

Abstract

The 2′-hydroxyl groups within RNA contribute in essential ways to RNA structure and function. Previously, we designed an atomic mutation cycle (AMC) that uses ribonucleoside analogues bearing different C-2′-substituents, including −OCH3, −NH2, −NHMe, and −NMe2, to identify hydroxyl groups within RNA that donate functionally significant hydrogen bonds. To enable AMC analysis of the nucleophilic guanosine cofactor in the Tetrahymena ribozyme reaction and at other guanosines whose 2′-hydroxyl groups impart critical functional contributions, we describe here the syntheses of 2′-methylamino-2′-deoxyguanosine (GNHMe) and 2′-N,N-dimethylamino-2′-deoxyguanosine (GNMe2) and their corresponding phosphoramidites. The key step in obtaining the nucleosides involved SN2 displacement of 2′-β-triflate from an appropriate guanosine derivative by methylamine or dimethylamine. We readily obtained the GNMe2 phosphoramidite and incorporated it into RNA. However, the GNHMe phosphoramidite posed a significantly greater challenge due to lack of a suitable -2′-NHMe protecting group. After testing several strategies, we established that allyloxycarbonyl (Alloc) provided suitable protection for 2′-N-methylamino group during the phosphoramidite synthesis and the subsequent RNA synthesis. This work enables AMC analysis of guanosine’s 2′-hydroxyl group within RNA.
Introduction
Folded RNAs rely upon their 2′-OH groups to confer stability, often through hydrogen bond interactions.1 For many catalytic RNAs, including the group I and II introns, the ribosome, and the spliceosome, multiple 2′-OH groups on the substrates and ribozyme make important energetic contributions to function, as 2′-H substitution at these positions results in a substantial loss of activity.2 However, defining the physiochemical role of these relevant 2′-OH groups poses significant challenges and frequently requires the design and synthesis of chemically modified nucleosides in conjunction with their application in functionally meaningful ways.3
To investigate hydrogen bond donation by 2′-OH groups within RNA, we previously developed atomic mutation cycle (AMC) analysis (Figure 1).4 This approach requires the synthesis and functional characterization of three analogues bearing modifications at the 2′-position: –OCH3, –NH2, and –NHMe. When the energetic penalty for the 2′-OH to 2′-OCH3 substitution (ΔΔGOH→OCH3) exceeds that for the 2′-NH2 to 2′-NHCH3 substitution) (ΔΔGNH2→NHCH3), we attribute the difference to the absence of a hydrogen atom (ΔGH removal) on the 2′-OCH3 analogue and infer that the 2′-hydroxyl group under investigation imparts function by donating a hydrogen bond. A nucleoside bearing a 2′-N(CH3)2 substitution enables a further test of this conclusion, based on the prediction that the absence of a hydrogen atom on the 2′-amine would engender a greater energetic penalty than the 2′-NHCH3 derivative.
Figure 1.
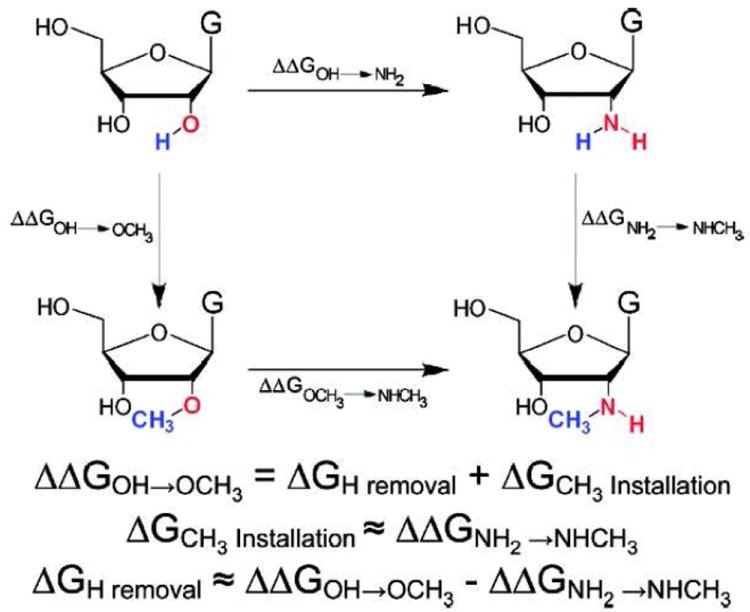
Atomic mutation cycle for analysis of the guanosine nucleophile 2’-hydroxyl
As part of our effort to conduct AMC analysis on the guanosine nucleophile 2′-hydroxyl group in the Tetrahymena ribozyme reaction,5 we herein describe the syntheses of 2′-N-methylamino-2′-deoxyguanosine and 2′-N,N-dimethylamino-2′-deoxyguanosine (herein referred to as GNHMe and GNMe2 respectively) and their incorporation into RNA. The availability of these two novel nucleosides expands the arsenal of modified nucleosides to better understand the role of the RNA’s 2′-OH group.
Result and Discussion
1. Syntheses of 2′-N,N-dimethylamino-2′-deoxyguanosine (4) and 2′-N-methylamino-2′-deoxyguanosine (5)
Unlike GNH2 and its phosphoramidites, whose syntheses and incorporation into oligonucleotides are well documented6, there are no reports describing the syntheses of GNHMe and GNMe2 and their phosphoramidites. Since GNH2 has been successfully synthesized from 2′-N-trifluoroacetylamido-2′-deoxyuridine by transglycosylation,6a we first attempted to synthesize GNHMe in the analogous manner. Thus, 2′-N-methyl-N-trifluoroacetylamido-2′-deoxyuridine was synthesized from 2′-amino-2′-deoxyuridine by trifluoroacetylation followed by methylation with methyl iodide. However, subsequent transglycosylation with N2-palmitoylguanine in the presence of BSA and TMSOTf afforded only a by-product, 2′-N-methylamino-2′-deoxyuridine, indicating loss of the 2′-N-trifluoroacetyl (TFA) protecting group. Premature deacylation from 2′-N-methyl-N-trifluoroacetylamido-2′-deoxyuridine but not 2′-N-trifluoroacetylamido-2′-deoxyuridine under the same reaction conditions suggests that the presence of the N-methyl group increases the liability of the trifluoroacetamide derivative.
As an alternative, we turned to our previously reported strategy to prepare substituted guanosine derivatives by SN2 reaction from the 2′-O-triflate derivative 17 (Scheme 1). Treatment of 1 with dimethylamine (2M in THF) or methylamine (2M in THF) overnight at 60 °C in a sealed pressure tube generated the corresponding silyl protected 2′-N,N-dimethylamino-2′-deoxyguanosine (2) and 2′-N-methylamino-2′-deoxyguanosine (3) in 55% and 60% yield, respectively. In both cases, we observed by-product resulting from elimination. Treatment of 2 and 3 with ammonium fluoride in MeOH gave free nucleosides 4 and 5, respectively, which were purified by reverse-phase column chromatography eluting with water.
Scheme 1.
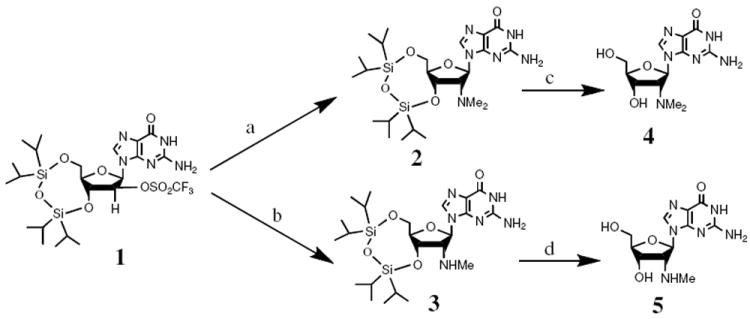
Syntheses of 2′-N-methylamino-2′-deoxyguanosine (4) and 2′-N,N-dimethylamino-2′-deoxyguanosine (5). (a) 2M Me2NH in THF, 60 °C, 55%. (b) 2M MeNH2 in THF, 60 °C, 60%. (c) 0.5 M NH4F in MeOH, 60 °C, 74%. (d) 0.5 M NH4F in MeOH, 60 °C, 72%.
2. Synthesis of the phosphoramidite of 2′-N,N-dimethylamino-2′-deoxyguanosine and its incorporation into RNA
We proceeded with synthesis of the phosphoramidite for GNMe2 (4) without protection of the tertiary amine (Scheme 2). Treatment of 2 with DMF-DMA in methanol to protect the exocyclic amino group generated intermediate 6 in 88% yield. Removal of the 3′, 5′-silyl protecting group with TBAF provided intermediate 7 in 78% yield. 4,4′-Dimethoxyltritylation of 7 gave 8 in 86% yield and subsequent phosphitylation of 8 under standard conditions produced phosphoramidite 9 in 82% yield. Incorporation of 9 into an RNA sequence CUCGmA (Gm = GNMe2) by solid phase synthesis occurred as efficiently as that of the commercial guanosine phosphoramidite. After standard deprotection and reverse phase HPLC purification, the structure of the oligonucleotide was confirmed by MALDI-TOF MS ([M-H]- = 1554).
Scheme 2.
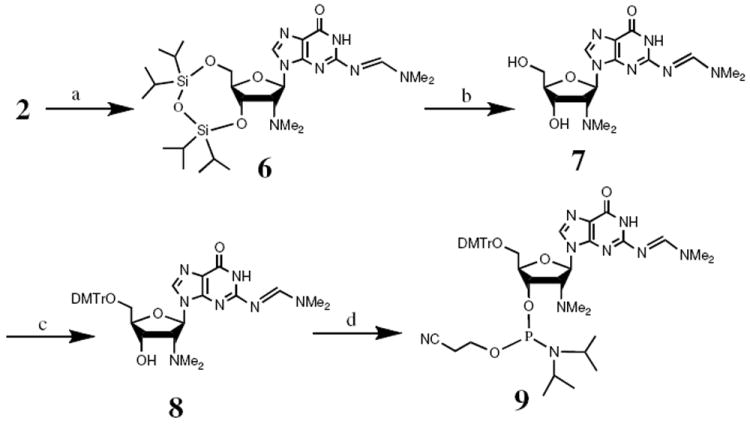
Synthesis of the phosphoramidite of 2′-N,N-dimethylamino-2′-deoxyguanosine (9). (a) DMF-DMA, MeOH, 88%. (b) 1.0 M TBAF in THF, 78%. (c) DMTr-Cl, Py, 86%. (d) (i-Pr)2NP(Cl)OCH2CH2CN, (i-Pr)2NEt/CH2Cl2, 82%.
3. Synthesis of the phosphoramidite for 2′-N-methyl-N-phenoxyacetylamido-2′-deoxyguanosine and its incorporation into an oligonucleotide (14)
In contrast to the phosphoramidite 9, the synthesis of a phosphoramidite suitable for GNHMe incorporation into RNA presented significant challenges due to the need to establish an appropriate protecting group for the 2′-methylamino group (2′-NHMe). For the synthesis of the phosphoramidite of GNH2, Eckstein et al. protected the 2′-amino group (2′-NH2) as a trifluoroacetamide (TFA), which remains stable during phosphoramidite and oligonucleotide synthesis but releases the free amine after oligonucleotide synthesis upon exposure to ethanolic ammonia.8 Intending to follow the analogous strategy for GNHMe, we generated the trifluoroacetamide derivative of 3 (Figure 2) by treatment with 1-(trifluoroacetyl)imidazole9 in pyridine followed by protection of the exocyclic amino group as the isobutyrylamide to give I-1 in 75% yield as a 1:5 mixture of isomers (cis and trans 2′-deoxy-2′-N-methyl amides).10 However, attempts to remove the silyl protecting group from I-1 with TBAF or ammonium fluoride also removed the TFA protecting group to yield only byproduct II-1 (Figure 2). In contrast, the corresponding derivative of GNH2, which lacks the methyl group, retains the TFA group under the same desilylation conditions. The greater liability of the N-methyltrifluoracetamide parallels our observations from the transglycosylation reaction noted above.
Figure 2.
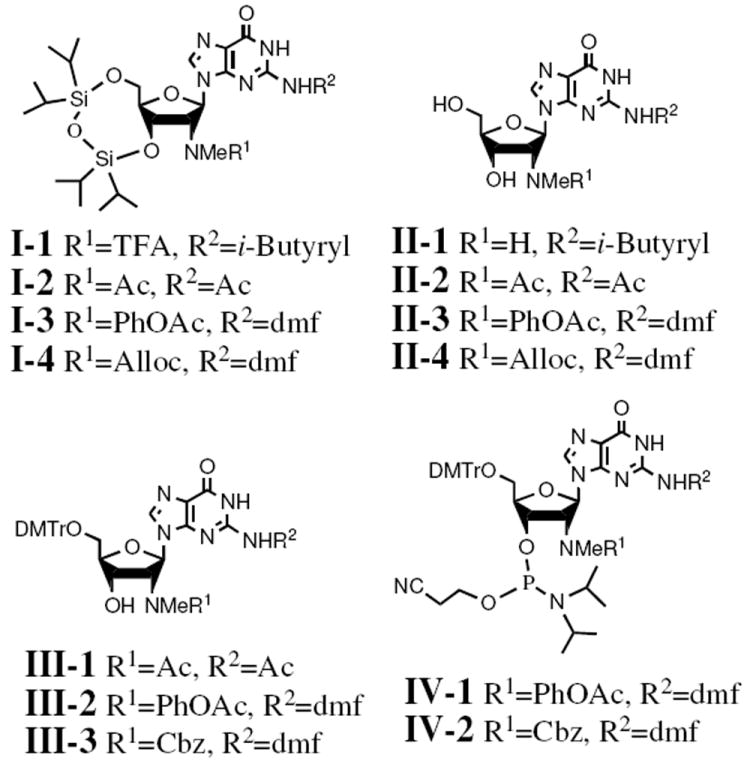
The structures of some intermediates with TFA, Ac, PhOAc or Cbz as 2’-NHMe protecting group
As an alternative protection strategy for the methylamino group, we considered acetylation. Acetylation is less useful for protection of 2′-aminonucleosides because postsynthetic deacetylation from the 2′-amine occurs too slowly.11 We anticipated that the greater stability of the acetamide relative to the trifluoracetamide could balance the liability conferred by the presence of the methyl group, allowing the N-methylacetamide to remain intact during solid phase synthesis but undergo postsynthetic deacetylation smoothly. To test this hypothesis, we treated 3 with excess acetyl anhydride in pyridine and isolated a bisacetamide I-2 bearing an acetyl group on both 2′-NHMe and exocyclic amine (Figure 2). TBAF treatment removed the 3′,5′-silyl protecting group readily while retaining the acetyl groups to generate II-2. When II-2 was treated with ammonium hydroxide at 55 °C, the acetyl group on the exocyclic amino group was readily removed within 2 h, but the removal of the acetyl group on the 2′-methylamino required 48 h to reach completion. Subsequent 4,4′-dimethoxytritylation of the 5′-OH afforded III-1 (Figure 2), but its polar character and poor solubility in dicholoromethane made the corresponding phosphoramidite difficult to isolate. We abandoned this strategy and turned our attention the phenoxyacetyl group (PhOAc).
Two features of PhOAc protection made it seem attractive: (1) the electron-withdrawing character of the phenoxy group causes the PhOAc ester to undergo hydrolysis 50 times faster than the corresponding Ac ester;12 (2) the large, hydrophobic phenyl ring could reduce the polarity and thus improve the solubility of the DMTr derivative in dichloromethane, thereby facilitating synthesis of the phosphoramidite.
PhOAc has been used to protect the exocyclic amines of adenosine and guanosine in phosphoramidite chemistry13 but not the 2′-amino group. To test the suitability of PhOAc as a protecting group for the 2′-amine, we first treated 5 with DMF-DMA in methanol to protect the exocyclic amine with dimethylaminomethylene group. Subsequent treatment with phenoxyacetyl chloride in pyridine generated I-3 (Figure 2). Desilylation with TBAF gave II-3. Unlike the N-methyltrifluoroacetamide, II-3, the 2′-N-methylphenoxyacetamide remained intact during this treatment. To test whether the ethanolic ammonia treatment that follows oligonucleotide synthesis would remove the PhOAc, we performed a control reaction by treating II-3 with ethanolic ammonia at 55 °C. Both dimethylaminomethylene and PhOAc groups were cleanly removed within 4 hours to give 5. Thus, we prepared the corresponding phosphoramidite IV-1 by treating II-3 with DMTr-Cl in pyridine to generate III-1 followed by 3′-O-phosphitylation (Figure 2). Under standard coupling conditions, phosphoramidite IV-1 was incorporated into a RNA sequence CUCGmA as efficiently as commercial guanosine phosphoramidite. After ethanolic ammonia (55 °C, 4h) and fluoride treatment, we purified the modified oligonucleotide by HPLC. MALDI-TOF MS analysis ([M-H]- = 1674) indicated that the oligonucleotide retained the PhOAc group, however.
We suspected that loss of the PhOAc group from the nucleoside but not the oligonucleotide upon ethanolic ammonia treatment might reflect participation of the free 3′-OH (Figure 3). Possibly the 3′-OH group attacks the carbonyl directly to form a five-member ring tetrahedral intermediate. Subsequent expulsion of the nitrogen would generate the 3′-O-acetyl ester, which would undergo deacylation. The absence of a free 3′-OH in the oligonucleotide may allow the PhOAc group to survive ethanolic ammonia treatment. We also attempted to remove the PhOAc group using harsher conditions (stronger base methylamine at 65 °C, 4 hours), but MALDI TOF MS indicated that no desired deprotected oligonucleotide formed.
Figure 3.
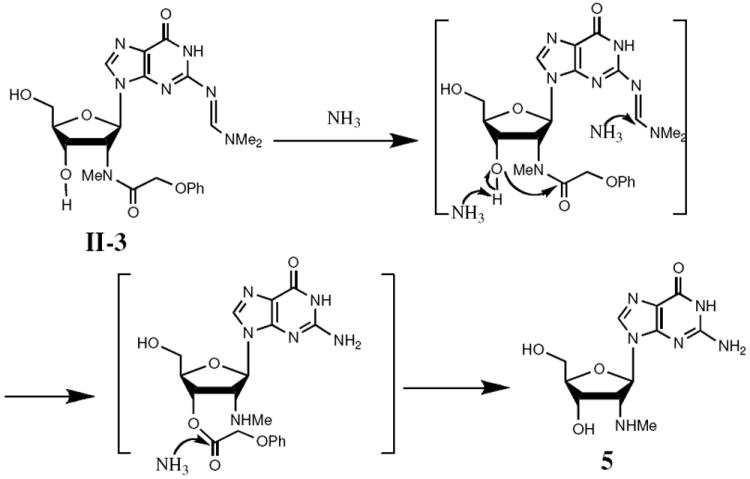
Possible mechanism for removal of PhOAc from I-3
4. Synthesis of 2′-N-methyl-N-benzyloxycarbonylamido- and 2′-N-methyl-N-alloxycarbonylamido-2′-deoxyguanosine phosphoramidites (IV-2 and 14)
Having encountered problems with each of the amide protecting groups (TFA, Ac, and PhOAc), we decided to test carbamate protecting groups for 2′-NHMe, which utilize hydrogenation for removal. Both benzyloxycarbonyl (Cbz)14 and allyloxycarbonyl (Alloc)15 groups have been used for protection of the exocyclic amines of the nucleobases in phosphoramidites of dA, dG and dC. Following DNA synthesis and cleavage of the oligonucleotide from the solid support by ammonium hydroxide treatment, the Cbz groups are removed by Pd-C catalyzed hydrogenation while Alloc groups are removed using a soluble palladium catalyst.
Treatment of 3 with DMF-DMA in methanol gave intermediate 10, which was converted to 11 and I-4 by treatment with allyloxycarbonyloxybenzotriazolyl16 or benzyloxycarbonyl chloride in pyridine, respectively (Scheme 3 and Figure 2). TBAF treatment removed the silyl protecting groups without affecting Cbz or Alloc groups to give 12 and II-4, which were converted to the corresponding phosphoramidites 14 and IV-2, respectively, using standard tritylation and phosphitylation reactions (Scheme 3 and Figure 2).
Scheme 3.
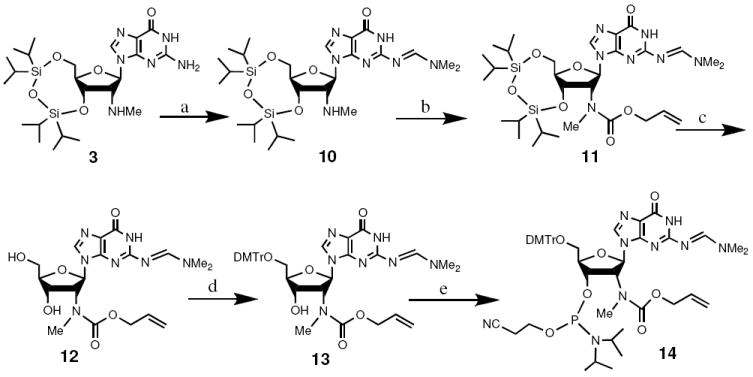
Synthesis of the 2′-N-methyl-N-allyloxycarbonyl-amido 2′-deoxyguanosine phosphoramidite (14). (a) DMF-DMA, MeOH, 88%. (b) allyloxycarbonyloxybenzotriazolyl, i-Pr2NEt, THF, 86%. (c) TBAF, THF, 84%. (d) DMTr-Cl, Py, 81%. (e) (i-Pr)2NP(Cl)OCH2CH2CN, (i-Pr)2NEt, 1-methylimidazole, CH2Cl2, 78%.
During solid phase synthesis of the RNA sequence CUCGmA (Gm= GNHMe), 13 and IV-4 coupled under standard conditions as efficiently as commercial guanosine phosphoramidite. However, attempts to remove the Cbz protecting group from the oligonucleotide by Pd/C catalyzed hydrogenation failed, either with the oligonucleotide attached to the CPG or removed from the CPG (with 2′-TBDMS groups still present or removed).17
In contrast to Cbz, removal of Alloc groups on the nucleobases requires only a solution-phase catalyst. For example, Hayakawa et al. reported that tris(dibenzylideneacetone)-dipalladium(0)-chloroform complex [Pd2(dba)3-CHCl3] catalyzes removal of Alloc protecting groups from CPG-supported DNA oligonucleotides (32-60mer).18 We treated 5′-CUCGmA-CPG similarly followed by standard ethanolic ammonia and fluoride. MS analysis of the resulting oligonucleotide showed only partial removal of the Alloc group (~50%). We tested other conditions and identified two ways to remove the Alloc group more effectively: Pd(Ph3P)4 in the presence of HCO2H/Et3N19 or Pd(Ph3P)4 in the presence of AcOH and Bu3SnH20. Both conditions removed the Alloc group completely to allow access to the desired oligonucleotide.21
Summary and Implications
We have synthesized two guanosine analogues, GNHMe and GNMe2, with the key reaction involving SN2 displacement of suitably protected 2′-β-triflate derivatives of guanosine with methylamine and dimethylamine, respectively. The tertiary amine of GNMe2 required no protection, allowing straightforward synthesis of the phosphoramidite. The synthesis of the corresponding GNHMe phosphoramidite proved to be more challenging due to the need to identify a suitable protecting group for the 2′-N-methylamino group. After testing several strategies, we established that the allyloxycarbonyl moiety provided suitable protection for 2′-N-methylamino group, being easily introduced, stable during the phosphoramidite preparation and subsequent RNA synthesis, and readily removed from the synthetic oligonucleotide by reduction using a soluble palladium catalyst. Both GNMe2 and GNHMe phosphoramidites enabled successful synthesis of oligonucleotides containing these analogues.
The availability of GNHMe and GNMe2, in conjunction with the other guanosine analogues required for AMC analysis (Figure 1), permits further investigation of functionally important guanosine 2′-hydroxyl groups within structured RNAs. Additionally, GNHMe and GNMe2 may serve as useful analogues to define the functional contribution of hydroxyl groups by application of Quantitative Structure Activity Relationship (QSAR) analysis2g or to evaluate the packing density around individual hydroxyl groups.4a,22.
Experimental
2′-Dimethylamino-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-2′-deoxyguanosine (2)
To a pressure tube (35 mL) under argon was added 2′-deoxy-2′-β-triflate-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-guanosine11a (1, 623 mg, 0.95 mmol) and dimethylamine in THF (2 M, 20 mL). The mixture was stirred overnight at 60 °C. After cooling to rt, the reaction mixture was concentrated to dryness under reduced pressure. The residue was purified by silica gel chromatography, eluting with 5-7% methanol in dichloromethane, to give 2 (287 mg, 55%) as white solid. 1H NMR (500.1 MHz) (DMSO-d6) δ: 10.58 (br. 1H), 7.78 (s, 1H), 6.38 (br., 2H), 5.84 (d, J=6.0 Hz, 1H), 4.56 (m, 1H), 3.90 (m, 1H), 3.80 (m, 2H), 3.54 (m, 1H), 2.35 (s, 6H), 0.85-0.97 (m, 28H). 13C NMR (125.8 MHz) (DMSO-d6) δ: 158.1, 155.3, 152.3, 136.5, 118.0, 85.6, 83.8, 75.0, 69.4, 64.5, 43.9, 18.8, 18.67, 18.65, 18.61, 18.43, 18.41, 18.35, 18.34. HRMS calcd. for C24H45N6O5Si2, [MH]+ 553.2990 (calcd.), 553.2967 (found).
2′-Methylamino-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-2′-deoxyguanosine (3)
To a flask (100 mL) under argon was added 2′-deoxy-2′-β-triflate-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-guanosine (1, 580 mg, 0.88 mmol) and methylamine in THF (2 M, 18 mL). The mixture was stirred overnight at 60 °C. After cooling to rt, the reaction mixture was concentrated to dryness under reduced pressure. The residue was purified by silica gel chromatography, eluting with 5-8% methanol in dichloromethane, to give 3 (284 mg, 60%) as white solid. 1H NMR (500.1 MHz) (DMSO-d6) δ: 10.55 (br. 1H), 7.71 (s, 1H), 6.34 (br., 2H), 5.52 (d, J=3.5 Hz, 1H), 4.44 (m, 1H), 3.90 (m, 1H), 3.81 (m, 1H), 3.26 (m, 2H), 2.30 (s, 3H), 0.90-0.99 (m, 28H). 13C NMR (125.8 MHz) (DMSO-d6) δ: 158.1, 155.2, 152.2, 135.9, 118.1, 87.2, 84.2, 71.2, 67.3, 63.2, 36.2, 18.8, 18.64, 18.60, 18.58, 18.4, 18.3, 18.25, 18.24, 14.2, 14.1, 14.0, 13.9. HRMS calcd. for C23H43N6O5Si2, [MH]+ 539.2834 (calcd.), 539.2811 (found).
2’-N,N-Dimethylamino-2′-deoxyguanosine (4)
To a solution of 2′-dimethylamino-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-2′-deoxyguanosine (2, 110 mg, 0.2 mmol) in MeOH (8 mL) was added ammonium fluoride (0.5 M solution in MeOH, 0.1 mL) under argon. The mixture was heated to 60 °C for 16 h. After removing the solvent under reduced pressure, the residue was co-evaporated with deionized water (5×30 mL). The residue was purified by C18 reverse-phase column chromatography, eluting with water to give 4 (46 mg, 74%) as white solid. 1H NMR (500.1 MHz) (DMSO-d6) δ: 10.75 (br., 1H), 8.08 (s, 1H), 6.60 (br., 2H), 6.05 (d, J= 7.8 Hz, 1H), 5.16 (br., 2H), 4.33 (m, 1H), 3.96 (m, 1H), 3.59 (m, 2H), 3.32 (m, 1H), 2.22 (s, 6H). 13C NMR (125.8 MHz) (DMSO-d6) δ: 157.1, 154.2, 151.4, 135.8, 116.7, 87.0, 84.0, 72.0, 70.4, 62.2, 43.7. HRMS calcd. for C12H19N6O4, [MH]+ 311.1468 (calcd.), 311.1480 (found).
2’-N-Methylamino-2’-deoxyguanosine (5)
To a solution of 2′-methylamino-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-2′-deoxyguanosine (3, 82 mg, 0.17 mmol) in MeOH (8 mL) was added ammonium fluoride (0.5 M solution in MeOH, 0.1 mL) under argon. The mixture was heated to 60 °C for 16 h. After removing the solvent under reduced pressure, the residue was co-evaporated with deionized water (5×30 mL). The residue was purified by C18 reverse-phase column chromatography, eluting with water to give 5 (36.5 mg, 72%) as white solid. 1H NMR (500.1 MHz) (DMSO-d6) δ: 10.96 (br., 1H), 7.92 (s, 1H), 6.72 (br., 2H), 5.55 (d, J= 7.5 Hz, 1H), 5.52 (br., 1H), 5.12 (br., 1H), 4.38 (m, 1H), 3.91 (m, 1H), 3.54 (m, 2H), 3.46 (m, 1H), 2.24 (s, 3H). 13C NMR (125.8 MHz) (DMSO-d6) δ: 157.2, 154.4, 151.7, 135.8, 117.0, 86.9, 85.9, 68.8, 66.1, 62.0, 34.9. HRMS calcd. for C11H17N6O4, [MH]+ 297.1311 (calcd.), 297.1302 (found).
2′-Dimethylamino-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-N2-dimethylaminomethylene-2′-deoxyguanosine (6)
To a solution of 2′-dimethylamino-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-2′-deoxyguanosine (2, 242 mg, 0.44 mmol) in methanol (10 mL) was added DMF-DMA (1 mL). After stirring at rt for 3 hours, all the volatile components were removed under reduced pressure. The residue was purified by silica gel chromatography, eluting with 3-5% methanol in dichloromethane, to give 6 (234 mg, 88%) as white solid. 1H NMR (500.1 MHz) (CDCl3) δ: 10.27 (s, 1H), 8.48 (s, 1H), 7.83 (s, 1H), 6.17 (s, 1H), 4.74 (m, 1H), 4.23 (m, 1H), 4.06 (s, 1H), 3.39 (m, 1H), 3.17 (s, 3H), 3.05 (s, 3H), 2.65 (s, 6H), 1.06 (m, 28H). 13C NMR (125.8 MHz) (CDCl3) δ: 158.2, 158.0, 156.9, 149.6, 135.4, 120.2, 84.9, 83.5, 72.4, 70.3, 61.9, 43.3, 41.4, 35.0, 17.4, 17.29, 17.26, 17.22, 17.0, 16.94, 16.88, 13.3, 13.02, 12.96, 12.5. HRMS calcd. for C27H50N7O5Si2, [MH]+ 608.3412 (calcd.), 608.3414 (found).
2′-Dimethylamino-N2-dimethylaminomethylene-2′-deoxyguanosine (7)
2′-Dimethylamino-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-N2-dimethylaminomethylene-2′-deoxyguanosine (6) (220 mg, 0.36 mmol) in THF (10 mL) was added TBAF (1M, 72 μL) and the mixture was stirred at rt for 15 min. All the volatile components were removed under reduced pressure. The residue was purified by silica gel chromatography, eluting with 0-12% methanol in dichloromethane, to give 7 (103 mg, 78%) as white solid. 1H NMR (500.1 MHz) (MeOD) δ: 8.65 (s, 1H), 8.22 (s, 1H), 6.26 (d, J=8.5 Hz, 1H), 4.42 (d, J=5.0 Hz, 1H), 4.10 (t, J=5.0 Hz, 1H), 3.73 (m, 2H), 3.44 (m, 1H), 3.19 (s, 3H), 3.10 (s, 3H), 2.21 (s, 6H). 13C NMR (125.8 MHz) (MeOD) δ: 158.6, 158.4, 157.9, 150.4, 137.5, 119.0, 87.2, 85.5, 72.2, 71.5, 62.2, 42.9, 40.0, 33.8. HRMS calcd. for C15H24N7O4, [MH]+ 366.1890 (calcd.), 366.1899 (found).
2′-Dimethylamino-5′-O-4,4′-dimethoxytrityl-N2-dimethylaminomethylene-2′-deoxyguanosine (8)
2′-Dimethylamino-N2-dimethylaminomethylene-2′-deoxyguanosine (7, 88 mg, 0.24 mmol) was dissolved in pyridine (3 mL), and 4,4′-dimethoxytrityl chloride (98 mg, 0.29 mmol, 1.2 equiv.) was added while stirring the solution. After being stirred overnight at rt, the reaction mixture was diluted with MeOH (1 mL), and stirred for an additional 5 min. The reaction mixture was concentrated to dryness under vacuum. Water was added to the resulting residue, and the mixture was extracted with CH2Cl2. The organic phase was washed consecutively with 5% sodium bicarbonate, water, and brine, and then dried over sodium sulfate. After the organic phase was concentrated to dryness, the residue was purified by silica gel chromatography, eluting with 4% MeOH in CH2Cl2 containing 0.2% Et3N, to give 8 (143 mg, 86%) as white foam. 1H NMR (500 MHz) (CD3CN) δ: 10.05 (br., 1H), 8.65 (s, 1H), 7.76 (s, 1H), 7.51 (d, J=7.5 Hz, 2H), 7.31-7.46 (m, 7H), 6.91 (m, 4H), 6.27 (d, J=8.5 Hz, 1H), 4.38 (d, J=5.0 Hz, 1H), 4.21 (m, 1H), 3.82 (s, 6H), 3.46 (m, 2H), 3.28 (m, 1H), 3.13 (s, 3H), 3.08 (s, 3H), 2.26 (s, 6H). 13C NMR (125.8 MHz) (CD3CN) δ: 158.6, 158.4, 157.9, 157.6, 150.3, 144.9, 135.9, 135.7, 135.6, 130.0, 127.9, 127.8, 126.8, 119.8, 113.0, 86.3, 84.8, 84.2, 71.7, 71.0, 64.4, 54.8, 43.1, 40.6, 34.3. HRMS calcd. for C36H41N7O6Na, [MNa]+ 690.3011 (calcd.), 690.3022 (found).
2′-Dimethylamino-5′-O-4,4′-dimethoxytrityl-N2-dimethylaminomethylene-2′-deoxyguanosine 3′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (9)
2′-Dimethylamino-5′-O-4,4′-dimethoxytrityl-N2-dimethylaminomethylene-2′-deoxyguanosine (8) (120 mg, 0.17 mmol) was dissolved in dry CH2Cl2 (5 mL) and 1-methylimidozale (2.90 mg, 35.0 μmol). N,N-diisopropylethylamine (126 mg, 0.68 mmol) was added to the stirring solution followed by 2-cyanoethyl N,N-(diisopropylchloro)phosphoramidite (2.0 equiv.). After being stirred at room temperature for 1 h, the reaction mixture was diluted with CH2Cl2 (50 mL) and washed with 5% NaHCO3 and brine. After dried over sodium sulfate, and filter, the filtrate was concentrated to dryness. The residue was purified by silica gel chromatography, eluting with 12% acetone in CH2Cl2 containing 0.2% Et3N, to give 9 (115 mg, 78%) as a colorless oil. 1H NMR (500 MHz) (CD3CN) δ: 10.03 (br., 1H), 8.62 (m, 1H), 7.78 (m, 1H), 7.50 (m, 2H), 7.35-7.40 (m, 7H), 6.91 (m, 4H), 6.36 (m, 1H), 4.92 (m, 1H), 4.01-4.42 (m, 2H), 3.82 (m, 6H), 3.55 (m, 2H), 3.50 (m, 1H), 3.24 (m, 1H), 3.09-3.06 (m, 6H), 2.78 (m, 1H), 2.72 (m, 1H), 2.32 (m, 6H), 1.30 (m, 12H). 31P NMR (202.4 MHz) (CD3CN) δ: 141.3, 141.5. HRMS calcd. for C45H59N9O7P, [MH]+ 868.4275 (calcd.), 868.4268 (found).
2′-Methylamino-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-N2-dimethylaminomethylene-2′-deoxyguanosine (10)
To a solution of 2′-methylamino-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-2′-deoxyguanosine (3, 264 mg, 0.50 mmol) in methanol (10 mL) was added DMF-DMA (1 mL). After stirred overnight at rt, all the volatile components were removed under reduced pressure. The residue was purified by silica gel chromatography, eluting with 5% methanol in dichloromethane, to give 10 (234 mg, 88%) as white solid. 1H NMR (500.1 MHz) (CDCl3) δ: 9.94 (br., 1H), 8.63 (s, 1H), 7.94 (s, 1H), 6.02 (s, 1H), 4.65 (m, 2H), 4.20 (m, 1H), 4.08 (m, 1H), 3.23 (r. 1H), 3.20 (s, 3H), 3.14 (s, 3H), 2.70 (s, 3H), 1.02-1.16 (m, 28H). 13C NMR (125.8 MHz) (CDCl3) δ: 158.0, 158.0, 156.7, 149.2, 135.5, 120.6, 86.9, 82.3, 68.7, 67.8, 60.6, 41.4, 35.1, 17.4, 17.3, 17.2, 17.0, 16.95, 16.88, 16.8, 13.3, 12.91, 12.86, 12.47. HRMS calcd. for C26H47N7O5Si2, [MH]+ 594.3272 (calcd.), 594.3256 (found).
2′-N-Methyl-N-alloxycarbonyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-N2-dimethylaminomethylene-2′-deoxyguanosine (11)
To a solution of 2′-methylamino-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-N2-dimethylaminomethylene-2′-deoxyguanosine (10, 220 mg, 0.37 mmol) in THF (10 mL) was added allyloxycarbonyloxybenzotriazolyl (122 mg, 1.5 equiv.) and N,N-diisopropylethylamine (0.2 mL). The mixture was stirred at rt for 1 h and MeOH (0.5 mL) was added to quench the reaction. All the volatile components were removed under reduced pressure. The residue was purified by silica gel chromatography, eluting with 4% methanol in dichloromethane, to give 11 (216 mg, 86%). 1H NMR (500.1 MHz) (CDCl3) δ: 9.75 (br., 1H), 8.61 (s, 0.75H), 8.50 (s, 0.25H), 8.04 (s, 1H), 6.16 (m, 1H), 5.86 (m, 0.75H), 5.75 (m, 0.25H), 5.42 (m, 0.75H), 5.26 (m, 0.25H), 5.15 (m, 2H), 4.62 (m, 2H), 4.57 (m, 1H), 4.09 (m, 2H), 3.82 (m, 0.25H), 3.80 (m, 0.75), 3.17 (s, 2.25H), 3.12 (s, 0.75H), 3.07 (s, 6H), 0.93-1.03 (m, 28H). HRMS calcd. for C30H51N7O7Si2, [MH]+ 678.3467 (calcd.), 678.3458 (found).
2′-N-Methyl-N-alloxycarbonyl-N2-dimethylaminomethylene-2′-deoxyguanosine (12)
2′-N-Methyl-N-alloxycarbonyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-N2-dimethylaminomethylene-2′-deoxyguanosine (11) (190 mg, 0.28 mmol) in THF (10 mL) was added TBAF (1M, 0.28 mL) and the mixture was stirred at rt for 15 min. All the volatile components were removed under reduced pressure. The residue was purified by silica gel chromatography, eluting with 0-10% methanol in dichloromethane, to give 12 (112 mg, 92%) as white solid. 1H NMR (500.1 MHz) (DMSO-d6) δ: 11.35 (br., 1H), 8.55 (s, 1H), 8.03 (s, 1H), 6.31 (m, 1H), 5.85 (br., 1H), 5.78 (br., 1H), 5.20 (m, 1H), 5.14 (m, 2H), 4.44 (m, 2H), 4.34 (m, 1H), 3.97 (m, 1H), 3.62 (m, 2H), 3.34 (m, 1H), 3.14 (s, 3H), 3.01 (s, 6H). 13C NMR (125.8 MHz) (DMSO-d6) δ: 158.6, 158.5, 157.9, 157.8, 150.1, 149.5, 137.2, 133.5, 120.4, 120.2, 117.3, 116.4, 88.0, 87.5, 87.0, 82.7, 77.1, 71.1, 66.6, 65.9, 62.1, 61.6, 61.2, 49.0, 41.1, 35.0, 32.9, 30.2. 13C NMR (125.8 MHz) (CDCl3) δ: 158.4, 158.0, 157.9, 157.0, 156.5, 156.0, 150.0, 135.8, 134.9, 132.6, 132.2, 120.8, 120.6, 118.1, 117.5, 85.8, 85.2, 83.7, 72.6, 71.9, 66.5, 66.4, 64.6, 64.0, 62.3, 61.1, 41.3, 35.1, 33.2, 32.7, 17.4, 17.3, 17.23, 17.21, 16.90, 16.86, 16.78, 13.21, 13.16, 12.8, 12.5. HRMS calcd. for C18H26N7O6, [MH]+ 436.1945 (calcd.), 436.1940 (found).
2′-N-Methyl-N-alloxycarbonyl-N2-dimethylaminomethylene-5′-O-4,4′-dimethoxytrityl-2′-deoxyguanosine (13)
2′-N-Methyl-N-alloxycarbonyl-N2-dimethylaminomethylene-2′-deoxyguanosine (12) (100 mg, 0.23 mmol) was co-evaporated with anhydrous pyridine (10 mL × 20) and then dissolved in pyridine (8 mL), and 4,4′-dimethoxytrityl chloride (116 mg, 0.34 mmol, 1.5 equiv.) was added. After being stirred overnight at room temperature, the reaction mixture was diluted with MeOH (1 mL), and stirred for an additional 5 min. The reaction mixture was concentrated to dryness under vacuum and co-evaporated with toluene (20 mL × 2). Water was added to the resulting residue, and the mixture was extracted with CH2Cl2. The organic phase was washed consecutively with 5% sodium bicarbonate, water, and brine, and then dried over sodium sulfate. After the organic phase was concentrated to dryness, the residue was purified by silica gel chromatography, eluting with 0-3% MeOH in CH2Cl2 containing 0.2% Et3N, to give 13 (137 mg, 81%) as white foam. 1H NMR (500.1 MHz) (CD3CN) δ: 13C NMR (125.8 MHz) (CD3CN) δ: 10.06 (br., 1H), 8.57 (s, 1H), 7.73 (m, 1H), 7.42 (m, 2H), 7.15-7.30 (m, 7H), 6.79 (m, 4H), 6.41 (m, 1H), 5.85 (br., 1H), 4.955.30 (m, 3H), 4.53 (m, 2H), 4.44 (m, 1H), 4.20 (m, 1H), 3.72 (s, 6H), 3.35 (m, 1H), 3.27 (m, 1H), 3.08 (s, 3H), 3.00 (s, 3H), 2.96 (s, 3H). 13C NMR (125.8 MHz) (CD3CN) δ: 158.6, 158.5, 158.3, 157.9, 157.4, 150.4, 144.9, 135.70, 135.67, 133.2, 130.0, 129.9, 127.9, 127.8, 126.8, 120.1, 116.4, 113.0, 86.2, 85.5, 83.1, 65.9, 64.2, 54.8, 40.6, 34.2, 32.7. HRMS calcd. for C39H44N7O8, [MH]+ 738.3251 (calcd.), 738.3232 (found).
2′-N-Methyl-N-alloxycarbonyl-N2-dimethylaminomethylene-5′-O-4,4′-dimethoxytrityl-2′-deoxyguanosine -(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (14)
2′-N-Methyl-N-alloxycarbonyl-N2-dimethylaminomethylene-5′-O-4,4′-dimethoxytrityl-2′-deoxyguanosine (13, 120 mg, 0.16 mmol) was dissolved in dry CH2Cl2 (5 mL) and 1-methylimidazole (2.90 mg, 35.0 μmol). N,N-diisopropylethylamine (120 mg, 0.64 mmol) was added to the stirring solution followed by 2-cyanoethyl N,N-(diisopropylchloro)phosphoramidite (2.0 equiv.). After being stirred at room temperature for 1 h, the reaction mixture was diluted with dichloromethane (50 mL) and the mixture was washed with 5% aqueous sodium carbonate and brine, dried over sodium sulfate, and concentrated. The residue was purified by silica gel chromatography, eluting with 12% acetone in CH2Cl2 containing 0.2% Et3N, to give 14 (119 mg, 78%) as a colorless oil. 31P NMR (202.4 MHz) (CD3CN) δ: 149.95 and 149.66, 149.34. HRMS calcd. for C48H60N9O9P, [M]+ 937.4252 (calcd.), 937.4288 (found).
2′-N-Methyl-N-phenoxyacetyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-N2-dimethylaminomethylene-2′-deoxyguanosine (I-3)
To a solution of 2′-methylamino-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-N2-dimethylaminomethylene-2′-deoxyguanosine (10, 178 mg, 0.30 mmol) in CH2Cl2 (10 mL) and pyridine (10 mL) at 0 °C under argon were added phenoxyacetyl chloride (47 μL, 1.1 equiv.) and N,N-diisopropylethylamine (0.2 mL). The mixture was stirred at 0 °C for 1 h and MeOH (0.5 mL) was added to quench the reaction. All the volatile components were removed under reduced pressure. The residue was co-evaporated with toluene. CH2Cl2 was added to the resulting residue, and the mixture was washed consecutively with 5% sodium bicarbonate, water, and brine, and then dried over sodium sulfate. After the organic phase was concentrated to dryness, the residue was purified by silica gel chromatography, eluting with 0-4% MeOH in CH2Cl2, to give I-3 (164 mg, 75%) as white foam. 1H NMR (500.1 MHz) (CDCl3) δ: 9.86 (br., 1H), 8.67 (s, 0.75H), 8.40 (s, 0.25H), 7.78 (s, 0.25H), 7.76 (s, 0.75H), 7.30 (m, 2H), 7.25 (m, 1H), 7.00 (m, 1.5H), 6.86 (m, 0.5H), 6.20 (m, 1H), 6.05 (m, 0.75H), 5.25 (m, 0.25H), 4.74-4.80 (m, 3H), 4.25 (m, 1H), 4.18 (m, 1H), 4.05 (m, 0.25H), 3.90 (m, 0.75H), 3.08-3.30 (m, 9H), 1.01-1.06 (m, 28H). 13C NMR (125.8 MHz) (CDCl3) δ: 158.8, 157.9, 157.8, 157.6, 157.4, 157.0, 149.9, 149.6, 136.0, 135.9, 129.6, 129.4, 121.7, 121.5, 120.8, 114.3, 114.0, 86.3, 85.8, 84.6, 84.0, 72.5, 72.3, 68.0, 66.9, 64.5, 63.9, 62.3, 58.8, 41.3, 35.1, 33.0, 32.1, 17.4, 17.3, 17.2, 17.1, 16.97, 16.95, 16.8, 13.21, 13.19, 12.8, 12.5, 12.4. HRMS calcd. for C34H53N7O7Si2, [MH]+ 728.3618 (calcd.), 728.3584 (found).
2′-N-Methyl-N-phenoxyacetyl-N2-dimethylaminomethylene-2′-deoxyguanosine (II-3)
2′-N-Methyl-N-phenoxyacetyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-N2-dimethylaminomethylene-2′-deoxyguanosine (I-3) (150 mg, 0.21 mmol) in THF (10 mL) was added TBAF in THF (1.0 M, 0.21 mL) and the mixture was stirred at rt for 15 min. Silica gel (1.0 g) was added and the volatile components were removed under reduced pressure. The residue was purified by silica gel chromatography, eluting with 0-9% methanol in dichloromethane, to give II-3 (95 mg, 95%) as white solid. 1H NMR (500.1 MHz) (CD3OD) δ: 8.59 (s, 0.75H), 8.40 (s, 0.25H), 8.20 (s, 0.25H), 8.18 (s, 0.75H), 7.16-7.22 (m, 2H), 6.91 (m, 1H), 6.79 (d, J=8.0 Hz, 1.5H), 6.73 (d, J=8.5 Hz, 0.5H), 5.75 (m, 0.75H), 5.25 (m, 0.25), 4.85 (m, 2H), 4.75 (m, 1H), 4.62 (m, 1H), 4.22 (m, 1H), 3.83 (m, 2H), 2.98-3.30 (m, 9H). 13C NMR (125.8 MHz) (CD3OD) δ: 170.6, 169.8, 158.6, 158.1, 157.7, 157.6, 150.3, 150.0, 137.8, 129.1, 128.9, 121.1, 120.9, 119.8, 119.5, 114.1, 114.0, 113.8, 87.6, 87.4, 85.0, 83.4, 71.0, 66.6, 65.9, 64.4, 62.5, 61.9, 61.5, 60.0, 58.0, 40.0, 33.81, 33.75, 32.3, 31.2. HRMS calcd. for C22H27N7O6, [MH]+ 486.2101 (calcd.), 486.2092 (found).
2′-N-Methyl-N-phenoxyacetyl-N2-dimethylaminomethylene-5′-O-4,4′-dimethoxytrityl-2′-deoxyguanosine (III-2)
2′-N-Methyl-N-phenoxyacetyl-N2-dimethylaminomethylene-2′-deoxyguanosine (II-3) (95 mg, 0.20 mmol) was co-evaporated with anhydrous pyridine (10 mL × 20) and then dissolved in pyridine (8 mL), and 4,4′-dimethoxytrityl chloride (99 mg, 0.29 mmol, 1.5 equiv.) was added. After being stirred overnight at room temperature, the reaction mixture was diluted with MeOH (1 mL), and stirred for an additional 5 min. The reaction mixture was concentrated to dryness under vacuum and co-evaporated with toluene (20 mL × 2). Water was added to the resulting residue, and the mixture was extracted with CH2Cl2. The organic phase was washed consecutively with 5% sodium bicarbonate, water, and brine, and then dried over sodium sulfate. After the organic phase was concentrated to dryness, the residue was purified by silica gel chromatography, eluting with 0-3% MeOH in CH2Cl2 containing 0.2% Et3N, to give III-2 (136 mg, 88%) as white foam. 1H NMR (500.1 MHz) (CD3CN) δ: 9.91 (br., 1H), 8.54 (s, 0.75H), 8.37 (s, 0.25H), 7.88 (s, 0.25H), 7.76 (s, 0.75H), 7.48 (m, 1.5H), 7.42 (m, 0.5H), 7.22-7.36 (m, 7H), 6.83 (m, 4H), 6.47 (m, 0.75H), 6.42 (m, 0.25H), 5.72 (m, 0.75H), 5.15 (m, 0.25H), 4.82 (s, 1.5H), 4.75 (s, 0.25H), 4.67 (m, 1H), 4.28 (m, 2H), 3.78 (s, 6H), 3.38 (m, 2H), 3.22 (s, 2.25H), 3.18 (s, 0.75H), 2.97-3.01 (m, 6H). HRMS calcd. for C43H45N7O8, [MNa]+ 810.3222 (calcd.), 810.3248 (found).
2′-N-Methyl-N-phenoxyacetyl-N2-dimethylaminomethylene-5′-O-4,4′-dimethoxytrityl-2′-deoxyguanosine -(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (IV-1)
2′-N-Methyl-N-phenoxyacetyl-N2-dimethylaminomethylene-5′-O-4,4′-dimethoxytrityl-2′-deoxyguanosine (III-2, 120 mg, 0.15 mmol) was dissolved in dry CH2Cl2 (5 mL). N,N-diisopropylethylamine (104 μL, 0.60 mmol) was added to the stirring solution followed by 2-cyanoethyl N,N-(diisopropylchloro)phosphoramidite (2.0 equiv.). After being stirred at room temperature for 1 h, the reaction mixture was diluted with dichloromethane (50 mL) and the mixture was washed with 5% aqueous sodium carbonate and brine, dried over sodium sulfate, and concentrated. The residue was purified by silica gel chromatography, eluting with 12% acetone in CH2Cl2 containing 0.2% Et3N, to give IV-1 (114 mg, 75%) as a colorless oil. 31P NMR (202.4 MHz) (CD3CN) δ : 152.2, 151.1, 150.51 and 150.46. HRMS calcd. for C52H62N9O9P, [MNa]+ 1010.4300 (calcd.), 1010.4344 (found).
2′-N-Methyl-N-benzyloxycarbonyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-N2-dimethylaminomethylene-2′-deoxyguanosine (I-4)
To a solution of 2′-methylamino-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-N2-dimethylaminomethylene-2′-deoxyguanosine (10, 210 mg, 0.35 mmol) in anhydrous THF (10 mL) under argon was added N,N-diisopropylethylamine (122 μL, 2.0 equiv.) and benzyl chloroformate (68 μL, 1.2 equiv.). The mixture was stirred at rt for 2 h and MeOH (0.5 mL) was added to quench the reaction. All the volatile components were removed under reduced pressure. CH2Cl2 (50 mL) was added to the resulting residue, and the mixture was washed consecutively with 5% sodium bicarbonate, water, brine, and then dried over sodium sulfate. After the organic phase was concentrated to dryness, the residue was purified by silica gel chromatography, eluting with 0-4% MeOH in CH2Cl2, to give I-4 (216 mg, 85%) as white foam. 1H NMR (500.1 MHz) (CD3CN) δ: 9.95 (br., 1H), 8.68 (s, 0.63H), 8.35 (s, 0.37H), 7.73 (s, 0.63H), 7.68 (s, 0.37H), 7.25 (m, 5H), 7.01 (s, 0.37H), 6.12 (d, J=5.5 Hz, 0.63H), 5.56 (m, 1H), 5.05 (m, 2H), 4.61 (m, 1H), 4.06 (s, 1H), 3.84 (m, 2H), 3.08-3.17 (m, 9H), 0.89-1.03 (m, 28H). 13C NMR (125.8 MHz) (CDCl3) δ: 158.0, 157.8, 156.9, 156.3, 149.7, 136.4, 136.0, 128.6, 128.4, 128.2, 127.9, 127.8, 121.1, 98.5, 86.1, 85.3, 84.4, 72.8, 72.1, 67.8, 67.6, 67.4, 64.8,64.1, 61.2, 41.8, 35.8, 33.3, 32.9, 23.6, 17.6, 17.44, 17.40, 17.08, 17.04, 16.9, 13.3, 13.0, 12.6. HRMS calcd. for C34H53N7O7Si2, [MH]+ 728.3623 (calcd.), 728.3609 (found).
2′-N-Methyl-N-phenoxyacetyl-N2-dimethylaminomethylene-2′-deoxyguanosine (II-4)
2′-N-Methyl-N-benzyloxycarbonyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-N2-dimethylaminomethylene-2′-deoxyguanosine (I-4) (198 mg, 0.27 mmol) in THF (10 mL) was added TBAF in THF (1 M, 0.27 mL) and the mixture was stirred at rt for 10 min. Silica gel (1.0 g) was added and volatile components were removed under reduced pressure. The residue was purified by silica gel chromatography, eluting with 0-8% MeOH in CH2Cl2, to give II-4 (126 mg, 96%) as white solid. 1H NMR (500.1 MHz) (DMSO-d6) δ: 11.38 (br., 1H), 8.57 (s, 0.63H), 8.42 (s, 0.37H), 8.04 (s, 1H), 7.09 (m, 5H), 6.33 (m, 1H), 5.13 (m, 2H), 5.02 (m, 2H), 4.35 (m, 1H), 4.12 (m, 1H), 3.62 (m, 1H), 3.57 (m, 1H), 2.99-3.34 (m, 9H). 13C NMR (125.8 MHz) (DMSO-d6) δ: 158.7, 158.1, 158.0, 156.7, 150.3, 137.3, 128.9, 128.3, 127.9, 126.9, 120.5, 88.1, 87.7, 87.2, 77.3, 71.4, 67.0, 66.8, 62.3, 61.8, 61.2, 58.1, 35.15, 33.12, 30.39. C22H27N7O6, [MH]+ 486.2101 (calcd.), 486.2092 (found).
2′-N-Methyl-N-benzyloxycarbonyl -N2-dimethylaminomethylene-5′-O-4,4′-dimethoxytrityl-2′-deoxyguanosine (III-3)
2′-N-Methyl-N-benzyloxycarbonyl-N2-dimethylaminomethylene-2′-deoxyguanosine (II-4) (120 mg, 0.25 mmol) was co-evaporated with anhydrous pyridine (10 mL × 20) and then dissolved in pyridine (8 mL), and 4,4′-dimethoxytrityl chloride (125 mg, 0.37 mmol, 1.5 equiv.) was added. After being stirred overnight at room temperature, the reaction mixture was diluted with MeOH (1 mL), and stirred for an additional 5 min. The reaction mixture was concentrated to dryness under vacuum and co-evaporated with toluene (20 mL × 2). CH2Cl2 was added and the mixture was washed consecutively with 5% sodium bicarbonate, water, and brine, and then dried over sodium sulfate. After the organic phase was concentrated to dryness, the residue was purified by silica gel chromatography, eluting with 0-3% MeOH in CH2Cl2 containing 0.2% Et3N, to give III-3 (160 mg, 82%) as white foam. 1H NMR (500.1 MHz) (CD3CN) δ: 13C NMR (125.8 MHz) (CD3CN) δ: 9.70 (br., 1H), 8.55 (m, 1H), 7.71 (m, 1H), 7.11-7.42 (m, 14H), 6.78 (m, 4H), 6.38 (m, 1H), 5.09-5.25 (m, 3H), 4.58 (m, 1H), 4.16 (m, 1H), 3.73 (m, 6H), 3.30 (m, 2H), 2.98-3.08 (m, 9H). HRMS calcd. for C43H45N7O8, [MNa]+ 810.3227 (calcd.), 810.3216 (found).
2′-N-Methyl-N-benzyloxycarbonyl-N2-dimethylaminomethylene-5′-O-4,4′-dimethoxytrityl-2′-deoxyguanosine -(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (IV-2)
2′-N-Methyl-N-benzyloxycarbonyl-N2-dimethylaminomethylene-5′-O-4,4′-dimethoxytrityl-2′-deoxyguanosine (III-3, 95 mg, 0.12 mmol) was dissolved in dry CH2Cl2 (5 mL). N,N-diisopropylethylamine (83 μL, 0.48 mmol) was added to the stirring solution followed by 2-cyanoethyl N,N-(diisopropylchloro)phosphoramidite (2.0 equiv.). After being stirred at room temperature for 1 h, the reaction mixture was diluted with dichloromethane (50 mL) and the mixture was washed with 5% aqueous sodium carbonate and brine, dried over sodium sulfate, and concentrated. The residue was purified by silica gel chromatography, eluting with 12% acetone in CH2Cl2 containing 0.2% Et3N, to give IV-2 (92 mg, 78%) as a colorless oil. 31P NMR (202.4 MHz) (CD3CN) δ :150.5, 150.0, 149.8 and 149.5. HRMS calcd. for C52H62N9O9P, [MNa]+ 1010.4300 (calcd.), 1010.4321 (found).
RNA synthesis and deprotection
RNA Oligonucleotide synthesis was performed on an Expedite Nucleic Acid Synthesis System using standard RNA synthesis conditions (scale: 1 μM). Phosphoramidites for A, C, G, U and CPG carriers were obtained from Glen Research. The terminal DMTr protecting group was removed from the oligonucleotides by using the DMTr off mode. After oligo synthesis, the resin containing oligos was transferred to a 2 mL vial. For oligo containing GNMe2 modification, the resin was treated with 2.0 mL 3:1 concentrated NH4OH:EtOH overnight at 55 °C. The supernant was collected and concentrated to dryness using speedvac. The residue was then treated with the mixture of N-methyl pyrrolidinone, triethylamine and triethylamine trihydrofluoride (0.3 mL, 6:3:4 v/v/v) at 65 °C for 90 min. After cooled to rt, the mixture was desalted and purified by HPLC. It structure was further confirmed by MALDI-TOF MS (SI, [M-H]- = 1554, [MNa-2H]- = 1576). For oligo containing GNHMe modification, the extra step treatment of resin is needed. the resin was treated with Pd(Ph3P)4 in the presence of HCO2H/Et3N or Pd(Ph3P)4 in the presence of AcOH and Bu3SnH and washed with acetone for three time. The rest of the procedure is the same as oligo containing GNMe2 modification. It structure was also further confirmed by MALDI-TOF MS (SI, [M-H]- = 1540).
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health (AI081987) to J.A.P. Q.D., a Senior Research Associate, was supported by the SPARK Award from the Chicago Biomedical Consortium with support from The Searle Funds at The Chicago Community Trust. We thank the following members of the Piccirilli laboratory for critical comments on the manuscript: Dr. N.-S. Li, N. Tuttle, K. Sundaram and N. Suslov. We also thank Dr. N.-S. Li for oligonucleotide synthesis.
Footnotes
Supporting Information Available 1H and 13C NMR spectra for compounds 2-8, 10-14 and 31P NMR spectra for compounds 9 and 14. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Auffinger P, Westhof E. J Mol Biol. 1997;274:54. doi: 10.1006/jmbi.1997.1370. [DOI] [PubMed] [Google Scholar]
- 2.(a) Pyle AM, Cech TR. Nature. 1991;350:628. doi: 10.1038/350628a0. [DOI] [PubMed] [Google Scholar]; (b) Pyle AM, Murphy FL, Cech TR. Nature. 1992;358:123. doi: 10.1038/358123a0. [DOI] [PubMed] [Google Scholar]; (c) Moore MJ, Sharp PA. Science. 1992;256:992. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]; (d) Herschlag D, Eckstein F, Cech TR. Biochemistry. 1993;32:8299. doi: 10.1021/bi00083a034. [DOI] [PubMed] [Google Scholar]; (e) Abramovitz DL, Friedman RA, Pyle AM. Science. 1996;271:1410. doi: 10.1126/science.271.5254.1410. [DOI] [PubMed] [Google Scholar]; (f) Ortoleva-Donnelly L, Szewczak AA, Gutell RR, Strobel SA. RNA. 1998;4:498. doi: 10.1017/s1355838298980086. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Weinger JS, Parnell KM, Dorner S, Green R, Strobel SA. Nat Struct Mole Biol. 2004;11:1101. doi: 10.1038/nsmb841. [DOI] [PubMed] [Google Scholar]
- 3.Das SR, Fong R, Piccirilli JA. Curr Opin Chem Biol. 2005;9:585. doi: 10.1016/j.cbpa.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 4.(a) Gordon PM, Fong R, Deb SK, Li NS, Schwans JP, Ye JD, Piccirilli JA. Chem Biol. 2004;11:237. doi: 10.1016/j.chembiol.2004.02.011. [DOI] [PubMed] [Google Scholar]; (b) Hougland JL, Deb SK, Maric D, Piccirilli JA. J Am Chem Soc. 2004;126:13578. doi: 10.1021/ja0469129. [DOI] [PubMed] [Google Scholar]
- 5.Hougland JL, Sengupta RN, Dai Q, Deb SK, Piccirilli JA. Biochemistry. 2008;47:7684. doi: 10.1021/bi8000648. [DOI] [PubMed] [Google Scholar]
- 6.(a) Imazawa M, Eckstein F. J Org Chem. 1979;44:2039. [Google Scholar]; (b) Greiner B, Pfleiderer W. Helv Chim Acta. 1998;81:1528. [Google Scholar]; (c) Benseler F, Williams DM, Eckstein F. Nucleosides Nucleotides. 1992;11:1333–51. [Google Scholar]; (d) Dai Q, Deb SK, Hougland JL, Piccirilli JA. Bioorg Med Chem. 2006;14:705. doi: 10.1016/j.bmc.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 7.(a) Schwans JP, Cortez CN, Olvera JM, Piccirilli JA. J Am Chem Soc. 2003;125:10012. doi: 10.1021/ja035175y. [DOI] [PubMed] [Google Scholar]; (b) Dai Q, Lea CR, Lu J, Piccirilli JA. Org Lett. 2007;9:3057. doi: 10.1021/ol071129h. [DOI] [PubMed] [Google Scholar]
- 8.Imazawa M, Eckstein F. J Org Chem. 1979;44:2039. [Google Scholar]
- 9.Khlebnicova TS, Isakova VG, Baranovsky AV, Borisov EV, Lakhvich FA. J Fluorine Chem. 2006;127:1564. [Google Scholar]
- 10.As indicated by 1H NMR and 19F NMR spectra.
- 11.Beigelman L, Karpeisky A, Matulic-Adamic J, Haeberli P, Sweedler D, Usman N. Nucleic Acids Res. 1995;23:4434. doi: 10.1093/nar/23.21.4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reese, et al. J Chem Soc Perkins I. 1975:934. doi: 10.1039/p19750000934. [DOI] [PubMed] [Google Scholar]
- 13.Schulhof JC, Molko D, Teoule R. Nucleic Acids Res. 1987;15:397. doi: 10.1093/nar/15.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watkins BE, Kiely John S, Rapoport H. J Am Chem Soc. 1982;104:5702. [Google Scholar]
- 15.(a) Hayakawa Y, Wakabayashi S, kato H, Noyori R. J Am Chem Soc. 1990;112:1691. [Google Scholar]; (b) Spinelli N, Meyer A, Hayakawa Y, Imbach J-L, Vasseur J-J. Eur J Org Chem. 2002;67:49. [Google Scholar]; (c) Hayakawa Y, Kawai R, Hirata A, Sugimoto J, Kataoka M, Sakakura A, Hirose M, Noyori R. J Am Chem Soc. 2001;123:8165. doi: 10.1021/ja010078v. [DOI] [PubMed] [Google Scholar]
- 16.Hayakawa Y, Kato H, Uchiyama M, Kajino H, Noyori R. J Org Chem. 1986;51:2400. [Google Scholar]
- 17.In both cases, MS data gave the major peak at 1674 Daton, suggesting that Cbz survived the hydrogenation reaction on the solid catalyst, Pd/C or Pd black
- 18.Experimental details: Treatment at 50 °C for 0.5-1 h with a THF solution of triphenylphosphine and a large excess of formic acid/n-butylamine (1:1) followed by washing with THF, acetone and an aqueous solution of sodium N,N-diethyldithiocarbamate at pH=9.7.
- 19.Kanda Y, Arai H, Ashizawa T, Morimoto M, Kassi M. J Med Chem. 1992;35:2781. doi: 10.1021/jm00093a010. [DOI] [PubMed] [Google Scholar]
- 20.Dangles O, Guibe F, Balavoine G, lavielle S, Marquet A. J Org Chem. 1987;52:4984. [Google Scholar]
- 21.MS data show that the peaks of CUCGmA with Alloc protected (1609) completely disappeared while the peaks corresponding to deprotected CUCGmA (1526) appeared.
- 22.Schwans JP, Li NS, Piccirilli JA. Angew Chem Int Ed. 2004;43:3033. doi: 10.1002/anie.200353575. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.