

Abstract
Aims
Rho kinases (ROCKs) are the best characterized effectors of the small G-protein RhoA, and play a role in enhanced vasoconstriction in animal models of congestive heart failure (CHF). This study examined if ROCK activity is increased in CHF and how it is associated with the outcome in CHF.
Methods and results
Patients admitted with CHF (n =178), disease controls (n =31), and normal subjects (n =30) were studied. Baseline ROCK activity was measured by phosphorylation of themyosin-binding subunit in peripheral leucocytes. The patients were followed up for 14.4 ± 7.2 months (range 0.5–26 months) or until the occurrence of cardiac death. The ROCK activity in CHF patients (2.93 ± 0.87) was significantly higher than that of the disease control (2.06 ± 0.38, P < 0.001) and normal control (1.57 ± 0.43, P < 0.001) groups. Similarly, protein levels of ROCK1 and ROCK2 as well as the activity of RhoA in CHF were significantly higher than in disease controls and normal controls (all P < 0.05). Dyspnoea at rest (β =0.338, P < 0.001), low left ventricular ejection fraction (β = –0.277, P < 0.001), and high creatinine (β =0.202, P =0.006) were independent predictors of the baseline ROCK activity in CHF. Forty-five patients died within 2 years follow-up (25.3%). Combining ROCK activity and N-terminal pro brain natriuretic peptide (NT-proBNP) had an incremental value (log rank χ2 =11.62) in predicting long-term mortality when compared with only NT-proBNP (log rank χ2 =5.16, P < 0.05).
Conclusion
ROCK activity is increased in CHF and it might be associated with the mortality in CHF. ROCK activity might be a complementary biomarker to CHF risk stratification.
Keywords: Rho kinases, Congestive heart failure, Mortality
Introduction
Cardiac hypertrophy leading to congestive heart failure (CHF) is a major cause of morbidity and mortality worldwide. Cardiac hypertrophy is an adaptive response of the heart to pressure or volume overload. This initial adaptative response becomes maladaptative, switching the heart from a compensated to decompensated state and finally leading to heart failure. The molecular response to pressure overload is complex and may include modulation of various intracellular signal pathways. Furthermore, pressure overload leads to the secretion of vasoactive peptides, such as angiotensin II and endothelin 1, which play pivotal roles in the induction of these hypertrophic responses.1 Recent studies suggest that the hypertrophic process is also mediated, in part, by an increase in myocardial oxidative stress.2 In the myocardium, Ras, Rho, and Rac are involved in the hypertrophic response.3 On the other hand, vasoconstrictor neurohumoral systems, such as the renin–angiotensin–aldosterone system and the sympathetic nervous system, are important in the pathophysiology of CHF.4 Studies have also revealed the importance of the Rho proteins and their associated kinases, Rho kinases (ROCKs), in the regulation of the vascular tone of various blood vessels, including the renal vasculature.5
Rho-associated kinase is a serine/threonine kinase that mediates some of the downstream signalling of RhoA.6 Currently, there are two isoforms of ROCK, ROCK1 and ROCK2. Pharmacological inhibition of ROCK suggests that it plays an important role in the pathogenesis of diverse cardiovascular diseases such as cerebral and coronary vasospasm, hypertension, vascular inflammation, ischaemia/reperfusion injury,6 left ventricular remodelling after myocardial infarction,6 and atrial natriuretic factor expression.6 Activation of ERK1/2 and of the cardiac transcription factor GATA-4 identify them as downstream nuclear mediators of ROCKs during myocardial cell hypertrophy.7
These findings suggest that ROCK-dependent signalling pathways may potentially contribute to the mechanism of systemic and renal vasoconstriction and cardiac hypertrophy in CHF. Indeed, cardiac-specific overexpression of RhoA in mice resulted in a lethal form of heart failure, characterized by atrial enlargement, conduction defects, contractile failure, and generalized oedema.8 Similarly, Kobayashi et al.9 demonstrated the importance of ROCK pathways in the induction of cardiac dysfunction and remodelling in the failing hearts of Dahl salt-sensitive rats with CHF, and Kishi et al.10 proved that ROCK is involved in the increased forearm vascular resistance and impaired vasodilatation in patients with heart failure. However, there is little information on ROCK activity in patients with CHF and, therefore, the purpose of this study was to clarify the involvement of ROCK in CHF patients and whether ROCK activity predicts long-term mortality.
Methods
Study subjects
Consecutive patients (52% men; aged 74 ± 12 years) admitted to a university teaching hospital (the Prince of Wales Hospital in Hong Kong) for CHF were enrolled between December 2007 and January 2009. A total of 178 patients were recruited. CHF was diagnosed based on the ACC/AHA guideline.11 All the patients were followed up until 1 February 2010 (14.4 ±7.2, 0.5–26 months) or until the occurrence of cardiac death. Sixty-one volunteers were subdivided into the disease control group (n =31) (76% men; aged 69 ±8 years) and the healthy control group (n =30) (67% men; aged 67 ±9 years) depending on the presence or absence of hypertension or smoking status which have been proved to influence ROCK activity.12 All disease control subjects had normal epicardial coronary arteries on angiography. The effect of statins was statistically adjusted. Written informed consentwas obtained from all subjects. The study was approved by the Institution's Ethics Committee.
Analysis of Rho kinase activity, ROCK1, ROCK2, and RhoA activity
Leucocytes were isolated from 10 mL of peripheral blood at admission following a validated and standardized protocol.13 The leucocytes were frozen and stored at –80°C until all samples were collected. The ROCK assays were performed on all leucocyte samples at the same time. The resulting samples were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and bound proteins were detected by immunoblotting. The samples were analysed using the rabbit anti-myosin-binding subunit (MBS) polyclonal antibody (Covance, Princeton, NJ, USA), anti-ROCK1 monoclonal antibody, anti-ROCK2 monoclonal antibody (BD Biosciences, San Jose, CA, USA), anti-actin monoclonal antibody (Sigma), anti-phospho-specific Ser854-MBS13 polyclonal antibody [kindly provided by Professor J.K. Liao (Boston, MA)], or anti-RhoA antibody (Bioword kit). Rho kinase activity was expressed as the ratio of phosphorylation levels of the MBS (pMBS) in each sample per pMBS in each positive control divided by the MBS in each sample per MBS in each positive control. RhoA activity was determined by glutathione S-transferase (GST) pull-down assay. RhoA-GTP was collected using GST–Rhotekin RBD (Cytoskeleton, Inc. catalogue number BK036).
Statistical analysis
Categorical variables are expressed as percentages of the corresponding population and continuous variables as means ±standard deviation. Values of P < 0.05 were considered to indicate statistical significance. One-way analysis of variance (ANOVA) was used for comparison of the mean values of continuous variables among groups, and post-hoc analysis was performed by Scheffe's test to examine for intergroup differences. ROCK activity was adjusted for age between different groups as healthy control subjects were inevitably younger than other diseased controls. Univariate linear regression (Pearson and Spearman's correlation) models were used to assess the relationship between parametric clinical variables and ROCK activity. All variables with a significant association but which did not exhibit excessive collinearity with each other were evaluated for inclusion in a stepwise multiple regression analysis model using ROCK activity as the dependent variable. Receiver operating characteristics (ROC) analysis was performed to determine the best cut-off value of ROCK activity and N-terminal pro brain natriuretic peptide (NT-proBNP) for following up outcomes in the patient cohort. Multivariate Cox regression analysis was performed to investigate for independent predictors of death outcomes. Event-free survival (days alive) was estimated by the Kaplan–Meier method and compared between groups by the log-rank test. All statistical analyses were conducted with the SPSS statistical package for Vista version 15.0 (SPSS Inc., Chicago, IL, USA).
Results
Baseline characteristics
Baseline clinical features and biochemical profiles are summarized in Table 1. A comparison of baseline clinical and biochemical parameters between the heart failure and disease control groups did not show any statistically significant differences in terms of age, gender, smoking status, and medical history of hypertension. The normal controls showed no risk factors, and were aged 67 ±9 years, which matched with the heart failure (74 ±12 years) and disease controls (69 ±8 years). Compared with disease and normal controls, the heart failure patients had a lower left ventricular ejection fraction (LVEF) (47.5 ±14.3% vs. 64.2 ±4.1% and 68.5 ±3.5%, P < 0.001), total cholesterol (TC) (4.3 ±1.3 mmol/L vs. 4.8 ±0.8 and 5.1 ±0.5 mmol/L, P =0.003) and LDL cholesterol (LDL-C) (2.4 ±1.1 mmol/L vs. 2.9 ±1.0 and 3.0 ±0.3 mmol/L, P =0.011). Heart failure patients had higher systolic blood pressure (SBP) (148 ±31 mmHg vs. 129 ±22 mmHg, P =0.002), fasting glucose (6.5 ±1.8 mmol/L vs. 5.2 ±0.3, mmol/L, P =0.002), and white blood cells (WBC) (9.2 ±4.0 ×109/L vs. 5.5 ±1.8 ×109/L, P =0.004) than normal controls.
Table 1.
Baseline characteristics of congestive heart failure, disease control, and normal control subjects
| Baseline characteristic | CHF (n = 178) | Disease controls (n = 31) | Normal controls (n = 30) | ANOVA P-value |
|---|---|---|---|---|
| Age (years) | 74 ±12 | 69 ±8 | 67 ±9 | <0.001 |
| Gender(male) | 92 (52%) | 16 (76%) | 20 (67%) | 0.003 |
| Current smoker | 18 (10%) | 4 (19%) | 0 | NA |
| Medical history | ||||
| Hypertension | 113 (63%) | 17 (81%) | 0 | NA |
| Diabetes mellitus | 71 (40%) | 0 | 0 | NA |
| Hyperlipidaemia | 46 (26%) | 0 | 0 | NA |
| Chronic renal failure | 36 (20%) | 0 | 0 | NA |
| Medication | ||||
| Diuretics | 84 (47%) | 0 | 0 | NA |
| ACEI | 71 (40%) | 5 (16%) | 0 | NA |
| Losartan | 13 (7%) | 0 | 0 | NA |
| CCB | 49 (27%) | 7 (23%) | 0 | NA |
| Beta-blocker | 65 (36%) | 0 | 0 | NA |
| Clinic | ||||
| LVEF (%) | 47.5 ±14.3‡ | 64.2 ±4.1 | 68.5 ±3.5 | <0.001 |
| SBP (mmHg) | 148 ±31§ | 143 ±20 | 129 ±22 | 0.003 |
| DBP (mmHg) | 79 ±20 | 82 ±10 | 77 ±13 | 0.622 |
| HR (b.p.m.) | 88 ±24 | 69 ±13 | 87 ±11 | 0.08 |
| BMI | 24.8 ±4.1 | 25.3 ±8.5 | 23.6 ±3.2 | 0.375 |
| Laboratory | ||||
| Fasting glucose (mmol/L) | 6.5 ±1.8‡ | 5.2 ±0.8 | 5.2 ±0.3 | 0.007 |
| TC (mmol/L) | 4.3 ±1.3§ | 4.8 ±0.8 | 5.1 ±0.5 | 0.003 |
| LDL-C (mmollL) | 2.4 ±1.1§ | 2.9 ±1.0 | 3.0 ±0.3 | 0.011 |
| TG (mmol/L) | 1.6 ±1.5 | 1.7 ±1.9 | 1.3 ±0.2 | 0.317 |
| HDL-C (mmol/L) | 1.3 ±0.6 | 1.4 ±0.4 | 1.5 ±0.6 | 0.204 |
| WBC (109/L) | 9.2 ±4.0§ | 8.4 ±3.7 | 5.5 ±1.8 | 0.052 |
| Creatinine (μmol/L) | 173 ±148 | 91 ±13 | 71 ±19 | 0.086 |
ACEI, angiotensin-converting enzyme inhibitor; ANOVA, analysis of variance; BMI, body mass index; CCB, calcium channel blocker; CHF, congestive heart failure; DBP, diastolic blood pressure; HDL-C, high-density lipoprotein cholesterol; HR, heart rate; LDL-C, low-density lipoprotein cholesterol; LVEF, left ventricular ejection fraction; NA, not applicable; SBP, systolic blood pressure; TC, total cholesterol; TG. triglycerides; WBC, white blood cells.
‡P < 0.001 vs. disease control and normal control.
§P < 0.05 vs normal control.
Rho kinase activity, ROCK1, ROCK2, and RhoA activity in congestive heart failure and control groups
The ROCK activity (2.93 ±0.87, n =178) in the CHF group was significantly higher than that of the disease control (2.06 ±0.38, n =31, P < 0.001) and normal control groups (1.57 ±0.43, n =30, P < 0.001) (Figure 1). Western blot analysis showed increased ROCK1 and ROCK2 protein levels in heart failure subjects (vs. disease and normal control, all P < 0.05) (Figure 2A, B, D, E). Similarly, RhoA activity was found to be increased in heart failure (vs. disease and normal control, all P < 0.05) (Figure 2C, F). Also, RhoA activity in the disease controls was significantly higher than that in normal controls (Figure 2C, F). However, no significant differences between ROCK1 and ROCK2 were found between disease and normal subjects (Figure 2). The heart failure patients with ischaemic heart disease had higher ROCK activity (3.10 ±1.01, n =80) than other patients with hypertensive (2.69 ±0.75, n =37) or valvular diseases (2.92 ±1.11, n =12) and dilated cardiomyopathy (2.94 ±0.82, n =11) (P < 0.05). In the CHF cohort, patients were divided into four subgroups according to LVEF on admission: severe (<25%), moderate (25–40%), mild (40–55%), and normal EF (>55%). ROCK activity in the severe EF group (3.48 ±1.36, n =15) was significantly higher than that in the mild (2.88 ±0.74, n =49; P = 0.003) and normal EF (2.75 ±0.85, n =67; P =0.016) groups. Patients with moderate EF (3.09 ±0.65, n =37) had higher ROCK activity than those with normal EF (P =0.050) (Figure 3). In addition ROCK activity was greater in systolic heart failure (3.12 ±0.13, n =86) than in diastolic heart failure (2.74 ±0.10, n =82) (P < 0.05). ROCK activity was also highest in those heart failure patients with New York Heart Association (NYHA) class IV [3.51 ±1.09, n =41 vs. 3.06 ±0.71, n =56 (NYHA III), 2.62 ±0.67, n =46 (NYHA II), and 2.35 ±0.84, n =25 (NYHA I)] (P < 0.001). Since NYHA class is relatively subjective mainly based on the patient's description, ‘heart failure status (acute)’, ‘dyspnoea at rest’, and ‘6 minhallwalk’ were used to assess the severity of the heart failure.
Figure 1.

Comparison of congestive heart failure (CHF), disease, and normal control groups. (A) Western blotting results of the group with CHF, and disease and normal control groups. (B) Rho kinase (ROCK) activity in different groups. DC, disease control; NC, normal control; p-MBS, phosphorylated myosin-binding subunit.
Figure 2.
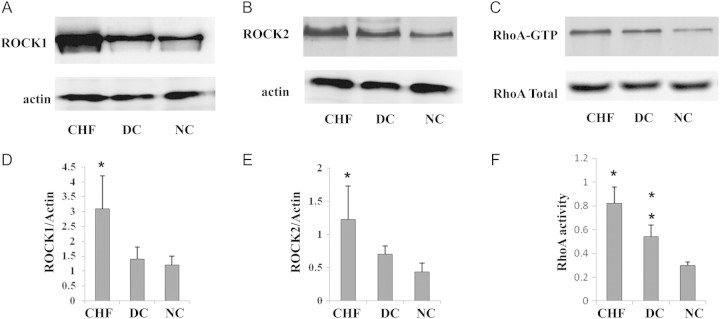
Western blot analysis of ROCK1, ROCK2, and RhoA activity in heart failure, disease control, and normal control groups. ROCK1 and ROCK2 increased significantly in heart failure compared with the disease and normal control groups (*P < 0.05; A, B, D, E). Similarly, RhoA activity increased in heart failure when compared with the other groups (*P < 0.05; C, F). RhoA activity in the disease control group was significantly higher than that in the normal control group (**P < 0.05; C, F). CHF, congestive heart failure; DC, disease control; NC, normal control.
Figure 3.

Comparison of Rho kinase (ROCK) of different congestive heart failure groups with severe (<25%), moderate (25–40%), mild (40–55%), and normal ejection fraction (EF) (>55%).
Predictors of Rho kinase activity in congestive heart failure measured at baseline
Univariate association analysis showed that among the factors associated with ROCK activity, heart failure status (r =0.443, P < 0.001), dyspnoea at rest (r =0.411, P < 0.001), heart failure with myocardial ischaemia (MI) on admission (r =0.423, P < 0.001), history of ischaemic heart disease (r =0.254, P =0.030) or heart failure (r =0.182, P =0.020), heart rate (r =0.298, P =0.001), urea (r =0.176, P =0.019), and creatinine (r =0.360, P < 0.001) were all positively associated with increased levels of ROCK activity. In contrast, LVEF (r = –0.587, P < 0.001) and sodium (r = –0.169, P =0.025) were negatively associated with ROCK activity. Multivariate regression models (stepwise) showed that dyspnoea at rest (β =0.338, P < 0.001), low LVEF (β = –0.277, P < 0.001), and high creatinine (β =0.202, P =0.006) predict baseline ROCK activity in CHF (Table 2).
Table 2.
Prediction of Rho kinase activity in univariate and multivariate regression models
| Variables | Univariate |
Multivariate (stepwise) |
||
|---|---|---|---|---|
| Coefficient | P-value | β | P-value | |
| Heart failure status (acute or stable) | 0.443 | <0.001 | – | – |
| Dyspnoa at rest | 0.411 | <0.001 | 0.338 | <0.001 |
| Heart failure with MI on admission | 0.423 | <0.001 | – | – |
| History of ischaemic heart disease | 0.254 | 0.030 | – | – |
| History of heart failure | 0.182 | 0.020 | – | – |
| History of renal failure | 0.321 | 0.028 | – | – |
| Left ventricular ejection fraction (%) | –0.587 | <0.001 | –0.277 | <0.001 |
| Heart rate (b.p.m.) | 0.298 | 0.001 | – | – |
| Sodium (mmol/L) | −0.169 | 0.025 | - | – |
| Urea (mmol/L) | 0.176 | 0.019 | – | – |
| Creatinine (μmol/L) | 0.361 | <0.001 | 0.202 | 0.006 |
MI, myocardial ischaemia.
Clinical outcome and predictors of long-term event-free survival
The mean duration of follow-up was 14.4 ( ±7.2) months (range 0.5–26 months). A total of 112 patients (82%) were followed up for > 1 year and 45 patients (25.3%) reached the primary endpoint of death. Accordingly, event-free survival was from 382 to 783 days. Further investigation was performed by Cox regression survival analysis for a long-term event outcome including the following baseline variables which were significantly correlated with death: age (r =0.177, P =0.019), serum sodium (r = –0.180, P =0.018), heart rate (r =0.183, P = 0.020), creatinine concentration (r =0.263, P < 0.001), blood urea (r =0.275, P < 0.001), ROCK activity (r =0.178, P =0.019), and NT-proBNP (r =0.352, P =0.005). Of all variables tested, age [hazard ratio (HR) 1.038, 95% confidence interval (CI) 1.001–1.076, P =0.044], sodium level (HR 0.894, 95% CI 0.825–0.969, P =0.007), heart rate at admission (HR 1.020, 95% CI 1.006–1.034, P =0.006), and NT-proBNP level (HR 1.200, 95% CI 1.000–1.002, P =0.038) were the independent predictors for long-term mortality (Table 3).
Table 3.
Multivariate Cox regression of baseline variables to long-term mortality
| Covariate | Multivariate (forward) |
||
|---|---|---|---|
| HR | 95% CI | P-value | |
| Age (years) | 1.038 | 1.001–1.076 | 0.044 |
| Sodium (mmol/L) | 0.894 | 0.825–0.969 | 0.007 |
| Heart rate (b.p.m.) | 1.02 | 1.006–1.034 | 0.006 |
| Creatinine (μmol/L) | – | – | 0.195 |
| Urea (mmol/L) | – | – | 0.126 |
| ACEI usage | – | – | 0.267 |
| Beta-blocker usage | – | – | 0.315 |
| ROCK activity | – | – | 0.563 |
| NT-proBNP (pg/mL) | 1.2 | 1.000–1.002 | 0.038 |
ACEI, angiotensin-converting enzyme inhibitor; CI, confidence interval; HR, hazard ratio; NT-proBNP, N-terminal pro brain natriuretic peptide; ROCK, Rho kinase.
In our study, the best cut-off value for ROCK activity to predict long-term mortality was 3.015, with sensitivity and specificity rates of 58% and 60%, respectively. The area under the curve (AUC) was 0.61, P =0.037. The best cut-off value for NT-proBNP to predict long-term mortality was 3788 pg/mL, with sensitivity and specificity rates of 65% and 59%, respectively. The AUC was 0.59, P =0.012. CHF patients were separated into four subgroups according to these two cut-off points. Patients with >3788 pg/mL NT-proBNP and 3.015 ROCK activity had 44% mortality over 2 years, which is significantly higher than that of the group with <3788 pg/mL NT-proBNP and 3.015 ROCK activity (12%, P =0.005). The mortality of the group with low NT-proBNP but high ROCK activity (22%) was also significantly lower than that of the group with high NT-proBNP and high ROCK activity (44%, P =0.049) (Figures 4 and 5).
Figure 4.

Comparison of mortality in different congestive heart failure (CHF) subgroups with different levels of N-terminal pro brain natriuretic peptide (NT-proBNP) and Rho kinase (ROCK) activity.
Figure 5.

The upper figure is the Kaplan–Meier curves for event-free survival within 2 years according to N-terminal pro brain natriuretic peptde (NT-proBNP) (log rank χ2 =5.16, P =0.023). The lower figure is the Kaplan–Meier curves for event-free survival within 2 years according to NT-proBNP combined with Rho kinase (ROCK) activity (log rank χ2 =11.62, P =0.009).
Furthermore, although only NT-proBNP has prognostic value, combining both NT-proBNP and ROCK activity was significantly superior in predicting mortality when compared with only a single factor (P < 0.05) (Figure 6).
Figure 6.

Incremental predictive value of combining the N-terminal pro brain natriuretic pepide (NT-proBNP) and Rho kinase (ROCK) activity for long-term mortality on top of the individual NT-proBNP predictor.
Discussion
In this study, we have shown for the first time that circulating ROCK activity in leucocytes is higher in CHF subjects than in disease control and normal control groups. Furthermore, higher baseline ROCK activity was associated with several features of CHF, such as more severe symptoms on admission (heart failure status, dyspnoea at rest), MI on admission, history of ischaemic heart disease, heart failure, or renal failure, poor systolic cardiac function (LVEF and heart rate), and lower renal function (sodium, urea, and creatinine). In addition, dyspnoea at rest, low LVEF, and high creatinine predicted the baseline ROCK activity in CHF. Combining ROCK activity and NT-proBNP had an incremental value in predicting long-term mortality. These findings indicate that ROCK activity might be a risk marker for CHF and suggest that ROCK activity may have a part to play in the pathopysiology and progression of CHF.
Increased Rho kinase activity in congestive heart failure
It has been shown that ROCK may act as a downstream effector in the intracellular signalling of several G protein-coupled receptors, including those of angiotensin II, norepinephrine, and endothelin-1, the activities of which are known to be elevated in CHF.14 In addition, the ROCK system has been implicated in the mediation of endothelin-1- and mechanical stress-induced hypertrophic responses in cardiac myocytes.15 These findings suggest that ROCK-dependent signalling pathways may potentially contribute to the mechanism of systemic and renal vasoconstriction and cardiac hypertrophy in CHF.
Although there are few studies on ROCK activity in humans with CHF, many studies in animal models have confirmed the potential involvement of Rho/ROCK in heart failure. In a dog model of tachypacing-induced heart failure, Y-27632, which is an inhibitor of ROCK, attenuates this response without a significant change in intracellular Ca2+ concentrations in vascular smooth muscle cells (VSMCs), suggesting that the Rho/ROCK pathway is involved in the increased vasoconstrictor response in heart failure.14 Transgenic mice that overexpress RhoA in the heart develop loss of systolic function and dilated cardiomyopathy.8 Long-term inhibition of ROCK by fasudil treatment reduces the angiotensin II-induced cardiomyocyte hypertrophy in wild-type as well as in apoE-KO mice.16 In addition, ROCK inhibition improves cardiac function by preventing the angiotensin II-induced decrease in ventricular contractility, cardiac output, and cardiac stoke volume.16 In Dahl salt-sensitive hypertensive rats, the ventricular hypertrophy and function is significantly ameliorated by ROCK inhibition.9,17 It has been suggested that the cardioprotective effect of ROCK inhibition involved up-regulation of the down-regulated endothelial nitric oxide synthase (eNOS) and the reduction of oxidative stress through the inhibition of NAD(P)H oxidase and lectin-like oxidized LDL receptor-1 expression.17 Recently, ROCK1 was proven to play an essential role in the transition from cardiac hypertrophy to cardiac failure in mice.18 Fasudil could suppresses isoproterenol-induced heart failure in rats via JNK and ERK1/2 pathways.19 In patients with heart failure, intra-arterial infusion of fasudil causes a preferential increase in forearm blood flow as compared with control subjects, suggesting an involvement of the Rho/Rho kinase pathway in the increased peripheral vascular resistance in heart failure in humans.10 The long-term effects of fasudil as a vasodilator therapy in the treatment of heart failure remain to be examined.
Interestingly, poor renal function also predicted high ROCK activity. In previous studies, ROCK has been shown to be constitutionally active in the renal circulation. Thus, ROCK inhibition by Y-27632 and fasudil dilates basal tone of afferent as well as efferent arterioles in in vitro20 and in vivo hydronephrotic kidney models.21 Furthermore, both Y-27632 and fasudil reverse the angiotensin II-induced vasoconstriction of afferent and efferent arterioles.20A recent study found that inhibition of the Rho/ROCK pathway could attenuate cyclosporine-induced kidney injury through the suppression of the induction of inflammation and apoptosis.22 Although these in vitro observations strongly suggest substantial roles for ROCK in mediating the progression of renal injury, only a couple of studies have been conducted that provided direct in vivo evidence for the contribution of ROCK to the development of renal disease.
In this study, we found that ROCK1 and ROCK2 protein levels were significantly different between heart failure and disease or normal controls, as was RhoA activity. The same observation was found in an ischaemia/reperfusion injury animal model.23 However, no obvious increase in ROCK1 and ROCK2 was found between disease and normal control groups, which was similar to findings in previous studies.24,25 Thus in acute heart failure, the ROCK activity increase is probably due to the elevated ROCK itself, as well as the increase in the protein level. Meanwhile, in smokers or those with hypertension, who are in a relatively stable state, the increase in ROCK activity is most probably the result of the activation of the kinase itself. Noma et al.12 demonstrated that not only endothelial dysfunction but also activated ROCK in VSMCs were found in healthy young male smokers compared with non-smokers. This suggests that smoking is involved in not only endothelial dysfunction but also activation of ROCK in VSMCs in the forearm circulation. In our study we did not find any relationship between smoking and ROCK activity in CHF. This might be because the combined effects of impaired systolic cardiac function and renal failure in severe heart failure outweigh any effect on ROCK activity due to smoking.
Heart failure is characterized by a chronic inflammatory status.26 Circulating C-reactive protein and pro-inflammatory cytokines are increased at all stages of renal failure. In fact, numerous studies demonstrated that the Rho/ROCK pathway is involved in the inflammatory process. ROCK1 mediates leucocyte recruitment and neointima formation following vascular injury.27 ROCK is also important in mediating the adhesion and transmigration of monocytes.28 Peripheral elevated ROCK activity could be used as a biomarker of vascular injury and to indicate endothelial activation. Thus, the peripheral leucocyte ROCK activity could represent the severity of heart failure or renal failure. In our study, white blood cells were not significantly correlated with ROCK activity. This may be due to other factors playing a more important role in elevating inflammatory mediators such as the ROCK protein level in heart failure. However, it is not clear whether this relationship represents cause or effect. The Rho/ROCK pathway is also involved in smooth muscle cell contraction and hypertrophy, which are important pathological processes in heart failure. Therefore, ROCK-dependent signalling pathways may potentially contribute to the mechanisms of systemic and renal vasoconstriction and cardiac hypertrophy in CHF.
Subjects with high baseline N-terminal pro brain natriuretic peptide and high Rho kinase activity have worse long-term outcome
In this study, we demonstrated for the first time the prognostic value of ROCK and NT-proBNP in patients with CHF. In those who subsequently died, ROCK activity and NT-proBNP were significantly higher than in the survivors. NT-proBNP is a well-established marker for the diagnosis of heart failure29 and can also be used as a prognostic tool30 and for monitoring treatment.31 In our CHF cohort, NT-proBNP was an independent predictor of long-term mortality whereas ROCK activity was not. A previous study has proved that patients with high NT-proBNP were more likely to be admitted to hospital and to the intensive medical unit (IMC)/intensive care unit (ICU).32 In this study, the combination of ROCK activity and NT-proBNP was more useful for predicting mortality in patients with CHF than NT-proBNP alone. This suggests that although NT-proBNP is a more sensitive biomarker than ROCK activity in CHF, ROCK is more representative of the inflammation, endothelial dysfunction, and vasoconstriction.
Recently, besides NT-proBNP, other molecular biomarkers have been evaluated in heart failure, such as mid-regional pro-atrial natriuretic peptide, mid-regional pro-adrenomedullin, C-terminal pro-endothelin-1, and C-terminal pro-vasopressin.33,34 Some studies have shown an incremental benefit of measuring other biomarkers in addition to NT-proBNP in heart failure. Such combinations with NT-proBNP include cardiac troponin I in systolic heart failure,35 cardiac troponin T in decompensated heart failure,36 estimated glomerular filtration rate (eGFR) after acute myocardial infarction predicting a heart failure event,37 and copeptin in chronic heart failure.38 Therefore, simultaneous measurement of ROCK activity and NT-proBNP could provide complementary information, and a simple multimarker strategy that categorizes the patients with advanced CHF based on the number of elevated biomarkers may provide rapid risk stratification. There may be a therapeutic aspect to ROCK as Winaver et al. demonstrated the possible beneficial antihypertrophic properties of ROCK in rats with heart failure.39
Finally, we suggest that in clinical practice the evaluation of a change of ROCK levels during admission is probably more helpful than a single pre-discharge ROCK absolute value, just like NT-proBNP. Further studies are required to prove this and evaluate whether selective ROCK antagonists may be useful as an additional treatment modality to attenuate cardiac hypertrophy in CHF.
Limitations
We have not measured ROCK activity in cardiac tissue because biopsy material is difficult to obtain in our locality. The pathological changes of circulating white blood cells sometimes are used to reflect the tissue condition and metabolic effects in many situations. Additional functional changes in the leucocytes, such as cytokine production, should be tested.
Conclusion
RhoA kinase activity is elevated in CHF. Dyspnoea at rest, low LVEF, and high creatinine predict baseline ROCK activity in CHF. In addition, ROCK activity combined with NT-proBNP was a good predictor for long-term event-free survival in CHF. Further studies would be helpful to elucidate the correlation of other inflammatory factors with ROCK activity in patients with CHF.
Funding
A research grant from the University Grants Committee of Hong Kong (RGC Co1laborative Research Fund 2010/11: CUHK9/CRF/10).
Conflict of interest: none declared.
References
- 1.Yamazaki T, Komuro I, Kudoh S, Zou Y, Shiojima I, Mizuno T, Takano H, Hiroi Y, Ueki K, Tobe K, Kadowaki T, Nagai R, Yazaki Y. Angiotensin II partly mediates mechanical stress-induced cardiac hypertrophy. Circ Res. 1995;77:258–265. doi: 10.1161/01.res.77.2.258. [DOI] [PubMed] [Google Scholar]
- 2.Pracyk JB, Tanaka K, Hegland DD, Kim KS, Rovira Sethi R, II, Blazina DR, Lee L, Bruder JT, Kovesdi I, Goldshmidt-Clermont PJ, Irani K, Finkel T. A requirement for the rac1 GTPase in the signal transduction pathway leading to cardiac myocyte hypertrophy. J Clin Invest. 1998;102:929–937. doi: 10.1172/JCI2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thorburn J, Xu S, Thorburn A. MAP kinase- and Rho-dependent signals interact to regulate gene expression but not actin morphology in cardiac muscle cells. EMBO J. 1997;16:1888–1900. doi: 10.1093/emboj/16.8.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dzau VJ. Renal and circulatory mechanisms in congestive heart failure. Kidney Int. 1987;31:1402–1415. doi: 10.1038/ki.1987.156. [DOI] [PubMed] [Google Scholar]
- 5.Wettschureck N, Offermanns S. Rho/Rho-kinase mediated signaling in physiology and pathophysiology. J Mol Med. 2002;80:629–638. doi: 10.1007/s00109-002-0370-2. [DOI] [PubMed] [Google Scholar]
- 6.Dong M, Yan BP, Liao JK, Lam YY, Yip GW, Yu CM. Rho-kinase inhibition: a novel therapeutic target for the treatment of cardiovascular diseases. Drug Discov Today. 2010;15:622–629. doi: 10.1016/j.drudis.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yanazume T, Hasegawa K, Wada H, Morimoto T, Abe M, Kawamura T, Sasayama S. Rho/ROCK pathway contributes to the activation of extracellular signal-regulated kinase/GATA-4 during myocardial cell hypertrophy. J Biol Chem. 2002;277:8618–8625. doi: 10.1074/jbc.M107924200. [DOI] [PubMed] [Google Scholar]
- 8.Sah VP, Minamisawa S, Tam SP, Wu TH, Dorn GW, 2nd, Ross J, Jr, Chien KR, Brown JH. Cardiac-specific overexpression of RhoA results in sinus atrioventricular nodal dysfunction contractile failure. J Clin Invest. 1999;103:1627–1634. doi: 10.1172/JCI6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobayashi N, Horinaka S, Mita S, Nakano S, Honda T, Yoshida K, Kobayashi T, Matsuoka H. Critical role of Rho-kinase pathway for cardiac performance and remodeling in failing rat hearts. Cardiovasc Res. 2002;55:757–767. doi: 10.1016/s0008-6363(02)00457-1. [DOI] [PubMed] [Google Scholar]
- 10.Kishi T, Hirooka Y, Masumoto A, Ito K, Kimura Y, Inokuchi K, Tagawa T, Shimokawa H, Takeshita A, Sunagawa K. Rho-kinase inhibitor improves increased vascular resistance and impaired vasodilation of the forearm in patients with heart failure. Circulation. 2005;111:2741–2747. doi: 10.1161/CIRCULATIONAHA.104.510248. [DOI] [PubMed] [Google Scholar]
- 11.Surawicz B, Childers R, Deal BJ, Gettes LS, Bailey JJ, Gorgels A, Hancock EW, Josephson M, Kligfield P, Kors JA, Macfarlane P, Mason JW, Mirvis DM, Okin P, Pahlm O, Rautaharju PM, van Herpen G, Wagner GS, Wellens H American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part III: intraventricular conduction disturbances: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society: endorsed by the International Society for Computerized Electrocardiology. Circulation. 2009;119:e235–e240. doi: 10.1161/CIRCULATIONAHA.108.191095. [DOI] [PubMed] [Google Scholar]
- 12.Noma K, Goto C, Nishioka K, Hara K, Kimura M, Umemura T, Jitsuiki D, Nakagawa K, Oshima T, Chayama K, Yoshizumi M, Higashi Y. Smoking, endothelial function, and Rho-kinase in humans. Arterioscler Thromb Vasc Biol. 2005;25:2630–2635. doi: 10.1161/01.ATV.0000189304.32725.bd. [DOI] [PubMed] [Google Scholar]
- 13.Liu PY, Liao JK. A method for measuring Rho kinase activity in tissues and cells. Methods Enzymol. 2008;439:181–189. doi: 10.1016/S0076-6879(07)00414-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hisaoka T, Yano M, Ohkusa T, Suetsugu M, Ono K, Kohno M, Yamada J, Kobayashi S, Kohno M, Matsuzaki M. Enhancement of Rho/Rho-kinase system in regulation of vascular smooth muscle contraction in tachycardia-induced heart failure. Cardiovasc Res. 2001;49:319–329. doi: 10.1016/s0008-6363(00)00279-0. [DOI] [PubMed] [Google Scholar]
- 15.Clerk A, Sugden PH. Small guanine nucleotide-binding proteins and myocardial hypertrophy. Circ Res. 2000;86:1019–1023. doi: 10.1161/01.res.86.10.1019. [DOI] [PubMed] [Google Scholar]
- 16.Wang YX, da CV, Martin-McNulty B, Vincelette J, Li W, Choy DF, Halks-Miller M, Mahmoudi M, Schroeder M, Johns A, Light DR, Dole WP. Inhibition of Rho-kinase by fasudil attenuated angiotensin II-induced cardiac hypertrophy in apolipoprotein E deficient mice. Eur J Pharmacol. 2005;512:215–222. doi: 10.1016/j.ejphar.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 17.Mita S, Kobayashi N, Yoshida K, Nakano S, Matsuoka H. Cardioprotective mechanisms of Rho-kinase inhibition associated with eNOS and oxidative stress–LOX-1 pathway in Dahl salt-sensitive hypertensive rats. J Hypertens. 2005;23:87–96. doi: 10.1097/00004872-200501000-00017. [DOI] [PubMed] [Google Scholar]
- 18.Shi J, Zhang YW, Yang Y, Zhang L, Wei L. ROCK1 plays an essential role in the transition from cardiac hypertrophy to failure in mice. J Mol Cell Cardiol. 2010;49:819–828. doi: 10.1016/j.yjmcc.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.J Wang N, Guan P, Zhang JP, Li YQ, Chang YZ, Shi ZH, Wang FY, Chu L. Fasudil hydrochloride hydrate, a Rho-kinase inhibitor, suppresses isoproterenol-induced heart failure in rats via JNK and ERK1/2 pathways. J Cell Biochem. 2011;112:1920–1929. doi: 10.1002/jcb.23112. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura A, Hayashi K, Ozawa Y, Fujiwara K, Okubo K, Kanda T, Wakino S, Saruta T. Vessel- and vasoconstrictor-dependent role of rho/rho-kinase in renal microvascular tone. J Vasc Res. 2003;40:244–251. doi: 10.1159/000071888. [DOI] [PubMed] [Google Scholar]
- 21.Cavarape A, Endlich N, Assaloni R, Bartoli E, Steinhausen M, Parekh N, Endlich K. Rho-kinase inhibition blunts renal vasoconstriction induced by distinct signaling pathways in vivo. J Am Soc Nephrol. 2003;14:37–45. doi: 10.1097/01.asn.0000039568.93355.85. [DOI] [PubMed] [Google Scholar]
- 22.Park JW, Park CH, Kim IJ, Bae EH, Ma SK, Lee JU, Kim SW. Rho kinase inhibition by fasudil attenuates cyclosporine-induced kidney injury. J Pharmacol Exp Ther. 2011;338:271–279. doi: 10.1124/jpet.111.179457. [DOI] [PubMed] [Google Scholar]
- 23.Zhang J, Bian HJ, Li XX, Liu XB, Sun JP, Li N, Zhang Y, Ji XP. ERK–MAPK signaling opposes rho-kinase to reduce cardiomyocyte apoptosis in heart ischemic preconditioning. Mol Med. 2010;16:307–315. doi: 10.2119/molmed.2009.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hidaka T, Hata T, Soga J, Fujii Y, Idei N, Fujimura N, Kihara Y, Noma K, Liao JK, Higashi Y. Increased leukocyte rho kinase (ROCK) activity and endothelial dysfunction in cigarette smokers. Hypertens Res. 2010;33:354–359. doi: 10.1038/hr.2010.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu PY, Chen JH, Lin JL, Liao JK. Increased Rho kinase activity in a Taiwanese population with metabolic syndrome. J Am Coll Cardiol. 2007;49:1619–1624. doi: 10.1016/j.jacc.2006.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Picano E, Morales MA, del Ry S, Sicari R. Innate inflammation in myocardial perfusion and its implication for heart failure. Ann NY Acad Sci. 2010;1207:107–115. doi: 10.1111/j.1749-6632.2010.05724.x. [DOI] [PubMed] [Google Scholar]
- 27.Noma K, Rikitake Y, Oyama N, Yan G, Alcaide P, Liu PY, Wang H, Ahl D, Sawada N, Okamoto R, Hiroi Y, Shimizu K, Luscinskas FW, Sun J, Liao JK. ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J Clin Invest. 2008;118:1632–1644. doi: 10.1172/JCI29226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Honing H, van den Berg TK, van der Pol SM, Dijkstra CD, van der Kammen RA, Collard JG, de Vries HE. RhoA activation promotes transendothelial migration of monocytes via ROCK. J Leukoc Biol. 2004;75:523–528. doi: 10.1189/jlb.0203054. [DOI] [PubMed] [Google Scholar]
- 29.Tang WH, Francis GS, Morrow DA, Newby LK, Cannon CP, Jesse RL, Storrow AB, Christenson RH, Apple FS, Ravkilde J, Wu AH National Academy of Clinical Biochemistry Laboratory Medicine. National Academy of Clinical Biochemistry Laboratory Medicine practice guidelines: clinical utilization of cardiac biomarker testing in heart failure. Circulation. 2007;116:e99–e109. doi: 10.1161/CIRCULATIONAHA.107.185267. [DOI] [PubMed] [Google Scholar]
- 30.Miller WL, Hartman KA, Burritt MF, Grill DE, Rodeheffer RJ, Burnett JC, Jr, Jaffe AS. Serial biomarker measurements in ambulatory patients with chronic heart failure: the importance of change over time. Circulation. 2007;116:249–257. doi: 10.1161/CIRCULATIONAHA.107.694562. [DOI] [PubMed] [Google Scholar]
- 31.Jourdain P, Jondeau G, Funck F, Gueffet P, Le Helloco A, Donal E, Aupetit JF, Aumont MC, Galinier M, Eicher JC, Cohen-Solal A, Juillière Y. Plasma brain natriuretic peptide-guided therapy to improve outcome in heart failure: the STARS-BNP Multicenter Study. J Am Coll Cardiol. 2007;49:1733–1739. doi: 10.1016/j.jacc.2006.10.081. [DOI] [PubMed] [Google Scholar]
- 32.Luchner A, Möckel M, Spanuth E, Möcks J, Peetz D, Baum H, Spes C, Wrede CE, Vollert J, Müller R, Katus H, Giannitsis E. N-terminal pro brain natriuretic peptide in the management of patients in the medical emergency department (PROMPT): correlation with disease severity, utilization of hospital resources, and prognosis in a large, prospective, randomized multicentre trial. Eur J Heart Fail. 2012;14:259–267. doi: 10.1093/eurjhf/hfr171. [DOI] [PubMed] [Google Scholar]
- 33.Masson S, Latini R, Carbonieri E, Moretti L, Rossi MG, Ciricugno S, Milani V, Marchioli R, Struck J, Bergmann A, Maggioni AP, Tognoni G, Tavazzi L GISSI-HF Investigators. The predictive value of stable precursor fragments of vasoactive peptides in patients with chronic heart failure: data from the GISSI-heart failure (GISSI-HF) trial. Eur J Heart Fail. 2010;12:338–347. doi: 10.1093/eurjhf/hfp206. [DOI] [PubMed] [Google Scholar]
- 34.Yandle TG, Troughton RW. Improving risk stratification in heart failure: a role for new biomarkers? Eur J Heart Fail. 2010;12:338–347. doi: 10.1093/eurjhf/hfq030. [DOI] [PubMed] [Google Scholar]
- 35.Tsutamoto T, Kawahara C, Nishiyama K, Yamaji M, Fujii M, Yamamoto T, Horie M. Prognostic role of highly sensitive cardiac troponin I in patients with systolic heart failure. Am Heart J. 2010;159:63–67. doi: 10.1016/j.ahj.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 36.Perna ER, Macin SM, Cimbaro Canella JP, Szyszko A, Franciosi V, Vargas Morales W, Bayol AP, Kriskovich JO, Medina F, Gonzalez Arjol B, Brizuela M. Importance of early combined N-terminal pro-brain natriuretic peptide and cardiac troponin T measurements for long-term risk stratification of patients with decompensated heart failure. J Heart Lung Transplant. 2006;25:1230–1240. doi: 10.1016/j.healun.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 37.Palmer SC, Yandle TG, Frampton CM, Troughton RW, Nicholls MG, Richards AM. Renal and cardiac function for long-term (10 year) risk stratification after myocardial infarction. Eur Heart J. 2009;30:1486–1494. doi: 10.1093/eurheartj/ehp132. [DOI] [PubMed] [Google Scholar]
- 38.Neuhold S, Huelsmann M, Strunk G, Stoiser B, Struck J, Morgenthaler NG, Bergmann A, Moertl D, Berger R, Pacher R. Comparison of copeptin, B-type natriuretic peptide, and amino-terminal pro-B-type natriuretic peptide in patients with chronic heart failure: prediction of death at different stages of the disease. J Am Coll Cardiol. 2008;52:266–272. doi: 10.1016/j.jacc.2008.03.050. [DOI] [PubMed] [Google Scholar]
- 39.Winaver J, Ovcharenko E, Rubinstein I, Gurbanov K, Pollesello P, Bishara B, Hoffman A, Abassi Z. Involvement of Rho kinase pathway in the mechanism of renal vasoconstriction and cardiac hypertrophy in rats with experimental heart failure. Am J Physiol Heart Circ Physiol. 2006;290:H2007–H2014. doi: 10.1152/ajpheart.00600.2005. [DOI] [PubMed] [Google Scholar]