

Abstract
CCG-1423 (1) is a novel inhibitor of Rho/MKL1/SRF-mediated gene transcription that inhibits invasion of PC-3 prostate cancer cells in a Matrigel model of metastasis. We recently reported the design and synthesis of conformationally restricted analogs (e.g. 2) with improved selectivity for inhibiting invasion vs acute cytotoxicity. In this study we conducted a survey of aromatic substitution with the goal of improving physicochemical parameters (e.g. ClogP, MW) for future efficacy studies in vivo. Two new compounds were identified that attenuated cytotoxicity even further, and were 4-fold more potent than 2 at inhibiting PC-3 cell migration in a scratch wound assay. One of these (8a, CCG-203971, IC50 = 4.2 μM) was well tolerated in mice for 5 days at 100 mg/kg/day i.p., and was able to achieve plasma levels exceeding the migration IC50 for up to 3 hours.
Metastasis is the major driver in cancer-related deaths. Metastases involve dysregulation of numerous cellular processes that allow malignant cells to escape their point of origin and establish themselves in distant sites. Altered programs of gene transcription play a key role in these processes, and recent evidence points to an important role for RhoA/C-stimulated gene transcription1, 2. The small G proteins in the RhoA subfamily are almost never mutated in cancer; rather, their activity is increased by overexpression or by increases in the activity of upstream activators. Studies of human cancer cell lines selected for high metastatic character in xenograft models show upregulation of RhoC expression3. Upregulation of RhoC in human cancers is also associated with poor clinical outcome4. In breast and prostate cancer models, blockade of RhoC by toxins or dominant negative approaches prevents cellular invasion5, 6. Importantly, a RhoC knockout mouse shows complete suppression of in vivo metastasis of polyoma T-antigen-induced mammary tumors1. Signals downstream of RhoC have been implicated in the invasiveness of aggressive prostate cancers6, 7 and in the in vivo metastasis of breast cancer1 and melanoma3. RhoC activation induces transcriptional responses through the actin-regulated cytosolic-to-nuclear translocation of MegaKaryocytic Leukemia 1 protein (MKL1), which binds to the serum-response factor (SRF), and the resulting complex activates transcription of serum response element (SRE) regulated target genes8, 9. Recently, both MKL1 and SRF have been shown to play important roles in metastasis of melanoma and breast cancer in vivo10, and SRF has been shown to be clinically associated with castration-resistant prostate cancer11. Thus transcriptional signals, rather than actin cytoskeletal changes per se, are critical for the role of RhoC in metastasis. Few drugs are known to target such transcriptional signals.
In 2007, Evelyn et al reported the identification of small molecule CCG-1423 (1, Figure 1) as an inhibitor of SRE-driven luciferase (SRE.L) gene transcription initiated by the RhoA/C signaling pathways.12 Discovered using a high-throughput screen in the University of Michigan Center for Chemical Genomics (CCG), 1 possessed considerable potency (<1 μM), but also significant cytotoxicity. Therefore, we carried out molecular modifications of 1 with the aim of improving its potency and/or selectivity while mitigating cytotoxicity. Through replacement of the potentially labile N-O bond and conformational restriction of the flexible tether region between the two aromatic rings, we arrived at our lead compound, nipecotic bis(amide) 2 (Figure 1), which offered an improved biological profile over compound 1.13 Although there was a 10-fold decrease in potency, 2 retained inhibition of Gα12QL-stimulated SRE.L expression with similar efficacy to 1, but with greatly reduced cytotoxicity. Unfortunately, compound 2 exhibits poor physicochemical properties, including low solubility (<10 μg/mL) and apparently low permeability (Table 4).
Figure 1.

Table 4.
Physicochemical property data for selected new analogs
| Cmpd No |
Solubility (μg/mL)a |
CLogPb | Log Peff (cm/s)c |
|---|---|---|---|
| 2 | 7.99-4.50 (LOW) | 5.55 | −10 ± 0.00 (LOW) |
| 8a | 4.26-3.09 (LOW) | 4.88 | −8.3 ± 2.3 (LOW) |
| 8u | 2.41-1.36 (LOW) | 5.37 | −10 ± 0.00 (LOW) |
| 12a | 4.30-3.22 (LOW) | 6.68 | −5.1 ± 0.40 (MOD-LOW) |
| 12b | 3.85-1.89 (LOW) | 6.65 | −4.7 ± 0.05 (MOD) |
| 19 | 3.45-2.21 (LOW) | 5.40 | −10 ± 0.00 (LOW) |
We elected to further explore diversity at each of the terminal aromatic rings of 2 with the goals of improving its potency and/or physicochemical parameters (e.g. reduced lipophilicy and/or molecular weight). Our initial survey of aromatic substitution13 indicated that some degree of lipophilicity on the aromatic rings is necessary for activity, likely due to the need for cell permeability. Therefore, we designed libraries 3 to explore diversity on the aromatic rings while modestly reducing lipophilicity with the goal of increasing solubility (Figure 1).
We first explored ring A of 2 (Figure 1). We utilized MScreen14 to select commercially available m-benzoic acids with calculated logP (ClogP) values less than that of 3,5-bis(trifluoromethyl)benzoic acid, and which would produce final analogs with total molecular weights below 550. We initially chose only meta-substitutions, as they have greater rotational freedom to interact with various areas in the binding site than do ortho- or para-substitutions. Similarly, we explored ring B (Figure 1), again using MScreen to select commercially available meta-substituted anilines with ClogPs less than that of 4-chloroaniline and that would result in molecular weights of 550 or less. In choosing only starting acids and anilines with lower lipophilicities than those in 2, we hoped to decrease the overall lipophilicities of our new analogs to enhance solubility.
The synthetic routes to new analogs of 2 are presented in Schemes 1 and 2. For the acid library (Scheme 1), the starting nipecotic acid 4 was BOC protected, and the resulting acid 5 was coupled to 4-chloroaniline with EDC/DMAP to afford intermediate amide 6. Following deprotection, the resulting common piperidine intermediate 7 was coupled with diverse m-substituted benzoic acids, again using EDC/DMAP.15 The resulting compounds were purified using acidic Amberlyst-15 resin to remove excess 7, followed by a water wash to remove residual DMF, affording library analogs 8a-t. This synthesis afforded compounds with overall yields ranging between 39-100% and purities greater than 90%. For the aniline library (Scheme 2), ethyl nipecotate 9 was acylated with 3,5-bis(trifluoromethyl)benzoyl chloride followed by saponification to afford free acid 11. The acid was then coupled with diverse m-anilines using EDC/DMAP. Compounds were purified by aqueous workup and, if necessary, chromatographed to afford aniline library analogs 12a-w. This synthesis afforded compounds with yields ranging between 32-100% and with purities between 81 to greater than 95%.
Scheme 1.

Reagents and conditions: (a) BOC2O, NaOH, H2O, Dioxane; (b) 4-ClPhNH2, EDC, DMAP, DCM; (c) TFA, DCM, −10 °C; (d) i. m-R1PhCO2H, EDC, DMAP, DCM/DMF; ii. Amberlyst-15, DCM.
Scheme 2.

Reagents and conditions: (a) 3,5-bis(CF3)PhCOCl, DIPEA, DCM; (b) LiOH, EtOH; (c) m-R2PhNH2, EDC, DMAP, DCM.
The effects on Rho-mediated gene transcription and cytotoxicity of all newly synthesized analogs were determined in PC-3 cells transiently transfected with a luciferase reporter gene driven by the the SRE promoter (SRE.L) at an initial concentration of 100 μM.13 For compounds showing greater than 50% inhibition at 100 μM, a full dose-response curve (DRC) was generated. Table 1 summarizes the effects of benzamide analogs 8. Also included in the table are efficacies at the maximum concentration (100 μM) as we expect that both potency and overall efficacy will be important indicators of therapeutic potential. In general, the new analogs demonstrated little to no cytotoxicity, with the exception of 8d. A majority of analogs, however, had lower potency than lead 2, with the exception of analogs 8a and 8b that had equivalent or slighly better potency and similar maximal effiacies.
Table 1.
Effects of Ring A substitution on transcription and cytotoxicity in transfected PC-3 cellsa
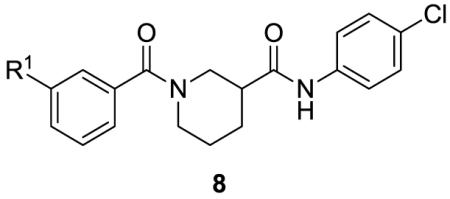
| Cmpd No. |
R1 | IC50 SRE.L (μM)b |
% inh SRE.L (100 μM)b |
% inh WST-1 (100 μM)c |
|---|---|---|---|---|
| 2 | 3,5-bis(CF3) | 9.8 | 78 | 14 |
| 8a | Furan-2-yl | 6.4 | 87 | 0 |
| 8b | PhCO | 9.9 | 75 | 0 |
| 8d | Thiazol-2-yl | 27 | 92 | 32 |
| 8e | t-BuCH2CONH | 26 | 69 | 0 |
| 8f | CH2=CH | 33 | 84 | 0 |
| 8g | 2-Me-thiazol-4-yl | 33 | 81 | 1 |
| 8h | Me | 35 | 63 | 0 |
| 8i | MeS | 35 | 76 | 0 |
| 8j | Oxazol-5-yl | 43 | 72 | 0 |
| 8k | Et2NSO2 | 51 | 69 | 0 |
| 8l | MeO2C | 67 | 64 | 0 |
| 8m | 5-Me-1,2,4-oxadiazol-3- yl |
78 | 59 | 0 |
| 8n | MeO | ND | 38 | 0 |
| 8o | MeOCH2 | ND | 32 | 0 |
| 8p | MeSO2NH | ND | 32 | 3 |
| 8q | −CN | ND | 20 | 0 |
| 8r | MeSO2 | ND | 17 | 2 |
| 8s | −NO2 | ND | 15 | 0 |
| 8t | MeCONH− | ND | 8 | 4 |
For assay descriptions, see Ref. 12.
Inhibition of Rho-pathway selective serum response element-luciferase reporter. Values are mean of n ≥ 3 independent experiments. ND: not determined.
Inhibition of mitochondrial reduction of WST-1. Values are mean of n ≥ 3 independent experiments.
Selected structure-activity relationships (SAR) suggest that lipophilicity and/or topological polar surface area (tPSA) may be correlated with activity. Oxazole 8j adds a nitrogen to the furan ring of 8a, resulting in diminished activity. This substitution increases the tPSA (from 59 to 71 Å2) and decreases the CLogP from 4.88 to 3.46. A similar trend is observed when comparing sulfide 8i with sulfone 8r, styrene 8f with methoxy 8n, and oxazole 8j with oxadiazole 8m. In fact, overall we found that potency correlated weakly with decreasing tPSA (R2 = 0.32, data not shown), and moderately with increasing CLogP (R2 = 0.78, data not shown). In summary, the best analog from the carboxylic acid library was furan 8a, which had both improved potency and efficacy in the SRE.L assay, as well as lower acute cytotoxicity. Also of interest was analog 8b, which incorporates a photoactivatable benzophenone group without loss of activity. This suggests that photoaffinity probes based on 8b may be of possible use in identifying the unknown molecular target16.
The effects of aromatic substitution on ring B are summarized in Table 2. As with the ring A library, most compounds demonstrated little to no cytotoxicity, with the exception of amine 12p, which had significant cytotoxicity of 51% at 100 μM. The best analogs, 12a and 12b (not included as part of the original library design, but later as analogs of 8b), showed a nearly 10-fold increase in potency with slight decreases in efficacy, perhaps due to poor solubility at higher concentrations. Pyridine 12c showed a similar increase in potency while maintaining better efficacy. Analogs 12d-12i all showed similar or slight increases in potency.
Table 2.
Effects of Ring B substitution on transcription and cytotoxicity in transfected PC-3 cellsa
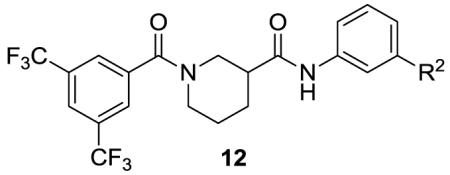
| Cmpd No. |
R2 | IC50 SRE.L (μM) |
% inh SRE.L (100 μM) |
% inh WST-1 (100 μM) |
|---|---|---|---|---|
| 2 | 4-Cl | 9.8 | 78 | 14 |
| 12a | PhO | 1.5 | 59 | 1 |
| 12b | PhCH2 | 1.6 | 59 | 0 |
| 12c | 2-Pyr-CH2CH2O | 2.3 | 82 | 5 |
| 12d | pyrrolidin-1-yl-SO2 | 5.6 | 70 | 0 |
| 12e | 2-Me-thiazol-4-yl | 6.1 | 73 | 1 |
| 12f | Et | 8.1 | 66 | 0 |
| 12g | CH2=CH | 11 | 53 | 1 |
| 12h | oxazol-5-yl | 12 | 87 | 9 |
| 12i | MeS | 12 | 60 | 0 |
| 12j | MeO2C | 26 | 63 | 0 |
| 12k | MeO | 30 | 51 | 0 |
| 12l | −CN | 32 | 77 | 0 |
| 12m | morpholin-1-yl-CH2 | 32 | 73 | 0 |
| 12n | −NO2 | 66 | 65 | 9 |
| 12o | MeCO | 76 | 73 | 0 |
| 12p | pyrrolidin-1-yl-CH2 | ND | 94 | 51 |
| 12q | 2-CO2Me-furan-4-yl | ND | 39 | 0 |
| 12r | CH2=CHCH2O | ND | 38 | 0 |
| 12s | PhCO | ND | 38 | 0 |
| 12t | 4-methyl-1,2,4-triazol-3-yl | ND | 36 | 0 |
| 12u | 2-oxopyrrolidin-1-yl | ND | 35 | 0 |
| 12v | 3-Cl-4-CF3-pyridin-2-yl-O | ND | 25 | 0 |
| 12w | tBuO2C | ND | 24 | 0 |
Assays and abbreviations defined in Table 1.
Interestingly, there was no correlation overall between potency and either tPSA or CLogP in this library (R2 < 0.1). This perhaps suggests that local polarity/dipole changes on ring A are actually responsible for the SAR, rather than overall physical properties. However, some interesting comparisons can still be made between similar compounds. Inactive analog 12s differs from highly active analog 12b only in the presence of a ketone between the aromatic rings, suggesting that conformation could be important for activity. Replacement of the methyl ester of 12j with t-butyl ester in 12w resulted in complete loss of activity, as did replacing the methoxy of 12k with allyloxy in 12r, both suggesting some steric limitations. However, these latter results are not consistent with the favorable activity of bulky analogs 12a-c, possibly indicative of multiple binding modes. Overall, however, it must be emphasized that any interpretation of SAR needs to be undertaken with the caveat that activity in the cell-based SRE.L assay will be dependent on multiple variables in addition to intrinsic activity, including cell permeability, distribution inside the cell, and perhaps even metabolism.
Based on the results from the acid- and aniline-subsituted libraries, additional analogs were examined, as illustrated in Table 3. Replacement of the furan of 8a with a thiophene (8u) slightly increased potency, but introduced some detectable cytotoxicity. Movement of the furan of 8a from the meta- to the para-position (13) caused a loss in potency with introduction of slight cytotoxicity. 12x moved the furan from ring A to ring B, which led to a loss of both potency and efficacy. Compounds 12y and 12z shortened the pyridyl tether of 12c by one and two carbons, respectively. Both had inferior potency. Analogs 15 and 16 were prepared to determine if a pyridine B-ring could improve the solubility of 2. Unfortunately all showed unacceptable losses in potency.
Table 3.
Effects of additional substitutions on transcription and cytotoxicity in transfected PC-3 Cellsa
| Cmpd No |
Structure | IC50 SRE.L (μM)a |
% inh SRE.L (100 μM)a |
% inh WST-1 (100 μM)a |
|---|---|---|---|---|
| 8u |
|
4.7 | 81 | 4 |
| 13 |
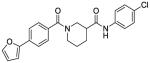
|
9.2 | 80 | 6 |
| 12x |
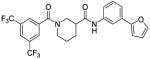
|
21 | 63 | 0 |
| 12y |
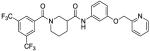
|
7.4 | 79 | 15 |
| 12z |
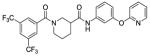
|
10 | 60 | 0 |
| 14 |
|
10 | 63 | 0 |
| 15 |
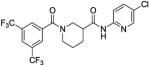
|
ND | 37 | 0 |
| 16 |
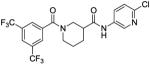
|
31 | 88 | 25 |
| 17 |
|
ND | 21 | 0 |
| 18 |
|
ND | 21 | 0 |
Assays and abbreviations defined in Table 1.
Next, we examined “hybrid” analogs 17 and 18 of our most promising compounds, combining the A-ring furan moiety of 8a with the B-ring moieties of 12a and 12b. Surprisingly, neither hybrid retained any activity. To determine if this reflects a steric intolerance for aromatic substitution on both rings, we prepared hybrid 14 containing the B-ring thioether of 12i. In this case activity was not lost, but there also was no improvement.
A small subset of compounds was selected for evaluation of the impact of structure on kinetic solubility17 and passive permeability as measured by the PAMPA Explorer Kit (pION) (Table 4). For each parameter, compounds were binned into one of three groups: high, medium or low (described in Table 4 legend). Unfortunately none of the new analogs had solubilities exceeding that of leads 1 or 2; all were binned Low solubility. Marginal improvement in permeability was observed with furan analog 8a relative to 2, but it did not rise above the Low bin. Significant improvements in permeability were only realized with the most lipophilic analogs 12a and 12b, which may account for their increased potency in the cell-based SRE-luc assay. One interesting observation from this dataset is that the superior activity of conformationally restrained analog 2 vs acyclic analog 19 (Figure 1, IC50 = 38 uM13) is likely not due to increased passive permeability and therefore probably reflects a true increase in affinity for the target.
We previously demonstrated that 2 can inhibit invasion into Matrigel by cultured PC-3 cells.13 To compare our best new analogs with 2 for their ability to inhibit PC-3 cell migration, we employed a scratch wound assay. PC-3 cells (5.0 × 105) were plated in DMEM containing 10% FBS and grown to confluence in a 12-well plate. After 24 hours, a scratch was made using a 200 μL pipette tip. Medium was replaced with DMEM containing 0.5% FBS and either test compounds (10 μM) or 0.1% DMSO control. Images of the wounds were taken at 0 hours using a bright-field inverted microscope (Leica DM IRB) at 2.5× magnification. After 24 hours the cells were fixed (10% formalin) and stained (0.5% crystal violet) to obtain high contrast images. Area quantification of the wounds was determined computationally using ImageJ ® software (NIH). The extent of migration was determined by subtracting the area of the wound after 24 hours from the initial area of the wound. The percent inhibition was plotted by normalizing the compound treated cells to the DMSO control. Results are summarized in Figure 2. In this assay, lead compound 2 only inhibited PC-3 cell migration by 22%. Acyclic analog 19 was included as a negative control with poor SRE.L activity, and showed minimal (<10%) inhibition of migration. Surprisingly, none of the most potent new B-ring analogs (12a-d) performed significantly better than 2 in the scratch assay, all inhibiting less than 30% at 10 uM. In fact, the only B-ring analog that afforded any improvement was thiazole 12e (42% inhibition). On the other hand, A-ring analogs 8a and 8u, despite their more modest SRE.L potency, were much more effective in this migration assay, both inhibiting about 70% at 10 μM. Dose response curves were acquired for the most effective compounds and are depicted in Figure 3. Both 8a (IC50 = 4.2 μM) and 8u (IC50 = 4.5 μM) had potencies significantly better than 2 (IC50 = 17 μM) or the Rho-kinase (ROCK) inhibitor Y-27632 (IC50 = 28 μM), which was included for comparison because of its role in Rho-mediated signaling and reported effects on cancer cell migration18-21.
Figure 2.

Effects of new compounds on PC-3 cell migration using a scratch wound assay. A) Example images of the wounds after 24 hours taken with a bright-field inverted microscope (Leica DM IRB, 2.5× magnification). B) Percent inhibition of migration into wound area. Area quantification of the wounds was determined computationally using ImageJ® software (NIH).
Figure 3.

Effects of new compounds on PC-3 cell migration. Compounds were tested at various concentrations in a scratch wound assay. IC50 values are in μM.
Upon the initial discovery of 1, we also observed that it had an effect on the RhoC-overexpressing melanoma cell lines A375M2 and SK-Mel-147.12 Therefore, we also tested 8a in the RhoC-SRE.L assay using transiently transfected SK-Mel-147 cells. We observed similar results to the PC-3 cell line, with 8a displaying an IC50 of 5.3 μM and no WST-1 cytotoxicity, exhibiting its potential as an inhibitor of metastatic melanoma, all of which will be disclosed in a future publication.22
Mechanistic analysis of compound 1 has demonstrated that it acts downstream of RhoA and targets MKL/SRF-dependent transcriptional activation. However, the exact mechanism of action has not yet been deciphered. Our findings suggest that 1 could affect the functions of MKL1 in various ways, including: preventing its release from actin, blocking translocation from the cytoplasm to the nucleus, repressing transcription via increasing sumoylation, disrupting the interaction between MKL1 and its transcoactivator SRF, or inhibiting its coactivator function.12 Preliminary studies have in fact indicated that 8a blocks MKL1 nuclear localization.22
In preliminary in vivo tolerability studies, compound 1 demonstrated a level of toxicity that would preclude extended dosing in xenograft models (deaths observed with repeated dosing at 7.5 mg/kg intraperitoneally (IP)). Based on its favorable activity in the migtration assay and markedly lower acute cytotoxicity, we selected 8a for the next round of in vivo testing. A 5-day tolerability study was conducted at 10, 20, 50, and 100 mg/kg/day dosed IP using 3 mice per dose. Following sacrifice on the 8th day, the mice were weighed and blood samples were collected. Dissection was performed, and the internal organs were weighed and examined for abnormalities. Weight was maintained over the course of the study, with the only abnormality observed being a slight increase in liver weight. All mice survived the 5-day study without any noted abnormal behavior. In preliminary pharmacokinetic studies, a single IP dose of 100 mg/kg 8a resulted in plasma levels of drug exceeding the PC-3 cell migration IC50 for up to 3 hrs, indicating its potential use for further in vivo studies. More detailed studies will be reported in due course.
In summary, an SAR study of 2 focusing on aromatic ring diversity was undertaken with the goal of improving selectivity and/or potency, while attenuating cytotoxicity and improving drug-like properties. Although we were not successful at improving solubility, we did identify one analog (8a, CCG-203971) that has reduced acute cytoxicity and improved potency vs 2 with regard to inhibition of PC-3 cell migration (IC50 = 4.2 μM vs 16.6 μM), as well as reduced lipophilicity and molecular weight. Furthermore, preliminary tolerability studies in normal mice indicate that 8a is well tolerated up to doses of 100 mg/kg IP over 5 days, and possesses pharmacokinetic properties suitable for future xenograft studies.
Acknowledgments
This work was supported in part by a Pharmacological Sciences Training Program grant GM007767 from NIGMS (AJH). The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Hakem A, Sanchez-Sweatman O, You-Ten A, Duncan G, Wakeham A, Khokha R, Mak TW. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. 2005;19(17):1974–9. doi: 10.1101/gad.1310805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mees C, Nemunaitis J, Senzer N. Transcription factors: their potential as targets for an individualized therapeutic approach to cancer. Cancer Gene Ther. 2009;16(2):103–12. doi: 10.1038/cgt.2008.73. [DOI] [PubMed] [Google Scholar]
- 3.Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406(6795):532–5. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 4.Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer. 2002;2(2):133–42. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- 5.van Golen KL, Wu ZF, Qiao XT, Bao LW, Merajver SD. RhoC GTPase, a novel transforming oncogene for human mammary epithelial cells that partially recapitulates the inflammatory breast cancer phenotype. Cancer Res. 2000;60(20):5832–8. [PubMed] [Google Scholar]
- 6.Yao H, Dashner EJ, van Golen CM, van Golen KL. RhoC GTPase is required for PC-3 prostate cancer cell invasion but not motility. Oncogene. 2006;25(16):2285–96. doi: 10.1038/sj.onc.1209260. [DOI] [PubMed] [Google Scholar]
- 7.Evelyn CR, Wade SM, Wang Q, Wu M, Iniguez-Lluhi JA, Merajver SD, Neubig RR. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther. 2007;6(8):2249–60. doi: 10.1158/1535-7163.MCT-06-0782. [DOI] [PubMed] [Google Scholar]
- 8.Treisman R. The serum response element. Trends Biochem Sci. 1992;17(10):423–6. doi: 10.1016/0968-0004(92)90013-y. [DOI] [PubMed] [Google Scholar]
- 9.Cen B, Selvaraj A, Burgess RC, Hitzler JK, Ma Z, Morris SW, Prywes R. Megakaryoblastic leukemia 1, a potent transcriptional coactivator for serum response factor (SRF), is required for serum induction of SRF target genes. Mol Cell Biol. 2003;23(18):6597–608. doi: 10.1128/MCB.23.18.6597-6608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Medjkane S, Perez-Sanchez C, Gaggioli C, Sahai E, Treisman R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol. 2009;11(3):257–68. doi: 10.1038/ncb1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prencipe M, Madden SF, O’Neill A, O’Hurley G, Culhane A, O’Connor D, Klocker H, Kay EW, Gallagher WM, Watson WR. Identification of transcription factors associated with castration-resistance: Is the serum responsive factor a potential therapeutic target? The Prostate. 2013;73 doi: 10.1002/pros.22618. [DOI] [PubMed] [Google Scholar]
- 12.Evelyn CR, Wade SM, Wang Q, Wu M, Iniguez-Lluhi JA, Merajver SD, Neubig RR. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Molecular Cancer Therapeutics. 2007;6(8):2249–2260. doi: 10.1158/1535-7163.MCT-06-0782. [DOI] [PubMed] [Google Scholar]
- 13.Evelyn CR, Bell JL, Ryu JG, Wade SM, Kocab A, Harzdorf NL, Hollis Showalter HD, Neubig RR, Larsen SD. Design, synthesis and prostate cancer cell-based studies of analogs of the Rho/MKL1 transcriptional pathway inhibitor, CCG-1423. Bioorganic & Medicinal Chemistry Letters. 2010;20(2):665–672. doi: 10.1016/j.bmcl.2009.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacob RT, Larsen MJ, Larsen SD, Kirchhoff PD, Sherman DH, Neubig RR. MScreen: An Integrated Compound Management and High-Throughput Screening Data Storage and Analysis System. Journal of Biomolecular Screening. 2012;17(8):1080–1087. doi: 10.1177/1087057112450186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawrence RM, Biller SA, Fryszman OM, Poss MA. Automated Synthesis and Purification of Amides: Exploitation of Automated Solid Phase Extraction in Organic Synthesis. Synthesis. 1997;1997(5):553–558. [Google Scholar]
- 16.Leslie BJ, Hergenrother PJ. Identification of the cellular targets of bioactive small organic molecules using affinity reagents. Chem Soc Rev. 2008;37(7):1347–60. doi: 10.1039/b702942j. [DOI] [PubMed] [Google Scholar]
- 17.Kerns EH, Di L. Drug-like properties: concepts, structure design and methods: from ADME to toxicity optimization. Academic Press; Amsterdam; Boston: 2008. p. 292. [Google Scholar]
- 18.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389(6654):990–4. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 19.Somlyo AV, Bradshaw D, Ramos S, Murphy C, Myers CE, Somlyo AP. Rho-kinase inhibitor retards migration and in vivo dissemination of human prostate cancer cells. Biochem Biophys Res Commun. 2000;269(3):652–9. doi: 10.1006/bbrc.2000.2343. [DOI] [PubMed] [Google Scholar]
- 20.Vial E, Sahai E, Marshall CJ. ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell. 2003;4(1):67–79. doi: 10.1016/s1535-6108(03)00162-4. [DOI] [PubMed] [Google Scholar]
- 21.Jeong KJ, Park SY, Cho KH, Sohn JS, Lee J, Kim YK, Kang J, Park CG, Han JW, Lee HY. The Rho/ROCK pathway for lysophosphatidic acid-induced proteolytic enzyme expression and ovarian cancer cell invasion. Oncogene. 2012;31(39):4279–89. doi: 10.1038/onc.2011.595. [DOI] [PubMed] [Google Scholar]
- 22.Haak AJW,SM, Bell JL, Larsen SD, Verhaegen M, Lawlor ER, Neubig RR. Small molecule targeting of RhoC regulated gene transcription in metastatic, undifferentiated melanoma. Manuscript in preparation. [Google Scholar]