

Abstract
Disease causing mutations for heterotaxy syndrome were first identified in the X-linked laterality gene, ZIC3. Mutations typically result in males with situs ambiguus and complex congenital heart disease; however affected females and one male with isolated d-transposition of the great arteries (d-TGA) have been reported. We hypothesized that a subset of patients with heart defects common to heterotaxy but without laterality defects would have ZIC3 mutations. We also sought to estimate the prevalence of ZIC3 mutations in sporadic heterotaxy.
Patients with TGA (n=169), double outlet right ventricle (DORV) (n=89), common atrioventricular canal (CAVC) (n= 41) and heterotaxy (n=54) underwent sequencing of ZIC3 exons. We tested 90 patients with tetralogy of Fallot (TOF) to correlate genotype with phenotype. Three potentially disease-related missense mutations were detected: c.49G>T (Gly17Cys) in a female with isolated DORV, c.98C>T (Ala33Val) in a male with isolated d-TGA, and c.841C>T (His281Tyr) in a female with sporadic heterotaxy. We also identified a novel insertion (CPFP333ins) in a family with heterotaxy. All were absent in 200 control patients and the 1000 Genomes Project (n = 629). No significant mutations were found in patients with TOF. Functional studies demonstrated reduced transcriptional activity of the ZIC3 His281Tyr mutant protein.
ZIC3 mutations were rarely identified in isolated DORV and d-TGA suggesting that a subset of DORV and d-TGA may fall within the spectrum of laterality defects. ZIC3 mutations were found in 3.7% of patients with sporadic heterotaxy; therefore testing should be considered in patients with heterotaxy.
Keywords: Heterotaxy, Situs, Transposition of the great arteries, Double outlet right ventricle, ZIC3
INTRODUCTION
Heterotaxy, or situs ambiguus, is an uncommon disorder of abnormal organ arrangement along the left-right axis. It is estimated to occur in 1 in 10,000 births [Lin et al., 2000] and is associated with a vast array of congenital anomalies, including an exceptionally high incidence of congenital heart disease. Heterotaxy accounts for approximately 3% of patients with congenital heart disease [Ferencz et al., 1997] and these patients have increased morbidity and mortality compared with patients without laterality defects [Hashmi et al., 1998; Cohen, 2006; Cheung et al., 2002; Gilljam et al., 2000; Swisher et al., 2011].
Heterotaxy has classically been subdivided into two groups based on splenic anatomy and atrial/bronchial situs, namely asplenia/right atrial isomerism and polysplenia/left atrial isomerism. There is significant overlap in the cardiac phenotype between the two subgroups, including a high frequency of malposition of the great arteries as seen in transposition of the great arteries (TGA) or double outlet right ventricle (DORV), and common atrioventricular canal (CAVC) [Cohen, 2006; van Praagh et al., 2006]. Some lesions are considered characteristic of polysplenia/left atrial isomerism, for example interrupted inferior vena cava and complete heart block [Ferencz et al., 1997; Gilljam et al., 2000; van Praagh et al., 2006].
Heterotaxy is known to occur in families with various modes of inheritance. Familial disease-related mutations were first identified in the X-linked gene ZIC3 by Gebbia et al. in 1997 [Gebbia et al., 1997]. ZIC3, zinc finger in cerebellum 3, is a member of the ZIC family and GLI superfamily of genes. It encodes a nuclear transcription factor that contains five tandem C2H2 zinc finger motifs. ZIC3 mutations have subsequently been described in sporadic heterotaxy [Gebbia et al., 1997; Ware et al., 2004; Megarbane et al., 2000; Chhin et al., 2007; de Luca et al., 2010] and have been estimated to account for approximately 1% of total heterotaxy cases [Ware et al., 2004].
Overall, 13 mutations have been reported in 76 patients (excluding synonymous changes and polymorphisms), of which 36 (47%) are male. In males with ZIC3 mutations 86% (31/36) have heterotaxy with complex congenital heart disease [Gebbia et al., 1997; Ware et al., 2004; Megarbane et al., 2000; Chhin et al., 2007; de Luca et al., 2010; D’Alessandro et al., 2011], but one case of isolated transposition of the great arteries and two cases of incomplete penetrance have been reported [Megarbane et al., 2000; D’Alessandro et al., 2011]. In females with ZIC3 mutations, 83% (33/40) are asymptomatic carriers, although imaging studies to identify subclinical anomalies have not been consistently reported. Approximately 8% (3/40) of mutation-positive females have isolated congenital cardiac disease and no laterality defects, and all of these patients were detected due to the presence of heterotaxy in a family member [Gebbia et al., 1997; Ware et al., 2004; Megarbane et al., 2000; Chhin et al., 2007; de Luca et al., 2010]. One female case of heterotaxy [Chhin et al., 2007] and three female cases of situs inversus totalis have also been reported to have ZIC3 mutations [Gebbia et al., 1997].
We therefore hypothesized that isolated cardiovascular malformations commonly seen in heterotaxy, including TGA, DORV and CAVC, share a common genetic basis with laterality defects, such that a subset of patients with sporadic isolated lesions would have ZIC3 mutations. We also predicted that ZIC3 mutations would not be identified in patients with lesions that are not seen in the context of heterotaxy, such as tetralogy of Fallot (TOF).
METHODS
Study Population
The study protocol was approved by The Children’s Hospital of Philadelphia Institutional Review Board on Human Subjects. Patients were recruited from October 1991 through February 2008 after informed consent. Patients with TGA, DORV, CAVC, TOF and heterotaxy were included in the study (Table I). All available cardiac records and imaging studies were consulted to classify the congenital heart disease, including x-rays and ultrasounds, echocardiograms, cardiac catheterizations and operative notes. Any patient determined to have a chromosomal anomaly, 22q11.2 microdeletion, or known genetic syndrome was excluded from the study. Any patient with a laterality defect, including abnormal situs of any organ or a venous anomaly, was included in the heterotaxy cohort, regardless of intracardiac anatomy. Patients without heterotaxy were assigned to diagnostic groups first by conotruncal anatomy, and then by AV canal such that patients included in the CAVC group had normal conotruncal anatomy. For conotruncal defects, the aorta was assigned to the morphologic left ventricle (LV) if there was mitral to aortic valve fibrous continuity, and the pulmonary artery (PA) was assigned to the morphologic right ventricle (RV) if it was positioned over the RV by more than 50%. In the absence of mitral to aortic fibrous continuity, the aorta was assigned to the LV if it was positioned over the LV by more than 50%. TGA was therefore defined as the aorta assigned to the RV and the PA assigned to the morphologic left ventricle with subtypes defined by relative arterial position and ventricular loop. DORV was defined as both great arteries assigned to the RV (including pulmonic stenosis, pulmonary atresia, aortic stenosis, aortic atresia). TOF was defined as a conotruncal anomaly with normal ventriculoarterial alignment, fibrous continuity between the aortic and mitral valves and anterior malalignment of the conal septum, with subtypes by pulmonary valve morphology (pulmonic stenosis, pulmonary atresia, absent pulmonary valve). CAVC was defined as a single common valve annulus and included complete, partial and incomplete subtypes depending on the number of orifices and the presence of a ventricular communication. Where available, parental samples were collected and tested if a proband was found to carry a mutation. In one instance, after recruitment of a male affected with heterotaxy (BG-01-046A), a half-brother (BG-094A) with a different father was subsequently born with heterotaxy, and thus recruited. Maternal DNA was collected to assess for inheritance (BG-094B).
Table I.
Study Cohort
| Cardiac Anatomy | Number of Unrelated Patients Screened n = 443 | Number of Mutations (%) |
|---|---|---|
|
| ||
| Transposition of the Great Arteries | 160 | 1 (0.6%) |
| • D-TGA {S,D,D} with intact ventricular septum | 74 | 1 |
| • D-TGA {S,D,D} with ventricular septal defect* | 76 | |
| • TGA {S,D,L} | 8 | |
| • TGA not otherwise specified | 2 | |
|
| ||
| L-Transposition of the Great Arteries {S,L,L} | 9 | 0 |
| • With intact ventricular septum | 2 | |
| • With ventricular septal defect | 7 | |
|
| ||
| Double Outlet Right Ventricle | 89 | 1 (1.1%) |
| • With mitral atresia/stenosis and hypoplastic LV | 4 | |
| • With complete AV canal defect | 6 | |
| • Other (DILV) | 1 | |
| • Normal AV valves | 78 | |
|
| ||
| Common Atrioventricular Canal Defect | 41 | 0 |
|
| ||
| Heterotaxy (with CAVC) | 54 (34) | 2(3.7%) |
| • Normally related great vessels | 17 (14) | |
| • DORV | 21 (14) | 1 |
| • TGA | 7 (1) | 1 |
| • RV to Ao with PA | 9 (5) | |
|
| ||
| Tetralogy of Fallot | 90 | 0 |
| • Pulmonary stenosis | 76 | |
| • Pulmonary atresia | 9 | |
| • Absent pulmonary valve | 5 | |
AV – atrioventricular, CAVC – common atrioventricular canal defect, DILV – double inlet left ventricle, DORV – double outlet right ventricle, LV – left ventricle, RV-Ao with PA – right ventricle to aorta with pulmonary atresia, TGA – transposition of the great arteries, VSD – ventricular septal defect
23/76 also had other cardiac anomalies including tricuspid atresia with hypoplastic RV (n = 10), straddling tricuspid valve (n = 3), abnormal mitral valve (n = 3), CAVC (n = 1), DILV (n = 1) and RV-Ao with PA (n = 1)
Mutation Detection
Total genomic DNA was extracted from venous blood or a cell line for each patient. DNA was purified from lymphocyte pellets according to standard procedures using the PUREGENE kit (Gentra Systems, Minneapolis, MN).
Primer pairs were designed using Oligo primer design software (Molecular Biology Insights Inc., Cascade, CO) to amplify the ZIC3 exons, including 50-100 bp beyond each intron-exon boundary (Table II). Exon 1 was amplified in two overlapping segments. For each amplimer, the polymerase chain reaction (PCR) was performed using a GC-rich kit (Roach). Amplified products were separated by agarose gel electrophoresis and visualized by staining with ethidium bromide. Amplicons were purified using magnetic beads (Agencourt, Beverly, MA) and sequenced using BigDye Terminator version 3.1 on an Applied Biosystems sequencer (ABI3730, Genetic Analyzer; Applied Biosystems, Foster City, CA). Sequences were aligned with known reference genomic sequences available via the UCSC genome browser using Sequencher (Gene Codes, Ann Arbor, MI). Cases were compared with race-matched control patients (Coriell), the NCBI SNP database Build 137 for Human [Sherry et al., 2001], and data from the NHLBI GO Exome Sequencing Project Exome Variant Server [Exome Variant Server, 2012]. Cases were also compared with 629 individuals from the 1000 Genomes Project 20100804 sequence and alignment release [The 1000 Genomes Project Consortium, 2010]. Data from the 1000 Genomes Project was converted with MUTALYZER 2.0 beta-21 [Wildeman et al., 2008]. In-silico predictions of the effect of missense mutations on protein function were modeled using three different publically available software programs: PolyPhen-2 [Adzhubei et al., 2010], SIFT [Ng et al., 2001; Ng et al., 2002; Ng et al., 2003; Kumar et al., 2009] and PMut [Ferrer-Costa et al., 2005]. The degree of evolutionary conservation at mutation loci was analyzed with PhyloP [Cooper et al., 2005; Siepel et al., 2006].
Table II.
Primers for ZIC3
| Exon | Forward Primer | Reverse Primer |
|---|---|---|
| 1-AB | 5′-GCAGCCCCTGGTAGCGCCTTGG-3′ | 5′-GCTTGATAGGCTGCCGCATATAACG-3′ |
| 1-CD | 5′-CCGAGCCCCCTAGCTACTTG-3′ | 5′-CCAATTAGTTCCTGAGACCGCAGAG-3′ |
| 2 | 5′-GCTGCTTGCCTCTGAGAAAC-3′ | 5′-ACGTGGAAGACAGAGGGTTG-3′ |
| 3 | 5′-CACTGGGCGGTCTTGTTGTTACA-3′ | 5′-TCCTGAGAAAAGGGCATGGCTACT-3′ |
| Primers used to confirm insertion in exon 1 | 5′-CCGGGAGGGCAAGTCTTTCAAG-3′ | 5′-GGAACCCACGCCCGCCTGCTTTTT-3′ |
Mutagenesis and Plasmid
A 1462 basepair bc113393 fragment of human ZIC3 cDNA including the entire open reading frame was subcloned in frame into a pcDNA3.1 vector using a standard protocol (Invitrogen, Grand Island, NY). ZIC3 missense mutations were introduced into the pcDNA3.1-ZIC3 fragment via Quick-Change site-directed mutagenesis (Stratagene). All the mutations and wild-type ZIC3+pcDNA3.1 were confirmed by sequencing.
Transcriptional Assay
NIH3T3 cells were maintained in Dulbecco’s modified Eagle medium with 10% fetal calf serum and 2% L-glutamate. Fugene (Roach) was used for transfection according to the manufacturer’s protocol. Cotransfections were performed using pcDNA3.1-ZIC3 constructs and a SV40 luciferase reporter plasmid (pGL3 promoter vector, Promega). The results were normalized to the promoterless luciferase vector (pGL3 basic, Promega). Cells were harvested 48h after transfection, and luciferase activity was determined using the Dual Luciferase Reported Assay System (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity. All results represent a minimum of three experiments.
RESULTS
In total, 443 unrelated individuals were studied for ZIC3 mutations including 169 with TGA, 89 with DORV, 41 with CAVC and 54 with heterotaxy (Table I). We also tested 90 patients with TOF to correlate genotype with phenotype. Forty percent (179/443) were female and 60% (264/443) were male. Seventy-eight percent (344/443) were Caucasian, 11.5% (51/443) African American, 4.3% (19/443) Asian and 6.6% (29/443) were of unknown or other race.
ZIC3 Mutations in Patients
Three non-synonymous mutations and one insertion mutation were identified in four unrelated individuals from the study cohort (Tables III, IV shaded rows). Patient 796, a Caucasian female with isolated DORV, was found to have a missense mutation c.49G>T resulting in a cysteine substituted for glycine at amino acid position 17 (Gly17Cys) (Figs 1-3). The mutation was maternally inherited. Patient 356, a Caucasian male with isolated d-TGA, had a missense mutation c.98C>T resulting in valine substituted for alanine at amino acid position 33 (Ala33Val) (Figs 1-3). Parental genetic information was not available. Patient 767, a Caucasian female with heterotaxy, had a missense mutation c.841C>T resulting in substitution of histidine for tyrosine at amino acid position 281 (His281Tyr), located in the first zinc finger domain of the protein (Figures 1-3). This was a de novo mutation. All three missense mutations were absent in 100 Caucasian and 100 African American control patients and the 1000 Genomes Project (n = 629) [The 1000 Genomes Consortium et al., 2010]. The Ala33Val and His281Tyr mutations were also not found in the NCBI SNP database [Sherry et al., 2011] and the NHLBI Exome Variant Server (EVS) [Exome Variant Server, 2012]. The Gly17Cys mutation was listed in the NCBI SNP database under rs147232392 in reference to a submission through the NHLBI EVS [Exome Variant Server, 2012]. The EVS dataset reports this mutation in 19 heterozygous females and 12 hemizygous males out of 6467 individuals (4053 females and 2414 males) sequenced for this region of ZIC3 with a minor allele frequency of 0.002947. No homozygous females were identified. No additional phenotypic information is available for the mutation-positive individuals.
Table III.
ZIC3 Mutations in Cases
| Base Pair Change | Amino Acid Change | Primary Diagnosis | Patients
|
Caucasian Controls n = 100 | African American Controls n = 100 | 1000 Genomes n = 629 | dbSNP ID | Poly-Phen2 | SIFT | PMut | PhyloP | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient ID | Gender | Ethnicity | n = 443* | |||||||||||
|
| ||||||||||||||
| 49G>T | Gly17Cys | DORV | 796 | F | C | 1 | 0 | 0 | 0 | rs147232392 | Probably damaging | Damaging | Neutral | 0.525 |
|
| ||||||||||||||
| 98C>T | Ala33Val | dTGA | 356 | M | C | 1 | 0 | 0 | 0 | - | Benign | Tolerated | Pathological | -0.400 |
|
| ||||||||||||||
| 162CGCins | Ala55ins | dTGA | 359 | M | C | 1 | 0 | 0 | 0 | - | - | - | - | 0.427 |
|
| ||||||||||||||
| 162CGCCGCCGCins | AAA55ins | Heterotaxy | 195 | F | As | 1 | 0 | 1 | 0 | - | - | - | - | 0.427 |
|
| ||||||||||||||
| 649C>G | Pro217Ala | DORV | 630 | M | AA | 4 | 0 | 2 | 5+ | rs104894963 | Benign | Damaging | Pathological | 0.597 |
| L-TGA | 369 | M | AA | |||||||||||
| TOF | 367 | F | C | |||||||||||
| TOF | 687 | F | AA | |||||||||||
|
| ||||||||||||||
| 841C>T | His281Tyr | Heterotaxy | 767 | F | C | 1 | 0 | 0 | 0 | - | Probably Damaging | Damaging | Neutral | 0.483 |
|
| ||||||||||||||
| 999ATGCCCCTTCCCins | CPFP333ins | Heterotaxy | BG-01-046A | M | C | 1** | 0 | 0 | 0 | - | - | - | - | 0.596 |
| Heterotaxy | BG-094A | M | C | |||||||||||
AA – African American, As – Asian, C – Caucasian, F – female, M – male
Total number of unrelated individuals screened. See Table I for distribution by cardiac lesion.
Mutation detected in BG-01-046A and BG-094A who are half-brothers; only BG-01-046A is included in the total count of unrelated individuals.
Reported as a frequency 0.0082, n = 629 therefore approximately 5 patients.
Table IV.
Clinical Characteristics of Patients with ZIC3 Mutations
| Patient | Primary Diagnosis | Situs | Segments | Veins | AV Valves | Ventricles | Great Arteries | Other | Extracardiac Malformations |
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| 796 | DORV | Solitus | {S,D,D} | Normal SV return | Normal | Hypoplastic RV malalignment VSD | SubAS | Single (right) coronary ostium | None |
| Normal PV return | LAA NB | ||||||||
| CoA with arch hypoplasia | |||||||||
|
| |||||||||
| 356 | d-TGA | Solitus | {S,D,D} | Normal SV return | Normal | Normal size/morphology Intact septum | d-TGA Arch branching not specified | Pyloric stenosis | |
| Normal PV return | |||||||||
|
| |||||||||
| 359 | d-TGA | Solitus | {S,D,D} | Normal SV return | Normal | Normal size/morphology Intact septum | d-TGA Arch branching not specified | None | |
| Normal PV return | |||||||||
|
| |||||||||
| 195 | Heterotaxy | Ambiguus: dextrocardia atrial situs inversus right-sided stomach | {I,D,D} | RSVC to LA | UCAVC to left | Hypoplastic RV Single VSD | LAA aberrant RSCA (retroesophageal) DAo on the right PS, severe subPS | Single (right) coronary ostium AP collaterals | None |
| LSVC to RA | |||||||||
| LIVC to RA | |||||||||
| HV normal | |||||||||
| PV normal | |||||||||
|
| |||||||||
| 767 | Heterotaxy | Ambiguus: Levocardia Atrial situs ambiguus Right-sided stomach Asplenia* | {A,D,D} | RSVC to RA | Normal | Normal size/morphology Single inlet-type VSD | Transposed great arteries PS and subPS LAA NB | Severe LPA stenosis with LPA supplied by L-PDA | None |
| LSVC to CS | |||||||||
| LIVC to left-sided atrium | |||||||||
| HV to right-sided atrium | |||||||||
| Supracardiac TAPVC to SVC/RA junction | |||||||||
|
| |||||||||
| BG-01-046A | Heterotaxy | Ambiguus: Levocardia Atrial situs ambiguus Left-sided liver Right-sided stomach Asplenia Malrotation | {A,D,D} | RSVC to RA | UCAVC to right | Mildly hypoplastic LV | DORV RAA with mirror image branching DAo to right | Small LPA L-PDA to LPA | 2 vessel cord Ear pits Hiatal hernia |
| LSVC to LA | |||||||||
| I-IVC with azygos continuation | |||||||||
| HV to left-sided atrium | |||||||||
| PV to left-sided atrium | |||||||||
|
| |||||||||
| BG-094A | Heterotaxy | Ambiguus: Levocardia Atrial situs ambiguus Midline liver Midline stomach Asplenia Malrotation | {I,L,X} | RSVC to RA | UCAVC to right | Severely hypoplastic LV | RV to Ao with pulmonary atresia LAA NB DAo to right | R-PDA innominate to PAs | Hiatal hernia |
| LSVC to LA | |||||||||
| LIVC to left-sided RA HV to left-sided RA infradiaphragmatic | |||||||||
| TAPVR | |||||||||
AP – aortopulmonary, CS – coronary sinus, CoA – coarctation of the aorta, DAo – descending aorta, DORV – double outlet right ventricle, d-TGA – d-transposition of the great arteries, HV – hepatic veins, I-IVC – interrupted inferior vena cava, LA – left atrium, LAA – left aortic arch, LIVC – left inferior vena cava, L-PDA – left patent ductus arteriosus, LPA – left pulmonary artery, LSVC – left superior vena cava, NB – normal branching, PA – pulmonary artery, PFO – patent foramen ovale, PS – pulmonary stenosis, PV – pulmonary veins, RA – right atrium, RAA – right aortic arch, R-PDA – right patent ductus arteriosus, RSCA – right subclavian artery, RSVC – right superior vena cava, RV – right ventricle, subAS – subaortic stenosis, subPS – subpulmonary stenosis, SV – systemic venous, TAPVC – total anomalous pulmonary venous connection, UCAVC – unbalanced complete atrioventricular canal defect, VSD – ventricular septal defect
Patient on prophylactic amoxicillin for presumed functional asplenia however splenic anatomy unconfirmed.
Figure 1.
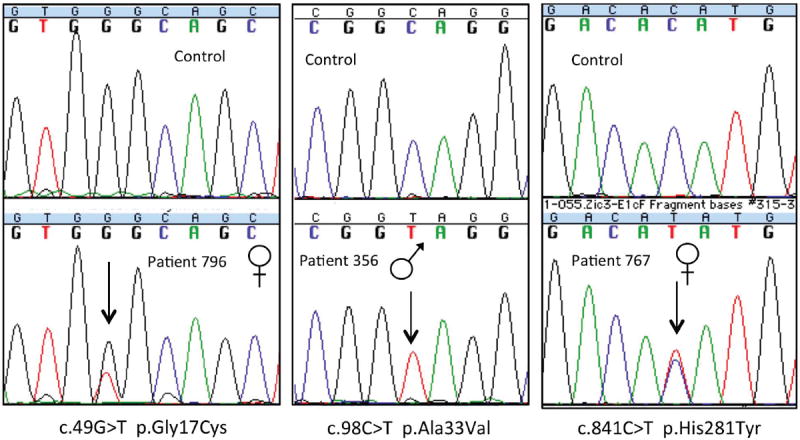
Potential disease-related mutations in cases. Sequences from three cases with unique missense mutations are presented in the lower panels with the corresponding sequence from a normal control above. The gender of each case is noted. The arrow points to the nucleotide change from wild-type.
Figure 3.

ZIC3 amino acid sequence including the polyalanine expansion in exon 1 (darkly-shaded box) and the five zinc finger domains (lightly-shaded boxes). The Gly17Cys (*), Ala33Val (+), His281Tyr (ˆ) mutations are shown (non-shaded boxes) as well as the position of the CPFP333ins mutation (↓) demonstrating conservation of these amino acids between species.
All three mutations are predicted by at least one algorithm to be damaging to ZIC3 function (Table III) [Adzhubei et al., 2010; Ng et al., 2001; Ng et al., 2002; Ng et al., 2003; Kumar et al., 2009; Ferrer-Costa et al., 2005]. The conservation scores generated by PhyloP were 0.525 for the Gly17Cys mutation, -0.400 for the Ala33Val mutation and 0.483 for the His281Tyr mutation (Table III, Fig 3). [Cooper et al., 2005; Siepel et al., 2006]
Two half-brothers with heterotaxy were found to have a novel 12 base pair insertion resulting in insertion of four amino acids at position 333 in a highly conserved third zinc finger domain (Tables III, IV; Figs 2-4). Their mother was found to be an asymptomatic carrier of the insertion; imaging to identify sub-clinical features has not been performed.
Figure 2.
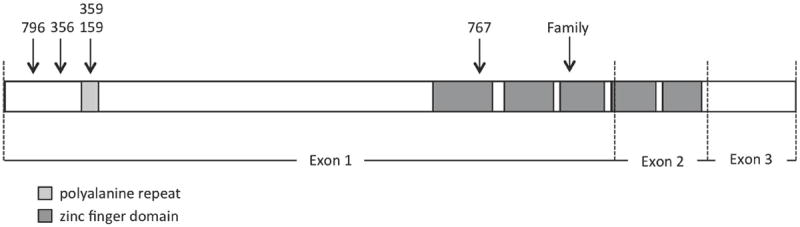
ZIC3 open reading frame structure including 5 highly conserved tandem regions coding for zinc finger domains and a polyalanine repeat. The locations of reported ZIC3 mutations are shown.
Figure 4.
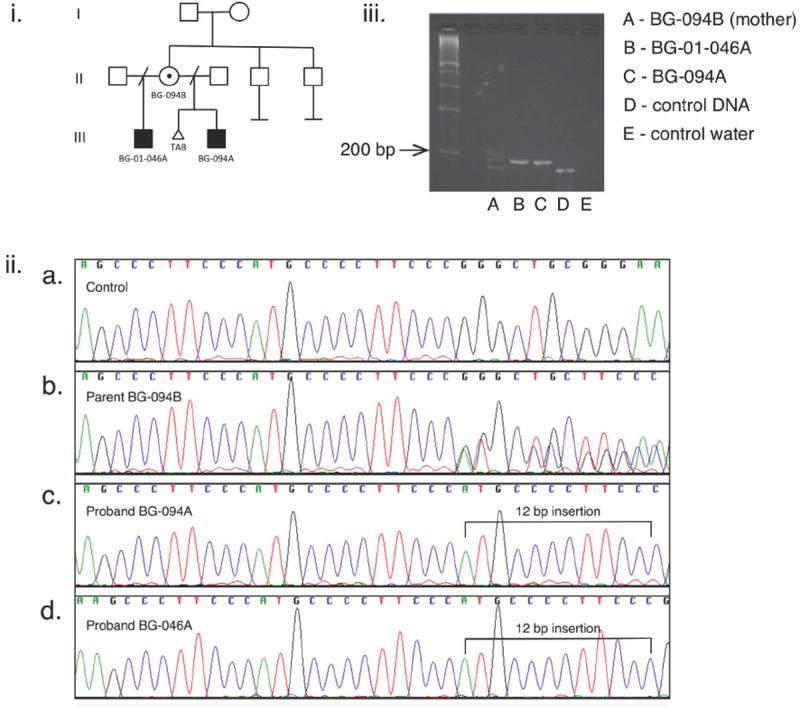
i. Pedigree of family with inherited heterotaxy with two affected sons and an asymptomatic carrier mother.
ii. Chromatograms demonstrate: a. the reference sequence, b. the maternal sequence (BG-094B) with heterozygosity beginning at the position of the insertion (c.999) consistent with both the reference sequence and a 12-bp insertion (c.999ATGCCCCTTCCCins), c. patients BG094A and d. BG-046A both with a 12-base pair insertion which is hemizygous in both male patients.
iii. The insertion was confirmed by gel electrophoresis using primers (Table II) designed to amplify a 184 bp sequence containing the insertion in exon 1. Column D shows control DNA with one band 184 bp in size. Column A shows the heterozygous maternal DNA with the wild-type 184 bp band, a slightly larger band consistent with the insertion (196 bp) and a third band consistent with a heteroduplex. Columns B and C show BG094-A and BG046-A both with the single 196 bp band confirming the insertion.
Two patients were found to have an expansion of the polyalanine repeat in exon 1 (c.123 to 153). Patient 359, a Caucasian male with isolated d-TGA, had a GCC insertion, expanding the region from 10 to 11 alanines. Patient 195, an Asian female with heterotaxy, had a 9 base pair insertion in the polyalanine repeat, expanding the repeat by 3 alanines (Tables III, IV; Figs 2, 3). Parental samples were not available for either patient. The 9 base pair insertion was also seen in a female African American control patient (1/100).
Four patients were found to have a c.649C>G resulting in substitution of alanine for proline at amino acid position 217 (Pro217Ala), a mutation previously reported to be disease-related [Ware et al., 2004] (Table III). All of our patients had isolated CHD; three were African American and one was Caucasian. However, we identified this missense mutation in 2 of 100 African American control patients, in the 1000 Genomes Project with a frequency of 0.0082 (~ 5/629 patients) [The 1000 Genomes Project Consortium et al., 2010], and in non-clinical submissions in the NCBI SNP database (rs104894963) [Sherry et al., 2001]. The EVS dataset reports this mutation in 2 homozygous females, 64 heterozygous females and 10 hemizygous males out of 6483 individuals sequenced for this region of ZIC3 with a minor allele frequency of 0.007465. [Exome Variant Server, 2012].
No clinically significant mutations were identified in patients with CAVC or TOF.
ZIC3 Mutations in Control Patients
As a means of assessing the frequency of nonsynonymous mutations in the general population and gauging the potential import of mutations identified in our patients, we examined control patients for the presence of additional mutations in the ZIC3 coding regions. Of the 200 control patients who underwent sequencing of the ZIC3 exons in our laboratory, one individual had a nonsynonymous mutation (c.75C>G His25Gln) previously reported in the NCBI SNP database as occurring in more than 75 apparently healthy control patients [Sherry et al., 2001], two individuals had the Pro217Ala mutation (as described above), and one individual had a polyalanine expansion mutation (as described above). Thirteen additional patients had synonymous mutations (Supplemental eTable I – see Supporting Information online). In the 1000 Genomes Project, the only mutation reported in the ZIC3 coding region was Pro217Ala among the data available from 629 controls. As noted above, this mutation occurred with a frequency of 0.0082, similar to that in our cohort [The 1000 Genomes Project Consortium et al., 2010]. No other mutations in the ZIC3 coding region were identified in 829 controls.
In the NHLBI Exome Variant Server, eight non-synonymous mutations were identified in 5992-6503 patients sequenced for the ZIC3 coding region, including the Gly17Cys and Pro217Ala mutations as described above (Supplemental eTable II – see Supporting Information online). To our knowledge, none of the six additional mutations have been described in clinical cohorts. The minor allele frequencies range from 0.000095 to 0.001058. None of the mutations lie in the zinc finger domains or the polyalanine expansion. Eight additional synonymous mutations were also detected [Exome Variant Server, 2012].
Functional Data
The effect of the ZIC3 missense mutations Gly17Cys, Ala33Val, and His281Tyr on in vitro transactivation of an SV40 luciferase was compared to wild-type ZIC3 (Figure 5). We also studied two previously reported mutations, Pro217Ala and Cys253Ser, which were reported to have increased and decreased transactivation respectively [Ware et al., 2004]. Compared with wild-type ZIC3, the Gly17Cys, Ala33Val and Pro217Ala mutants did not demonstrate altered transactivation. Both the newly identified His281Tyr and previously reported Cys253Ser mutants demonstrated reduced transactivation.
Figure 5.
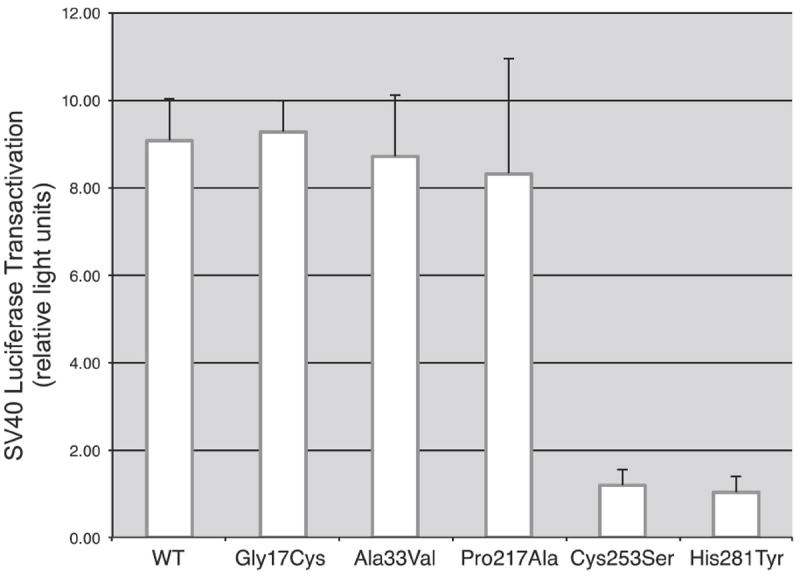
Effect of ZIC3 mutations on transactivation of an SV40 luciferase reporter gene. Transactivation for the missense mutations Gly17Cys and Ala33Val are not significantly different from wild-type ZIC3. Pro217Ala was previously reported as having increased transactivation, however in the present study was not significantly different from wild-type. The missense mutation His281Tyr shows decreased transactivation as compared to wild-type, similar to a previously reported mutation Cys253Ser.
DISCUSSION
We identified ZIC3 mutations in patients with sporadic isolated DORV and d-TGA, as well as those with sporadic and familial heterotaxy. The Ala33Val mutation identified in a case with dTGA and the His281Tyr mutation identified in a patient with sporadic heterotaxy are absent from 200 control patients, 629 controls in the 1000 Genomes Project [The 1000 Genomes Project Consortium et al., 2010] and the NCBI SNP database [Sherry et al., 2001], and therefore appear to be exceptionally rare.
Though likewise absent from 200 controls and 629 cases in the 1000 Genomes Project, the Gly17Cys mutation identified in a case with DORV has been reported infrequently in the NHLBI Exome Variant Server cohort of nearly 6500 subjects, in both hemizygous males (n = 12) and heterozygous females (n = 19) [Exome Variant Server, 2012]. The NHLBI EVS represents a compilation of cases from multiple adult cohorts enrolled in a variety of studies whose phenotypes are not individually available. Thus given the large study cohort, it is possible that cases with the Gly17Cys mutation have unrecognized or unreported congenital cardiac malformations, such as venous anomalies, that would place them on the mild heterotaxy spectrum. They may also be asymptomatic carriers or males with incomplete penetrance, as has been previously reported [Megarbane et al., 2000; D’Alessandro et al., 2011]. Overall even this mutation appears to be a very rare variant.
The Gly17Cys and His281Tyr mutations are predicted by two of three publically available software programs to be deleterious to ZIC3 protein function (Table III). The alterations are at loci with high conservation scores (0.525 and 0.483 respectively), which also suggests that a missense mutation is more likely to disrupt protein function at this locus. Additionally, the His281Tyr mutant demonstrated decreased transactivation in a luciferase assay. These data lend further support to the likely pathogenicity of the Gly17Cys and His281Tyr mutations. The Ala33Val mutant, though novel in a male patient and absent from control populations, was only predicted by 1 of 3 algorithms to be deleterious to the ZIC3 protein and had a negative conservation score. These results suggest that this mutant is less likely to be disease-related, however it cannot be ruled out, especially as it is present in a male patient. The familial 12 base pair mutation results in insertion of four amino acids at position 333 in the highly conserved third zinc finger domain. Although this mutant was not assessed with functional studies, it has previously been demonstrated that the zinc finger domains are necessary for binding with the cardiac α-actin promoter [Zhu et al., 2007], and interaction with GLI3 [Zhu et al., 2008], suggesting that this mutant is likely to confer functional changes and hence likely to be disease-related.
The Gly17Cys mutation and the familial 12 base pair insertion were each seen in asymptomatic maternal carriers. As imaging studies could not be performed on these two carriers, it is unclear whether they have sub-clinical features such as mild venous anomalies. Nonetheless, incomplete penetrance in female carriers has been commonly observed in this X-linked disease gene.
We did not identify ZIC3 mutations in cases with isolated CAVC, but the relatively small sample size may not have allowed detection of rare mutations with a low prevalence. We also did not identify significant mutations in the larger cohort of patients with TOF, suggesting that ZIC3 mutations may be specific to lesions commonly seen in heterotaxy, or at least those with malposed great arteries.
The Pro217Ala mutation was of interest as it was previously reported as a disease-causing mutation in a Caucasian male with isolated CHD that is not typical of heterotaxy syndrome (atrial septal defect and pulmonary stenosis) and not in ethnicity-matched controls or the isolated CHD cohort [Ware et al., 2004]. It was also previously reported to have increased transactivation on functional studies. In our study, this mutation was detected in four patients with isolated CHD including DORV, L-TGA, two patients with TOF, as well as two African American controls patients. The transactivation was not significantly different compared with wild-type ZIC3. It has since been reported in non-clinical submissions in the NCBI SNP database (rs104894963) [Sherry et al., 2001], in 1000 Genomes Project with a frequency of 0.0082 (n = 629) [The 1000 Genomes Project Consortium et al., 2010] and in the NHLBI EVS with a minor allele frequency of 0.007465, including in 2 females who are homozygous for the mutation. Despite the predicted deleterious affect of this mutation on protein function by two algorithms, the Pro217Ala mutant has been demonstrated to retain functions of wild-type Zic3, including physical and functional interaction with GLI3 [Zhu et al., 2008] and the ability to bind and inhibit the cardiac α-actin promoter [Zhu et al., 2007]. Therefore, an increasing body of data suggests that this mutation is most likely a benign polymorphism.
The significance of expansion of the polyalanine repeat in exon 1 is unclear. This finding was previously reported in a patient with a polyalanine expansion from 10 to 12 in a male patient with VACTERL association, dextroposition of the heart, and persistence of the left superior vena cava draining to the coronary sinus [Wessels et al., 2010]. It is unknown how expansion of the polyalanine repeat would alter the function of the ZIC3 protein, although several possibilities have been proposed [Wessels et al., 2010]. Polyalanine repeat expansion, most often within transcription factors, has been shown to be a mechanism of disease in other inherited disorders, such as holoproscencephaly with polyalanine expansion in ZIC2. The polyalanine expansion was also identified in one female control patient who was asymptomatic. However, very minor laterality defects may be undetected and female carriers have been reported. Therefore this finding in a female control patient does not rule out a disease-causing insertion in patients.
We examined our control patients, as well as the 1000 Genomes Project control patients and the NHLBI Exome Variant Server for additional mutations in the ZIC3 coding region as a means of assessing genetic burden. Of the 200 control patients who underwent sequencing, one individual had a previously identified polymorphism (His25Gln), two individuals had the Pro217Ala polymorphism (see above) and one individual had a polyalanine expansion mutation (see above). In the 1000 Genomes Project (n=629), one mutation in the ZIC3 coding region is reported, Pro217Ala, with a frequency of 0.0082, similar to that in our study (see above) [The 1000 Genomes Project Consortium et al., 2010]. In the NHLBI Exome Variant Server containing nearly 6500 individuals who have been sequenced for ZIC3, a total of 8 nonsynonymous mutations in the ZIC3 coding region were detected, including the Gly17Cys and Pro217Ala mutations. The Pro217Ala was the most frequently detected mutation, with a minor allele frequency of 0.0074654, and interestingly, it was homozygous in two females. As the phenotype of the Exome Variant Server patients is unknown, some of them may be affected, some may have subtle anomalies and be clinically asymptomatic, and some may be carriers or demonstrate incomplete penetrance. The paucity of mutations in this very large cohort, combined with the absence of rare, non-synonymous mutations predicted to be deleterious in our control patients renders the mutations identified in our patients with cardiac disease compelling.
This work, in combination with other studies, demonstrates that ZIC3 mutations are an important etiology in sporadic and familial heterotaxy. These results further support the hypothesis that a subset of patients with isolated malposition of the great vessels as manifest by DORV, and possibly dTGA, share a common genetic basis with heterotaxy. The mouse ortholog, Zic3 is expressed bilaterally in early embryogenesis and has been shown to play a role in the development of the anterior-posterior axis, which precedes and is requisite for left-right axis formation [Ware et al., 2006]. A Zic3 null mouse model demonstrates a comparable spectrum of phenotypic anomalies to those seen in humans with ZIC3 mutations, including 20% with incomplete penetrance for heterotaxy. Interestingly, when pulmonary anatomy was examined, both left and right bronchial isomerism were seen, consistent with reports of both asplenia- and polyspenia-type heterotaxy reported in humans with ZIC3 mutations [Purandare et al., 2002]. Zic3-deficient mice fail to maintain Nodal expression after the two-somite stage and exhibit randomization of expression of Nodal and Pitx2 in the lateral plate mesoderm, confirming that Zic3 is necessary for maintenance of molecular asymmetry that precedes organ asymmetry in the developing embryo [Purandare et al., 2002].
Unfortunately, only a small group of patients with ZIC3 mutations have been reported in the literature and few of them with completely characterized phenotypes leaving many questions unanswered. Nonetheless it is important to recognize that patients with isolated DORV and dTGA secondary to an underlying ZIC3 mutation are at an increased risk of having a child with heart disease of potentially wide-ranging severity, including heterotaxy and its associated anomalies. If the mutation is inherited, the same applies to sibling recurrence risk. It has been shown that patients with heterotaxy of all etiologies have increased post-surgical mortality, increased ECMO requirement, tracheostomy and prolonged ventilation compared with controls with cardiac disease of the same surgical severity score [Swisher et al., 2011]. The basis for increased morbidity is unclear but it is conceivable that this risk also extends to patients with ZIC3 mutations and isolated cardiac disease. Our findings highlight the importance of additional studies designed to correlate genotype with phenotype so we can better understand the potential implications relating to outcome, prognosis and recurrence risk.
Supplementary Material
Acknowledgments
We thank the children and families who participated in this study.
We also thank Sharon Edman for database management, Petra Werner for assistance with data analysis, our genetic counselors Jennifer Garbarini and Stacy Woyciechowski for patient recruitment, and Matthew Deardorff for his advice and assistance.
The authors would like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010).
FUNDING SOURCES
Supported by NHLBI Award HL51533 (EG), P50-HL74731 (EG). Additionally the project described was supported by the National Center for Research Resources, Grant UL1RR024134, and is now at the National Center for Advancing Translational Sciences, Grant UL1TR000003. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Contributor Information
Lisa C. A. D’Alessandro, Email: lisac.dalessandro@sickkids.ca.
Brande C. Latney, Email: latney@email.chop.edu.
Prasuna C. Paluru, Email: paluru@email.chop.edu.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A Method and Server for Predicting Damaging Missense Mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung YF, Cheng VY, Chau AK, Chiu CSW, Yung TC, Leung MP. Outcome of infants with right atrial isomerism: is prognosis better with normal pulmonary venous drainage? Heart. 2002;87:146–152. doi: 10.1136/heart.87.2.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhin B, Hatayama M, Bozon D, Ogawa M, Schon P, Tohmonda T, Sassolas F, Aruga J, Valard AG, Chen SC, Bouvagnet P. Elucidation of penetrance variability of a ZIC3 mutation in a family with complex heart defects and functional analysis of ZIC3 mutations in the first zinc finger domain. Hum Mutat. 2007;28:563–570. doi: 10.1002/humu.20480. [DOI] [PubMed] [Google Scholar]
- Cohen MS. Clarifying anatomical complexity: Diagnosing heterotaxy syndrome in the fetus. Prog Pediatr Cardiol. 2006;22:61–70. [Google Scholar]
- Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A NISC Comparative Sequencing Program. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro LCA, Casey B, Siu VM. Situs Inversus Totalis and a Novel ZIC3 Mutation in a Family with X-linked Heterotaxy. Congenit Heart Dis. 2011 doi: 10.1111/j.1747-0803.2011.00602.x. EPub ahead of print. [DOI] [PubMed] [Google Scholar]
- de Luca A, Sarkozy A, Consoli F, Ferese R, Guida V, Dentici ML, Mingarelli R, Bellacchio E, Tuo G, Limongelli G, Digilio MC, Marino B, Dallapiccola B. Familial transposition of the great arteries caused by multiple mutations in laterality genes. Heart. 2010;96:673–677. doi: 10.1136/hrt.2009.181685. [DOI] [PubMed] [Google Scholar]
- Exome Variant Server. NHLBI GO Exome Sequencing Project (ESP) Seattle, WA: [August 2012]. (URL: http://evs.gs.washington.edu/EVS/) [Google Scholar]
- Ferencz C, Correa-Villasenor A, Loffredo CA, Wilson PD. Genetic and environmental risk factors of major cardiovascular malformations. New York: Futura; 1997. [Google Scholar]
- Ferrer-Costa C, Gelpi JL, Zamakola L, Parraga I, de la Cruz X, Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21:3176–3178. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- Gebbia M, Ferrero GB, Pilia G, Bassi MT, Aylsworth AS, Penman-Splitt M, Bird LM, Bamforth JS, Burn J, Schlessinger D, Nelson DL, Casey B. X-linked situs abnormalities result from mutations in ZIC3. Nat Genet. 1997;17:305–308. doi: 10.1038/ng1197-305. [DOI] [PubMed] [Google Scholar]
- Gilljam T, McCrindle BW, Smallhorn JF, Williams WG, Freedom RM. Outcomes of left atrial isomerism over a 28-year period at a single institution. J Am Coll Cardiol. 2000;36:908–916. doi: 10.1016/s0735-1097(00)00812-3. [DOI] [PubMed] [Google Scholar]
- Hashmi A, Abu-Sulaiman R, McCrindle BW, Smallhorn JF, Williams WG, Freedom RM. Management and outcomes of right atrial isomerism: a 26-year experience. J Am Coll Cardiol. 1998;31:1120–1126. doi: 10.1016/s0735-1097(98)00062-x. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Lin AE, Ticho BS, Houde K, Westgate MN, Holmes LB. Heterotaxy: associated conditions and hospital-based prevalence in newborns. Genet Med. 2000;2:157–172. doi: 10.1097/00125817-200005000-00002. [DOI] [PubMed] [Google Scholar]
- Mégarbané A, Salem N, Stephan E, Ashoush R, Lenoir D, Delague V, Kassab R, Loiselet J, Bouvagnet P. X-linked transposition of the great arteries and incomplete penetrance among males with a nonsense mutation in ZIC3. Eur J Hum Genet. 2000;8:704–708. doi: 10.1038/sj.ejhg.5200526. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting Deleterious Amino Acid Substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Accounting for Human Polymorphisms Predicted to Affect Protein Function. Genome Res. 2002;12:436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purandare SM, Ware SM, Kwan KM, Gebbia M, Bassi MT, Deng JM, Vogel H, Behringer RR, Belmont JW, Casey B. A complex syndrome of left-right axis, central nervous system and axial skeleton defects in Zic3 mutant mice. Development. 2002;129:2293–2302. doi: 10.1242/dev.129.9.2293. [DOI] [PubMed] [Google Scholar]
- Siepel A, Pollard KS, Haussler D. New methods for detecting lineage-specific selection. Proceedings of the 10th International Conference on Research in Computational Molecular Biology (RECOMB); 2006. pp. 190–205. [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swisher M, Jonas R, Tian X, Lee ES, Lo CW, Leatherbury L. Increased postoperative and respiratory complications in patients with congenital heart disease associated with heterotaxy. J Thorac Cardiovasc Surg. 2011;141:637–644. doi: 10.1016/j.jtcvs.2010.07.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praagh S. In: NADAS’ Pediatric Cardiology. 2. Keane J, Lock J, Flyer D, editors. Philadelphia: 2006. pp. 675–696. [Google Scholar]
- Wessels MW, Kuchinka B, Heydanus R, Smit BJ, Dooijes D, de Krijger RR, Lequin MH, de Jong EM, Husen M, Willems PJ, Case B. Polyalanine expansion in the ZIC3 gene leading to X-linked heterotaxy with VACTERL association: a new polyalanine disorder? J Med Genet. 2010;47:351–355. doi: 10.1136/jmg.2008.060913. [DOI] [PubMed] [Google Scholar]
- Ware SM, Peng J, Zhu L, Fernbach S, Colicos S, Casey B, Towbin J, Belmont JW. Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am J Hum Genet. 2004;74:93–105. doi: 10.1086/380998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware SM, Harutyunyan KG, Belmont JW. Zic3 is critical for early embryonic patterning during gastrulation. Dev Dyn. 2006;235:776–785. doi: 10.1002/dvdy.20668. [DOI] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1016–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildeman M, van Ophuizen E, Den Dunnen JT, Taschner PE. Improving sequence variant descriptions in mutation databases and literature using the MUTALYZER sequence variation nomenclature checker. Hum Mutat. 2008;29:6–13. doi: 10.1002/humu.20654. [DOI] [PubMed] [Google Scholar]
- Zhu L, Zhou G, Poole S, Belmont JW. Characterization of the Interactions of Human ZIC3 Mutants With GL3. Hum Mutat. 2007;29:99–105. doi: 10.1002/humu.20606. [DOI] [PubMed] [Google Scholar]
- Zhu L, Harutyunyan KG, Pen JL, Wang J, Schwartz RJ, Belmont JW. Identification of a novel role of ZIC3 in regulating cardiac development. Hum Mol Genet. 2007;16:1649–1660. doi: 10.1093/hmg/ddm106. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.