

Fecal inflammatory markers at diagnosis correlate with diarrhea persistence and treatment failure in Clostridium difficile infection, whereas C. difficile fecal bacterial burden does not. C. difficile bacterial concentration decreases similarly in patients treated with metronidazole and vancomycin.
Keywords: Clostridium difficile, cytokines, inflammation, IL-8, CXCL-5
Abstract
Background. Clostridium difficile is a leading hospital-acquired infection. Many patients remain symptomatic for several days on appropriate antibiotic therapy. To assess the contribution of ongoing infection vs persistent inflammation, we examined the correlation between fecal cytokine levels, fecal C. difficile burden, and disease outcomes in C. difficile infection (CDI).
Methods. We conducted a prospective cohort study in Barnes Jewish Hospital between June 2011 and May 2012 of hospitalized adults with CDI. We determined fecal interleukin 8 (IL-8) and lactoferrin protein concentrations by enzyme immunoassay. We used real-time polymerase chain reaction (PCR) to measure relative fecal IL-8 and CXCL-5 RNA transcript abundances, and quantitative PCR to enumerate C. difficile burden.
Results. Of 120 study subjects, 101 (84%) were started on metronidazole, and 33 of those (33%) were subsequently given vancomycin. Sixty-two (52%) patients had diarrhea persistent for 5 or more days after starting CDI therapy. Initial fecal CXCL-5 messenger RNA (mRNA), IL-8 mRNA, and IL-8 protein correlated with persistent diarrhea and use of vancomycin. Time to diarrhea resolution was longer in patients with elevated fecal cytokines at diagnosis. Fecal cytokines were more sensitive than clinical severity scores in identifying patients at risk of treatment failure. Clostridium difficile burden did not correlate with any measure of illness or outcome at any point, and decreased equally with metronidazole and vancomycin.
Conclusions. Persistent diarrhea in CDI correlates with intestinal inflammation and not fecal pathogen burden. These findings suggest that modulation of host response, rather than adjustments to antimicrobial regimens, might be a more effective approach to patients with unremitting disease.
(See the Editorial Commentary by Yacyshyn and Yacyshyn on pages 1722–3.)
Clostridium difficile is among the most prevalent hospital-associated infections [1]. Its illness spectrum ranges from asymptomatic carriage to severe diarrhea, toxic megacolon, and death [2, 3]. The incidence and severity of C. difficile infection (CDI) have increased in recent years [4–6], and even after 10–15 days of treatment, antibiotic therapy fails in 22% of patients [7, 8]. An even higher proportion of patients have ongoing symptoms at 5–6 days of therapy, when many clinicians consider altering antimicrobial therapy [9, 10]. Multiple severity scores have attempted to identify patients at risk of severe disease (intensive care unit admission, colectomy, or death) [9, 11–15], but few focus on identifying patients at risk of poor response to therapy or prolonged morbidity [16]. We aimed to detect biomarkers that would categorize patients most at risk of treatment failure or persistent disease. We were specifically interested in determining if persistent symptoms correlated with antimicrobial therapy failure or, rather, were a consequence of persistent intestinal inflammation.
The pathophysiology of C. difficile disease is incompletely understood [17, 18]. The best-studied virulence factors are the 2 toxins TcdA and TcdB, encoded by tcdA and tcdB, respectively. These toxins are thought to be responsible for the clinical manifestations of CDI by injuring intestinal epithelial cells [19]. Toxin exposure in vitro elicits proinflammatory cytokine release [20, 21], with prominent neutrophil activation and recruitment, leading to an intense inflammatory response [22, 23]. Fecal interleukin 8 (IL-8) and lactoferrin are also elevated in vivo in patients with severe CDI [24, 25]. In addition, CXCL-5 (epithelial neutrophil activating peptide 78) is hyperexpressed in the colons of C. difficile–infected mice, and levels correlate with outcome severity (Y. S. Chen et al, unpublished data). CXCL-5 belongs to the family of CXC chemokines, is 22% homologous to IL-8, binds to the IL-8 receptor 2, recruits and activates neutrophils [26], and is upregulated in patients with inflammatory bowel disease [27].
Here, we study intestinal inflammation, as defined by fecal expression levels of inflammatory cytokines, and C. difficile concentration, to correlate host and microbial biology with disease severity in hospitalized patients with symptomatic CDI.
METHODS
Study Population
The study was conducted in Barnes Jewish Hospital, St Louis, Missouri, and approved by the Institutional Review Board of the Washington University School of Medicine. We approached all hospitalized patients who had diarrhea (either on admission or during their hospitalization) and a positive C. difficile toxin A/B enzyme immunoassay performed in the clinical microbiology laboratory between 24 June 2011 and 10 May 2012, and obtained written informed consent. We collected specimens daily thereafter when available.
Clinical Data
For all subjects, we collected information on demographic characteristics, clinical presentation (with special emphasis on the diarrhea: onset, number of stools, consistency according to the Bristol stool chart [28], associated symptoms), medications, and healthcare exposures. We also reviewed charts and recorded vital signs, laboratory, and imaging findings. We assigned to all patients on study admission 3 severity scores reported to predict poor outcome (HINES VA, UPMC1, and CSI [11, 13, 14, 16]; Supplementary Table 1). We then recorded symptoms and CDI therapy during daily visits or, if patients were discharged, by telephone interview within 10 days of treatment initiation. Pertinent outcomes were time to diarrhea resolution, diarrhea persistence (defined as ≥3 bowel movements with a Bristol stool score ≥6, 5 days or more after initiation of CDI therapy), CDI treatment, and severe disease (defined as intensive care unit admission, toxic megacolon, colectomy, or death). We used 5 days of therapy as a clinically relevant time-point [10, 16] to distinguish persistent from short-duration illness. Barnes Jewish Hospital physicians generally use metronidazole as first-line treatment for mild-to-moderate disease, and reserve vancomycin for severe cases or if there is concern with treatment failure.
Stool Collection
We retrieved the initial stool specimen submitted to the microbiology laboratory for testing within 48 hours of submission. We subsequently collected daily stool specimens when available from the patients during their hospitalization. Stools were aliquoted and stored at −80°C upon receipt.
Laboratory Assays
Enzyme Immunoassay
Stools were suspended in Protease Inhibitor Lysis Buffer (Roche Diagnostics, Indianapolis, Indiana; 1/100 w/v). We quantified IL-8 protein levels using 50 µL of lysate and the Human CXCL8/IL-8 Quantikine kit (R&D Systems, Minneapolis, Minnesota), according to the manufacturer's instructions. For fecal lactoferrin concentrations, we used 100 µL of lysate with the IBD-Scan kit (Techlab, Blacksburg, Virginia), per the manufacturer's instructions.
Nucleic Acid Extraction
Total nucleic acid was extracted using the NucliSENS EasyMAG automated system software 1.0.2 specific A protocol (bioMérieux, Marcy L'Etoile, France), according to the manufacturer's instructions. 500 μg of stool was lysed in 1 mL of lysis buffer (bioMérieux, St Louis, Missouri). 200 μL of the stool eluate was then added to 2 mL of lysis buffer, followed by 100 µL of magnetic silica for a final volume of 110 µL of nucleic acid extract.
Nucleic Acid Amplification
Cytokine Polymerase Chain Reaction
Real-time polymerase chain reaction (PCR) was performed using 7500 Fast Real-Time PCR System and Path-ID Multiplex One-Step RT-PCR mix (Applied Biosystems, Foster City, California). The reaction mixture included 50–100 ng nucleic acid, 10 µL buffer, 2 µL enzyme mix, 2 µL water, 1 µL TaqMan probe ×20 for IL-8 (Hs00174103_m1) or CXCL-5 (Hs00171085_m1), and 1 µL glyceraldehyde 3-phosphate dehydrogenase (GAPDH) ×20 internal control (catalogue number 4308313, Applied Biosystems). The PCR conditions included 10 minutes of incubation at 48°C and 10 minutes at 95°C, followed by 50 cycles of 95°C for 15 seconds and 60°C for 45 seconds. Results are reported as ΔCT values, denoting the normalization of the cycle threshold (CT) of the detection of the cytokine to the CT of the internal control (GAPDH) (ΔCT = CT GAPDH – CT cytokine).
Quantitative tcdB DNA PCR
SYBR Green–based real-time PCR was performed using 7500 Fast Real-Time PCR System (Applied Biosystems). PCR conditions were modified from Wroblewski et al [29], by including 10 µL of fast SYBR Green Master Mix (Applied Biosystems), 0.5 µM of each primer TcdBF1 (5′-ACGGACAAGCAGTTGAATATAGTGGTTTAGTTAGAG-3′) and TcdBR1 (5′-ATTAATACCTTTGCATGCTTTTTTAGTTTCTGGATTGAA-3′) [29], 5 µL (50–100 ng) of nucleic acid, and water for a final reaction volume of 20 μL. The PCR conditions included an initial 5-minute incubation step at 95°C, followed by 50 cycles of 15 seconds at 95°C, 30 seconds at 55°C, and 45 seconds at 68°C. After amplification, the melting curve was performed with an initial denaturation step at 95°C for 15 seconds, followed by 60 seconds at 60°C and continuous heating at 0.1°C per second to 95°C. We reported CT values if the melting temperature (Tm) ranged between 70°C and 72.5°C.
We validated the linearity of the tcdB PCR using tcdB cloned into plasmid pHIS1525 (MoBiTec Inc, Boca Raton, Florida) as a DNA standard. Linear regression allows calculations of C. difficile burden using the following formula: C. difficile concentration (colony-forming units [CFU]/mL) = 7000 × 10(CT-32.4)/−5.2 (data not shown). However, we chose to report CT values because their distribution is symmetrical, whereas expression as CFU/mL skews the data. Control experiments demonstrated that incubating the stools for up to 24 hours at room temperature did not affect tcdB DNA quantification or cytokine messenger RNA (mRNA) relative abundance (data not shown).
Statistical Analysis
We used a nonparametric Wilcoxon rank-sum test to assess the significance of correlations of mRNA transcripts and tcdB CT with outcomes. We used the χ2test and, when appropriate, Fisher exact test, for categorical data comparison, as well as for dichotomized IL-8 and lactoferrin protein results. We fitted multivariate binary logistic regression models to determine the predictors of poor clinical outcomes, assigned a priori. We used a backward selection process to find the significant predictors. Variables offered in the regression models included high CXCL-5 mRNA (ΔCT CXCL-5 mRNA >0), high IL-8 mRNA (ΔCT IL-8 mRNA >0), positive IL-8 protein, the 3 severity scores, white blood cell count (WBC) >15 000 cells/µL, and serum creatinine concentration ≥1.5 times baseline (all defined as binomial data), tcdB CT and age (defined as continuous variables). We used Kaplan-Meier survival (log-rank test) analysis to interrogate the statistical significance of the time to diarrhea resolution in patients with elevated fecal cytokines on admission compared to those with no elevation. Proportional hazard assumptions were assessed visually from the plots. Repeated measures mixed models and generalized linear models assessed daily fecal cytokines and C. difficile bacterial burden in relation to diarrhea and day of therapy adjusting for within subject correlation. Two-sided P values of <.05 were considered significant.
RESULTS
Study Population
Of 250 inpatients identified with CDI, we excluded 54 because of altered consciousness or dementia and no availability of a family member for consent by proxy, and 49 because they were discharged before enrollment. Of the 147 patients approached, 131 consented and were enrolled. Stools were available on 121. We excluded 1 patient who had consistently negative tcdB PCR in our laboratory from all samples provided. Demographic characteristics of the final cohort of 120 patients are presented in Table 1. We excluded from analysis the initial tcdB CT values on 6 patients because the tcdB PCR did not amplify adequately on those initial samples, whereas subsequent samples from these patients amplified as expected.
Table 1.
Baseline Characteristics of the Study Patients
| Variable | Subjects (N = 120) |
|---|---|
| Age, y, mean ± SD | 61.1 ± 14.9 |
| Male sex | 64 (53) |
| Cancer | 54 (45) |
| Diagnosis at admission to the hospital | |
| Malignancy | 36 (30) |
| Diarrhea | 25 (21) |
| Infections | 15 (12) |
| Gastrointestinal disease | 12 (10) |
| Cardiovascular disease | 12 (10) |
| Renal disease | 5 (4) |
| Respiratory disease | 5 (4) |
| Surgery | 5 (4) |
| Other | 5 (4) |
| CDI in the past 90 d | 15 (12) |
| Hospitalization in the past 30 d | 68 (57) |
| Antibiotic use in the past 90 d | 112 (93) |
| Cephalosporins | 75 (62) |
| Carbapenems | 22 (18) |
| Other β-lactams | 27 (22) |
| Vancomycin, intravenous | 52 (43) |
| Clindamycin | 4 (3) |
| Fluoroquinolones | 41 (34) |
| Macrolides | 10 (8) |
| Metronidazole | 8 (7) |
| Immunosuppression | 62 (52) |
| Steroids in the past 90 d | 58 (48) |
| Other immunosuppressants in the past 90 d | 48 (40) |
| Acid suppression in the past 90 d | 99 (82) |
| Proton pump inhibitors | 87 (72) |
| H2 blockers | 28 (23) |
| Nonsteroidal anti-inflammatory drugs in the past 90 d | 17 (14) |
| Fecal IL-8 mRNA ΔCT, median (IQR) | 1.03 (−1.29, 3.68) |
| Fecal CXCL-5 mRNA ΔCT, median (IQR) | −2.84 (−5.63, 0.75) |
| Positive fecal IL-8 protein, concentration >0.75 ng/mL | 61 (51) |
| Positive fecal lactoferrin protein, concentration >7.25 μg/mL | 72 (60) |
Data are No. (%) unless otherwise specified.
Abbreviations: ΔCT, difference of cycle thresholds between internal control and cytokine tested; CDI, Clostridium difficile infection; CT, cycle threshold; IL-8, interleukin 8; IQR, interquartile range; mRNA, messenger RNA; SD, standard deviation.
Of these 120 patients, 19 (16%) were started on oral vancomycin and 101 (84%) on metronidazole. However, vancomycin was added to, or replaced, metronidazole in 33 (33%) of those patients started on metronidazole, suggesting a judgment of treatment failure by their physicians. Diarrhea persisted for at least 5 days in 62 (52%) patients, and duration was not determinable for 12 (10%) patients who were not available for follow-up. Eleven (9%) patients had severe disease (Table 2). There was no difference in clinical presentation, CDI therapy, diarrhea persistence, or time to diarrhea resolution between immunosuppressed (receiving steroids or other immunosuppressives) and nonimmunosuppressed patients.
Table 2.
Outcomes of Patients With Clostridium difficile Infection
| Outcome | No. (%) |
|---|---|
| Clostridium difficile treatment at diagnosis | |
| Metronidazole (intravenous or oral) | 101 (84) |
| Vancomycin oral | 19 (16) |
| Switched from metronidazole to vancomycin | 33/100 (33) |
| Diarrhea lasting ≥5 d after initiation of therapy | 62 (52) |
| Creatinine ≥1.5 × baseline | 8 (7) |
| WBC count >15 × 103 cells/µL | 17 (14) |
| Severe disease: admission to the intensive care unit, toxic megacolon, colectomy or death | 11 (9) |
| HINES VA score severe (≥3) at study admission | 13 (11) |
| UPMC1 score severe (≥2) at study admission | 35 (29) |
| CSI score severe (≥2) at study admission | 28 (23) |
Abbreviations: WBC, white blood cell.
Cytokine Levels and Bacterial Burden
Fecal CXCL-5 mRNA, IL-8 mRNA and IL-8 protein levels at the time of CDI diagnosis correlated with persistent diarrhea (P = .03, P = .005, and P = .05, respectively) as well as treatment with vancomycin (P = .007, P = .006, and P = .02, respectively). We found no difference between cytokine levels in patients started on vancomycin and those who had vancomycin added to their therapy during their illness course. Interestingly, tcdB CT at the time of diagnosis did not correlate with diarrhea persistence or choice of CDI therapy (Figure 1). We identified 15 patients with comorbidities that might have complicated assessment of the diarrhea (inflammatory bowel disease, 3; gastrointestinal graft-vs-host disease, 4; irritable bowel syndrome, 2; pancreatitis, 4; intestinal bleeding and short bowel syndrome, 1 each). After excluding these patients, fecal CXCL-5 mRNA and IL-8 mRNA remained associated with persistent diarrhea (P = .047 and P = .029, respectively) and therapy with vancomycin (P = .028 and P = .039, respectively).
Figure 1.
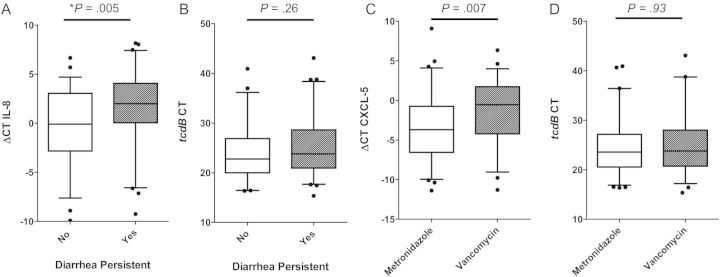
Initial cytokines and tcdB cycle threshold (CT) in patients with Clostridium difficile infection (CDI). A, Initial interleukin 8 ΔCT levels in patients with diarrhea persistent 5 days after starting CDI therapy (dashed bar) are higher than those whose diarrhea resolved (clear bar). B, Initial tcdB CT values in patients with persistent diarrhea 5 days after starting CDI therapy (dashed bar) and patients with resolved diarrhea (clear bar) do not differ. C, Initial CXCL-5 ΔCT levels in patients treated with metronidazole (clear bar) are lower than those started on vancomycin or had vancomycin added to their therapy during their hospitalization (dashed bar). D, Initial tcdB CT values in patients treated with metronidazole (clear bar) and patients started on vancomycin or had vancomycin added to their therapy during their hospitalization (dashed bar) do not differ. Boxes represent median ± 25th–75th percentiles; whiskers represent 5th–95th percentiles; dots represent outliers. P, P values of the 2-tailed Wilcoxon rank-sum test; *P < .05. Abbreviations: ΔCT, difference of cycle thresholds between internal control and cytokine tested; CT, cycle threshold; IL-8, interleukin 8.
CXCL-5 and IL-8 mRNA levels were higher in nonimmunosuppressed patients with CDI compared to those with immunosuppression (P = .015 and P = .016, respectively). Interestingly, CXCL-5 mRNA was only associated with persistent diarrhea and vancomycin therapy in nonimmunosuppressed patients (P = .017 and P = .001, respectively). Patients with CDI in the prior 90 days had more frequent bowel movements (P = .045) and were more likely to receive vancomycin therapy during their hospitalization (P < .001). They also had higher relative abundances of CXCL-5 and IL-8 mRNA on admission than those who had no history of CDI (P = .011 and P = .028, respectively).
Initial fecal IL-8 mRNA and lactoferrin protein concentrations were higher in patients with a severe HINES VA score (P = .003 and P = .002, respectively); IL-8 protein concentrations were elevated in patients with a severe UPMC1 score (P = .015). Fecal bacterial burden at the time of diagnosis did not correlate with any severity score or any other clinical measure such as number of stools, Bristol stool score, or pain. Although UPMC1 scores correlated with persistent diarrhea and vancomycin therapy (P = .02 and P = .05, respectively), and HINES VA correlated with vancomycin therapy (P = .05), inflammatory markers were much more sensitive than these scores in identifying patients who failed treatment (Supplementary Table 2). HINES VA, CSI, and leukocytosis (WBC >15 000 cells/µL) correlated with the presence of severe disease (P = .002, P = .02, and P = .03, respectively).
A backward logistic regression model of predictors of diarrhea persistence identified elevated CXCL-5 mRNA (odds ratio [OR] = 3.705; 95% confidence interval [CI], 1.124–12.215; P = .031), severe UPMC1 (OR = 4.223; 95% CI, 1.219–14.631; P = .023), and WBC >15 000 cells/µL (OR = 0.234; 95% CI, .057–.962; P = .044). When patients on immunosuppressive therapy were excluded, the only predictor of persistent diarrhea was an elevated CXCL-5 mRNA (OR = 4.33; 95% CI, .937–20.03; P = .061), although this did not reach statistical significance. The only variable that correlated with vancomycin therapy was an elevated CXCL-5 mRNA (OR = 5.49; 95% CI, 1.86–16.16; P = .002). Predictors of severe disease included a severe HINES VA score (OR = 14.44; 95% CI, 2.6–80.24; P = .002) and creatinine ≥1.5 × baseline (OR = 8.67; 95% CI, 1.16–64.5; P = .035).
Time to diarrhea resolution was longer in patients with high CXCL-5 mRNA or IL-8 mRNA at study entry (log-rank test, P = .009 and P = .03, respectively; Figure 2). A subanalysis revealed that these findings were only applicable to nonimmunosuppressed patients (Supplementary figure). When patients were segregated into quartiles based on their initial tcdB CT, there was no difference in the time to diarrhea resolution among the 4 groups (Figure 2). We did not find a correlation between the number of symptomatic days prior to CDI diagnosis and fecal inflammatory markers, suggesting that the length of symptoms prior to treatment initiation is not related to the level of intestinal inflammation.
Figure 2.

Time to diarrhea resolution in patients with elevated markers on admission compared to those with low markers. A, Time to diarrhea resolution is more prolonged in patients with high CXCL-5 (ΔCT CXCL-5 messenger RNA >0) at diagnosis (CXCL-5+, green line; median 12 days) compared to those with low CXCL-5 mRNA levels (CXCL-5−, blue line; median 4 days). B, Time to diarrhea resolution is more prolonged in patients with high interleukin 8 (IL-8) mRNA (ΔCT IL-8 mRNA >0) at diagnosis (IL-8+, green line; median 6 days) compared to those with negative IL-8 mRNA levels (IL-8−, blue line; median 4 days). C, Kaplan-Meier survival curve comparing patients with a tcdB CT in the lower quartile (CT<20.7, red line) to those with tcdB CT in the second quartile (20.71< CT <23.6, blue line), the third quartile (23.61< CT < 27.1, orange line), and the higher quartile (CT > 27.11, green line). There was no difference in the time to diarrhea resolution among the 4 groups. “+” represents censored values, and P is the P value of the log-rank (Mantel-Cox) test. *P < .05. Abbreviations: ΔCT, difference of cycle thresholds between internal control and cytokine tested; CDI, Clostridium difficile infection; CT, cycle threshold; IL-8, interleukin 8; mRNA, messenger RNA.
When examining the disease progression, CXCL-5 mRNA levels did not change significantly during therapy, whereas IL-8 mRNA levels decreased after starting therapy (P = .004). Elevated CXCL-5 and IL-8 mRNA levels correlated with diarrhea throughout the patients’ hospital course (P = .04 and P = .03, respectively). As expected, bacterial burden decreased significantly with day of therapy (P < .001), independent of the therapy used (P = .38), and at no point correlated with diarrhea (P = .40). Figure 3 delineates daily cytokines and tcdB CT levels in the different treatment groups. Figure 4 compares these levels in patients with persistent and resolved diarrhea.
Figure 3.
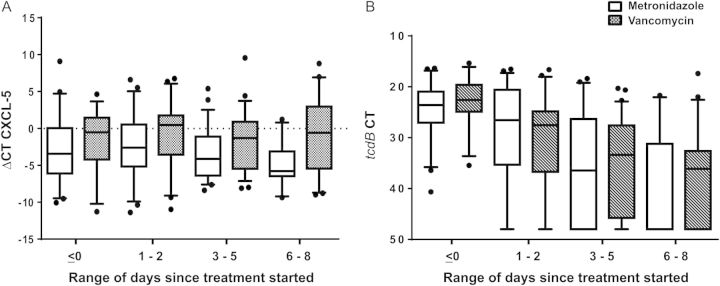
Daily CXCL-5 and tcdB cycle threshold (CT) levels in patients treated with vancomycin compared to those treated with metronidazole. A, CXCL-5 ΔCT levels in patients treated with vancomycin (dashed bars) are persistently higher than levels of those treated with metronidazole (clear bars). Levels do not decrease significantly with time. B, tcdB CT values in patients treated with vancomycin (dashed bars) are similar to those of patients treated with metronidazole (clear bars). Bacterial burden drops at the same rate with both therapies. Boxes represent median ± 25th–75th percentiles; whiskers represent 5th–95th percentiles; dots represent outliers. Abbreviations: ΔCT, difference of cycle thresholds between internal control and cytokine tested; CT, cycle threshold.
Figure 4.
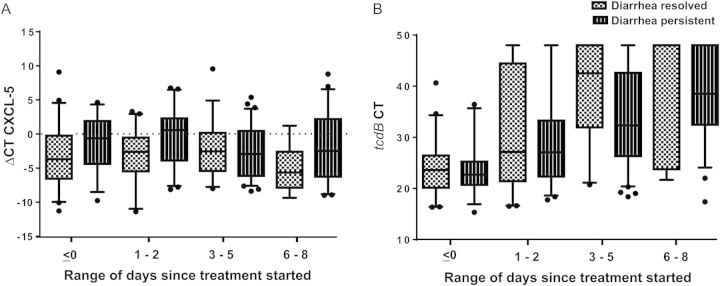
Daily CXCL-5 and tcdB cycle threshold (CT) levels in patients with persistent diarrhea compared to those whose diarrhea resolved with 5 days of therapy. A, CXCL-5 ΔCT levels in patients with persistent diarrhea (dashed bars) are persistently higher than levels of those with resolved diarrhea (dotted bars). Levels do not decrease significantly with time. B, tcdB CT values in patients with persistent diarrhea (dashed bars) are similar to those of patients with resolved diarrhea (dotted bars). Bacterial burden drops at the same rate in the 2 groups. Boxes represent median ± 25th–75th percentiles; whiskers represent 5th–95th percentiles; dots represent outliers. Abbreviations: ΔCT, difference of cycle thresholds between internal control and cytokine tested; CT, cycle threshold.
DISCUSSION
We identified biologically relevant markers that correlate with disease severity and outcome in patients with CDI. Interestingly, bacterial burden at the time of CDI diagnosis did not correlate with clinical measures or outcome, and the rate at which it decreased on therapy did not differ between those with persistent or resolved diarrhea. Furthermore, we found no statistically significant difference in the microbiologic response of C. difficile to metronidazole and vancomycin between these groups of subjects. In contrast, elevated fecal inflammatory makers at the time of CDI diagnosis, particularly in patients on no immunosuppressive therapy, were associated with poor outcomes, and remained elevated in those with persistent disease.
Our data indirectly contradict those of Al-Nassir et al, who reported that fecal C. difficile concentration was higher in patients treated with metronidazole, attributing the failure of metronidazole therapy to a slower and less consistent microbiological response than oral vancomycin [30]. Although other reports describe higher rates of treatment failure with metronidazole [8], a recent Cochrane systematic review did not identify superiority of vancomycin over metronidazole in CDI symptomatic or bacteriologic cure [31].
Clostridum difficile is a major healthcare burden; its case severity has increased in recent years [5, 8], and its costs are staggering [6]. Despite appropriate antibiotic therapy, symptomatic cure is not achieved in 20%–30% of cases [31], and up to 22% of patients with CDI remain symptomatic beyond 10 days of treatment [7]. Efforts have been directed into identifying patients at risk of severe disease [9, 11–15]; however, few studies have attempted to identify factors associated with treatment failure or symptom duration. In the current study, fecal cytokines were more sensitive in identifying patients at risk of persistent diarrhea and correlated better with treatment failure than did any clinical parameter or score. It is interesting to note that HINES VA severity score, although designed to identify patients at risk of treatment failure [16], better predicted severe disease than persistence of symptoms in our cohort. It was surprising to find that an elevated WBC seemed to be protective against persistent diarrhea, because it is one marker used to predict severe disease [9, 16, 32]. This might be attributable to the high proportion of patients with immunodeficiencies in our study. When we excluded these patients from the analysis, the only predictor of persistent diarrhea was a high CXCL-5 mRNA level.
Whether or not strain type correlates with severe disease in CDI is unresolved. Initial reports suggested that the NAP1 strain is associated with more severe outcomes [33, 34], whereas some recent reports dispute that finding [35–38]. Regardless of which strain accounts for the patient's symptoms, antimicrobial options are the same, and therefore delineating the strain type has limited clinical utility for individual patients at this time. Our results suggest that the character of the intestinal inflammatory response determines clinical severity, and that initial host response should be evaluated prospectively as a clinically relevant prognostic parameter and potentially modifiable contributor to disease outcome.
CDI is characterized by tissue injury and an acute intestinal inflammatory response characterized by neutrophil infiltration [22]. Both pathogen and host play a major role in the disease pathogenesis. Clostridium difficile toxins cause direct injury to the intestinal epithelium, which is associated with a robust host inflammatory response with cytokine release leading to neutrophil activation and recruitment and subsequently to a worsening intestinal injury [20–22]. Inhibiting this acute inflammatory response might attenuate intestinal injury [22]. Indeed, anti-inflammatory agents diminish intestinal injury in experimental CDI [22, 39, 40].
In summary, fecal inflammatory markers at diagnosis of CDI correlate with outcome. Such profiling at the time of diagnosis of CDI might identify patients at risk for treatment failure. Additionally, our findings suggest that it is not bacterial persistence that causes prolonged CDI symptoms, but rather a sustained host response to a waning population of C. difficile. Once the inflammatory response is initiated, the choice of antibiotic does not appear to alter the illness course. Strategies to reduce the bacterial burden are unlikely to achieve a better response in acute disease than the existing antimicrobials. Instead, mitigation of the host response might be a more fruitful strategy for such patients.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online (http://cid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank Malick Ndao, MD, and Nurmohammad Shaikh, PhD, for providing advice on cytokine PCR methods; Carey-Ann Burnham, PhD, for providing advice on tcdB PCR methods; Gregory Storch, MD, and Erik Dubberke, MD, MSPH, for advice on the study design; and William Shannon, PhD, for his help with the statistical analysis.
Financial support. This work was supported by a Washington University–Pfizer Biomedical Agreement and by funds from the Midwest Regional Centers of Excellence in Biodefense (U54-AI057160) to D. B. H. P. I. T. is supported by the Melvin E. Carnahan Professorship in Pediatrics and by the Washington University Digestive Diseases Research Core Center (Biobank grant number 5P30 DK052574).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.McFarland LV. Antibiotic-associated diarrhea: epidemiology, trends and treatment. Future Microbiol. 2008;3:563–78. doi: 10.2217/17460913.3.5.563. [DOI] [PubMed] [Google Scholar]
- 2.Johnson S, Gerding DN. Clostridium difficile–associated diarrhea. Clin Infect Dis. 1998;26:1027–34. doi: 10.1086/520276. quiz 35–6. [DOI] [PubMed] [Google Scholar]
- 3.Dubberke ER, Haslam DB, Lanzas C, et al. The ecology and pathobiology of Clostridium difficile infections: an interdisciplinary challenge. Zoonoses Public Health. 2011;58:4–20. doi: 10.1111/j.1863-2378.2010.01352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dubberke ER, Reske KA, Yan Y, Olsen MA, McDonald LC, Fraser VJ. Clostridium difficile–associated disease in a setting of endemicity: identification of novel risk factors. Clin Infect Dis. 2007;45:1543–9. doi: 10.1086/523582. [DOI] [PubMed] [Google Scholar]
- 5.DuPont HL, Garey K, Caeiro JP, Jiang ZD. New advances in Clostridium difficile infection: changing epidemiology, diagnosis, treatment and control. Current Opinion Infect Dis. 2008;21:500–7. doi: 10.1097/QCO.0b013e32830f9397. [DOI] [PubMed] [Google Scholar]
- 6.Lucado J, Gould C, Elixhauser A. Clostridium difficile infections (CDI) in hospital stays, 2009: statistical brief 124. Healthcare Cost and Utilization Project (HCUP) statistical briefs. Rockville, MD. 2009. [PubMed]
- 7.Musher DM, Aslam S, Logan N, et al. Relatively poor outcome after treatment of Clostridium difficile colitis with metronidazole. Clin Infect Dis. 2005;40:1586–90. doi: 10.1086/430311. [DOI] [PubMed] [Google Scholar]
- 8.Vardakas KZ, Polyzos KA, Patouni K, Rafailidis PI, Samonis G, Falagas ME. Treatment failure and recurrence of Clostridium difficile infection following treatment with vancomycin or metronidazole: a systematic review of the evidence. Int J Antimicrob Agents. 2012;40:1–8. doi: 10.1016/j.ijantimicag.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Belmares J, Gerding DN, Tillotson G, Johnson S. Measuring the severity of Clostridium difficile infection: implications for management and drug development. Expert Rev Anti Infect Ther. 2008;6:897–908. doi: 10.1586/14787210.6.6.897. [DOI] [PubMed] [Google Scholar]
- 10.Teasley DG, Gerding DN, Olson MM, et al. Prospective randomised trial of metronidazole versus vancomycin for Clostridium-difficile-associated diarrhoea and colitis. Lancet. 1983;2:1043–6. doi: 10.1016/s0140-6736(83)91036-x. [DOI] [PubMed] [Google Scholar]
- 11.Fujitani S, George WL, Murthy AR. Comparison of clinical severity score indices for Clostridium difficile infection. Infect Control Hosp Epidemiol. 2011;32:220–8. doi: 10.1086/658336. [DOI] [PubMed] [Google Scholar]
- 12.Louie TJ, Peppe J, Watt CK, et al. Tolevamer, a novel nonantibiotic polymer, compared with vancomycin in the treatment of mild to moderately severe Clostridium difficile-associated diarrhea. Clin Infect Dis. 2006;43:411–20. doi: 10.1086/506349. [DOI] [PubMed] [Google Scholar]
- 13.Lungulescu OA, Cao W, Gatskevich E, Tlhabano L, Stratidis JG. CSI: a severity index for Clostridium difficile infection at the time of admission. J Hosp Infect. 2011;79:151–4. doi: 10.1016/j.jhin.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 14.McEllistrem MC, Carman RJ, Gerding DN, Genheimer CW, Zheng L. A hospital outbreak of Clostridium difficile disease associated with isolates carrying binary toxin genes. Clin Infect Dis. 2005;40:265–72. doi: 10.1086/427113. [DOI] [PubMed] [Google Scholar]
- 15.Rubin MS, Bodenstein LE, Kent KC. Severe Clostridium difficile colitis. Dis Colon Rectum. 1995;38:350–4. doi: 10.1007/BF02054220. [DOI] [PubMed] [Google Scholar]
- 16.Belmares J, Gerding DN, Parada JP, Miskevics S, Weaver F, Johnson S. Outcome of metronidazole therapy for Clostridium difficile disease and correlation with a scoring system. J Infect. 2007;55:495–501. doi: 10.1016/j.jinf.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 17.Sebaihia M, Wren BW, Mullany P, et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet. 2006;38:779–86. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- 18.Loo VG, Bourgault AM, Poirier L, et al. Host and pathogen factors for Clostridium difficile infection and colonization. N Engl J Med. 2011;365:1693–703. doi: 10.1056/NEJMoa1012413. [DOI] [PubMed] [Google Scholar]
- 19.Hatheway CL. Toxigenic clostridia. Clin Microbiol Rev. 1990;3:66–98. doi: 10.1128/cmr.3.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hippenstiel S, Soeth S, Kellas B, et al. Rho proteins and the p38-MAPK pathway are important mediators for LPS-induced interleukin-8 expression in human endothelial cells. Blood. 2000;95:3044–51. [PubMed] [Google Scholar]
- 21.Ishida Y, Maegawa T, Kondo T, et al. Essential involvement of IFN-gamma in Clostridium difficile toxin A-induced enteritis. J Immunol. 2004;172:3018–25. doi: 10.4049/jimmunol.172.5.3018. [DOI] [PubMed] [Google Scholar]
- 22.Kelly CP, Kyne L. The host immune response to Clostridium difficile. J Med Microbiol. 2011;60(Pt 8):1070–9. doi: 10.1099/jmm.0.030015-0. [DOI] [PubMed] [Google Scholar]
- 23.Savidge TC, Pan WH, Newman P, O'Brien M, Anton PM, Pothoulakis C. Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine. Gastroenterology. 2003;125:413–20. doi: 10.1016/s0016-5085(03)00902-8. [DOI] [PubMed] [Google Scholar]
- 24.Steiner TS, Flores CA, Pizarro TT, Guerrant RL. Fecal lactoferrin, interleukin-1beta, and interleukin-8 are elevated in patients with severe Clostridium difficile colitis. Clin Diagn Lab Immunol. 1997;4:719–22. doi: 10.1128/cdli.4.6.719-722.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wren MW, Kinson R, Sivapalan M, Shemko M, Shetty NR. Detection of Clostridium difficile infection: a suggested laboratory diagnostic algorithm. Br J Biomed Sci. 2009;66:175–9. doi: 10.1080/09674845.2009.11730269. [DOI] [PubMed] [Google Scholar]
- 26.Walz A, Burgener R, Car B, Baggiolini M, Kunkel SL, Strieter RM. Structure and neutrophil-activating properties of a novel inflammatory peptide (ENA-78) with homology to interleukin 8. J Exp Med. 1991;174:1355–62. doi: 10.1084/jem.174.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Z'Graggen K, Walz A, Mazzucchelli L, Strieter RM, Mueller C. The C-X-C chemokine ENA-78 is preferentially expressed in intestinal epithelium in inflammatory bowel disease. Gastroenterology. 1997;113:808–16. doi: 10.1016/s0016-5085(97)70175-6. [DOI] [PubMed] [Google Scholar]
- 28.Lewis SJ, Heaton KW. Stool form scale as a useful guide to intestinal transit time. Scand J Gastroenterol. 1997;32:920–4. doi: 10.3109/00365529709011203. [DOI] [PubMed] [Google Scholar]
- 29.Wroblewski D, Hannett GE, Bopp DJ, et al. Rapid molecular characterization of Clostridium difficile and assessment of populations of C. difficile in stool specimens. J Clin Microbiol. 2009;47:2142–8. doi: 10.1128/JCM.02498-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Nassir WN, Sethi AK, Nerandzic MM, Bobulsky GS, Jump RL, Donskey CJ. Comparison of clinical and microbiological response to treatment of Clostridium difficile-associated disease with metronidazole and vancomycin. Clin Infect Dis. 2008;47:56–62. doi: 10.1086/588293. [DOI] [PubMed] [Google Scholar]
- 31.Nelson RL, Kelsey P, Leeman H, et al. Antibiotic treatment for Clostridium difficile-associated diarrhea in adults. Cochrane Database Syst Rev. 2011:CD004610. doi: 10.1002/14651858.CD004610.pub4. [DOI] [PubMed] [Google Scholar]
- 32.Cohen SH, Gerding DN, Johnson S, et al. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA) Infect Control Hosp Epidemiol. 2010;31:431–55. doi: 10.1086/651706. [DOI] [PubMed] [Google Scholar]
- 33.Miller M, Gravel D, Mulvey M, et al. Health care-associated Clostridium difficile infection in Canada: patient age and infecting strain type are highly predictive of severe outcome and mortality. Clin Infect Dis. 2010;50:194–201. doi: 10.1086/649213. [DOI] [PubMed] [Google Scholar]
- 34.Pepin J. Vancomycin for the treatment of Clostridium difficile infection: for whom is this expensive bullet really magic? Clin Infect Dis. 2008;46:1493–8. doi: 10.1086/587656. [DOI] [PubMed] [Google Scholar]
- 35.Cloud J, Noddin L, Pressman A, Hu M, Kelly C. Clostridium difficile strain NAP-1 is not associated with severe disease in a nonepidemic setting. Clin Gastroenterol Hepatol. 2009;7:868–73 e2. doi: 10.1016/j.cgh.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 36.Morgan OW, Rodrigues B, Elston T, et al. Clinical severity of Clostridium difficile PCR ribotype 027: a case-case study. PloS One. 2008;3:e1812. doi: 10.1371/journal.pone.0001812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sirard S, Valiquette L, Fortier LC. Lack of association between clinical outcome of Clostridium difficile infections, strain type, and virulence-associated phenotypes. J Clin Microbiol. 2011;49:4040–6. doi: 10.1128/JCM.05053-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walk ST, Micic D, Jain R, et al. Clostridium difficile ribotype does not predict severe infection. Clin Infect Dis. 2012;55:1661–8. doi: 10.1093/cid/cis786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim H, Kokkotou E, Na X, et al. Clostridium difficile toxin A-induced colonocyte apoptosis involves p53-dependent p21(WAF1/CIP1) induction via p38 mitogen-activated protein kinase. Gastroenterology. 2005;129:1875–88. doi: 10.1053/j.gastro.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 40.Warny M, Keates AC, Keates S, et al. p38 MAP kinase activation by Clostridium difficile toxin A mediates monocyte necrosis, IL-8 production, and enteritis. J Clin Invest. 2000;105:1147–56. doi: 10.1172/JCI7545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.