

Abstract
During cell competition fitter cells take over the tissue at the expense of viable, but less fit, cells, which are eliminated by induction of apoptosis or senescence. This probably acts as a quality-control mechanism to eliminate suboptimal cells and safeguard organ function. Several experimental conditions have been shown to trigger cell competition, including differential levels in ribosomal activity or in signalling pathway activation between cells, although it is unclear how those differences are sensed and translated into fitness levels. Many of the pathways implicated in cell competition have been previously linked with cancer, and this has led to the hypothesis that cell competition could play a role in tumour formation. Cell competition could be co-opted by cancer cells to kill surrounding normal cells and boost their own tissue colonization. However, in some cases, cell competition could have a tumour suppressor role, as cells harbouring mutations in a subset of tumour suppressor genes are killed by wild-type cells. Originally described in developing epithelia, competitive interactions have also been observed in some stem cell niches, where they play a role in regulating stem cell selection, maintenance and tissue repopulation. Thus competitive interactions could be relevant to the maintenance of tissue fitness and have a protective role against aging.
Introduction
Cell competition occurs when cells with different fitness levels confront one another. It results in the elimination of the weaker population through apoptosis or senescence, whereas the stronger population survives and proliferates. Originally described in developing epithelia, competitive interactions have been linked with tissue homoeostasis, organ size control and stem cell maintenance. Recent work also suggests that they may play a role in tissue regeneration and in cancer development. In the present chapter we will introduce the pathways implicated in initiating competition and report on our current understanding of the mechanisms involved in this process.
Pathways of cell competition
Cell competition was discovered in 1975, through characterization of the growth defects of Minute heterozygous mutations in Drosophila wing imaginal discs [1]. Minute (M) genes encode ribosomal subunits. Thus homozygous mutations are lethal due to a lack of functional ribosomes; however, heterozygous animals are viable and display just a developmental delay and minor morphological abnormalities. These early studies from Morata, Ripoll and Simpson [1–3] showed that when Minute heterozygous (M/ +) and wild-type cells were present in the same tissue, wild-type cells would take over, whereas slow-growing M/+ cells were eliminated and their contribution to the adult wing was reduced. This suggested that competition could act as a surveillance system to actively remove mutant defective cells from the tissue. In 2004, Oliver et al. [4] reproduced these findings in a mouse Minute mutation (Belly Spot and Tail), providing the first indication that the phenomenon of cell competition seen in Drosophila also occurs in mice.
Over 20 years after these first observations were made, Johnston et al. [5] reported that cells with differing levels of the transcription factor dMyc could also initiate competition. Cells with low levels of dMyc, because of a mutation in the corresponding gene, were lost in the presence of wild-type cells, but were viable when surrounded by the same cells. Moreno and Basler [6] and de la Cova et al. [7] later showed that mutant clones overexpressing dMyc could outcompete wild-type cells, suggesting that it was the relative levels of dMyc that decided the outcome of competition. In other words, the outcome of competition is context-dependent and cells become winners or losers depending on the fitness of their neighbours. This work confirmed the concept of supercompetitors: cells capable of outcompeting normal wild-type cells [8]. The discovery of supercompetitors established the initial link between cell competition and cancer. The human homologue of dmyc is an established proto-oncogene, controlling the expression of many other genes involved in growth and proliferation and is frequently overexpressed in tumours [9]. Thus it was proposed that, similarly to what had been observed in Drosophila, cancer cells with high Myc levels could cause the elimination of surrounding normal cells, creating space in which to expand.
This view was further strengthened when mutations in tumour suppressors also gave rise to supercompetitors. The Hippo pathway modulates cell survival and proliferation and thus safeguards against neoplastic growth. Inactivation of this pathway through yorkie overexpression or mutations in fat, expanded or warts enables cells to eliminate surrounding wild-type tissue [10,11]. Similarly, Vincent et al. [12] showed that, in Drosophila, relative differences in Wnt signalling induce competition. In this study, cells that cannot transduce the Wnt signal or cells that overactivate the pathway (APC or Axin mutant) were juxtaposed to wild-type cells. In both cases, those cells with relatively lower Wnt signalling levels were eliminated. Wnt signalling is overactivated in a number of cancers and Axin and APC are frequently mutated tumour suppressor genes [13]. Thus cell competition could also play a role in Wnt-induced cancers.
Tumour-suppressor-based mechanisms of cell competition have also been studied in vivo in a mammalian system. The transcription factor p53 is one of the best-known and most studied tumour suppressor genes [14]. Bondar and Medzhitov [15] characterized a form of cell competition induced by stress and mediated by p53. Doing repopulation assays in lethally irradiated bone marrow, they found that in the mouse haemopoietic stem cell, niche cell competition selects for the least damaged cells by comparing levels of p53 activity and selecting those cells with relatively lower p53 levels. This work, carried out with mouse HSPCs (haemopoietic stem and progenitor cells), shows that competition is not restricted to epithelial tissues. In addition to this study, the occurrence of cell competition in stem cell niches has also been reported in the Drosophila ovary and testis [16–19]. Moreover, cell competition has been shown during liver repopulation assays in rats [20].
The above tumour suppressor mutations induce a supercompetitive behaviour; however, this is not always the case, loss-of-function of the tumour suppressor genes scrib (scribble) or lgl (lethal giant larvae) leads to their elimination by the surrounding wild-type cells, both in Drosophila mosaic discs and mammalian cells [21–24]. Thus not all tumour-promoting mutations lead to a competitive advantage, and in some cases precancerous cells can be eliminated by cell competition. How do mutations alter the competitive status of a cell and what cellular events take place during competition? We review the current knowledge in the following sections.
Sensing cellular fitness
By far the most mysterious aspect of the cell competition process is to understand how cells sense and compare fitness levels across tissues. What are the signals and mechanisms that cells use to compare fitness and how are less fit cells identified and earmarked as losers?
Several studies have highlighted a correlation between differential proliferation rates and cell competition. For example, slow-growing Minute cells behave as losers next to normally growing wild-type cells; in turn, wild-type cells behave as losers next to cells that grow faster, e.g. because of high Myc activity [1–3,6,7]. Thus it has been suggested that, during competition, information about growth rates is translated into fitness levels and accounts for the acquisition of the loser/winner status. However, not all manipulations that increase growth rates appear to be sufficient to change the competitive status of a cell. For example boosting cellular growth by overactivating the insulin pathway is not sufficient to induce a super-competitor status [7]. Therefore it is possible that cells do not compare growth rates; rather they sense differences in a parameter, whose changes mirror differences in growth rates in some instances.
Another clue in understanding how cells sense relative fitness levels comes from the many reports indicating that disparities in some signalling pathways often lead to cell competition. This has been described for the Fat/Hippo [11,22,25] and Wnt signalling pathways [12]. In addition it has been reported that, during Minute competition, loser cells display reduced Dpp [Drosophila TGF-β (transforming growth factor β) superfamily member] signalling and that increasing the levels of signalling is sufficient to rescue them during both Minute- and Myc-induced competition [6,26]; however, this view has been questioned in subsequent work [7,10,27]. Thus, although it is unclear how signalling levels are translated into fitness levels, it appears that several signalling pathways are able to modulate the levels of cell fitness that are compared during cell competition.
A recent breakthrough in the study of cell competition has come from the identification of a trio of proteins as important components of the fitness-sensing process. Performing transcriptional profiling of competing cells, Moreno and co-workers [28] showed that three differently spliced isoforms of the flower gene become differentially expressed in several contexts where competition is induced: the Flowerubi isoform is down-regulated in loser cells, whereas two other Flower isoforms, FlowerLoseA and FlowerLoseB, are exclusively up-regulated in loser cells. Importantly, expression of either FlowerLose isoform is necessary and sufficient for the elimination of loser cells [28]. The mechanism of action of the Flower proteins and how they induce death in losers is entirely unknown at present. Flower has been described in another study as a calcium transporter [29]; however, it is unclear whether this function is required for its role in competition. Since they are transmembrane proteins, it is likely that Flower proteins are involved in extracellular recognition events between winners and losers. However, it is worth noting that, since FlowerLose isoforms are not present at the onset of competition and become expressed only as competition is triggered, additional molecules must be responsible for the early sensing events that lead to the differential expression of the Flower isoforms.
The identification of membrane proteins as integral components of the fitness-sensing process indicates that some aspects of cell competition require cell–cell contact [28]. Moreover, additional studies have shown that competitive interactions are observed at a short range (ranging from direct proximity [30] to a few cell diameters away [7]). However, independent experiments in cell culture show that cell competition can still happen if winner and loser cells share the same culture medium but are not allowed direct contact [31]. Therefore it is possible that multiple independent molecular pathways initiate competition and that some of those are mediated by soluble factors. The nature of these factors remains to be identified: they could be signalling proteins or by-products of cellular metabolism. Interestingly, in the context of competition induced by Wnt signalling, high-Wnt cells release Notum, a diffusible Wnt inhibitor that is required for the outcompetition of low-Wnt cells [12]. Notum does not take part in the fitness-sensing process, but contributes to competition by lowering the Wnt signalling levels (and hence fitness) of surrounding wild-type cells.
Elimination of loser cells
As described in the Introduction, a key feature of cell competition is the non-autonomous induction of death in the weaker population. How are loser cells instructed to die by neighbouring winner cells? With one exception, in which loser outcompetition happens via induction of senescence rather than death (discussed below), loser cells are killed by apoptosis (Figure 1). However, while this is a common denominator, several independent upstream pathways have been shown to trigger the apoptotic event during cell competition. Their involvement seems to be context-dependent and influenced by the specific type of cell competition studied and possibly by the tissue studied (Figure 1A).
Figure 1. Mechanisms involved in the elimination of loser cells.
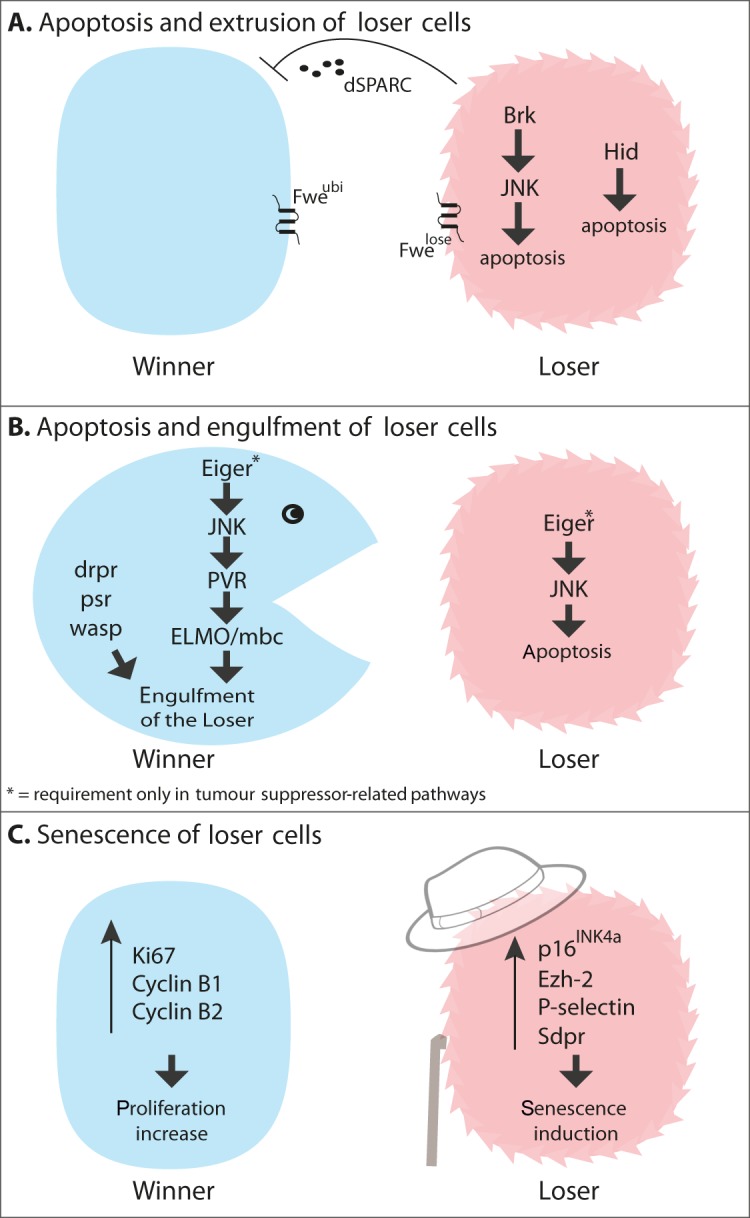
(A) Apoptosis and extrusion of loser cells. Two mechanisms of apoptosis induction have been identified. In the first, JNK is activated via Brk (Brinker), whereas in the second activation of the pro-apototic gene Hid leads to death. Different Fwe (Flower) isoforms label winner and loser cells. dSPARC is secreted from loser cells and acts as a temporary shield to protect them from apoptosis. (B) Apoptosis and engulfment of loser cells. In some contexts, winner cells engulf neighbouring loser cells, via activation of genes involved in phagocytosis (listed in the Figure). This is regulated by Eiger/JNK activation, which promotes apoptosis in the losers and engulfment in the winners. (C) Senescence of loser cells. Competition among HSPCs results in the activation of senescence-related genes in the loser cells (listed in the Figure). Conversely, winner cells activate proliferation markers (listed in the Figure).
Activation of the JNK (c-Jun N-terminal kinase) pathway, a pro-apoptotic pathway frequently activated in response to stress [32], is quite a common event during competition, having been observed during Minute [26] and Myc [6] competition (Figure 1A) and during competition induced by mutations in polarity-linked tumour suppressors [33,34]. Is JNK activation required to induce death in loser cells? With respect to Minute and Myc competition there is some controversy. Some studies report that blocking JNK in loser cells inhibits their apoptosis and rescues them from competition [6,26]; however, independent reports found that inactivating the JNK pathway had little or no effect on cell competition [7,10]. In the case of competition induced by mutations in polarity-linked tumour suppressors, the data instead rather unequivocally shows that JNK signalling plays a key role [22,34,35] (Figure 1B). In scrib−/− cells, altered endocytic trafficking leads to hyperactivation of Eiger [Drosophila TNF (tumour necrosis factor) superfamily member] signalling, which results in JNK activation [34]. Importantly, in this context Eiger/JNK signalling has a dual function. In addition to being active within scrib−/− cells, where it promotes apoptosis, Eiger/JNK signalling becomes activated in a row of wild-type cells just surrounding the mutant scrib−/− tissue. There, JNK activation has quite the opposite effect, since it is required for these cells to eliminate their scrib−/− neighbours [33]. Interestingly, a recent study shows that, in a mammalian cell culture model of scrib cell competition, scrib−/− cells do not activate JNK, but activate p38 MAPK (mitogen-activated protein kinase), another kinase activated by cellular stress. In this context, p38 activation is required for the apoptosis of scrib−/− cells [36].
Interestingly, Eiger is not an obligate upstream activator of JNK during competition. For example, during Minute competition, Eiger function has no obvious effects on the elimination of Minute cells [33], whereas JNK inhibition rescues, at least partially, the outcompetition of loser cells [6,7]. This would suggest that Eiger is dispensable for JNK activation. Consistent with this, JNK activation during Minute competition requires Brinker, a transcriptional repressor that is up-regulated in competing Minute cells and also becomes required for their apoptosis [26].
As a consequence of cell competition a tissue may be presented with a substantial increase in the amount of apoptotic cells. What is the fate of such apoptotic bodies and how are they cleared from the tissue? Initial observations reported that, in wing imaginal discs apoptotic cells are extruded from the epithelial layer and accumulate basally [6] where they are probably cleared by macrophages. It was later found that during Minute and Myc competition, apoptotic bodies are engulfed by surrounding winner cells [30]. In other words, during competition fitter cells eat their less-fit neighbours (Figure 1B). This has also been observed recently for scrib−/− cells, which are engulfed by surrounding wild-type neighbours [33]. In both studies it was found that molecular components normally involved in phagocytosis are required for cell engulfment [30,33]. In a surprising turn, both studies further reported that cell engulfment is not simply a secondary event that clears cellular debris from the tissue; rather it is an active and essential component of the cell competition process. This conclusion is on the basis of the observation that inhibiting the function of proteins required for engulfment actually leads to inhibition of competition. Since both studies confirmed this finding by targeting several components of the phagocytic machinery, the evidence for a requirement of engulfment in competition is strong [30,33]; however alternative explanations are possible. For example, removing the function of these genes could reduce cellular fitness. Alternatively, mutations in these genes could affect endocytic trafficking, which has been shown to modulate cell competition [6,34].
Regardless of whether they are simply extruded or actively engulfed, loser cells do not die without putting up a fight. A recent report shows that, during the early stages of competition, loser cells express dSPARC (Drosophila secreted protein acidic and rich in cysteine) [37], a secreted matricellular glycoprotein involved in extracellular matrix remodelling and in modulating the activity of multiple signalling pathways [38]. dSPARC expression temporarily protects loser cells from competition by delaying the induction of apoptosis, although the exact mechanism is not presently understood. It is proposed that dSPARC acts as a shield to delay the effect of cell competition. This would avoid unnecessary elimination of cells that are able to recover from transient damage [37].
Recent work studying cell competition in the HSPC niche found that outcompeted cells may actually survive [15]. In this context, the outcome of cell competition is induction of senescence in outcompeted stem cells (Figure 1C). Gene expression profiling shows that, during competition, loser cells up-regulate the senescence-related genes p16(INK4a) and Ezh-2, and have high levels of P-selectin and Sdpr, which are normally up-regulated in aged HSPCs [15]. Ultimately, since senescent cells are permanently cell-cycle-arrested, the outcome is not dissimilar from other modes of competition; although they remain alive, loser cells are inhibited from repopulating the niche.
Tissue colonization by winner cells
During cell competition, despite the intense elimination of loser cells, organ size and tissue growth are unaffected and overall cell number is conserved. This is possible because, as loser cells die, winner cells display a corresponding increase in the rate of proliferation. This has been observed during Minute competition [30] (although it has been questioned [27]) and in other competitive contexts. Thus high-Myc cells overproliferate as a consequence of competition [6,31] and HSPCs with relatively low levels of p53 up-regulate markers of cell proliferation under competitive conditions, such as Ki67, cyclin B1 and cyclin A2 [15] (Figure 1C). Thus cell competition can, at least in some cases, increase winner cell proliferation. Importantly, in one instance where this was investigated, growth stimulation appeared to be induced at a short-range. Elegant genetic experiments conducted by Simpson and Morata [3] showed that, during Minute competition, clones of winner cells display increased growth if they are in the proximity of loser cells, but not if they are further away from the site of competition. However, in Drosophila cell culture models of competition, where cell–cell contact is not required for apoptosis induction, winner cell overproliferation is also triggered in the absence of cell contact [31].
Cell competition and disease
A quality-control mechanism
Cell-autonomous apoptosis is activated in cells that sustain major functional damage, but how are viable, but suboptimal, cells removed? During development, cell competition acts as a quality-control mechanism that eliminates suboptimal cells before they contribute to the adult organism (Figure 2A). Is there also a role for cell competition in maintaining organ fitness in the adult? Adult tissues, as a consequence of environmental stress or damage, accumulate suboptimal cells. One way to neutralize them is through the induction of cellular senescence, which leads to a permanent cell proliferation arrest [39]. However, this appears not to be ideal, as a recent study shows that the accumulation of senescent cells during aging contributes to the insurgence of age-related pathologies [40]. Thus it could be proposed that cell competition eliminates damaged cells before the activation of senescence. By selecting fitter cells and reducing the number of senescent cells, cell competition could contribute to maintaining organ fitness and delaying tissue aging. However, a recent study indicates that cell competition itself could lead to the accumulation of senescent cells in HSPCs [15]. This could be a peculiarity of the HSPC niche. In addition, it is unclear whether in this system a proportion of loser cells are also eliminated by cell death during competition.
Figure 2. Potential links between cell competition and disease.

(A) Quality control in normal and disease conditions. Suboptimal cells are eliminated from epithelial tissues through apoptosis, whereas the fitter cells show a corresponding increase in proliferation. (B) Tumour suppressor role. Cell competition could act as a protective mechanism to remove potentially dangerous cells from a tissue, thus preventing tumour formation. Tumour promoting role. By contrast, cell competition could be exploited by mutant cells to promote tumour formation. Mutant supercompetitor cells with precancerous lesions could outcompete and cull surrounding wild-type cells, leading to the generation of cancerization fields. (C) Regeneration. On the basis of experiments in liver repopulation assays, it has been proposed that cell competition could be exploited in regenerative medicine.
Cell competition could be relevant to a number of human diseases. In a rare sporadic skin disorder (ichthyosis with confetti) it is observed that patients develop patches of healthy skin within abnormal skin areas [41]. Each patch originates from clonal expansion of a single revertant stem cell which has lost the dominant mutation in keratin-10. In some patients more than a thousand revertant clones are seen, suggesting that these stem cells produce healthy skin cells able to outcompete surrounding diseased tissue. Competition could also be involved in non-random X-chromosome inactivation, seen in various X-linked immunodeficiencies [42]. In female mammals random inactivation of one of the two X-chromosomes occurs in every somatic cell during early development. Once X-inactivation has taken place all clonal descendents will have the same X inactivated. In a number of X-linked genetic diseases, preferential tissue-specific inactivation of one X-chromosome is seen. In these contexts, cells inactivating the mutant allele could gain an advantage over cells in which the normal allele is inactivated. Although not proven, removal of mutated cells could be occurring via a mechanism similar to cell competition.
Cancer
Cell competition has been linked to cancer, as many of the genes involved are also known vertebrate oncogenes or tumour suppressors. Initially cell competition was thought of as a mechanism that would favour the growth of tumour cells [6]. The discovery of genes which could transform cells into supercompetitors led to the hypothesis that cells with precancerous lesions could overcolonize the tissue by eliminating the surrounding cells (Figure 2B) [6–8,11]. This expanded population of cells would then be more likely to undergo further mutations, finally allowing the cells to overcome the restraints of the tissue and develop into a tumour. This theory is consistent with the model of field cancerization, which was proposed to explain the development of multiple concomitant primary tumours in the same tissue, together with the observation that abnormal tissue often surrounds the tumour [43,44]. It was thus proposed that early precancerous lesions could allow cells to expand and colonize the tissue. In this field, further independent genetic hits at multiple tissue locations would give rise to tumours that share a monoclonal origin [43,44]. This model is indeed supported by the fact that tumour-associated genetic mutations are frequently present in biopsies taken from the macroscopically normal mucosa adjacent to the tumour [43,44]. Competition could also be involved in helping tumour cells establish themselves at secondary sites during formation of metastasis.
More recently it has been shown that cell competition could also play an opposite role during cancer formation. In fact there are now observations suggesting that surrounding normal cells can suppress the proliferation of tumour cells. Cells mutated in the tumour suppressors scrib and lgl are eliminated by wild-type cells, indicating that cell competition could act as a tumour suppressor mechanism eliminating potentially harmful cells from the tissue [21–24].
Manipulating cell competition during cancer formation could provide an alternative new strategy to fight cancer. The emergence of new in vitro cell culture competition assays means it is now possible to study competition between normal and transformed cells in mammalian systems [35]. The discovery of competition-specific genes will be essential in establishing these new therapies and in identifying novel biomarkers for early detection.
Tissue regeneration
The established role of cell competition in controlling cell proliferation suggests that competition could be relevant for regenerative medicine (Figure 2C). In 2006 Oertel et al. [20] demonstrated that the process of cell competition could be used to replace functional tissue in the adult liver by fetal liver progenitors. They observed that younger highly proliferative fetal cells replaced slower growing adult cells by inducing apoptosis. This strategy could serve to design effective treatments of regenerative medicine for a wide variety of disorders.
Conclusions
Since the original discovery of cell competition over 35 years ago, the field has made tremendous advances. In particular, the list of experimental conditions that trigger cell competition continues to grow, suggesting that this is a frequently occurring phenomenon. Most importantly, there is now ample evidence that this is also a mammalian event. However, several unanswered questions remain. Although some progress has been made, the molecular mechanisms of cell competition are still largely unknown. Furthermore, we are still to fully grasp its physiological relevance in health and disease, and to realise its potential for biomedical applications. The next decade will undoubtedly be an exciting time for this field of research.
Summary
-
•
Many experimental conditions have been shown to result in cell competition, including defects in ribosomal proteins and alterations in growth factor signalling levels. It is likely that multiple independent pathways initiate competition.
-
•
Differences in cellular fitness may be induced by differential growth rates, signalling pathway levels or expression of extracellular proteins.
-
•
Loser cells are eliminated through apoptosis or senescence. Several independent pathways trigger these events depending on the context of competition.
-
•
Outcompeted loser cells can be expelled from epithelial tissues through extrusion or active engulfment.
-
•
Organ growth and tissue size are maintained during competition: as loser cells die the winners show a corresponding increase in proliferation.
-
•
Cell competition acts as a quality-control mechanism to eliminate suboptimal cells during development and possibly also in the adult.
-
•
Cell competition has been implicated in liver repopulation assays and it has been proposed that it could therefore be used in regenerative medicine.
References
- 1.Morata G., Ripoll P. Minutes: mutants of Drosophila autonomously affecting cell division rate. Dev. Biol. 1975;42:211–221. doi: 10.1016/0012-1606(75)90330-9. [DOI] [PubMed] [Google Scholar]
- 2.Simpson P. Parameters of cell competition in the compartments of the wing disc of Drosophila. Dev. Biol. 1979;69:182–193. doi: 10.1016/0012-1606(79)90284-7. [DOI] [PubMed] [Google Scholar]
- 3.Simpson P., Morata G. Differential mitotic rates and patterns of growth in compartments in the Drosophila wing. Dev. Biol. 1981;85:299–308. doi: 10.1016/0012-1606(81)90261-x. [DOI] [PubMed] [Google Scholar]
- 4.Oliver E.R., Saunders T.L., Tarle S.A., Glaser T. Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute. Development. 2004;131:3907–3920. doi: 10.1242/dev.01268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnston L.A., Prober D.A., Edgar B.A., Eisenman R.N., Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98:779–790. doi: 10.1016/s0092-8674(00)81512-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moreno E., Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117:117–129. doi: 10.1016/s0092-8674(04)00262-4. [DOI] [PubMed] [Google Scholar]
- 7.de la Cova C., Abril M., Bellosta P., Gallant P., Johnston L.A. Drosophila myc regulates organ size by inducing cell competition. Cell. 2004;117:107–116. doi: 10.1016/s0092-8674(04)00214-4. [DOI] [PubMed] [Google Scholar]
- 8.Abrams J.M. Competition and compensation: coupled to death in development and cancer. Cell. 2002;110:403–406. doi: 10.1016/s0092-8674(02)00904-2. [DOI] [PubMed] [Google Scholar]
- 9.Donaldson T.D., Duronio R.J. Cancer cell biology: Myc wins the competition. Curr. Biol. 2004;14:R425–R427. doi: 10.1016/j.cub.2004.05.035. [DOI] [PubMed] [Google Scholar]
- 10.Tyler D.M., Li W., Zhuo N., Pellock B., Baker N.E. Genes affecting cell competition in Drosophila. Genetics. 2007;175:643–657. doi: 10.1534/genetics.106.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziosi M., Baena-Lopez L.A., Grifoni D., Froldi F., Pession A., Garoia F., Trotta V., Bellosta P., Cavicchi S. dMyc functions downstream of Yorkie to promote the supercompetitive behavior of hippo pathway mutant cells. PLoS Genet. 2010;6:e1001140. doi: 10.1371/journal.pgen.1001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vincent J.P., Kolahgar G., Gagliardi M., Piddini E. Steep differences in wingless signaling trigger Myc-independent competitive cell interactions. Dev. Cell. 2011;21:366–374. doi: 10.1016/j.devcel.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 14.Vazquez A., Bond E.E., Levine A.J., Bond G.L. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat. Rev. Drug Discovery. 2008;7:979–987. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 15.Bondar T., Medzhitov R. p53–mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6:309–322. doi: 10.1016/j.stem.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rhiner C., Diaz B., Portela M., Poyatos J.F., Fernandez-Ruiz I., Lopez-Gay J.M., Gerlitz O., Moreno E. Persistent competition among stem cells and their daughters in the Drosophila ovary germline niche. Development. 2009;136:995–1006. doi: 10.1242/dev.033340. [DOI] [PubMed] [Google Scholar]
- 17.Issigonis M., Tulina N., de Cuevas M., Brawley C., Sandler L., Matunis E. JAK-STAT signal inhibition regulates competition in the Drosophila testis stem cell niche. Science. 2009;326:153–156. doi: 10.1126/science.1176817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin Z., Kirilly D., Weng C., Kawase E., Song X., Smith S., Schwartz J., Xie T. Differentiation-defective stem cells outcompete normal stem cells for niche occupancy in the Drosophila ovary. Cell Stem Cell. 2008;2:39–49. doi: 10.1016/j.stem.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheng X.R., Brawley C.M., Matunis E.L. Dedifferentiating spermatogonia outcompete somatic stem cells for niche occupancy in the Drosophila testis. Cell Stem Cell. 2009;5:191–203. doi: 10.1016/j.stem.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oertel M., Menthena A., Dabeva M.D., Shafritz D.A. Cell competition leads to a high level of normal liver reconstitution by transplanted fetal liver stem/progenitor cells. Gastroenterology. 2006;130:507–520. doi: 10.1053/j.gastro.2005.10.049. [DOI] [PubMed] [Google Scholar]
- 21.Brumby A.M., Richardson H.E. Scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003;22:5769–5779. doi: 10.1093/emboj/cdg548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menendez J., Perez-Garijo A., Calleja M., Morata G. A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc. Natl. Acad. Sci. U.S.A. 2010;107:14651–14656. doi: 10.1073/pnas.1009376107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grzeschik N.A., Amin N., Secombe J., Brumby A.M., Richardson H.E. Abnormalities in cell proliferation and apico-basal cell polarity are separable in Drosophila lgl mutant clones in the developing eye. Dev. Biol. 2007;311:106–123. doi: 10.1016/j.ydbio.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Froldi F., Ziosi M., Garoia F., Pession A., Grzeschik N.A., Bellosta P., Strand D., Richardson H.E., Grifoni D. The lethal giant larvae tumour suppressor mutation requires dMyc oncoprotein to promote clonal malignancy. BMC Biol. 2010;8:33. doi: 10.1186/1741-7007-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neto-Silva R.M., de Beco S., Johnston L.A. Evidence for a growth-stabilizing regulatory feedback mechanism between Myc and Yorkie, the Drosophila homolog of. Yap. Dev. Cell. 2010;19:507–520. doi: 10.1016/j.devcel.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreno E., Basler K., Morata G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature. 2002;416:755–759. doi: 10.1038/416755a. [DOI] [PubMed] [Google Scholar]
- 27.Martin F.A., Herrera S.C., Morata G. Cell competition, growth and size control in the Drosophila wing imaginal disc. Development. 2009;136:3747–3756. doi: 10.1242/dev.038406. [DOI] [PubMed] [Google Scholar]
- 28.Rhiner C., Lopez-Gay J.M., Soldini D., Casas-Tinto S., Martin F.A., Lombardia L., Moreno E. Flower forms an extracellular code that reveals the fitness of a cell to its neighbors in Drosophila. Dev. Cell. 2010;18:985–998. doi: 10.1016/j.devcel.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 29.Yao C.K., Lin Y.Q., Ly C.V., Ohyama T., Haueter C. M., Moiseenkova-Bell V.Y., Wensel T.G., Bellen H.J. A synaptic vesicle-associated Ca2+ channel promotes endocytosis and couples exocytosis to endocytosis. Cell. 2009;138:947–960. doi: 10.1016/j.cell.2009.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li W., Baker N.E. Engulfment is required for cell competition. Cell. 2007;129:1215–1225. doi: 10.1016/j.cell.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 31.Senoo-Matsuda N., Johnston L.A. Soluble factors mediate competitive and cooperative interactions between cells expressing different levels of Drosophila. Myc. Proc. Natl. Acad. Sci. U.S.A. 2007;104:18543–18548. doi: 10.1073/pnas.0709021104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bogoyevitch M.A., Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 2006;70:1061–1095. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohsawa S., Sugimura K., Takino K., Xu T., Miyawaki A., Igaki T. Elimination of oncogenic neighbors by JNK-mediated engulfment in Drosophila. Dev. Cell. 2011;20:315–328. doi: 10.1016/j.devcel.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 34.Igaki T., Pastor-Pareja J.C., Aonuma H., Miura M., Xu T. Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev. Cell. 2009;16:458–465. doi: 10.1016/j.devcel.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamori Y., Bialucha C.U., Tian A.G., Kajita M., Huang Y.C., Norman M., Harrison N., Poulton J., Ivanovitch K., Disch L., et al. Involvement of Lgl and Mahjong/VprBP in cell competition. PLoS Biol. 2010;8:e1000422. doi: 10.1371/journal.pbio.1000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Norman M., Wisniewska K.A., Lawrenson K., Garcia-Miranda P., Tada M., Kajita M., Mano H., Ishikawa S., Ikegawa M., Shimada T., Fujita Y. Loss of Scribble causes cell competition in mammalian cells. J. Cell Sci. 2012;125:59–66. doi: 10.1242/jcs.085803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Portela M., Casas-Tinto S., Rhiner C., Lopez-Gay J. M., Dominguez O., Soldini D., Moreno E. Drosophila SPARC is a self-protective signal expressed by loser cells during cell competition. Dev. Cell. 2010;19:562–573. doi: 10.1016/j.devcel.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 38.Clark C.J., Sage E.H. A prototypic matricellular protein in the tumor microenvironment: where there's SPARC, there's fire. J. Cell. Biochem. 2008;104:721–732. doi: 10.1002/jcb.21688. [DOI] [PubMed] [Google Scholar]
- 39.Kuilman T., Michaloglou C., Mooi W. J., Peeper D.S. The essence of senescence. Genes Dev. 2010;24:2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baker D.J., Wijshake T., Tchkonia T., LeBrasseur N.K., Childs B.G., van de Sluis B., Kirkland J.L., van Deursen J.M. Clearance of p16Ink4a–positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choate K.A., Lu Y., Zhou J., Choi M., Elias P.M., Farhi A., Nelson-Williams C., Crumrine D., Williams M.L., Nopper A.J., et al. Mitotic recombination in patients with ichthyosis causes reversion of dominant mutations in KRT10. Science. 2010;330:94–97. doi: 10.1126/science.1192280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puck J.M., Stewart C.C., Nussbaum R.L. Maximum-likelihood analysis of human T-cell X chromosome inactivation patterns: normal women versus carriers of X-linked severe combined immunodeficiency. Am. J. Hum. Genet. 1992;50:742–748. [PMC free article] [PubMed] [Google Scholar]
- 43.Slaughter D.P., Southwick H.W., Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963–968. doi: 10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 44.Braakhuis B.J., Tabor M.P., Kummer J.A., Leemans C.R., Brakenhoff R.H. A genetic explanation of Slaughter's concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727–1730. [PubMed] [Google Scholar]