

Abstract
The human DEK proto-oncogene is a nuclear protein with suspected roles in human carcinogenesis. DEK appears to function in several nuclear processes, including transcriptional regulation and modulation of chromatin structure. To investigate the clinicopathological significance of DEK in patients with non–small cell lung cancer (NSCLC), we analyzed DEK immunohistochemistry in 112 NSCLC cases. The results showed that DEK was overexpressed mainly in the nuclear compartment of tumor cells. In squamous cell carcinoma, DEK-positive expression occurred in 47.9% (23/48) of cases, and in lung adenocarcinoma, DEK-positive expression occurred in 67.2% (43/64) of cases and correlated with differentiation, p-TNM stage, and nodal status. Moreover, in lung adenocarcinoma, DEK expression was significantly higher compared with DEK expression in squamous cell carcinoma. Kaplan-Meier analysis showed that patients with low DEK expression had higher overall survival compared with patients with high DEK expression. Depleting DEK expression inhibited cellular proliferation and migration. Furthermore, in DEK-depleted NSCLC cells, we found that RhoA expression was markedly reduced; in conjunction, active RhoA-GTP levels and the downstream effector phosphorylated MLC2 were also reduced. Taken together, DEK depletion inhibited cellular migration in lung cancer cell lines possibly through inactivation of the RhoA/ROCK/MLC signal transduction pathway.
Keywords: DEK, proliferation, apoptosis, migration, RhoA, oncogenic
Non–small cell lung carcinoma (NSCLC) is a major cause of cancer-related death worldwide (Campillos et al. 2003). Despite recent advancements in early tumor detection, surgical treatment, radiochemotherapy, and targeted therapy, the overall survival of patients with NSCLC remains poor (Schiller et al. 2002; Reed et al. 2004; Farray et al. 2005). Due to the intricate biological functions of the disease, the prognosis of lung cancer remains very poor (Rooney and Sethi 2011). Hence, it is vital to uncover the biological functions of the disease for the sake of identifying useful biomarkers and novel therapeutic targets.
The DEK proto-oncogene, located on chromosome 6p22.3, was initially identified in 1992 as a fusion protein with the nucleoporin CAN in patients with a subtype of acute myeloid leukemia (Soekarman et al. 1992; von Lindern et al. 1992). The ability of DEK to bind nucleic acids has led to functional associations with several cellular processes, including chromatin remodeling, transcriptional regulation, replication, messenger RNA (mRNA) splicing, and DNA repair (Alexiadis et al. 2000; Campillos et al. 2003; Soares et al. 2006; Kavanaugh et al. 2011). Importantly, DEK plays a critical role in chromatin organization and the maintenance of genome stability in vivo (Sawatsubashi et al. 2010; Kappes et al. 2011). Therefore, when DEK is upregulated, as observed in numerous types of cancer, perturbations to the normal genome architecture and integrity are likely contributors to oncogenesis (Carro et al. 2006). DEK depletion can result in cell death and impaired DNA double-strand break repair (Privette Vinnedge et al. 2011). Therefore, cellular DEK expression is tightly controlled to maintain proper cell function and viability. In addition to epigenetics and chromatin integrity, the DEK oncogene has also been implicated in regulating the expression, phosphorylation, and/or activity of several important signaling molecules and pathways. For example, in breast cancer cell lines, DEK has been shown to enhance cellular invasion by stimulating β-catenin activity (Privette Vinnedge et al. 2011). Furthermore, DEK has been shown to inhibit p53-dependent and p53-independent apoptosis and mediate the apoptotic response to clastogenic chemotherapeutic agents such as doxorubicin and cisplatin (Wise-Draper et al. 2006).
Despite the growing evidence that DEK is a crucial factor in human cancers, the protein expression of DEK in primary lung cancer and its relationship to clinicopathological significance have not been examined. In addition, the biological role of DEK in lung cancer cells is still unclear. To address the above questions, we examined DEK expression in NSCLC tissues by immunohistochemistry. Furthermore, we also examined the role of DEK in cellular proliferation and migration in several lung cancer cell lines.
Materials and Methods
Patients and Specimens
This study was conducted with the approval of the local institutional review board at the China Medical University. Paraffin specimens (n=112; 72 men and 40 women) were obtained from patients with lung cancer who underwent surgery or biopsy at the First Affiliated Hospital of China Medical University. None of these patients had received chemotherapy or radiotherapy before surgical resection, and all patients were treated with routine chemotherapy after the operation. The mean age of the patients was 61 years (range, 36–76 years). The histological diagnosis and grade of differentiation of the tumors were defined by evaluation of the hematoxylin and eosin–stained tissue sections, according to the World Health Organization guidelines of classification. All 112 specimens were reevaluated with respect to their histological subtypes, differentiation status, and tumor stages. For NSCLC samples, squamous cell carcinoma and adenocarcinoma were identified in 48 and 64 of the 112 cases, respectively. The p-TNM staging system of the International Union Against Cancer was used to classify specimens as stages I (n=55), II (n=33), and III (n=24).
Cell Lines
A549, H1299, H157, LH7, and BE1 cell lines were obtained from American Type Culture Collection (Manassas, VA). SPC, LTE, and LK2 cell lines were purchased from the Shanghai Cell Bank of the Chinese Academy of Science. The cells were cultured in RPMI 1640 (Invitrogen; Carlsbad, CA) containing 10% fetal calf serum (Invitrogen), 100 IU/ml penicillin (Sigma; St. Louis, MO), and 100 µg/ml streptomycin (Sigma). Cells were grown on sterile tissue culture dishes and passaged every 2 days using 0.25% trypsin (Invitrogen).
Immunohistochemistry
Surgically excised tumor specimens were fixed with 10% neutral formalin and embedded in paraffin, and 4-µm-thick sections were prepared. Immunostaining was performed using the avidin-biotin-peroxidase complex method (Ultra Sensitive TM; Maixin, Fuzhou, China). The sections were deparaffinized in xylene, rehydrated in graded alcohol series, and boiled in 0.01 M citrate buffer (pH 6.0) for 2 min in an autoclave. Endogenous peroxidase activity was blocked using hydrogen peroxide (0.3%), which was followed by incubation with normal goat serum to reduce nonspecific binding. Tissue sections were incubated with DEK rabbit polyclonal antibody (1:200 dilution; Proteintech, Chicago, IL). Rabbit immunoglobulin was used as a negative control. Staining for all primary antibodies was performed at room temperature for 2 hr. Biotinylated goat anti–rabbit serum IgG (ready-to-use; Maixin) was used as the secondary antibody. After washing, the sections were incubated with horseradish peroxidase–conjugated streptavidin-biotin, followed by 3,3′-diaminobenzidine tetrahydrochloride to develop the peroxidase reaction. Counterstaining of the sections was done with hematoxylin, which were then dehydrated in ethanol before mounting.
Two independent investigators examined all tumor slides randomly. Five views were examined per slide, and 100 cells were observed per view at ×400 magnification. Immunostaining of DEK was scored following a semi-quantitative scale by evaluating, in representative tumor areas, the intensity and percentage of cells showing higher immunostaining than the control cells. Nuclear staining of the tumor cells was considered positive immunostaining. The intensity of DEK nuclear staining was also scored as 0 (no staining), 1 (weak), or 2 (marked). Percentage scores were assigned as 1 (1–25%), 2 (26–50%), 3 (51–75%), and 4 (76–100%). The scores of each tumor sample were multiplied to give a final score of 0 to 8, and the total expression of DEK was determined as either negative or low expression (−) (score <4) or overexpression (+) (score ≥4).
Quantitative Real-Time PCR (SYBR Green Method)
Quantitative real-time PCR was performed using the SYBR Green PCR master mix (Applied Biosystems; Foster City, CA) in a total volume of 20 µl on a 7900HT Fast Real-Time PCR System (Applied Biosystems) as follows: 95C for 30 sec, 40 cycles of 95C for 5 sec, and 60C for 30 sec. A dissociation step was performed to generate a melting curve to confirm the specificity of the amplification. β-actin was used as the reference gene. The relative levels of gene expression were represented as ΔCt=Ct gene – Ct reference, and the fold change of gene expression was calculated by the 2−ΔΔCt method. Experiments were repeated in triplicate.
The primer sequences were as follows:
DEK: forward, 5′-CCGAGAAAGAACCCGAAATGC-3′; reverse, 5′-GTGCCTGGCCTGTTGTAAAGC-3′
β-Actin: forward, 5′-ATAGCACAGCCTGGATAGCAACGTAC-3′; reverse, 5′-CACCTTCTACAATGAGCTGCGTGTG-3′
RhoA: forward, 5′-CGACAGCCCTGATAGTTTA-3′; reverse, 5′-GTGCTCATCATTCCGAAGA-3′
RhoB: forward, 5′-CGACGAGCATGTCCGCAC-3′; reverse, 5′-AGCCGTTCTGGGAGCCGTAG -3′
RhoC: forward, 5′-GGAGGTCTACGTCCCTACTGT-3′; reverse, 5′-CGCAGTCGATCATAGTCTTCC-3′
Western Blot Analysis
Total proteins from cell lines were extracted in lysis buffer (Thermo Fisher Scientific; Rockford, IL) and quantified using the Bradford method. Then, 50 µg of protein was separated by SDS-PAGE (10%). After transferring, the polyvinylidene fluoride (PVDF) membranes (Millipore; Billerica, MA) were incubated overnight at 4C with the following antibodies: DEK (1:2000; Proteintech), GAPDH (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), cyclin A (1:1000), cyclin B (1:1000), and cyclin D1 (1:1000) (Cell Signaling Technology, Boston, MA). The Rho pathway was studied using specific antibodies against RhoA (1:500; Proteintech), ROCK1 (1:1000; Proteintech), MLC2 and phospho-MLC2 (1:500; Cell Signaling Technology). After incubation with peroxidase-coupled anti–mouse/rabbit IgG (Santa Cruz Biotechnology) at 37C for 2 hr, bound proteins were visualized using ECL (Thermo Fisher Scientific) and detected using BioImaging Systems (UVP; Upland, CA). The relative protein levels were calculated based on GAPDH as the loading control.
Transient Transfection Assay
A549 and H1299 cells were plated onto 6-cm2 cell culture dishes and grown to 30–50% confluence before transfection with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. The transfection efficiency was assessed by flow cytometry. Following transfection, the mRNA and protein levels were assessed 48 hr later. Myc-DDK–tagged DEK human complementary DNA (cDNA) open reading frame, cloned into pCMV6-Entry vector, was from OriGene (Rockville, MD). Small interfering (Si) RNA for DEK and RhoA were purchased from RiboBio (Guangzhou, China).
Cell Proliferation Test and Colony Formation Assay
Twenty-four hours after transfection, cells were plated in 96-well plates in medium containing 10% FBS at an approximate density of 3500 cells per well, and the quantitation of cell viability was determined by the MTT assay. Briefly, 20 µl of 5 mg/ml MTT (Sigma) solution was added to each well and incubated for 4 hr at 37C. The media was then removed from each well and the resultant MTT formazan was solubilized in 150 µl DMSO. The results were quantitated spectrophotometrically using a test wavelength of 490 nm. For the colony formation assay, cells were plated into 6-cm cell culture dishes (1000 per dish for A549 and H1299 cell lines) and incubated for 12 days. Plates were washed with PBS and stained with hematoxylin. The number of colonies with more than 50 cells was counted.
Apoptosis Analysis
For detection of apoptosis, adherent cells were collected and resuspended in cold PBS for analysis. Cells were stained with the Annexin V-FITC Apoptosis Kit (BD Pharmingen, San Jose, CA) to quantitate the number of apoptotic cells and propidium iodide (PI) to detect dead cells. Data were collected using BD systems. FACS-scan flow cytometer (Becton-Dickinson, San Jose, CA) and the percentage of cell apoptosis were analyzed using CellQuest analysis software (Becton-Dickinson).
Cell Migration Assay
Cell migration was assessed by the Transwell migration assay (Costar; Cambridge, MA). Forty-eight hours after transfection, cells were trypsinized and transferred to the upper Matrigel chamber at a density of 4 × 104 cells in 100 µl of serum-free medium and incubated for 24 hr. Medium supplemented with 10% FBS alone or containing 100 ng/ml epidermal growth factor (Invitrogen) was added to the lower chamber as the chemoattractant. After incubation, the cells on the upper membrane surface were removed with a cotton tip. The migrated cells on the lower surface of the inserts were fixed in 100% methanol and stained in hematoxylin before being mounted onto a glass slide. Ten randomly selected high-power fields were analyzed using light microscopy and the number of migrated cells scored. Each experiment was performed in duplicate inserts and the mean value expressed as a percentage from three independent experiments.
Rho-GTP Pull-Down Assay
The Rho-GTP pull-down assay was performed using an RhoA/Rac1/Cdc42 Activation Assay Combo Kit (STA-405; Cell Biolabs, San Diego, CA). Cells cultured to approximately 80% to 90% confluence, were rinsed twice with ice-cold PBS before being treated with ice-cold lysis buffer (0.5–1 mL per 100-mm tissue culture plate). The cell culture plates were subsequently placed on ice for 10 to 20 min. The cells were scraped from the wells and placed the lysate into microcentrifuge tubes for centrifugation. The cells were centrifugated at 14,000 × g for 10 min at 4C, and then 20 µl of 0.5 M EDTA was added to each sample. The positive and negative controls received 10 µl of 100× GTPγS and 10 µl of 100× GDP, respectively. All tubes were incubated with agitation for 30 min at 30C. Reactions were stopped using 65 µl of 1.0 M MgCl2. Rhotekin RBD or PAK PBD Agarose beads were added to the cell lysates and incubated for 1 hr at 4C. After centrifugation for 10 sec at 14,000 × g, the beads were washed with lysis buffer and Rho-GTP was eluted in Laemmli sample buffer. Western blotting was performed to analyze the amount of GTP-bound RhoA.
Statistical Analysis
SPSS version 16.0 for Windows (SPSS, Inc., an IBM Company, Chicago, IL) was used for all analyses. The Chi-squared test was used to examine possible correlations between DEK expression and clinicopathological factors. The Student’s t-test was used to compare other data. The Kaplan-Meier method was used to estimate the probability of patient survival, and differences in the survival of subgroups of patients were compared using Mantel’s log-rank test. The p value was based on the two-sided statistical analysis, and p<0.05 was considered to indicate statistical significance.
Results
Overexpression of DEK Protein in NSCLC Tissue
We performed immunohistochemistry for 112 cases of NSCLC and 38 cases of normal lung tissue and found that DEK expression levels were significantly higher in the malignant cells compared with that in normal lung epithelial cells. In normal bronchial epithelial cells, DEK exhibited negative staining (Fig. 1A, B). In 112 cases of NSCLC, DEK was overexpressed mainly in the nuclear compartment of the tumor cells (Fig. 1C, D). As shown in Table 1, positive DEK staining was observed in 58.9% (66/112) of cases. In squamous cell carcinoma, DEK-positive expression was found in 47.9% (23/48) of cases. In lung adenocarcinoma, DEK-positive expression was found in 67.2% (43/64) of cases and correlated with differentiation (p=0.001), p-TNM stage (p=0.017), and nodal status (p=0.025). Moreover, in lung adenocarcinoma, DEK expression was significantly higher compared with DEK expression in squamous cell carcinoma (p=0.040). Kaplan-Meier analysis showed that patients with low DEK expression had higher overall survival compared with patients with high DEK expression (p=0.021) (Fig. 1G). Overall, our data show a correlation between DEK overexpression and poor prognosis in patients with NSCLC.
Figure 1.

DEK expression in non–small cell lung cancer (NSCLC) specimens. (A) Negative staining in normal bronchial epithelium in non-cancerous lung tissue. (B) Negative staining in normal pneumocytes in the alveoli of non-cancerous lung tissue. (C) Positive DEK staining in a case of adenocarcinoma. (D) Positive DEK staining in a case of squamous cell carcinoma. (E) Negative control. (F) Positive control (DEK is strongly positive in invasive ductal carcinoma of breast). Scale bars: 50 um. (G) Survival curves of patients with positive and negative DEK expression.
Table 1.
Distribution of DEK Status in Non–Small Cell Lung Cancer According to Clinicopathological Characteristics.
| Characteristics | No. of Patients | DEK Negative, No. (%) | DEK Positive, No. (%) | χ2 Value | P Value |
|---|---|---|---|---|---|
| Age, y | |||||
| <60 | 51 | 22 (43.1) | 29 (56.9) | 0.165 | 0.684 |
| ≥60 | 61 | 24 (39.3) | 37 (60.7) | ||
| Sex | |||||
| Male | 72 | 28 (38.9) | 44 (61.1) | 0.397 | 0.529 |
| Female | 40 | 18 (45.0) | 22 (55.0) | ||
| Histology | |||||
| Adenocarcinoma | 64 | 21 (32.8) | 43 (67.2) | 4.209 | 0.040* |
| Squamous cell carcinoma | 48 | 25 (52.1) | 23 (47.9) | ||
| Differentiation | |||||
| Well | 41 | 16 (39.0) | 25 (61.0) | 13.373 | 0.001* |
| Moderate | 47 | 27 (57.4) | 20 (42.6) | ||
| Poor | 24 | 3 (12.5) | 21 (87.5) | ||
| TNM stage | |||||
| I | 55 | 28 (50.9) | 27 (49.1) | 8.130 | 0.017* |
| II | 33 | 14 (42.4) | 19 (57.6) | ||
| III | 24 | 4 (16.7) | 20 (83.3) | ||
| Tumor status | |||||
| T1 | 59 | 25 (42.4) | 34 (57.6) | 0.087 | 0.768 |
| T2, T3, and T4 | 53 | 21 (39.6) | 32 (60.4) | ||
| Lymph node metastasis | |||||
| N0 | 69 | 34 (49.3) | 35 (50.7) | 4.998 | 0.025* |
| N1, N2, and N3 | 43 | 12 (27.9) | 31 (72.1) | ||
p<0.05 was considered significant.
DEK Depletion Inhibits Proliferation of A549 and H1299 Cells
The expression of DEK was analyzed by Western blot in a panel of lung cancer cell lines (Fig. 2A, B). To elucidate the biological function of DEK in lung cancer, we employed siRNA to knock down DEK expression in H1299 and A549 cell lines, both of which showed moderate expression of DEK protein. DEK-specific siRNA substantially reduced DEK mRNA and protein expression levels after 48 hr of siRNA treatment (Fig. 2C, D). The proliferation rate was determined using the MTT assay. In A549 and H1299 cells transfected with DEK siRNA, the proliferation rate was significantly reduced compared with the negative control (Fig. 3A). Consistent with the MTT assay, depletion of DEK in A549 (control vs. DEK siRNA: 266 ± 22 vs 148 ± 15, p<0.05) and H1299 (control vs. DEK siRNA: 357 ± 35 vs 167 ± 23, p<0.05) cells led to a significant reduction in foci numbers as well as sizes (Fig. 3B, C). These data suggest that DEK modulates the proliferation of lung cancer cells. In addition, we evaluated the effect of DEK knockdown on cyclin A, cyclin B, and cyclin D1 levels. As shown in Fig. 4, Western blotting analysis revealed that knockdown of DEK resulted in decreased protein levels of cyclin A (p<0.05).
Figure 2.

DEK depletion in A549 and H1299 cell lines. (A, B) The expression level of DEK was analyzed by Western blot in a panel of lung cancer cell lines. (C) Western blot of DEK depletion efficiency in cancer cells. (D) Real-time PCR analyses of DEK depletion efficiency in cancer cells.
Figure 3.

DEK depletion impaired cancer cell proliferation. (A) The MTT assay was performed after DEK small interfering RNA (siRNA) treatment. A reduction in absorbance was observed (p<0.05). (B) Assessment of the clonogenic potential of DEK-depleted cancer cells. Scale bars: 50 um. (C) Numbers of colonies were counted. The number of colonies formed by cells treated with DEK siRNA was significantly less than that of control cells (p<0.05). *p<0.05.
Figure 4.
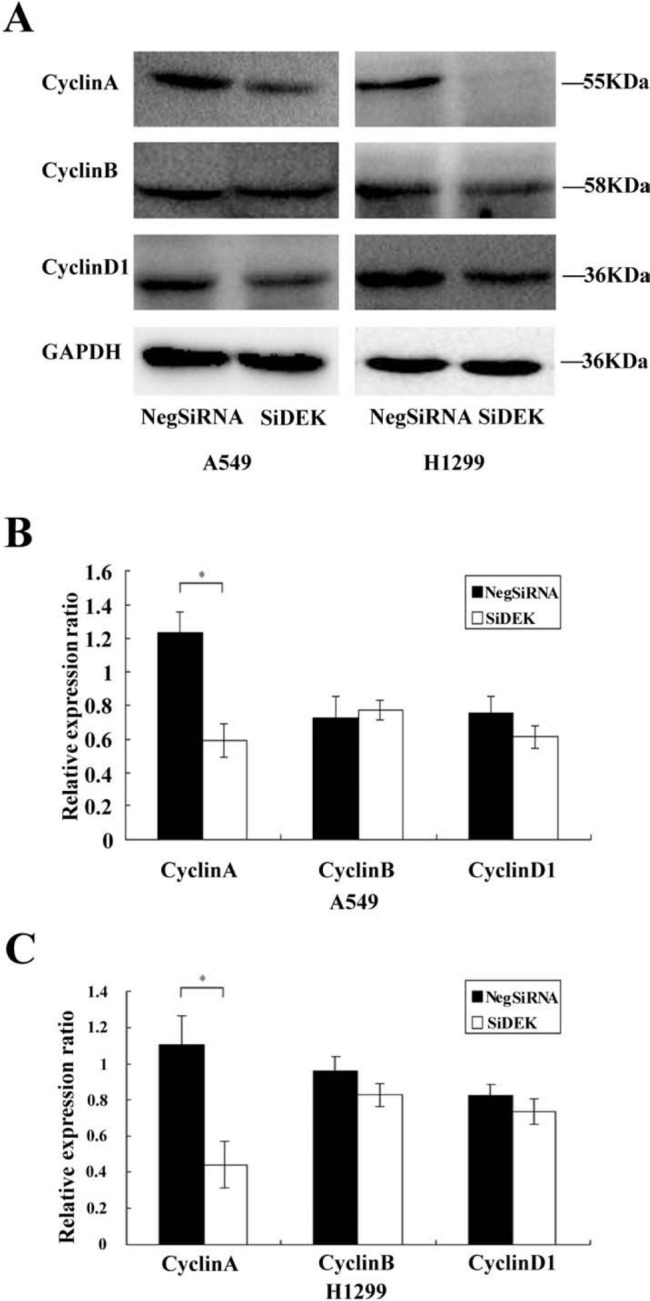
Decreased cell growth induced by DEK knockdown was associated with decreased cyclin A expression. (A) Western blot analysis of a series of cell cycle–related factors showed after silencing DEK in H1299 and A549 cells, protein levels of cyclin A were decreased (p<0.05), but no changes were observed in protein levels of cyclin B or cyclin D1 (p>0.05). (B, C) Densitometric analyses of protein levels normalized against GAPDH. *p<0.05.
Effect of DEK Depletion on Apoptosis in A549 and H1299 Cells
The Annexin V kit was employed to characterize the effect of DEK knockdown on apoptosis of H1299 and A549 cells. DEK knockdown did not show an effect on cellular apoptosis in A549 and H1299 cells relative to siControl-treated cells (Fig. 5A–C).
Figure 5.
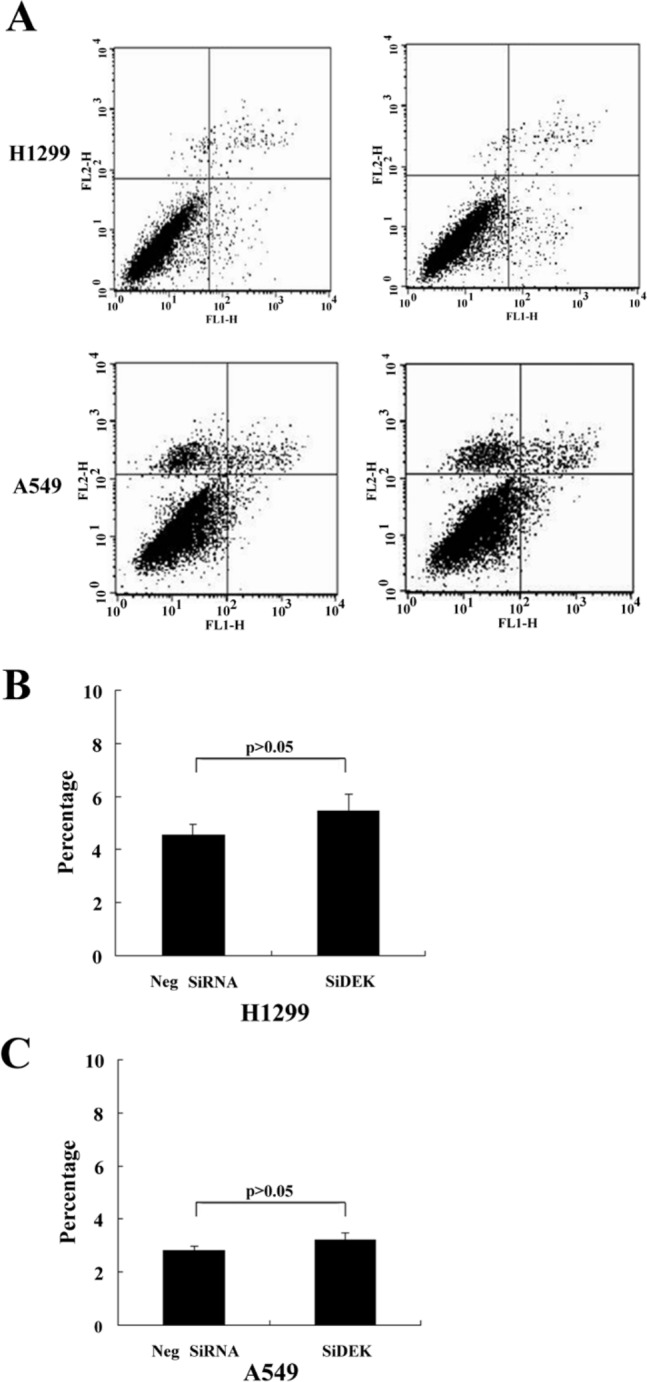
The effect of DEK knockdown on cell apoptosis in A549 and H1299 cells. (A–C) Lung cancer cells were transfected with DEK small interfering RNA (siRNA). Cells were harvested 48 hr after transfection and processed for Annexin V-FITC and propidium iodide (PI) staining and analyzed by flow cytometry. (A) Representative flow cytometry plots are shown. (B,C) Percentage of apoptotic cells.
Downregulation of DEK Expression in NSCLC Reduces Migration Activity
To determine whether DEK influences the migration of NSCLCs, we conducted Transwell migration assays. In siDEK-treated cells, cellular migration was significantly inhibited compared with the siControl-treated group (p<0.05) (Fig. 6A–C).
Figure 6.
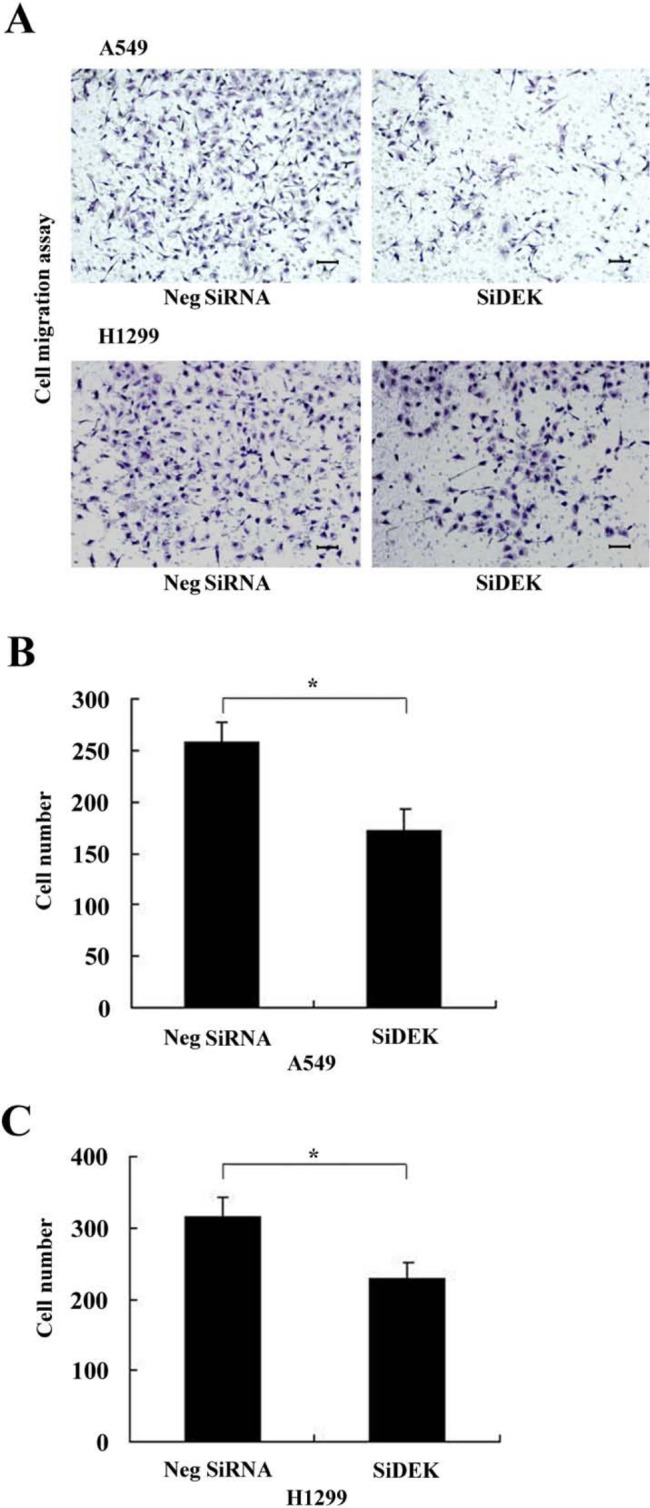
DEK depletion inhibited cancer cell migration. (A) DEK knockdown inhibited cell migration in both A549 and H1299 lines. Scale bars: 50 um. (B, C) Assessment of migratory ability of the DEK-depleted cancer cells. Numbers of cells migrating onto the lower surface were counted. The number of migratory cells after silencing DEK was significantly less than that of control cells in both A549 and H1299 lines (p<0.05). *p<0.05.
DEK Depletion Decreases RhoA Expression and Modulates Cell Migration through RhoA
To further explore the mechanisms by which DEK promotes NSCLC cell migration, we evaluated the expression of RhoA, RhoB, and RhoC before and after transfection of siRNA. As shown in Fig. 7, upon DEK depletion in A549 and H1299 cells, the RNA and protein levels of RhoA were significantly decreased (p<0.01, p<0.05). However, there was no change in RhoB or RhoC expression (p>0.05). Subsequently, a DEK expression plasmid was co-transfected with siRhoA in H1299 and A549 cell lines. The Transwell assay showed that RhoA siRNA significantly inhibited DEK-mediated cell migration (p<0.05) (Fig. 8A–E).
Figure 7.
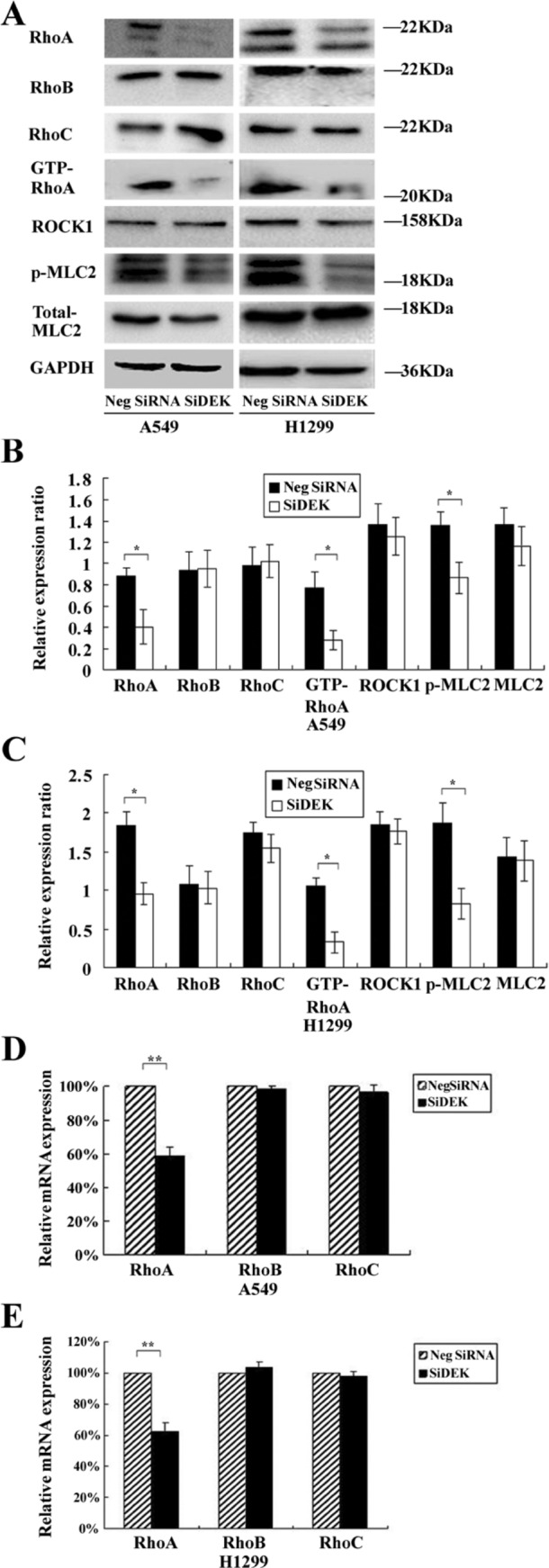
DEK depletion downregulated RhoA expression. (A) Western blot analyses of RhoA, RhoB, and RhoC expression in A549 and H1299 lines transfected with the DEK small interfering RNA (siRNA). (B, C) Densitometric analyses of protein levels normalized against GAPDH, which shows a significant reduction in RhoA (p<0.05), whereas the RhoB and RhoC levels were unaffected (p>0.05). (D, E) Real-time PCR analyses of RhoA, RhoB, and RhoC messenger RNA (mRNA) in A549 and H1299 lines. A significant reduction in the mRNA level of RhoA is also shown (p<0.01). *p<0.05. **p<0.01.
Figure 8.

RhoA small interfering RNA (siRNA) treatment significantly inhibited DEK-mediated cell migration. (A, B) Western blot analyses of RhoA and DEK expression in co-transfected cells. (C) RhoA knockdown significantly inhibited DEK-mediated cell migration in both A549 and H1299 lines. Scale bars: 50 um. (D, E) Assessment of migratory ability of co-transfected cancer cells. Numbers of cells migrating onto the lower surface were counted. The number of cells was significantly higher compared with control cells after DEK upregulation in both A549 and H1299 lines (p<0.05), but no significant changes in cell numbers after silencing RhoA in DEK-overexpressing H1299 and A549 lines. *p<0.05.
DEK Depletion Abolishes Activation of the RhoA Pathway
To determine whether knockdown of DEK could affect the GTP/GDP binding status of Rho GTPases, GTP-bound RhoA was investigated using the Rho-GTP pull-down assay. As shown in Fig. 7A–C, upon DEK depletion, the level of GTP-RhoA was significantly decreased in H1299 and A549 cells (p< 0.05). Hence, suppression of DEK could affect the level of active GTP-RhoA. Next, we hypothesized that the RhoA pathway could be inactivated upon DEK depletion. We investigated the key components in the RhoA signal transduction pathway—namely, the direct downstream effectors ROCK1 (Rho-associated serine/threonine protein kinase 1) and MLC2 (myosin light chain 2). Following the decrease in active GTP-RhoA in siDEK-treated A549 and H1299 cells, the phosphorylated active form of MLC2 was correspondingly found to be significantly decreased (p<0.05) (Fig. 7A–C), whereas total ROCK1 protein and total MLC2 levels were not statistically different between siDEK or siControl conditions (p>0.05) (Fig. 7A–C).
Discussion
The upregulation of DEK expression has been implicated in several human cancers such as retinoblastoma tumors (Grasemann et al. 2005; Orlic et al. 2006; Paderova et al. 2007), hepatocellular carcinoma (Kondoh et al. 1999), bladder cancer (Evans et al. 2004), brain malignant glioma (Kroes et al. 2000), melanoma (Khodadoust et al. 2009), and adult acute leukemias (Larramendy et al. 2002; Casas et al. 2003). However, the expression pattern of DEK as well as its correlation with clinical and pathological significance has not yet been defined in human lung cancer. In this study, we demonstrated that the expression of DEK protein in lung cancer tissues was higher compared with corresponding normal lung tissues. There was a correlation between the upregulation of DEK and pTNM stage, differentiation, and nodal status, which was in accord with previous data, suggesting that DEK may play an important role in lung cancer progression. To validate the potential role of DEK in lung cancer progression, we first examined DEK expression levels in several cell lines and selected the A549 and H1299 cell lines for further study because of their relatively moderate DEK levels. We employed siRNA to knock down DEK expression in the A549 and H1299 cell lines. We found an impaired proliferation capacity and colony formation ability for both A549 and H1299 cells after DEK knockdown. The decreased cell growth induced by DEK knockdown was associated with decreased cyclin A expression, which was in accord with a previous report using breast cancer cells (Privette Vinnedge et al. 2011). To determine if the impaired cell growth was due to changes in apoptosis, the Annexin V kit was employed to characterize apoptosis in the A549 and H1299 cells after DEK knockdown. DEK suppression did not show an apparent effect on cell apoptosis in A549 and H1299 cells relative to siControl-treated cells. In addition, we showed that DEK siRNA inhibited cell migration. To further explore the mechanism of DEK-mediated cell migration, we first examined several members of the Rho-GTPase family, including RhoA, RhoB, and RhoC. Both mRNA and protein levels of RhoA were significantly decreased upon DEK depletion in A549 and H1299 cells, whereas DEK depletion had no significant effect on the expression level of RhoB or RhoC. Furthermore, we determined that siRNA-mediated inhibition of RhoA reduced DEK-mediated cell migration, indicating that RhoA is essential for DEK-induced cell migration. The RhoA protein is a well-known member of the p21 Ras superfamily of small GTPases, which shuttles between an inactive GDP-bound state and an active GTP-bound state and exhibits intrinsic GTPase activities. RhoA regulates signal transduction from cell surface receptors to intracellular target molecules and is involved in a variety of biological processes, including cell morphology (Paterson et al. 1990), motility (Takaishi et al. 1993), smooth muscle contraction (Hirata et al. 1992), and tumor progression (Prendergast et al. 1995), and RhoA also acts as a molecular switch. Previous reports have shown that overexpression of RhoA facilitated the accumulation of RhoA protein in the cell membrane and led to further RhoA activation (Horiuchi et al. 2008). A similar result was also obtained using a rat liver cell model of MM1. Overexpression of RhoA resulted in increased levels of activated RhoA and enhanced tumor cell motility (Yoshioka et al. 1999). In other words, the inhibition of cell migration induced by DEK knockdown was associated with inactivation of the RhoA signal transduction pathway. The cellular migration of cancer cells requires Rho-GTPases and their associated signal transduction components, including ROCK and MLC. Active ROCK specifically phosphorylates myosin light chain 2 (MLC2) at Ser19; therefore, this ROCK-specific phosphorylation has been widely used as a surrogate marker of ROCK activity (Amano et al. 1996; Ueda et al. 2002; Wilkinson et al. 2005). Phosphorylation of MLC2 at Ser19 is important for the activity of myosin, which is responsible for actomyosin contractility and hence cell migration (Katoh et al. 2001). In this study, in DEK-depleted NSCLC cells, we found that RhoA RNA and protein levels were markedly reduced, in conjunction, the levels of active RhoA-GTP and the downstream effectors phosphorylating MLC2 were also reduced. It is therefore plausible that DEK-mediated cell motility may require at least in part the constitutive activation of the RhoA/ROCK/MLC signal transduction pathway, which promotes cytoskeletal dynamics and cell motility. Previous reports have shown that DEK is involved in transcriptional regulation. In this study, the data show that DEK is responsible for the transcription of RhoA and further affects the protein level of RhoA. It is plausible that overexpressed RhoA stimulates the translocation of RhoA from the cytosol to the membrane, activating RhoA protein on the membrane, followed by the stimulation of MLC2 phosphorylation, leading to cell migration in DEK-overexpressing cells.
Taken together, our findings are the first to report on the oncogenic activities of DEK in NSCLC and to define both the functional and molecular mechanisms for DEK in NSCLC pathogenesis. Furthermore, we report the significant and novel finding of a molecular mechanism for the migration phenotype, where DEK depletion inhibits migration activity in lung cancer cell lines through inactivation of the RhoA/ROCK/MLC signal transduction pathway. Naturally, it could be contended that other functional aspects of DEK contribute to the regulation of cellular migration, which needs further investigation. The next question that should be addressed is how DEK directly or indirectly regulates RhoA transcription in lung cancer cells.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (No. 30972967) and Specialized Research Fund for the Doctoral Program of Higher Education (No. 20092104110018) and Program for Liaoning Excellent Talents in University (NO.LR2011021).
References
- Alexiadis V, Waldmann T, Andersen J, Mann M, Knippers R, Gruss C. 2000. The protein encoded by the proto-oncogene DEK changes the topology of chromatin and reduces the efficiency of DNA replication in a chromatin-specific manner. Genes Dev. 14:1308–1312 [PMC free article] [PubMed] [Google Scholar]
- Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. 1996. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J Biol Chem. 271:20246–20249 [DOI] [PubMed] [Google Scholar]
- Campillos M, Garcia MA, Valdivieso F, Vazquez J. 2003. Transcriptional activation by AP-2alpha is modulated by the oncogene DEK. Nucleic Acids Res. 31:1571–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro MS, Spiga FM, Quarto M, Di Ninni V, Volorio S, Alcalay M, Müller H. 2006. DEK expression is controlled by E2F and deregulated in diverse tumor types. Cell Cycle. 5:1202–1207 [DOI] [PubMed] [Google Scholar]
- Casas S, Nagy B, Elonen E, Aventín A, Larramendy ML, Sierra J, Ruutu T, Knuutila S. 2003. Aberrant expression of HOXA9, DEK, CBL and CSF1R in acute myeloid leukemia. Leuk Lymphoma. 44:1935–1941 [DOI] [PubMed] [Google Scholar]
- Evans AJ, Gallie BL, Jewett MA, Pond GR, Vandezande K, Underwood J, Fradet Y, Lim G, Marrano P, Zielenska M, et al. 2004. Defining a 0.5-mb region of genomic gain on chromosome 6p22 in bladder cancer by quantitative-multiplex polymerase chain reaction. Am J Pathol. 164:285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farray D, Mirkovic N, Albain KS. 2005. Multimodality therapy for stage III non–small-cell lung cancer. J Clin Oncol. 23:3257–3269 [DOI] [PubMed] [Google Scholar]
- Grasemann C, Gratias S, Stephan H, Schüler A, Schramm A, Klein-Hitpass L, Rieder H, Schneider S, Kappes F, Eggert A, et al. 2005. Gains and overexpression identify DEK and E2F3 as targets of chromosome 6p gains in retinoblastoma. Oncogene. 24:6441–6449 [DOI] [PubMed] [Google Scholar]
- Hirata K, Kikuchi A, Sasaki T, Kuroda S, Kaibuchi K, Matsuura Y, Seki H, Saida K, Takai Y. 1992. Involvement of rho p21 in the GTP-enhanced calcium ion sensitivity of smooth muscle contraction. J Biol Chem. 267:8719–8722 [PubMed] [Google Scholar]
- Horiuchi A, Kikuchi N, Osada R, Wang C, Hayashi A, Nikaido T, Konishi I. 2008. Overexpression of RhoA enhances peritoneal dissemination: RhoA suppression with lovastatin may be useful for ovarian cancer. Cancer Sci. 12:2532–2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappes F, Waldmann T, Mathew V, Yu J, Zhang L, Khodadoust MS, Chinnaiyan AM, Luger K, Erhardt S, Schneider R, et al. 2011. The DEK oncoprotein is a Su(var) that is essential to heterochromatin integrity. Genes Dev. 25:673–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Kano Y, Amano M, Onishi H, Kaibuchi K, Fujiwara K. 2001. Rho-kinase–mediated contraction of isolated stress fibers. J Cell Biol. 153:569–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh GM, Wise-Draper TM, Morreale RJ, Morrison MA, Gole B, Schwemberger S, Tichy ED, Lu L, Babcock GF, Wells JM, et al. 2011. The human DEK oncogene regulates DNA damage response signaling and repair. Nucleic Acids Res. 39:7465–7476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodadoust MS, Verhaegen M, Kappes F, Riveiro-Falkenbach E, Cigudosa JC, Kim DS, Chinnaiyan AM, Markovitz DM, Soengas MS. 2009. Melanoma proliferation and chemoresistance controlled by the DEK oncogene. Cancer Res. 69:6405–6413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondoh N, Wakatsuki T, Ryo A, Hada A, Aihara T, Horiuchi S, Goseki N, Matsubara O, Takenaka K, Shichita M, et al. 1999. Identification and characterization of genes associated with human hepatocellular carcinogenesis. Cancer Res. 59:4990–4996 [PubMed] [Google Scholar]
- Kroes RA, Jastrow A, McLone MG, Colley P, Kersey DS, Yong VW, Mkrdichian E, Cerullo L, Leestma J, et al. 2000. The identification of novel therapeutic targets for the treatment of malignant brain tumors. Cancer Lett. 156:191–198 [DOI] [PubMed] [Google Scholar]
- Larramendy ML, Niini T, Elonen E, Nagy B, Ollila J, Vihinen M, Knuutila S. 2002. Overexpression of translocation associated fusion genes of FGFRI, MYC, NPMI, and DEK, but absence of the translocations in acute myeloid leukemia: a microarray analysis. Haematologica. 87:569–577 [PubMed] [Google Scholar]
- Orlic M, Spencer CE, Wang L, Gallie BL. 2006. Expression analysis of 6p22 genomic gain in retinoblastoma. Genes Chromosomes Cancer. 45:72–82 [DOI] [PubMed] [Google Scholar]
- Paderova J, Orlic-Milacic M, Yoshimoto M, da Cunha Santos G, Gallie B, Squire JA. 2007. Novel 6p rearrangements and recurrent translocation breakpoints in retinoblastoma cell lines identified by spectral karyotyping and mBAND analyses. Cancer Genet Cytogenet. 179:102–111 [DOI] [PubMed] [Google Scholar]
- Paterson HF, Self AJ, Garrett MD, Just I, Aktories K, Hall A. 1990. Microinjection of recombinant p21rho induces rapid changes in cell morphology. J Cell Biol. 111:1001–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast GC, Khosravi-Far R, Solski PA, Kurzawa H, Lebowitz PF, Der CJ. 1995. Critical role of Rho in cell transformation by oncogenic Ras. Oncogene. 10:2289–2296 [PubMed] [Google Scholar]
- Privette Vinnedge LM, McClaine R, Wagh PK, Wikenheiser-Brokamp KA, Waltz SE, Wells SI. 2011. The human DEK oncogene stimulates beta-catenin signaling, invasion and mammosphere formation in breast cancer. Oncogene. 30:2741–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed MF, Molloy M, Dalton EL, Howington JA. 2004. Survival after resection for lung cancer is the outcome that matters. Am J Surg. 188:598–602 [DOI] [PubMed] [Google Scholar]
- Rooney C, Sethi T. 2011. The epithelial cell and lung cancer: the link between chronic obstructive pulmonary disease and lung cancer. Respiration. 81:89–104 [DOI] [PubMed] [Google Scholar]
- Sawatsubashi S, Murata T, Lim J, Fujiki R, Ito S, Suzuki E, Tanabe M, Zhao Y, Kimura S, Fujiyama S, et al. 2010. A histone chaperone, DEK, transcriptionally coactivates a nuclear receptor. Genes Dev. 24:159–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, Zhu J, Johnson DH. 2002. Eastern Cooperative Oncology Group: comparison of four chemotherapy regimens for advanced non–small-cell lung cancer. N Engl J Med. 346:92–98 [DOI] [PubMed] [Google Scholar]
- Soares LM, Zanier K, Mackereth C, Sattler M, Valcarcel J. 2006. Intron removal requires proofreading of U2AF/3′ splice site recognition by DEK. Science. 312:1961–1965 [DOI] [PubMed] [Google Scholar]
- Soekarman D, von Lindern M, Daenen S, de Jong B, Fonatsch C, Heinze B, Bartram C, Hagemeijer A, Grosveld G. 1992. The translocation (6;9) (p23;q34) shows consistent rearrangement of two genes and defines a myeloproliferative disorder with specific clinical features. Blood. 79:2990–2997 [PubMed] [Google Scholar]
- Takaishi K, Kikuchi A, Kuroda S, Kotani K, Sasaki T, Takai Y. 1993. Involvement of rho p21 and its inhibitory GDP/GTP exchange protein (rho GDI) in cell motility. Mol Cell Biol. 13:72–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda K, Murata-Hori M, Tatsuka M, Hosoya H. 2002. Rho-kinase contributes to diphosphorylation of myosin II regulatory light chain in nonmuscle cells. Oncogene. 21:5852–5860 [DOI] [PubMed] [Google Scholar]
- von Lindern M, Fornerod M, van Baal S, Jaegle M, de Wit T, Buijs A, Grosveld G. 1992. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia-specific dek-can mRNA. Mol Cell Biol. 12:1687–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson S, Paterson HF, Marshall CJ. 2005. Cdc42-MRCK and Rho-ROCK signalling cooperate in myosin phosphorylation and cell invasion. Nat Cell Biol. 7:255–261 [DOI] [PubMed] [Google Scholar]
- Wise-Draper TM, Allen HV, Jones EE, Habash KB, Matsuo H, Wells SI. 2006. Apoptosis inhibition by the human DEK oncoprotein involves interference with p53 functions. Mol Cell Biol. 26:7506–7519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka K, Nakamori S, Itoh K. 1999. Overexpression of small GTP-binding protein RhoA promotes invasion of tumor cells. Cancer Res. 59:2004–2010 [PubMed] [Google Scholar]