

Abstract
Emerging single-molecule sequencing instruments can generate multi-kilobase sequences with the potential to dramatically improve genome and transcriptome assembly. However, the high error rate of single-molecule reads is challenging, and has limited their use to resequencing bacteria. To address this limitation, we introduce a novel correction algorithm and assembly strategy that utilizes shorter, high-identity sequences to correct the error in single-molecule sequences. We demonstrate the utility of this approach on Pacbio RS reads of phage, prokaryotic, and eukaryotic whole genomes, including the novel genome of the parrot Melopsittacus undulatus, as well as for RNA-seq reads of the corn (Zea mays) transcriptome. Our approach achieves over 99.9% read correction accuracy and produces substantially better assemblies than current sequencing strategies: in the best example, quintupling the median contig size relative to high-coverage, second-generation assemblies. Greater gains are predicted if read lengths continue to increase, including the prospect of single-contig bacterial chromosome assembly.
1 Introduction
Second-generation sequencing technologies, starting with 454 pyrosequencing1 in 2004, Illumina sequencing-by-synthesis2 in 2007 and others, have revolutionized DNA sequencing by reducing cost and increasing throughput exponentially over first-generation Sanger3 sequencing. Despite the great gains provided by second-generation instruments, they have several drawbacks. First, they require amplification of source DNA prior to sequencing, leading to amplification artifacts4 and biased coverage of the genome related to the chemical-physical properties of the DNA.5 Secondly, current second-generation technologies produce relatively short reads: typically 100 bp for Illumina (up to 150 bp) and ~700 bp for 454 (up to 1,000 bp). Short-reads make assembly and related analyses difficult, with theoretical modeling suggesting that decreasing read lengths from 1,000 bp to 100 bp can lead to a six-fold or more decrease in contiguity.6
Pacific Biosciences recently released their first commercial “third-generation” sequencing instrument, the PacBio RS: a real-time, single-molecule sequencer. It aims to address the problems outlined above by requiring no amplification and reducing compositional bias,7, 8 producing long sequences (e.g. median = 2,246, max = 23,000 bp using the latest PacBio chemistry),9 and supporting a short turn-around time (24 hrs sample to sequence).8, 10 The long read lengths would be beneficial for de novo genome and transcriptome assembly as they have the potential to resolve complex repeats and span entire gene transcripts. However, the instrument generates reads that average only 82.1%8–84.6%9 nucleotide accuracy, with uniformly distributed errors dominated by point insertions and deletions (Supplementary Fig S1). This high error rate obscures the alignments between reads and complicates analysis since the pairwise differences between two reads is approximately twice their individual error rate, and is far beyond the 5%–10% error rate1, 11, 12 that most genome assemblers can tolerate—simply increasing the alignment sensitivity of traditional assemblers is computationally infeasible (Supplementary Materials). Additionally, the PacBio technology utilizes hairpin adaptors for sequencing double stranded DNA, which can result in chimeric reads if the sequencing reaction processes both strands of the DNA (first in the forward and then reverse direction). While it is possible to generate accurate sequences on the PacBio RS by reading a circularized molecule multiple times (circular consensus or CCS), this approach reduces read length by a factor equal to the number of times the molecule is traversed, resulting in much shorter reads (e.g. median = 423 bp, max = 1,915 bp). Thus, there is a great potential advantage to the long, single-pass reads if the error rate can be algorithmically managed.
To overcome the limitations of single-molecule sequencing data and unlock its full potential for de novo assembly, we developed an approach that utilizes short, high-identity sequences to correct the error inherent in long, single-molecule sequences (Fig 1). Our pipeline PBcR (PacBio corrected Reads), implemented as part of the Celera Assembler,11 trims and corrects individual long-read sequences by first mapping short-read sequences to them and computing a highly accurate hybrid consensus sequence: improving read accuracy from as low as 80% to over 99.9%. The corrected, “hybrid” PBcR reads may then be de novo assembled alone, in combination with other data, or exported for other applications. As demonstrated below for several important genomes, including the previously unsequenced 1.2 Gbp genome of the parrot Melopsittacus undulatus, incorporation of PacBio data using this method leads to greatly improved assembly quality versus either first or second-generation sequencing, indicating the dawn of a “third generation” of sequencing and assembly.
Figure 1. The PBcR single-molecule read correction and assembly pipeline.
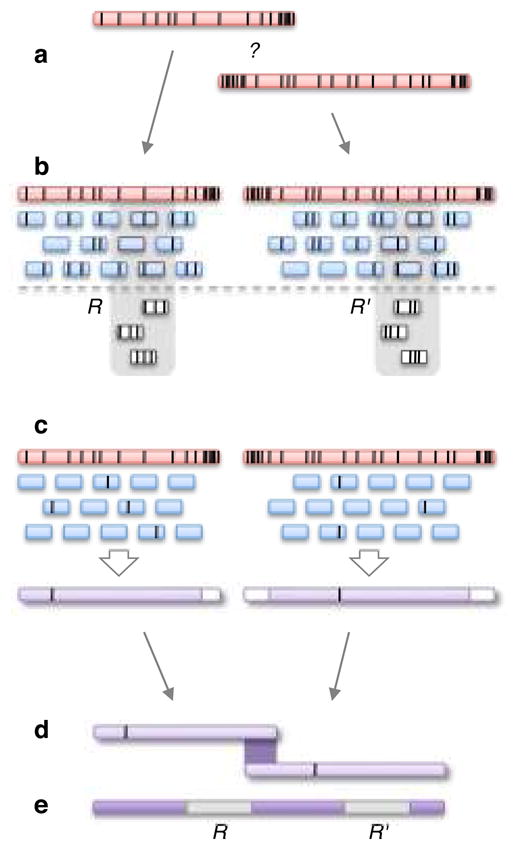
a) The high-error, indicated by black vertical bars, in single-pass PacBio RS sequences obscures overlaps. b) Given a high-accuracy sequence (~99% identical to the truth), the error between it and a PacBio RS sequence is half the error between two PacBio RS sequences. Therefore, accurate alignments can be computed. In this example, black bars in the short-reads indicate “mapping errors” that are a combination of the sequencing error in both the long and short reads. In addition, a two-copy inexact repeat is present (outlined in gray) leading to “pileups” of reads at each copy. To avoid mapping reads to the wrong repeat copy, the pipeline selects a cutoff, C, and only the top C hits for each short read are used. The spurious mappings (in white) are discarded. c) The remaining alignments are used to generate a new consensus sequence, trimming and splitting long reads whenever there is a gap in the short-read tiling. Sequencing errors, indicated in black, may propagate to the PBcR read in rare cases where sequencing error co-occurs. d) After correction, overlaps between long PBcR sequences can be easily detected. e) The resulting assembly is able to span repeats that are unresolvable using only the short reads.
2 Results
2.1 De novo assembly of long reads
Genome assembly is the computational problem of reconstructing a genome from sequencing reads.13, 14 It and the closely related problem of de novo transcriptome assembly are critical tools of genomics required to make order from a myriad of short fragments. The assembly problem is typically formulated as finding a traversal of a graph derived from sequencing reads using either the Overlap-Layout-Consensus (OLC or string graph) paradigm, where the graph is constructed from overlapping sequencing reads, or the de Bruijn graph formulation, where the graph is constructed from substrings of a given length k derived from the reads. Assembly graph complexity is determined by both sequencing error and repeats, but repeats are the single biggest impediment to all assembly algorithms and sequencing technologies.15 Under a de Bruijn graph formulation, repeats longer than k base-pairs form branching nodes that must be resolved by “threading” reads through the graph or by applying other constraints, such as mate-pair relationships.16 In contrast, only repeats longer than l = r − 2 × o cause unresolved branches in a string graph (where r is the read length and o is the minimum acceptable overlap length). For short-read sequences, k and l are very similar, so the corresponding graphs are nearly equivalent. However, for long reads, l may be substantially longer than feasible values of k. Therefore, long sequences have great potential to simplify the OLC assembly problem. In the extreme case, if all repeats are spanned by reads of greater length, OLC assembly of a genome into its constituent chromosomes and/or plasmids would be trivial. In practice, longer reads increase the probability of spanning repeats and detecting overlaps17, and thus produce better assemblies at lower sequencing coverage than short reads.
As a simple test, we evaluated the performance of multiple assemblers after error correcting lambda phage PacBio RS sequences with high-accuracy short-read sequencing technology (Supplementary Table S1, Fig S2, S3); only the OLC assembler produced a single contig. To test the benefits of increasing read lengths, we simulated error-free data of varying length from the Saccharomyces cerevisiae S228c genome and compared the resulting assemblies (Fig 2a). OLC assembly becomes progressively more powerful for longer reads, displaying a nearly linear increase in contig size as read lengths grow. In contrast, the de Bruijn assemblies plateau and cannot effectively utilize the long reads without increasing k beyond practical values due to the inherent limitations of the graph construction and the complexity of the read-threading problem.16, 18 Therefore, we developed a pipeline to correct and assemble PacBio RS sequences using an OLC approach.
Figure 2. Long-reads yield assembly improvements, even at low coverage.

a) Effect of PacBio corrected read length (PBcR) on contig size is measured for the OLC assembler Celera Assembler11 and the de Brujin assembler SOAPdenovo.49 Contig size, after breaking contigs at mis-joins, is measured using the standard N50 metric (N such that 50% of the genome is contained in contigs ≥ N). The baseline SOAPdenovo assembly (purple star) represents an assembly of 50X of real 76 bp Illumina paired-end (300 bp) reads from S. cerevisiae S228c. The effect of increasing PBcR read length was tested using 10X of simulated, error-free reads sampled from the S. cerevisiae genome. Read length was randomly sampled from actual length distributions of PBcR reads (from other genomes) to represent: the pre-release PacBio instrument (Q1, 2011), the first publicly available instrument (Q2, 2011), and the latest ”C2” chemistry upgrade (Q1, 2012). b). Effect of PBcR coverage is measured for Escherichia coli, sequenced with a combination of PacBio and second-generation sequencing. The benefit of the PBcR sequences is visible even below 5X, which leads to a 50%–100% increase in N50. Maximum contig N50 is reached by ~10X, where adding 10X of PBcR increases the N50 by up as much as 3.5-fold (250%). The larger gain versus the 454-only assembly is due the longer PBcR sequences available for E. coli JM221. The variation in N50 is due to random subsampling of sequencing data.
2.2 Correction accuracy and performance
We evaluated the PBcR correction and assembly algorithm on multiple short and long read datasets generated by Illumina, 454, and PacBio sequencing instruments, including three data sets with available reference sequences: Lambda NEB3011, Escherichia coli K12, and Saccharomyces cerevisiae S228c (Supplementary Table S2). The correction accuracy and assembly contiguity show diminishing returns after 50X of high-identity sequence and is recommended as a compromise between performance and accuracy (Supplementary Fig S4, S5). Using 50X of Illumina data to correct PacBio reads for each reference organism, the accuracy of the long reads improved from ~85% to over 99.9%, and chimeric and improperly trimmed reads measured < 2.5% and < 1% respectively (Table 1, Supplementary Fig S6). The concurrence of the corrected reads with their references is testament to the automated trimming process, which is necessary for the removal of adapter sequences that can be otherwise difficult to identify (Methods). As a result, the corrected reads are slightly shorter than the originals, but length is not drastically affected (e.g. median 848 pre-correction vs. 767 post-correction for E. coli K12). During correction, reads may also be discarded due to unusually low quality or short length, and the percentage of reads that are successfully corrected and output by the pipeline is termed throughput. The observed throughput is generally around 60%, but varies significantly depending on the quality of the individual runs. For example, throughput for the S. cerevisiae S228c reads appears unusually low, and is likely because much of this sequencing was performed using a pre-release PacBio RS instrument during testing at Cold Spring Harbor Laboratory. Nevertheless, in all cases the pipeline successfully identifies the usable data and outputs highly accurate long reads.
Table 1. PacBio correction results.
Corrected (PBcR) read accuracy as compared to reference sequence. Reads were mapped using Nucmer 3.23.50 For all statistics, only reads > 500 bp were included. %TP (Throughput): the percentage of raw uncorrected bases that are in non-chimeric, correctly trimmed sequences after correction. %Idy (Identity): average identity of good corrected reads to the reference. %Cov (Coverage): average coverage of good corrected reads by a single match to the reference. %Chimer: the percentage of corrected bases within reads with a split mapping to the reference. %Trim: the percentage of corrected bases within reads with a single match to the reference over less than 99.5% of their length. The corrected sequences remain above 99% identity and 99% trim within the repetitive regions of the genome (Supplementary Table S3).
| Organism | % TP | % Idy (Reads) | % Idy (Assembly) | % Cov | % Chimer | % Trim | Time (s) | Mem (GB) |
|---|---|---|---|---|---|---|---|---|
| Lambda NEB3011 | 74.03% | 99.90% | 100.00% | 100.00% | 1.82% | 0.10% | 121 | 0.12 |
| E. coli K12 | 57.46% | 99.99% | 99.99 % | 99.92% | 2.02% | 0.34% | 1580 | 2.10 |
| S. cerevisiae S228c | 21.86% | 99.90% | 99.97% | 99.93% | 1.46% | 0.33% | 4357 | 5.90 |
2.3 Hybrid de novo assembly
We evaluated the impact of PBcR reads on whole-genome assembly, either alone or in combination with the complementary reads. Two other assemblers are also reported to support PacBio reads: ALLPATHS-LG19 and ALLORA.9 However, neither program performs correction or de novo assembly from uncorrected reads. Instead, ALLPATHS-LG uses the raw reads to assist in scaffolding and gap closure of short-read de Bruijn assemblies. The downsides of this approach are that errors introduced in the short-read contigs may go uncorrected and this function is available only for genomes < 10 Mbp with both an Illumina paired-end library < 200 bp and a long-range Illumina jump library. Only the parrot genome presented here includes this required combination of Illumina and PacBio reads, but is larger than the size limit and could not be evaluated. ALLORA, a long-read assembler based on AMOS,20,21, 22 is computationally limited to small genomes and requires high-accuracy PacBio sequences, such as CCS, to operate. Inspired by our initial results, low-accuracy PacBio sequences from the 2011 German E. coli outbreak were manually corrected using our consensus module and iteratively assembled using ALLORA.9 We have now evaluated our automated correction and assembly pipeline on the same E. coli C227-11 genome, and have found it outperforms the previously published assembly (Table 2).
Table 2. PacBio assembly contiguity.
Organism: The genome being assembled. The median and max lengths of corrected PacBio sequences (PBcR) are given in parenthesis. The corrected length is shorter than original PacBio RS sequences due to trimming and splitting chimeric sequences. Supplementary Table S2 reports the original PacBio RS sequence lengths before correction. The three reference data sets (Lambda NEB3011, E. coli K12, and S. cerevisiae S228c) were generated using the pre-release PacBio RS, resulting in shorter read lengths. Technology: the read data used for assembly. Pair separation (if applicable) is listed immediately after the coverage. Reference bp: the assumed genome size used for the N50 calculation. Assembly bp: the total number of base pairs in all contigs (only contigs ≥ 10,000 bp are included in all results). #Contigs: The number of contigs comprising the assembly. Max Contig Length: The maximum contig length. N50: N such that 50% of the genome is contained in contigs of length ≥ N. Assemblies for next-gen (Illumina/454) were generated by Celera Assembler,11 SOAPdenovo,49 and ALLPATHS-LG19 (where possible). Only the best assembly (based on contiguity) in each case was reported.
| Organism | Technology | Reference bp | Assembly bp | # Contigs | Max Contig Length | N50 |
|---|---|---|---|---|---|---|
| Lambda NEB3011 | Illumina 100X 200bp | 48 502 | 48 492 | 1 | 48 492 / 48 492 | 48 492 / 48 492 (100%) * |
| (median: 727 max: 3 280) | PacBio PBcR 25X | 48 440 | 1 | 48 444 / 48 444 | 48 444 / 48 440 (100%) * | |
|
| ||||||
| E. coli K12 | Illumina 100X 500bp | 4 639 675 | 4 462 836 | 61 | 221 615 / 221 553 | 100 338/ 83 037 (82.76%) * |
| (median: 747 max: 3 068 ) | PacBio PBcR 18X | 4 465 533 | 77 | 239 058 / 238 224 | 71 479/ 68 309 (95.57%) * | |
| PacBio PBcR 18X + Illumina 50X 500bp | 4 576 046 | 65 | 238 272 / 238 224 | 93 048/ 89 431 (96.11%) * | ||
|
| ||||||
| S. cerevisiae S228c | Illumina 100X 300bp | 12 157 105 | 11 034 156 | 192 | 266 528 / 227 714 | 73 871/ 49 254 (66.68%) * |
| (median: 674 max: 5 994) | PacBio PBcR 13X | 11 110 420 | 224 | 224 478 / 217 704 | 62 898/ 54 633 (86.86%) * | |
| PacBio PBcR 13X + Illumina 50X 300bp | 11 286 932 | 177 | 262 846 / 260 794 | 82 543/ 59 792 (72.44%) * | ||
|
| ||||||
| E. coli C227-11 | PacBio CCS 50X | 5 504 407 | 4 917 717 | 76 | 249 515 | 100 322 |
| (median: 1 217 max: 14 901) | PacBio PBcR 25X (corrected by 25X CCS) | 5 207 946 | 80 | 357 234 | 98 774 | |
| PacBio PBcR 25X + CCS 25X | 5 269 158 | 39 | 647 362 | 227 302 | ||
| PacBio PBcR 50X (corrected by 50X CCS) | 5 445 466 | 35 | 1 076 027 | 376 443 | ||
| PacBio PBcR 50X + CCS 25X | 5 453 458 | 33 | 1 167 060 | 527 198 | ||
| Manually Corrected ALLORA Assembly9 | 5 452 251 | 23 | 653 382 | 402 041 | ||
|
| ||||||
| E. coli 17-2 | Illumina 100X 300bp | 5 000 000 | 4 975 331 | 62 | 226 141 | 74 940 |
| (median: 886 max: 10 069 ) | PacBio PBcR 50X | 4 981 368 | 58 | 318 969 | 143 307 | |
| PacBio PBcR 50X + Illumina 50X 300bp | 5 022 503 | 55 | 367 911 | 180 932 | ||
|
| ||||||
| E. coli JM221 | 454 50X | 5 000 000 | 4 714 344 | 66 | 308 063 | 106 034 |
| (median: 1 216 max: 12 552) | PacBio PBcR 25X | 5 005 429 | 30 | 631 286 | 314 500 | |
| PacBio PBcR 25X + 454 25X | 5 008 824 | 30 | 633 667 | 314 500 | ||
|
| ||||||
| Melopsittacus undulatus | Illumina 194X (220/500/800 paired-end 2/5/10Kb mate-pairs) | 1.23 Gbp | 1 023 532 850 | 24 181 | 1 050 202 | 47 383 |
| 454 15.4X (FLX + FLX Plus + 3/8/20Kbp paired-ends) | 999 168 029 | 16 574 | 751 729 | 75 178 | ||
| (median: 1 182 max: 14 596) | 454 15.4X + PacBio PBcR 3.83X (corrected by 15.4X 454) | 1 066 348 480 | 15 328 | 871 294 | 93 069 | |
| (median: 997 max: 13 079) | 454 15.4X + PacBio PBcR 3.75X (corrected by 54X Illumina) | 1 071 356 415 | 15 081 | 1 238 843 | 99 573 | |
For genomes with an available reference, the max and N50 contig is measured both before and after breaking contigs at assembly mis-joins. The percentages in parenthesis indicate the ratio between corrected and original N50. A higher ratio indicates a more correct assembly. Full assembly quality statistics are listed in Supplementary Table S4, following the GAGE assembly evaluation methodology.12
In all cases, from bacterial to eukaryotic, the incorporation of PBcR data produces substantially better assemblies than any other sequencing strategy tested—in the best cases, more than tripling the N50 contig size for equivalent depths of coverage (Table 2). These improvements also come without introducing additional assembly error, as measured against the three available reference genomes. The degree of improvement correlates with the median length of the corrected reads, with the newer, longer reads yielding the biggest gains and the older technology producing only modest gains (Table 2, Supplementary Fig S10). The observed gains are striking because they are entirely a result of resolving repeat structure rather than closing so-called “sequencing gaps” in the short-read coverage (Methods). This is due to the PBcR reads’ unique ability to close difficult gaps left by second generation technologies, such as interspersed, inverted, and complex tandem repeats (e.g. VNTRs and STRs) that can be difficult to assemble even with paired ends (Supplementary Fig S11).
Figure 3 summarizes the N50 results for various technologies and coverages for the E. coli genome. The three “short-read” alternatives of 50X 454, 50X PacBio CCS, and 100X Illumina paired-ends all produce similar assemblies. However, substituting half of the 454 coverage with corrected PacBio reads increases the N50 contig by 3 fold (e.g. 25X 454 + 25X PBcR); matching 50X short-read CCS coverage with 50X of PBcR reads results in a 5 fold increase. Because PacBio sequencing can be completed in just hours, this example provides a promising method for rapid genotyping and sequencing in time-critical situations, such as for an emerging disease outbreak.
Figure 3. Contig sizes for various combinations of sequencing technologies.

Assemblies are for E. coli C227-11 (assemblies including Illumina and PacBio CCS) and E. coli JM221 (assemblies including 454). Both genomes have similar repeat content, PacBio read length, and coverage. Assemblies of only second-generation data are comparable and average N50 ≈ 100 Kbp. By comparison, adding 25X or 50X of PBcR to these data sets increases N50 as much as 5 fold and pushes the maximum contig size greater than 1 Mbp (for the PBcR/CCS combination).
An assembly of PacBio and CCS reads also outperforms an assembly of simulated Illumina short and long pairs by 44%, with an N50 of 527,198 versus 364,181 (Supplementary Table S5). In addition, the combination of PacBio reads and Illumina short-range paired data produces an assembly nearly identical to the idealized Illumina long-range libraries (Supplementary Table S5). As Illumina short-range libraries double the sequencing time and long-range libraries are difficult to construct, these results suggest long, single-molecule sequencing is a practical alternative to both. This comparison is based on the second-generation PacBio chemistry, with an uncorrected median read length of ~2 Kbp. As read lengths increase, our simulations show that given adequate coverage of reads longer than around 5.5 Kbp (the size of the largest repeat), our pipeline can assemble the E. coli chromosome into a single closed contig, without the need for paired reads (Supplementary Materials).
2.4 Long-read coverage impact on assembly
Long reads are capable of producing better assemblies, even at greatly reduced coverages. A comparison of the literature shows that a 10–20X Sanger assembly is better than a 100X Illumina assembly, albeit with prohibitively greater sequencing costs using the older technology.19, 23 We found that for S. cerevisiae S228c, an assembly using 13X of PBcR data (corrected by 50X Illumina) is comparable to an assembly of 100X of paired-end Illumina data (Table 2). This is true despite the fact that sequencing was performed using a pre-release instrument. The corrected PacBio sequences also generate a more accurate assembly: while the 100X of Illumina produces a slightly longer raw N50, after splitting contigs at assembly errors, the N50 is larger for the PBcR assembly. Another striking example is E. coli JM221, for which the 25X PBcR assembly triples the N50 of the 50X 454 assembly.
Given the evident ability of PBcR reads to improve assemblies, the additive benefit of supplementing second-generation data was measured using E. coli. Between 1X to 50X of corrected PacBio data was added to the short read data for an existing assembly (Fig 2b). The large and rapid gains after the addition of long-read sequencing are readily apparent. At just 10X coverage, nearly the maximum N50 is reached for the second-generation/PBcR assembly. The N50 measures a 2.5 and 3.5-fold improvement over the Illumina and 454 assemblies, respectively. These results demonstrate significant improvements in continuity without the need for paired libraries and at relatively minor coverages. Thus, one might expect roughly double the N50 contig size with the addition of just 20X raw PacBio sequencing (assuming a throughput of > 50% during correction).
2.5 Assembling the parrot genome
Demonstrating applicability to vertebrate genomes, we successfully assembled the Melopsittacus undulatus genome using the PBcR pipeline. A total of 5.5X PacBio was corrected using 15.4X of 454 reads, producing 3.83X of sequences for a throughput of 69.62%. For comparison, the same PacBio RS sequences were corrected using 54X of Illumina, producing 3.75X of sequence. For the highest coverage dataset, the correction took 6.8 days (20K CPU hrs) to complete. For reference, an ALLPATHS-LG Illumina assembly and a Celera Assembler 454 assembly each took over one week to complete, with the Celera Assembler using the same number of cores as PBcR. Thus, the correction represents an approximate doubling of the total assembly time.
Because parrot is a novel genome without an available reference, correction accuracy was estimated by mapping PBcR reads to all parrot assemblies (except our own) submitted for the Assemblathon 2 (http://assemblathon.org).24 For this diploid genome, each assembly is a mosaic of the two haplotypes, so only the best mapping for each PBcR read was considered. Using this method, 99.9% of the 454-corrected PBcR reads have at least one mapping, and 97.0% map end-to-end with an average identity of 99.6%. Of the 3.0% of reads with fragmented mappings, 1.4% have breakpoints internal to a contig, which provides a rough estimate of chimerism. The remaining 1.6% map to contig boundaries and their accuracy cannot be determined. In contrast, the Illumina-corrected reads show a slightly increased rate of chimerism but maintain a similar identity (Supplementary Materials). Considering likely haplotype switching in the reference assemblies, these slight increases in estimated error are not unexpected, but are likely amplified for the shorter Illumina reads which are more difficult to uniquely map during correction. Nevertheless, in both cases the PBcR reads show good congruence with the independent assemblies, indicating that the pipeline succeeded for this difficult genome and can correct using both 454 or Illumina reads for complex vertebrate genomes, including human (Supplementary Fig S12).
The PBcR reads were then co-assembled with 15.4X of 454 reads, which included 3, 8, and 20 Kbp libraries to provide a diverse set of insert lengths, generating a PBcR-454 assembly and PBcR-454-Illumina assembly (where the Illumina data was used for correction only). For comparison, two additional assemblies were generated: one running Celera Assembler with identical parameters but on the 454 data only, and a second running ALLPATHS-LG on 194X of Illumina data, including 0.2, 0.5, 0.8, 2, 5, and 10 Kbp libraries. ALLPATHS-LG has been shown to be an effective short read assembler for large genomes,12, 24 and serves as an appropriate benchmark for assembling this genome using only Illumina data. A hybrid assembly of the 454 and Illumina data was not possible because Celera Assembler does not support high-coverage Illumina data and ALLPATHS-LG does not support 454. Interestingly, both the 454- and Illumina-corrected PBcR reads produce significantly better assemblies than the 454 data alone, demonstrating that the improvements are mostly attributable to the PacBio reads resolving repeats. To illustrate the effect of adding PBcR reads to an existing genome, the 454-corrected PBcR assembly is highlighted below. Full statistics for both PBcR assemblies are included in the Supplementary Materials.
The 454-PBcR assembly, with an N50 contig size of 93 Kbp is more continuous than the second generation assemblies in Table 2 and compares favorably with previous avian genome assemblies sequenced using the “gold-standard” Sanger method. The zebra finch (Taeniopygia guttata) was sequenced to 6X coverage using Sanger sequencing, generating a maximum contig of 424,635 and an N50 of 38,549.25 The chicken Gallus gallus was also sequenced using Sanger to 7.1X, resulting in a maximum contig of 624,663 and an N50 of 45,280.26 In contrast, for genomes assembled using only short-read sequencing, the N50 contig size rarely exceeds 30,000 bp.12, 19, 23 Much of the parrot genome continuity can be attributed to the long-read 454 data, including a mix of library sizes and the latest 454 FLX+ chemistry, but the addition of just 3.83X 454-PBcR sequences results in a 24% increase in N50, while the Illumina-corrected PBcR reads lead to an increase of 32% (Table 2). The increased continuity of the Illumina-assisted assembly is likely due to the complementary benefit of multiple technologies, with the Illumina reads correcting PacBio reads that fill coverage gaps in the 454 data.
In addition to improved continuity, the overall quality of the contigs remains high after the addition of the PBcR reads. Long-range accuracy is supported by satisfaction of both assembled 454 pairs and mapped Illumina mate-pairs (Supplementary Materials), which serve as an effective indicator of assembly quality.15,27 The percentage of bases not covered by satisfied 10 Kbp Illumina mates is virtually unchanged, and mate-pair coverage across the gaps closed by PBcR reads shows no observable deterioration (Supplementary Fig S13). Additionally, of the 33,881 scaffold gaps in the 454 assembly, the 16,251 gaps closed by the 454-PBcR reads closely match the corresponding gap size estimates from the 454 scaffolds (Supplementary Fig S14).
Completeness and correctness of the 454-PBcR assembly is also supported by mapped zebra finch mRNA transcripts, which align to the PBcR assemblies with slightly higher coverage and fewer chimeras than the 454 assembly(chimeric mappings: 81 454-PBcR, 86 454, 85 Illumina; mapped coding bases: 23.95 Mbp 454-PBcR, 23.78 Mbp 454, 24.26 Mbp Illumina; Supplementary Materials). Of the 15,275 finch mRNA sequences currently annotated in GenBank, approximately 95% are partially mappable to the parrot assemblies using the gmap spliced aligner.28 Despite its smaller contigs, ALLPATHS-LG appears very effective at assembling and scaffolding exons, with its scaffolds containing an additional 1–2% of the transcript bases than the other assemblies as a result of the high Illumina coverage. All assemblies also show similar identity to the mapped transcripts (89.17% 454-PBcR, 89.15% 454, 89.09% Illumina), but in terms of both exon coverage and identity the PBcR assemblies improve over the 454 assembly. For the 3,117 exons that are entirely contained in closed gaps, the average identity decreases slightly to 87.53%, and this 1.64% reduction from the average could be explained by limitations in the PBcR sequence accuracy or lowered sequencing depth across these difficult to sequence regions (Supplementary Materials).
However, despite similarity between all assemblies at the exon level, the PBcR assemblies excel at reconstructing the often repetitive non-coding sequence: in the case of 454-correction, splitting 22% and 7% less transcripts across contigs than the Illumina and 454 assemblies, and covering a greater fraction of each transcript with a single contig (Supplementary Fig S15, Supplementary Table S6). For example, 92% of the 454-PBcR closed gaps occur entirely outside of mapped finch exons, either within introns (18%) or between gene models (74%), and are enriched for extreme GC content (Supplementary Table S7). Such sequences are of particular importance for studying the parrot genome, because Warren et. al. report for zebra finch that “~40% of transcripts in the unstimulated auditory forebrain are non-coding and derive from intronic or intergenic loci”.25
Both the coding and non-coding sequences of genes with known relevance to vocal learning in birds are improved by the addition of PBcR reads. One striking example is the language and song associated FOXP2 gene,29–34 which is highly fractured in all but the PBcR assemblies (Supplementary Fig S16). Additional examples include the neurotransmitter glutamate receptors GRIK3, 35 GRIN 2A, and GRIN 2B,36 which contain intronic gaps closed only by the PBcR assemblies. The NAV337 and PLEXIN A438 axon guidance genes also show improved reconstruction in the PBcR assemblies, with the full PLEXIN A4 transcript recovered by both PBcR assemblies and only 20.8% and 47.5% by the 454 and Illumina assemblies, respectively. Lastly, the published zebra finch and chicken assemblies both contain gaps ~700 bp upstream of ERG1, a major immediate early gene that connects external stimuli to transcription in neurons.39, 40 The Illumina, 454, and 454-PBcR assemblies all contain a gap in this GC-rich (> 70% GC) promoter region as well, but the 454-PBcR-Illumina includes the full sequence. In this case, the combination of Illumina, 454, and PacBio succeeded where all independent assemblies failed (including Sanger). We note that there are other examples where the 454 and Illumina assemblies outperform the PBcR assemblies (Supplementary Table S6), and future work remains to best harness the complementary advantage of these multiple technologies.
2.6 Single-molecule RNA-Seq correction
Since the length of the single-molecule PacBio reads (ranging from a few hundred bases to several kilobases) from RNA-Seq experiments is within the size distribution of most transcripts, we expect many PacBio reads will represent full-length or near full-length transcripts. These long reads can therefore greatly reduce the need for transcript assembly, which requires complex algorithms for short reads,41 and confidently detect alternatively spliced isoforms. However, the predominance of indel errors makes analysis of the raw reads problematic. For example, in this study we generated 50,130 PacBio reads with a median size of 817 bp from a Zea mays B73 seedling mRNA sample, but only 11.6% (15,173) of the reads can be aligned to the reference genome by BLAT42 at >90% sequence identity. In contrast, for the corrected PBcR sequences, the percentage of sequences that align at > 90% identity increases dramatically to 99.1% (49,679 reads corrected in 3.6 days using 17.8X of Illumina data, Supplementary Materials). Consistent with the results reported above for genome assembly, the corrected RNA-Seq sequences have very low error rates, with only 0.06% insertion and 0.02% deletion rates.
As shown in Figure 4, many PacBio reads indeed represent close to full-length transcripts. However, the exon structure is not evident before the error correction by PBcR. The post-correction sequences have virtually no errors and precisely identify splicing junctions. As a result, two of the isoforms at the displayed reference locus in the reference annotation were confirmed by PacBio RNA-Seq reads. To systematically test the ability of PacBio reads to validate annotated gene structure, we aligned the PacBio reads to the reference genome and looked for PacBio reads that matched the exon structure over the entire length of the annotated transcripts. Before correction, only 41 (0.1%) of the PacBio reads exactly match the annotated exon structure. This number rises sharply after correction to 12, 065 (24.1%), suggesting that PBcR can greatly increase the usefulness of the PacBio RNA-Seq reads for transcript structure annotation or validation.
Figure 4. Error correction of RNA-Seq data provides more accurate mapping of transcripts.

A genome browser view of cDNA alignments using uncorrected (purple) and Illumina-corrected (green) PacBio reads generated from Zea mays B73 cDNAs. The splice-aware aligner, BLAT,42 was used for aligning PacBio reads to the genome. Long gaps in the alignment correspond to introns in the PacBio reads but not the reference genome, and short gaps (only visible in the pre-corrected PacBio reads) are putative indel errors. The read coverage of the Illumina reads used for correction is also shown, along with the current reference gene annotation for this locus. The corrected PBcR sequences match the reference annotations end-to-end and include two isoforms. The colored bars in read coverage are an artifact of the aligner, indicating reads that have overhangs across exon junctions. Genome coordinates for chr6 are shown from the RefGen v2 genome assembly (http://maizesequence.org/).
3 Discussion
Current de novo assemblers are unable to effectively use the long-read sequencing data generated by present single-molecule sequencing technologies primarily because of the considerable error rate. Our approach exploits this technology by complementing it with shorter, high-identity sequences resulting in long, accurate transcripts and improved assemblies. Since the average contig size produced by our approach correlates with read length, assembly results are expected to improve as the read lengths of the technology improve. This strategy also benefits from the complementarity of multiple technologies, which proved powerful when combining Sanger sequencing with second-generation data when it first became available.43 The result of our hybrid approach is higher quality assemblies with fewer errors and gaps, which will drive down the expensive cost of genome finishing and enable more accurate downstream analyses.
High-quality assemblies are critical for all aspects of genomics, especially genome annotation and comparative genomics. For example, many microbial genomic analyses depend on finished genomes,44 but producing finished sequence remains prohibitive with the cost of finishing proportional to the number of gaps in the original assembly. Eukaryotic genomics requires continuous assemblies to capture long, multi-exon genes and to determine genome organization and structural polymorphisms. In addition, recent work has suggested de novo assembly may be superior to read mapping approaches for discovering large structural variations, even when a reference genome is available.45 This is especially significant for understanding the genetic variations of cancer genomes and other human diseases such as autism that frequently contain gene fusions, copy number variations, and other large scale structural variations.46, 47 It is clear that higher quality assemblies, with long unbroken contigs, will have a positive impact on a wide range of disciplines.
Potential improvements to the PBcR pipeline include the addition of a gap closure routine to fill sequencing gaps in the short-read data using the PacBio reads and incorporation of the single-molecule base calls during consensus calling. This is particularly important for GC-rich sequences that tend to be underrepresented by second-generation sequencers, and for metagenomic and amplified samples that have severe coverage fluctuations. Non-uniform coverage will also require modifications to the repeat separation algorithm, since the current heuristic assumes uniform long-read coverage and error. This could include better utilization of paired-end information or variant clustering, which could also be applied to the problem of haplotype separation.
We have demonstrated that high error rates need not be a barrier to assembly. High-error, long reads can be efficiently assembled in combination with complementary short-reads to produce assemblies not possible with any prior technology, bringing us one step closer to the goal of “one chromosome, one contig.” The rapid turnaround time possible with PacBio and other technologies such as Ion Torrent48 will make it possible to produce high-quality genome assemblies at a fraction of the time once required. Future studies are needed to explore the relative costs and trade-offs of the available technologies, but from our results we anticipate future sequencing projects will consist of a combination of both long and short-read sequencing. Today this is particularly necessary for effective long-range scaffolding (≥ 9 Kbp pairs), for which the current PacBio reads provide limited assistance. However, if single-molecule technology continues to advance and reads begin to exceed the lengths of typical bacterial repeats (~6 Kbp) at reasonable cost and throughput, single-contig assemblies of some bacterial chromosomes will be possible without the need for expensive pair libraries. Additionally, we believe many long sought capabilities will be enabled, such as haplotype separation in eukaryotes, accurate transcriptome annotation, and true comparative genomics that extends beyond an exon-centric view to include the whole genome.
Methods
Our strategy consists of two phases: a long-read correction phase and an assembly phase. Both are implemented as part of the Celera Assembler,11 but the output of the correction phase can be used as input to any other analysis or assembler capable of utilizing long FastA sequences. The outline of the correction algorithm is as follows: 1) high-identity short-read sequences are simultaneously mapped to all long-read sequences, 2) repeats are resolved by placing each short-read sequence in its highest identity repeat copy, 3) chimera and trimming problems are detected and corrected within the long-read sequences, and 4) a consensus sequence is computed for each long-read sequence based on a multiple alignment of the short-read sequences. This approach was inspired by the intuition that while overlaps between single-pass PacBio reads average 31.6% pairwise differences (≈ 16.5% + 16.5%, Supplementary Materials), overlaps between long-read sequences and high-identity sequences would be lower and easier to detect. As most second-generation sequence overlaps are found below 3% error (Supplementary Fig S3a), the average overlap between PacBio reads and high-identity short-read sequences should be at most 17.5% (≈ 16.5% + 1%) (Supplementary Fig S3b).
The algorithm begins by computing all-vs-all overlaps between the low-accuracy single-pass (PacBio) long-read sequences and high-identity short-read sequences (Illumina, 454, PacBio CCS). The overlaps are computed only between fragments that have shared seed sequences of a pre-defined length (14 bp by default), and only short-read sequences aligned across their entire length to a long-read sequence are considered; support for partial overlaps to the ends of long reads is left for future versions. For efficiency, overlaps between reads of the same technology (e.g. short to short) are not computed during this phase.
Next, overlaps are converted into a tiling of short-read sequences along each long-read sequence. Each short-read sequence is permitted to map to more than one long-read sequence, since the long-read sequences are expected to cover the genome at more than 1X coverage. However, within a single long-read sequence, a short-read sequence is placed only in its highest identity location with ties broken randomly. In the case of repeats distributed across multiple long-read sequences, short-read sequences from all repeat copies will map to each copy of the repeat. To avoid tiling each repeat copy with the same set of reads, short-read sequences are separated into their appropriate copies by ranking their mappings by identity and permitting each short-read sequence to map only to its top C hits, where C is roughly defined to be the expected long-read sequencing depth. This effectively separates repeat copies when sequencing coverage and error is uniform. The value of C, a repeat threshold, is defined as follows:
Given a histogram and a threshold 0 ≤ 3 ≤ 1
Where ni is the number of long-read sequences a short-read sequence i maps to ∀i. Theoretically, the histogram H has a peak equal to the long-read depth of coverage. It can be expected that a unique short-read sequence will map to, on average, this many long-read sequences. Thus, a short-read sequence from a two-copy repeat will map to roughly double this number. The chosen repeat threshold is the point in the curve past this peak that includes at least T% of the high-identity reads (Supplementary Fig S7). In this way, each repeat copy will only be tiled by its best representative reads for correction. This approach can sometimes place reads in the wrong repeat copy. For instance, in cases where the error rate of two PacBio RS sequences from two separate repeat instances is significantly different, such that one is higher, Illumina sequences may preferentially map to the lower-error PacBio read. This would increase the mapped coverage of the low-error read by including some reads from the alternate copy, while decreasing the coverage of the high-error read. However, this problem should be alleviated as overall PacBio coverage is increased, because the read accuracy distribution in the different repeat copies will converge after a few fold redundancy. As evidence, systematic misplacement of reads in repeats, leading to inaccurate correction, coverage fluctuations, or decreased throughput, has not been observed in any of our experiments (e.g. Table 1, Supplementary Table S3, Supplementary Fig S8).
Finally, from the multiple-alignment of the tiled short-read sequences, the correction algorithm generates a new consensus sequence for each long-read sequence using the AMOS consensus module.20 In the consensus, if there is a gap in the layout between adjacent overlapping short reads, this is considered a irreconcilable discrepancy between the short and long-read sequences, especially since the reads are generated from the same biological sample and it is assumed there is sufficient coverage in the short sequences to tile each long-read sequence. Therefore, any gap in coverage is indicative of either improper trimming of the long-read sequence or chimera formation, and the long-read sequence is broken at this point. If instead there is merely insufficient coverage leading to a true sequencing gap for the short-read sequences, this will result in an unnecessary split. However, the correction algorithm errs on the side of caution. Future work remains to resolve any unnecessary gaps caused by the conservative trimming, such as by recognizing and filling these gaps during scaffolding.
The corrected, now high-identity, long-read sequences are provided in FastA format and can be assembled alone or co-assembled with other read types using standard OLC assembly techniques. To support de novo assembly using Celera Assembler we have increased the input size limitation, applying it successfully to sequences up to 30,000 bp in length.
Supplementary Material
Acknowledgments
Thank you to Pacific Biosciences, Roche 454, Illumina, BGI, and the Duke Genome Center for the generation and/or release of many of the datasets examined herein, and to the Assemblathon working group for the coordination and release of the parrot genome data.
This publication was developed and funded in part under Agreement No. HSHQDC-07-C-00020 awarded by the U.S. Department of Homeland Security for the management and operation of the National Biodefense Analysis and Countermeasures Center (NBACC), a Federally Funded Research and Development Center. The views and conclusions contained in this document are those of the authors and should not be interpreted as necessarily representing the official policies, either expressed or implied, of the U.S. Department of Homeland Security. The Department of Homeland Security does not endorse any products or commercial services mentioned in this publication. The work conducted by the U.S. Department of Energy Joint Genome Institute is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. This work was also funded in part by NIH R01-HG006677-12 (MCS), NIH 2R01GM077117-04A1 (BPW), the state of Maryland (DAR), NSF IOS-1032105 to WRM, and Howard Hughes Medical Institute and NIH Directors Pioneer Award to EDJ.
Footnotes
Competing Interests The authors declare that they have no competing financial interests.
Availability The latest version of the Celera Assembler (including PBcR) along with usage instructions is available at http://wgs-assembler.sourceforge.net. The software and data used for this manuscript are available at http://www.cbcb.umd.edu/software/PBcR.
Author Contributions SK and AMP conceived and designed the algorithm. SK implemented the algorithm and carried out the de novo assembly experiments. SK, MCS, and AMP drafted the manuscript, ran experiments, and contributed analysis. BPW modified the Celera Assembler to support long sequencing reads and developed the BOGART unitigger. JM and ZW sequenced Z. mays cDNA and performed analysis. JH, GG, and EDJ sequenced M. undulatus and performed analysis of vocal learning genes. DAR provided and sequenced E. coli strains. WRM sequenced S. cerevisiae S228c. All authors read and approved the final manuscript.
References
- 1.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bentley D. Whole-genome re-sequencing. Current Opinion in Genetics & Development. 2006;16:545–552. doi: 10.1016/j.gde.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 3.Sanger F, Nicklen S, Coulson A. DNA sequencing with chain-terminating inhibitors. PNAS. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niu B, Fu L, Sun S, Li W. Artificial and natural duplicates in pyrosequencing reads of metagenomic data. BMC bioinformatics. 2010;11:187. doi: 10.1186/1471-2105-11-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dohm J, Lottaz C, Borodina T, Himmelbauer H. Substantial biases in ultra-short read data sets from high-throughput dna sequencing. Nucleic Acids Research. 2008;36:e105. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kingsford C, Schatz M, Pop M. Assembly complexity of prokaryotic genomes using short reads. BMC bioinformatics. 2010;11:21. doi: 10.1186/1471-2105-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schadt EE, Turner S, Kasarskis A. A window into third-generation sequencing. Human Molecular Genetics. 2010;19:R227–R240. doi: 10.1093/hmg/ddq416. [DOI] [PubMed] [Google Scholar]
- 8.Chin CS, et al. The origin of the haitian cholera outbreak strain. New England Journal of Medicine. 2011;364:33–42. doi: 10.1056/NEJMoa1012928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rasko DA, et al. Origins of the E. coli strain causing an outbreak of hemolytic–uremic syndrome in Germany. New England Journal of Medicine. 2011;365:709–717. doi: 10.1056/NEJMoa1106920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eid J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 11.Miller JR, et al. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008;24:2818–2824. doi: 10.1093/bioinformatics/btn548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salzberg SL, et al. GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Research. 2011 doi: 10.1101/gr.131383.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pop M. Genome assembly reborn: recent computational challenges. Briefings in bioinformatics. 2009;10:354. doi: 10.1093/bib/bbp026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller J, Koren S, Sutton G. Assembly algorithms for next-generation sequencing data. Genomics. 2010;95:315–327. doi: 10.1016/j.ygeno.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phillippy A, Schatz M, Pop M. Genome assembly forensics: finding the elusive mis-assembly. Genome Biology. 2008;9:R55. doi: 10.1186/gb-2008-9-3-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pevzner PA, Tang H, Waterman MS. An Eulerian path approach to DNA fragment assembly. PNAS. 2001;98:9748–9753. doi: 10.1073/pnas.171285098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schatz MC, Witkowski J, McCombie WR. Current challenges in de novo plant genome sequencing and assembly. Genome Biology. 2011;13:243. doi: 10.1186/gb-2012-13-4-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagarajan N, Pop M. Parametric complexity of sequence assembly: theory and applications to next genera-tion sequencing. Journal of computational biology. 2009;16:897–908. doi: 10.1089/cmb.2009.0005. [DOI] [PubMed] [Google Scholar]
- 19.Gnerre S, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. PNAS. 2010 doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pop M, Phillippy A, Delcher AL, Salzberg SL. Comparative genome assembly. Briefings in Bioinformatics. 2004;5:237–248. doi: 10.1093/bib/5.3.237. [DOI] [PubMed] [Google Scholar]
- 21.Schatz MC, et al. Hawkeye and AMOS: visualizing and assessing the quality of genome assemblies. Briefings in Bioinformatics. 2011 doi: 10.1093/bib/bbr074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sommer D, Delcher A, Salzberg S, Pop M. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics. 2007;8:64. doi: 10.1186/1471-2105-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schatz MC, Delcher AL, Salzberg SL. Assembly of large genomes using second-generation sequencing. Genome Research. 2010;20:1165–1173. doi: 10.1101/gr.101360.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Earl DA, et al. Assemblathon 1: a competitive assessment of de novo short read assembly methods. Genome Research. 2011 doi: 10.1101/gr.126599.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warren WC, et al. The genome of a songbird. Nature. 2010;464:757–762. doi: 10.1038/nature08819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hillier L, et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- 27.Vezzi F, Narzisi G, Mishra B. Feature-by-Feature – evaluating de novo sequence assembly. PLoS ONE. 2012;7:e31002. doi: 10.1371/journal.pone.0031002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu TD, Watanabe CK. Gmap: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 29.Enard W, et al. Molecular evolution of FOXP2, a gene involved in speech and language. Nature. 2002;418:869–872. doi: 10.1038/nature01025. [DOI] [PubMed] [Google Scholar]
- 30.Enard W. FOXP2 and the role of cortico-basal ganglia circuits in speech and language evolution. Current Opinion in Neurobiology. 2011;21:415–424. doi: 10.1016/j.conb.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001;413:519–523. doi: 10.1038/35097076. [DOI] [PubMed] [Google Scholar]
- 32.Haesler S, et al. FoxP2 expression in avian vocal learners and non-learners. J Neuroscience. 2004;24:3164–3175. doi: 10.1523/JNEUROSCI.4369-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haesler S, et al. Incomplete and inaccurate vocal imitation after knockdown of FoxP2 in songbird basal ganglia nucleus Area X. PLoS Biology. 2007;5:e321. doi: 10.1371/journal.pbio.0050321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carroll SB. Evolution at two levels: on genes and form. PLoS Biology. 2005;3:e245. doi: 10.1371/journal.pbio.0030245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brose K, et al. Slit proteins bind Robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell. 1999;96:795–806. doi: 10.1016/s0092-8674(00)80590-5. [DOI] [PubMed] [Google Scholar]
- 36.Wada K, Sakaguchi H, Jarvis ED, Hagiwara M. Differential expression of glutamate receptors in avian neural pathways for learned vocalization. Journal of Comparative Neurology. 2004;476:44–64. doi: 10.1002/cne.20201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maes T, Barcelo A, Buesa C. Neuron navigator: a human gene family with homology to unc-53, a cell guidance gene from Caenorhabditis elegans. Genomics. 2002;80:21–30. doi: 10.1006/geno.2002.6799. [DOI] [PubMed] [Google Scholar]
- 38.Matsunaga E, Okanoya K. Vocal control area-related expression of neuropilin-1, plexin-A4, and the lig-and semaphorin-3A has implications for the evolution of the avian vocal system. Development, Growth, and Differentiation. 2009;51:45–54. doi: 10.1111/j.1440-169X.2008.01080.x. [DOI] [PubMed] [Google Scholar]
- 39.Morgan JI, Curran T. Stimulus-transcription coupling in neurons: role of cellular immediate-early genes. Trends in Neurosciences. 1989;12:459–462. doi: 10.1016/0166-2236(89)90096-9. [DOI] [PubMed] [Google Scholar]
- 40.Jarvis ED, Nottebohm F. Motor-driven gene expression. PNAS. 1997;94:4097–4102. doi: 10.1073/pnas.94.8.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kent WJ. Blat–the blast-like alignment tool. Genome Research. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goldberg S, et al. A sanger/pyrosequencing hybrid approach for the generation of high-quality draft assemblies of marine microbial genomes. PNAS. 2006;103 doi: 10.1073/pnas.0604351103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fraser CM, Eisen JA, Nelson KE, Paulsen IT, Salzberg SL. The value of complete microbial genome sequencing (you get what you pay for) J Bacteriol. 2002;184:6403–6405. doi: 10.1128/JB.184.23.6403-6405.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y, et al. Structural variation in two human genomes mapped at single-nucleotide resolution by whole genome de novo assembly. Nat Biotech. 2011;29:723–730. doi: 10.1038/nbt.1904. [DOI] [PubMed] [Google Scholar]
- 46.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 47.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rothberg JM, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475:348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 49.Li R, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Research. 2010;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome biology. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.