periods at room temperature prior to dispensing (Fig. 4C), which will insure that assay performance does not decay when long dispensing times are required for large screens. In addition, the AMP detection component of the assay is extremely stable under the final quenched reaction conditions, allowing more than 12 hours for plates to be read (Fig. 4B).
In summary, combining the Transcreener AMP Assay with coupling enzymes that convert SAH to AMP provides a generic, homogenous methyltransferase enzyme assay that is well suited for an automated HTS environment. The assay can accommodate SAM levels as low as 0.5 μM with initial velocity detection, and lower if higher substrate conversion levels are used, thus it should be useful for screening many methyltransferases using non-saturating SAM concentrations.
Acknowledgments
This work was funded in part by NIH grant R44GM073290. Thanks to Tim Wigle at Epizyme and Novartis colleagues, especially from CNIBR Shanghai, for many valuable discussions on methyltransferases and assay methods. Thanks to Bianca Heedmann for comparison with her TR-FRET based G9a results and thanks to Hans-Peter Gubler for explanations concerning robust statistics.
References
- 1.Cheng D, Yadav N, King RW, Swanson MS, Weinstein EJ, Bedford MT. Small molecule regulators of protein arginine methyltransferases. J Biol Chem. 2004;279:23892–23899. doi: 10.1074/jbc.M401853200. [DOI] [PubMed] [Google Scholar]
- 2.Collazo E, Couture JF, Bulfer S, Trievel RC. A coupled fluorescent assay for histone methyltransferases. Anal Biochem. 2005;342:86–92. doi: 10.1016/j.ab.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov. 2009;8:724–732. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- 4.Dhayalan A, Dimitrova E, Rathert P, Jeltsch A. A continuous protein methyltransferase (G9a) assay for enzyme activity measurement and inhibitor screening. J Biomol Screen. 2009;14:1129–1133. doi: 10.1177/1087057109345528. [DOI] [PubMed] [Google Scholar]
- 5.Dorgan KM, Wooderchak WL, Wynn DP, Karschner EL, Alfaro JF, Cui Y, Zhou ZS, Hevel JM. An enzyme-coupled continuous spectrophotometric assay for S-adenosylmethionine-dependent methyltransferases. Anal Biochem. 2006;350:249–255. doi: 10.1016/j.ab.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Ghoshal K, Bai S. DNA methyltransferases as targets for cancer therapy. Drugs Today (Barc) 2007;43:395–422. doi: 10.1358/dot.2007.43.6.1062666. [DOI] [PubMed] [Google Scholar]
- 7.Graves TL, Zhang Y, Scott JE. A universal competitive fluorescence polarization activity assay for S-adenosylmethionine utilizing methyltransferases. Anal Biochem. 2007 doi: 10.1016/j.ab.2007.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gravina GL, Festuccia C, Marampon F, Popov VM, Pestell RG, Zani BM, Tombolini V. Biological rationale for the use of DNA methyltransferase inhibitors as new strategy for modulation of tumor response to chemotherapy and radiation. Mol Cancer. 9:305. doi: 10.1186/1476-4598-9-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gubler H. Methods for Statistical Analysis, Quality Assurance and Management of Primary High-Throughput Screening Data. In: Hüser J, editor. High-Throughput Screening in Drug Discovery. GmbH Weinheim, Germany: Wiley-VCH; 2006. pp. 151–205. [Google Scholar]
- 10.Hendricks CL, Ross JR, Pichersky E, Noel JP, Zhou ZS. An enzyme-coupled colorimetric assay for S-adenosylmethionine-dependent methyltransferases. Anal Biochem. 2004;326:100–105. doi: 10.1016/j.ab.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 11.Horton JR, Sawada K, Nishibori M, Cheng X. Structural basis for inhibition of histamine N-methyltransferase by diverse drugs. J Mol Biol. 2005;353:334–344. doi: 10.1016/j.jmb.2005.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ibanez G, McBean JL, Astudillo YM, Luo M. An enzyme-coupled ultrasensitive luminescence assay for protein methyltransferases. Anal Biochem. 401:203–210. doi: 10.1016/j.ab.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 13.Janzen WP, Wigle TJ, Jin J, Frye SV. Epigenetics: Tools and Technologies. Drug Discov Today Technol. 7:e59–e65. doi: 10.1016/j.ddtec.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karijolich J, Kantartzis A, Yu YT. RNA modifications: a mechanism that modulates gene expression. Methods Mol Biol. 629:1–19. doi: 10.1007/978-1-60761-657-3_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kleman-Leyer KM, Klink TA, Kopp AL, Westermeyer TA, Koeff MD, Larson BR, Worzella TJ, Pinchard CA, van de Kar SA, Zaman GJ, Hornberg JJ, Lowery RG. Characterization and optimization of a red-shifted fluorescence polarization ADP detection assay. Assay Drug Dev Technol. 2009;7:56–67. doi: 10.1089/adt.2008.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 17.Kristin ED, Coan JO, Klumpp Martin. Non-stoichiometric inhibition in biochemical high-throughput screening. Exp Opin Drug Disc. 2011;6:405–417. doi: 10.1517/17460441.2011.561309. [DOI] [PubMed] [Google Scholar]
- 18.Kubicek S, O’Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, Rea S, Mechtler K, Kowalski JA, Homon CA, Kelly TA, Jenuwein T. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 19.Lowery RG, Kleman-Leyer K. Transcreener: screening enzymes involved in covalent regulation. Expert Opin Ther Targets. 2006;10:179–190. doi: 10.1517/14728222.10.1.179. [DOI] [PubMed] [Google Scholar]
- 20.McMillan FM, Archbold J, McLeish MJ, Caine JM, Criscione KR, Grunewald GL, Martin JL. Molecular recognition of sub-micromolar inhibitors by the epinephrine-synthesizing enzyme phenylethanolamine N-methyltransferase. J Med Chem. 2004;47:37–44. doi: 10.1021/jm0205752. [DOI] [PubMed] [Google Scholar]
- 21.Patnaik D, Chin HG, Esteve PO, Benner J, Jacobsen SE, Pradhan S. Substrate specificity and kinetic mechanism of mammalian G9a histone H3 methyltransferase. J Biol Chem. 2004;279:53248–53258. doi: 10.1074/jbc.M409604200. [DOI] [PubMed] [Google Scholar]
- 22.Rathert P, Cheng X, Jeltsch A. Continuous enzymatic assay for histone lysine methyltransferases. Biotechniques. 2007;43:602, 604, 606. doi: 10.2144/000112623. passim. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Staeben M, Kleman-Leyer KM, Kopp AL, Westermeyer TA, Lowery RG. Development and validation of a transcreener assay for detection of AMP- and GMP-producing enzymes. Assay Drug Dev Technol. 8:344–355. doi: 10.1089/adt.2009.0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vedvik KL, Eliason HC, Hoffman RL, Gibson JR, Kupcho KR, Somberg RL, Vogel KW. Overcoming compound interference in fluorescence polarization-based kinase assays using farred tracers. Assay Drug Dev Technol. 2004;2:193–203. doi: 10.1089/154065804323056530. [DOI] [PubMed] [Google Scholar]
- 26.Wigle TJ, Provencher LM, Norris JL, Jin J, Brown PJ, Frye SV, Janzen WP. Accessing protein methyltransferase and demethylase enzymology using microfluidic capillary electrophoresis. Chem Biol. 17:695–704. doi: 10.1016/j.chembiol.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye Y, Stivers JT. Fluorescence-based high-throughput assay for human DNA (cytosine-5)-methyltransferase 1. Anal Biochem. 401:168–172. doi: 10.1016/j.ab.2010.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 29.Zhu BT. Catechol-O-Methyltransferase (COMT)-mediated methylation metabolism of endogenous bioactive catechols and modulation by endobiotics and xenobiotics: importance in pathophysiology and pathogenesis. Curr Drug Metab. 2002;3:321–349. doi: 10.2174/1389200023337586. [DOI] [PubMed] [Google Scholar]