

Summary
According to the “free radical theory” of aging, normal aging occurs as the result of tissue damages inflicted by reactive oxygen species (ROS). ROS are known to induce cellular senescence, and senescent cells are believed to contribute to organismal aging. The molecular mechanisms that mediate the cellular response to oxidants remain to be fully identified. We have shown that oxidative stress induces cellular senescence through activation of the caveolin-1 promoter and upregulation of caveolin-1 protein expression. Here, we describe how reactive oxygen species activate the caveolin-1 promoter and how the signaling may be assayed. These approaches provide insight into the functional role of caveolin-1 and potentially allow the identification of novel ROS-regulated genes that are part of the signaling machinery regulating cellular senescence/aging.
Keywords: Caveolae, Caveolin-1, Cellular senescence, Free radicals, Tumor suppressor
1. Introduction
Several theories have been proposed in the past to explain why and how living organisms cannot escape aging. One of these is the “free radical theory” of aging, proposed by Denham Harman in the 1950s. According to this theory, normal aging occurs as the result of tissue damages inflicted by reactive oxygen species (ROS). In support of this theory, increased oxidative damage of DNA, proteins, and lipids has been reported in aged animals (1). Thus, endogenous and exogenous stimuli may significantly increase oxidant levels within the cell and induce a series of cellular damages.
Most cells cannot divide indefinitely due to a process termed cellular senescence (2-8). Growth arrest is associated with well-defined biochemical alterations. These include cell cycle arrest, increased p53 activity, increased p21Waf1/Cip1 and p16 protein expression, and hypo-phosphorylation of pRb (2-6). Interestingly, oxidative stress has been shown to induce premature senescence in fibroblasts in culture (9-11). Because a number of molecular changes that are observed in senescent cells occurs in somatic cells during the aging process, investigating the molecular mechanisms underlying oxidative stress induced premature senescence will allow us to better understand the more complicated aging process.
Caveolae are vesicular invaginations of the plasma membrane. Caveolin-1 is the structural component of caveolae. It has been proposed that caveolin-1 participates in vesicular trafficking events and signal transduction processes (12-14) by acting as a scaffolding protein (15) to organize and concentrate specific lipids (cholesterol and glyco-sphingolipids (16, 17)) and lipid-modified signaling molecules (Src-like kinases, H-Ras, eNOS, components of the p42/44 MAP kinase pathway, G-proteins, EGF-R, Neu, protein kinase A, and protein kinase C) within caveolar membranes (18-26). In addition to concentrating these signaling molecules within a specific region of the plasma membrane, caveolin-1 binding functionally inhibits the activity of caveolae-associated molecules.
We have shown that overexpression of caveolin-1 in fibro-blasts is sufficient to arrest the cells in the G0/G1 phase of the cell cycle and induce premature senescence. Consistent with these data, senescent human diploid fibroblasts have been shown to express higher levels of caveolin-1, as compared to younger human diploid fibroblasts (27). In addition, caveolin-1 has been shown to play an important role in senescence-associated morphological changes by regulating focal adhesion kinase activity and actin stress fiber formation in senescent cells (28). Finally, Park and colleagues have shown reentry of replicative senescent cells into cell cycle upon EGF stimulation after downregulation of caveolin-1 (29).
We have also demonstrated that oxidative stress induces cellular senescence in fibroblasts by stimulating caveolin-1 gene transcription through p38 MAPK/Sp1-mediated activation of two GC-rich promoter elements and upregulation of caveolin-1 protein expression (30). Interestingly, quercetin and vitamin E, two antioxidant agents, successfully prevent the premature senescent phenotype and the upregulation of caveolin-1 induced by hydrogen peroxide. Moreover, downregulation of caveolin-1 expression using an antisense-based approach inhibits oxidative stress-induced cellular senescence (31). Thus, caveolin-1 appears to play a major role in the signaling events linking oxidative stress to cellular senescence. Because oxidative stress induces cellular senescence and senescent cells are believed to contribute to organismal aging, studying ROS-mediated gene regulation will allow us to gain mechanistic insight into in vivo aging.
The assays described here were used to determine the transcription factor that mediates the oxidant-induced activation of the caveolin-1 promoter and include Luciferase-based Reporter assays, Electrophoretic Mobility Shift Assay (EMSA), and Chromatic Immunoprecipitation (ChIP) analysis. More precisely, luciferase-based reporter assays were used to define the sequence within the caveolin-1 promoter, which is responsive to free radical stimulation. Electrophoretic mobility shift assays were employed to determine whether the oxidant-responsive caveolin-1 promoter sequence identified with luciferase-based reporter assays formed a nucleoprotein complex after oxidative stress, which is indicative of binding of a transcription factor to the DNA. Finally, ChIP analysis was performed to pinpoint the transcription factor involved in the oxidant-induced activation of the caveolin-1 promoter.
2. Materials
2.1. Oxidative Stress
Cell culture: NIH 3 T3 Fibroblasts.
Cellular media: Dulbecco’s Modified Essential Medium supplemented with 10% Donor Bovine Calf Serum, Glutamine, and Antibiotics (Penicillin and Streptomycin).
150 μM Hydrogen peroxide (H2O2) diluted in cellular media.
Phosphate buffered saline (PBS).
2.2. Luciferase-Based Reporter Assay
GME Buffer (25 mM glycylglycine, 15 mM MgSO4, 4 mM EGTA, dissolved in H2O). GME Buffer must be stored at 4°C.
Z Buffer (100 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, pH 7.0). Right before using the solution, 50 mM β-mercaptoethanol should be added.
Extraction buffer (1% w/v Trition-X-100, 1 mM DTT in GME Buffer). Extraction buffer must be stored at 4°C. Additionally, 500 μL will be needed for each sample.
ATP mix (17 mM K Phosphate, 10 mM DTT, 2 mM ATP in GME Buffer). Mix must be kept at room temperature. Each sample needs 300 μL.
Luciferin solution (Add 1 ml of 1 mM luciferin and 50 μl of 1 M DTT to 4 ml of GME buffer). Do not add luciferin until right before using the solution. Solution can be kept at room temperature (see Note 1).
Calcium phosphate transfection reagents (CaCl2 and HeBs).
Caveolin-1 promoter luciferase reporter construct, luciferase reporter plasmid pTA-luc, and β-galactosidase-expressing construct.
PBS.
1 M Na2CO3.
4 mg/ml chlorophenol red-β-Dgalactopyranoside (CPRG) in ddH2O.
Luminometer reading at 562 nm and spectrometer reading at 574 nm.
2.3. Electrophoretic Mobility Shift Assay
NIH 3 T3 fibroblasts cultured in 10 cm dishes.
Cellular media: Dulbecco’s Modified Essential Medium supplemented with 10% Donor Bovine Calf Serum, Glutamine, and Antibiotics (Penicillin and Streptomycin).
Nuclear Extraction Buffer A (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 1 mM EDTA, and protease inhibitor tablet).
Nuclear Extraction Buffer B (20 mM HEPES pH 7.9, 25% glycerol, 0.43 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, and protease inhibitor tablet).
Phosphate buffered saline (PBS).
3′ end biotin-labeled double-stranded oligonucleotides containing a GC-rich box (in bold) (sequences are listed 5′ to 3′): Cav-1 (−244/−222): ggcactccccgccctctgctgcc; Cav-1 (−124/−101): cagccaccgccccccgccagcgc.
Annealing Buffer (10 mM Tris-Hcl, 0.5 mM EDTA, 0.5 mM trisodium phosphate, and 1 mM NaCl in sterile H2O).
10× Binding Buffer (100 mM Tris–HCl pH 8.0, 50% glycerol, 10 mM EDTA, 10 mM DTT, and 500 μg/mL) poly (Deoxyinosinic-deoxycytidylic acid).
5% nondenaturing polyacrylamide gel in 1× TBE along with appropriate running and gel transfer apparatus.
10× TBE Buffer (108 g Tris–base, 55 g Boric Acid, and 20 ml 0.5 M EDTA in 1 l of H2O; pH 8.0)
Positively charged Biodyne B nylon membrane.
Chemiluminescent Nucleic Acid Detection Module (Pierce Biotechnology, Illinois).
Film and developing cassettes.
2.4. Chromatin Immunoprecipitation Analysis
Cellular media: Dulbecco’s Modified Essential Medium supplemented with 10% Donor Bovine Calf Serum, Glutamine, and Antibiotics (Penicillin and Streptomycin).
Formaldehyde.
1.4 M glycine.
Chromatin IP buffer (50 mM HEPES KOH pH 8.0, 1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0, 140 mM NaCl, 10% glycerol, 0.5% IGEPAL, 0.25% Triton X-100, and protease inhibitor tablet).
Wash Buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0, 200 mM NaCl, and protease inhibitor tablet).
RIPA buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0, 140 mM NaCl, 1% Trition X-100, 0.1% Na-deoxycholate, 0.1% SDS, and protease inhibitor tablet).
Protein A Sepharose beads conjugated to salmon sperm DNA.
ChIP Dilution Buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris–HCl pH 8.1, and 167 mM NaCl).
Antibody of interest. For this protocol, Sp1 antibody was used.
LiCl Buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0, 250 mM LiCl, 1% Triton X-100, 1% Na-deoxycholate, and protease inhibitor tablet).
Elution Buffer (1% SDS and 0.1 M NaHCO3).
Proteinase K.
Qiagen PCR Purification Kit.
Polymerase chain reaction (PCR) primers for the Caveolin-1 gene promoter: Sense strand (5′ to 3′): caggctctcagctccccgcgc; antisense strand (5′ to 3′): gtatagaggggggaaaggcgc
PCR reagents (DNA template, primers, dNTPs, 10× reaction buffer, Taq enzyme, and H2O).
1.2% agarose DNA gel (with ethidium bromide) and TAE 6× DNA loading dye.
UV light gel documentation system.
3. Methods
3.1. Oxidative Stress
This section describes how to subject cells to oxidative stress using hydrogen peroxide. Hydrogen peroxide has been widely used as a source of free radicals and is shown by a number of groups to cause senescence (10, 11). Additionally, it is known to trigger the upregulation of caveolin-1 (30, 31).
H2O2 is diluted in cellular media to a concentration of 150 μM.
Media is removed from cell culture dishes, and H2O2 media is placed on cells (appropriate volume for dish size).
Cells are incubated for 2 h at 37°C.
Cells are washed twice in PBS to remove all traces of H2O2 media and are replated with fresh media (see Note 2).
3.2. Luciferase-Based Reporter Assay
This technique is commonly known as reporter gene assay. The firefly luciferase gene used in this assay is cloned downstream of the DNA promoter sequence under investigation in a promoterless DNA vector. The luciferase enzyme is synthesized after transient transfection of the DNA vector in cells only if the upstream DNA promoter sequence drives transcription of the luciferase gene. The ability of the cloned DNA promoter sequence to be activated by a certain stimulus is proportional to the amount of light produced by the oxidation of luciferin by luciferase in the presence of ATP in an in vitro reaction performed using cell exctracts. Cotransfection with a β-galactosidase expressing vector is commonly used to compensate for variations in transfection efficiency/sample manipulation. By generating a series of deletion mutants of the caveolin-1 promoter fused to the luciferase gene, we have identified two independent sequences within the caveolin-1 promoter that responded to oxidative stress.
Cells are seeded in 6-well plates with 270,000 cells per well. The following day, cells are transiently transfected with 2 μg of the caveolin-1 promoter-luciferase reporter constructs or the luciferase reporter construct alone and a β-galactosidase expressing vector by calcium phosphate precipitation method (see Note 3).
Twenty-four hours posttransfection, cells are rinsed with PBS, subjected to oxidative stress (see Subheading 29.3.1), and recovered in complete medium for 48 h.
Cells are washed twice with ice cold PBS and lysed in 500 μL of Extraction Buffer on a rocker at 4°C for 30 min.
During this incubation, place 300 μL of ATP Mix into cuvettes for luciferase assay and 600 μL of Z buffer into eppendorf tubes for the β-galactosidase assay.
Pipet 200 μL of sample into each cuvette for luciferase assay and 150 μL of sample into each eppendorf tube for the β-galactosidase assay. Vortex briefly to mix.
Luciferase activity is then measured by assessing light production at 562 nm using a luminometer, which inject 100 μl of luciferin solution into each sample.
For β-galactosidase activity, add 50 μl of CPRG to the Z buffer/sample mix, and let the incubation proceed for 30 min at 37°C. The reaction is stopped by adding 200 μL of 1 M Na2CO3. Light production is then measured at 574 nm using a spectrometer.
Variations in transfection efficiency/sample manipulation among the experimental points are compensated for by adjusting the luminometer readings for β-galactosidase readings (β-galactosidase activity is considered a nonvariable factor among the experimental groups so that variations of β-galactosidase readings reflect variations in transfection efficiency/sample manipulation).
Figure 29.1a shows that while Cav-1 (−1296/−1), Cav-1 (−800/−1), and Cav-1 (−372/−1) were activated by oxidative stress by ~15-fold, Cav-1 (−222/−1), and Cav-1 (−150/−1) showed instead only a fourfold induction upon hydrogen peroxide treatment (Fig. 29.1a). H2O2 did not activate the first 91 nucleotides of the caveolin-1 promoter (Fig. 29.1a). We concluded from these data that each of the nucleotides−372/−222 and −150/−91 of the caveolin-1 promoter contains an oxidative stress responsive element. Interestingly, both regions of the caveolin-1 promoter contain GC-rich boxes, which represent putative binding sites for the Sp1 transcription factor.
In Fig. 29.1b, we demonstrate that the two caveolin-1 promoter sequences containing GC-rich boxes (−244/−222 and −124/−101) were indeed oxidant-responsive elements. In fact, when these two sequences were fused to the luciferase gene, they were able to respond to hydrogen peroxide.
Fig. 29.1.

Oxidative stress activates the caveolin-1 promoter by acting through two GC-rich boxes. (a) Luciferase assay. Caveolin-1 promoter deletion mutant constructs were transiently transfected in NIH 3 T3 cells. pTA-luc alone was used as a control. Twenty-four hours after transfection, cells were treated with or without 150 μM H2O2 for 2 h. Cells were collected 48 h after oxidative stress and luciferase activity measured. Values represent means ±SEM. */#P < 0.001. (b) Luciferase assay. The caveolin-1 promoter −244/−222 and −124/−101 regions, both containing a GC-rich box, were cloned upstream of the luciferase gene in the pTA-luc vector. These constructs were transiently transfected in NIH 3 T3 cells. pTA-luc alone was used as a control. Twenty-four hours after transfection, cells were treated with or without 150 μM H2O2 for 2 h. Cells were collected 48 h after oxidative stress and luciferase activity measured. Values represent means ±SEM. *P < 0.001. Figure adapted from ref. 30.
3.3. Electrophoretic Mobility Shift Assay
Electromobility Shift Assay, or EMSA, allows for detection of sequence-specific DNA-transcription factor interactions. It is based on the premise that free DNA will run quicker than DNA bound to a protein when resolved on a polyacrylamide gel. When an antibody against the protein of interest is introduced into the mix, it further adds to the retardation of the complex in the gel matrix and allows the identification of the transcription factor, which binds the DNA sequence under investigation. This is deemed a supershift. Incubation with excess unlabeled doublestranded oligonucleotides representing the consensus sequence for the investigated transcription factor prevents the formation of the labeled DNA-protein complex and is often used to corroborate supershift data. Although 32P-labeled DNA has been the cornerstone for EMSA studies, there are alternative ways to label DNA, including the biotin-based method used in this study. Here, by using EMSA analysis, we found that oxidative stress promotes binding of nuclear proteins to Sp1 consensus elements within the Cav-1 promoter.
NIH 3 T3 cells are cultured in 10 cm dishes. They are treated with oxidative stress as described in Subheading 29.3.1. Control plates that are not treated are split to be the same confluency as the treated dishes at the time of the experiment.
Forty-eight hours after recovery from oxidative stress, nuclear extracts are prepared as follows: cells are washed twice with PBS and then collected in PBS. Cells are centrifuged at 1,500 × g at 4°C for 10 min. The pellet is resuspended in 400 μL ice cold Buffer A, and incubated on ice for 10 min. Samples are vortexed on high for 10 s and centrifuged again for 10 s at 12,000 × g. The supernatant is saved (see Note 4). Nuclear pellet is resuspended by pipetting up and down in 100 μL ice cold Buffer B and incubated on ice for 20 min at 4°C. Samples are centrifuged for 5 min at 4°C at 16,000 × g and the supernatant containing nuclear proteins is used for the experiment.
3′ end biotin-labeled double-stranded oligonucleotides containing a GC-rich box (see Subheading 2.3, item 6 for sequence) are resuspended in an eppendorf tube with Annealing Buffer. The oligonucleotides are serially diluted to a final concentration of 50 fmol/μl. Equal volumes (2 μl) of complementary single stranded oligonucleotides are incubated in an eppendorf tube, and the tube is placed into a beaker with 500 ml of water at 95°C. The beaker is then allowed to cool down slowly to room temperature.
5 μg of nuclear protein extracts from untreated and H2O2-treated cells are incubated with 100 fmol 3′ end biotin-labeled double-stranded oligonucleotides and 1× Binding buffer for 15 min at room temperature in a final volume of 20 μL.
See Note 5 for proper controls to run.
Samples are run on a 5% nondenaturing polyacrylamide gel in 1× TBE buffer. The gel is prerun at a constant voltage until the current no longer varies with time.
The gel is then transferred onto a positively charged Biodyne B nylon membrane at 380 mA for 60 min.
The DNA is then UV-light crosslinked to the membrane at 120 mJ/cm2.
DNA is visualized using Chemiluminescent Nucleic Acid Detection Module (Pierce) according to manufacturer’s instructions.
Membrane is exposed to film for 1–4 min depending on intensity of signal.
Figure 29.2a, b illustrate that oxidative stress promoted the formation of nucleoprotein complexes by the −244/−222 and −124/−101 caveolin-1 promoter sequences (Complex I and complex II, respectively). Because incubation with excess unlabeled double-stranded oligonucleotides representing the Sp1 consensus sequence totally prevented the formation of both complex I and complex II in Fig. 29.2a, b, these results suggest that Sp1 may indeed represent the transcription factor that mediates the response of the caveolin-1 promoter to oxidative stress.
Fig. 29.2.

Oxidative stress stimulates the binding of nuclear proteins to Sp1 consensus elements within the caveolin-1 promoter. (a) and (b) EMSA studies. Electrophoretic mobility shift assays were performed with nuclear extracts from untreated and H2O2-treated (150 μM for 2 h) NIH 3 T3 cells 48 h after oxidative stress. Nuclear extracts were incubated with either Cav-1 (−244/−222) (a) or Cav-1 (−124/−101) (b) biotin-labeled oligonucleotides. Lack of nuclear extract was used as a negative control. Note that two nucleoprotein complexes were identified in (a) (Complex I and III) and two in (b) (Complex II and IV). Incubation with excess unlabeled Sp1 consensus oligonucleotides was performed to show specificity of complex I and II. Figure adapted from ref. 30.
3.4. Chromatin Immunoprecipitation Assay
Chromatin immunoprecipitation (ChIP) assays can determine what transcription factors interact in vivo with a specific DNA sequence under certain conditions. Cells are exposed to a stimulus, and then, proteins that interact with the DNA are cross-linked to the DNA by formaldehyde. The DNA is broken into fragments by sonication, and antibodies precipitate the particular protein of interest attached to DNA. Reverse cross-linking and PCR amplification allow resolve whether the hypothesized protein-interaction takes place under the given conditions. This method is very efficient for determining transcription factors that bind to the Caveolin-1 promoter under oxidative stress. Using ChIP analysis, we identified that the transcription factor Sp1 binds to GC-rich elements within the Cav-1 promoter upon oxidative stress(30). Other groups have used ChIP assays to map transcription factor binding and epigenetic changes to the caveolin-1 promoter (32, 33).
Cells are subjected to oxidative stress as described in Subheading 3.1. Plates are washed twice with PBS and then incubated with regular media for 48 h. Untreated cells are used as controls.
Cells are crosslinked with 1% formaldehyde (final concentration), which is added to the cell plate containing 8 ml of growth media for 10 min at 37°C. The crosslinking is quenched by the addition of 0.8 ml of 1.4 M glycine to each plate for 5 min at 4°C. Media is promptly removed, and cells are washed twice with 4 ml of cold PBS, and are then scraped twice in 4 ml of PBS and placed into a 50 ml tube. An aliquot should be taken at this time to count the number of cells and the volume normalized for each sample so that there are approximately 5 million cells per time point for the assay.
Cells are spun down at 600 × g for 10 min at 4°C, and the pellet is resuspended in 500 μl of cold ChromatinIP buffer. Cells are rocked at 4°C for 10 min and then spun down at 600 × g at 4°C for 10 min.
The pellet is resuspended in 500 μl of cold Wash Buffer. Samples are rocked at 4°C for 10 min and then spun down at 600 × g at 4°C for 10 min.
The pellet is resuspended in 500 μl cold RIPA buffer. Samples are rocked at 4°C for 10 min.
Samples are then sonicated in polypropylene tubes at amplitude of 21%, 2 pulses on 1 pulse off, for 10 s with three cycles (so that the DNA is fragmented to ~250–1,000 bp sizes) (see Note 6).
Samples are transferred to eppendorf tubes and spun down at max speed for 15 min. The supernatant is transferred to a new eppendorf tube and the final volume brought to 500 μl in RIPA buffer + protease inhibitors.
ChIP lysate is precleared with Protein A Sepharose beads conjugated to salmon sperm DNA for 30 min at 4°C. Afterward, samples are spun down and the supernant saved. About 10% of the sample (50 μl) is taken as input. The inputs are reverse crosslinked immediately by adding 950 μl ChIP Dilution Buffer with 40 μl 5 M NaCl and incubated at 65°C for 4 h. Inputs are saved at −20°C.
ChIP lysate is combined with 70 μl of Protein A Sepharose beads conjugated to salmon sperm DNA in addition to 1–3 μg of antibody and rotated over night at 4°C.
Beads are centrifuged at 600 × g and washed twice with cold RIPA buffer for 5 min on rotation at 4°C, twice with cold RIPA buffer + 500 mM EGTA pH 8.0, and once with LiCl Buffer.
Complexes are eluted twice in 250 μl of fresh Elution Buffer.
Samples are reverse crosslinked by adding 20 μl of 5 M NaCl and incubating at 65°C for 4 h. To both ChIP samples and thawed inputs, 10 μl of 0.5 M EDTA, 20 μl of 1 M Tris–HCl pH 6.5, and 2 μl of 10 mg/ml Proteinase K are added, and samples are incubated at 45°C for 1 h.
Inputs and sample DNA are recovered by using Qiagen PCR purification kit according to manufacturer’s instructions and resuspended in 30 μl of H2O (see Note 7).
PCR was conducted using the following sequences for primers: sense (5′ to 3′) CAG GCT CTC AGC TCC CCG CCG. The antisense strand was (5′ to 3′) GTA TAG AGG GGG GAA AGG CGC. For both ChIP samples and inputs, PCR reactions were performed using 5 μl of sample DNA, 0.3 μM of sense and antisense primers, 0.3 mM dNTPs, 1× PCR Buffer, 1 unit of Taq enzyme, and H2O to a final volume of 50 μl. The PCR was run at (1) 94°C 5 min (2) 94°C 30 s (3) 58°C 30 s (4) 72°C 1 min (steps 2–4 were repeated 29 times) (5) 72°C 7 min and (6) 4°C 16 h.
8 μl of TAE 6× DNA loading dye is added to 40 μl of ChIP PCR, and run out on a 1.2% agarose gel with ethidium bromide. PRC products are visualized by UV light and documented.
In Fig. 29.3, a ChIP analysis was performed on chromatin from untreated and hydrogen peroxide-treated cells using an antibody probe specific for Sp1. We found that oxidative stress increased binding of Sp1 to the caveolin-1 promoter region containing the two GC-rich boxes. This result shows that free radicals stimulate direct binding of Sp1 to the GC-rich boxes of the caveolin-1 promoter in vivo.
Fig. 29.3.
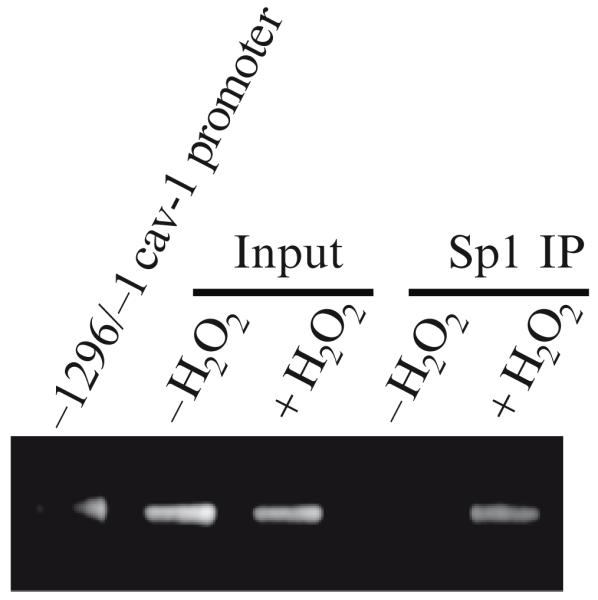
Oxidative stress promotes the binding of Sp1 to GC-rich elements within the caveolin-1 promoter. ChIP assay. Chromatin immunoprecipitation assay was done on chromatin derived from untreated or hydrogen peroxide-treated (150 μM for 2 h) NIH 3 T3 cells 48 h after oxidative stress using an antibody probe specific for Sp1. PCR was performed using primers surrounding the region of the caveolin-1 promoter containing the two GC-rich boxes. Amplification of input DNA from both untreated and H2O2-treated cells was performed before immunoprecipitation. A vector containing the entire caveolin-1 promoter sequence was used as a positive control for PCR. Figure adapted from ref. (30).
4. Notes
For the Luciferase assay, GME buffer and Z buffer (lacking β-mercaptoethanol) can be stored as stocks. Extraction buffer, ATP mix, and Luciferin solution should be made fresh on the day of the experiment. Luciferin is light sensitive and should be protected from light by wrapping container in foil.
Oxidative stress generated by hydrogen peroxide has been used on a number of cell lines. There can be slight differences in the concentrations needed to activate the caveolin-1 promoter and upregulate caveolin-1 protein expression, depending on the cell type used. Therefore, it is recommended to do a concentration dose curve to determine which dosage is needed for a particular cell line. For example, incubation of NIH 3 T3 murine fibroblasts with 150 μM H2O2 for 2 h is sufficient to upregulate caveolin-1 protein expression after 48–72 h of recovery from oxidative stress (31).
We have used the calcium phosphate precipitation method to transfect our cells. However, other transfection methods will work well, and should be used based on the efficiency with a particular cell line. The amount of DNA should be adjusted according to the particular transfection method used.
Supernatant can be run on a protein gel as an internal control.
Controls for EMSA assay. Run DNA only without nuclear extract. This will have biotin-labeled oligonucleotides without protein bound to it. This control will establish the nonshifted DNA band. Additionally, a competition assay should be done. Excess unlabeled double-stranded oligonucleotides containing an Sp1 consensus site should be added (in 200-fold molar excess) to the labeled oligonucleotides with nuclear extract. There should be competitive binding of the unlabeled oligonucleotides and a decrease in the bandshift intensity of the labeled oligo-nucleoprotein complex.
It is of utmost importance to have the DNA sheared to 200–1,000 base pairs in length. This must be worked out before the full experiment is conducted. The degree of sonication necessary to shear to this size will differ between cell lines. It is recommended to treat cells as would be called for by the experimental design, and fix them in 1% formaldehyde at 37°C for 10 min. Quench with glycine as described in Subheading 29.3.4, step 2. Wash and collect cells in PBS. Count cells and normalize to approximately 5 million for each experimental point. Pellet cells for 5 min at 600 × g at 4°C. Resuspend pellet in 200 μL of SDS Lysis Buffer and incubate 10 min on ice. Each experimental point can now be used to test different degrees of sonication. Add 8 μL of 5 M NaCl to lysate and incubate at 65°C to reverse crosslinks. Purify DNA by phenol/chloroform extraction, and run samples on an agarose gel with DNA ladder to determine fragmentation size.
Although we have had success with a Qiagen kit, there are other commercially available kits as well and should be used according to manufacturer’s instructions. Purification is needed to remove salts from the mixture.
Acknowledgments
Ferruccio Galbiati and this work were supported by Grant AG022548 from the National Institutes of Health (NIH). Janine N. Bartholomew was supported by a National Institute of Health Predoctoral Training Grant in Pharmacological Sciences (T32GM008424).
References
- 1.Chen QM. Replicative senescence and oxidant-induced premature senescence. Beyond the control of cell cycle checkpoints. Ann N Y Acad Sci. 2000;908:111–125. doi: 10.1111/j.1749-6632.2000.tb06640.x. [DOI] [PubMed] [Google Scholar]
- 2.Lundberg AS, Hahn WC, Gupta P, Weinberg RA. Genes involved in senescence and immortalization. Curr Opin Cell Biol. 2000;12:705–709. doi: 10.1016/s0955-0674(00)00155-1. [DOI] [PubMed] [Google Scholar]
- 3.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A bio-marker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Black EJ, Clark W, Gillespie DA. Transient deactivation of ERK signalling is sufficient for stable entry into G0 in primary avian fibroblasts. Curr Biol. 2000;10:1119–1122. doi: 10.1016/s0960-9822(00)00699-0. [DOI] [PubMed] [Google Scholar]
- 5.Sherr CJ, DePinho RA. Cellular senescence: mitotic clock or culture shock? Cell. 2000;102:407–410. doi: 10.1016/s0092-8674(00)00046-5. [DOI] [PubMed] [Google Scholar]
- 6.Wynford-Thomas D. Cellular senescence and cancer. J Pathol. 1999;187:100–111. doi: 10.1002/(SICI)1096-9896(199901)187:1<100::AID-PATH236>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 7.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 8.Lee SW, Reimer CL, Oh P, Campbel lDB, Schnitzer JE. Tumor cell growth inhibition by caveolin re-expression in human breast cancer cells. Oncogene. 1998;16:1391–1397. doi: 10.1038/sj.onc.1201661. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q, Ames BN. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc Natl Acad Sci U S A. 1994;91:4130–4134. doi: 10.1073/pnas.91.10.4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frippiat C, Chen QM, Zdanov S, Magalhaes JP, Remacle J, Toussaint O. Subcytotoxic H2O2 stress triggers a release of transforming growth factor-beta 1, which induces biomarkers of cellular senescence of human diploid fibroblasts. J Biol Chem. 2001;276:2531–2537. doi: 10.1074/jbc.M006809200. [DOI] [PubMed] [Google Scholar]
- 11.Chen QM, Bartholomew JC, Campisi J, Acosta M, Reagan JD, Ames BN. Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem J. 1998;332(Pt 1):43–50. doi: 10.1042/bj3320043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lisanti MP, Scherer P, Tang Z-L, Sargiacomo M. Caveolae, caveolin and caveolinrich membrane domains: A signalling hypothesis. Trends In Cell Biology. 1994;4:231–235. doi: 10.1016/0962-8924(94)90114-7. [DOI] [PubMed] [Google Scholar]
- 13.Couet J, Li S, Okamoto T, Scherer PS, Lisanti MP. Molecular and cellular biology of caveolae: Paradoxes and Plasticities. Trends Cardiovasc Med. 1997;7:103–110. doi: 10.1016/S1050-1738(97)00001-7. [DOI] [PubMed] [Google Scholar]
- 14.Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing “pre-assembled signaling complexes” at the plasma membrane. J Biol Chem (Mini-review) 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- 15.Sargiacomo M, Scherer PE, Tang Z-L, Kubler E, Song KS, Sanders MC, Lisanti MP. Oligomeric structure of caveolin: Implications for caveolae membrane organization. Proc Natl Acad Sci U S A. 1995;92:9407–9411. doi: 10.1073/pnas.92.20.9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li S, Song KS, Lisanti MP. Expression and characterization of recombinant caveolin: Purification by poly-histidine tagging and cholesterol-dependent incorporation into defined lipid membranes. J Biol Chem. 1996;271:568–573. [PubMed] [Google Scholar]
- 17.Murata M, Peranen J, Schreiner R, Weiland F, Kurzchalia T, Simons K. VIP21/caveolin is a cholesterol-binding protein. Proc Natl Acad Sci USA. 1995;92:10339–10343. doi: 10.1073/pnas.92.22.10339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song KS, Li S, Okamoto T, Quilliam L, Sargiacomo M, Lisanti MP. Copurification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent free purification of caveolae membranes. J Biol Chem. 1996;271:9690–9697. doi: 10.1074/jbc.271.16.9690. [DOI] [PubMed] [Google Scholar]
- 19.Garcia-Cardena G, Oh P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitric oxide synthase to endothelilal cell caveolae via palm-itoylation: implications for caveolae localization. Proc Natl Acad Sci USA. 1996;93:6448–6453. doi: 10.1073/pnas.93.13.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scherer PE, Lisanti MP, Baldini G, Sargiacomo M, Corley-Mastick C, Lodish HF. Induction of caveolin during adipogenesis and association of GLUT4 with caveolin-rich vesicles. J Cell Biol. 1994;127:1233–1243. doi: 10.1083/jcb.127.5.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mineo C, James GL, Smart EJ, Anderson RGW. Localization of EGF-stimulated Ras/Raf-1 interaction to caveolae membrane. J Biol Chem. 1996;271:11930–11935. doi: 10.1074/jbc.271.20.11930. [DOI] [PubMed] [Google Scholar]
- 22.Liu P, Ying Y, Anderson RG. Plateletderived growth factor activates mitogen-activated protein kinase in isolated caveolae. Proc Natl Acad Sci U S A. 1997;94:13666–13670. doi: 10.1073/pnas.94.25.13666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smart EJ, Foster D, Ying Y-S, Kamen BA, Anderson RGW. Protein kinase C acti-vators inhibit receptor-mediated potocytosis by preventing internalization of caveolae. J Cell Biol. 1993;124:307–313. doi: 10.1083/jcb.124.3.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smart EJ, Ying Y-S, Anderson RGW. Hormonal regulation of caveolae internalization. J Cell Biol. 1995;131:929–938. doi: 10.1083/jcb.131.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schnitzer JE, Liu J, Oh P. Endothelial caveolae have the molecular transport machinery for vesicle budding, docking, and fusion including VAMP, NSF, SNAP, annexins, and GTPases. J Biol Chem. 1995;270:14399–14404. doi: 10.1074/jbc.270.24.14399. [DOI] [PubMed] [Google Scholar]
- 26.Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA, Michel T. Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J Biol Chem. 1996;271:22810–22814. doi: 10.1074/jbc.271.37.22810. [DOI] [PubMed] [Google Scholar]
- 27.Park WY, Park JS, Cho KA, Kim DI, Ko YG, Seo JS, Park SC. Up-regulation of caveolin attenuates epidermal growth factor signaling in senescent cells. J Biol Chem. 2000;275:20847–20852. doi: 10.1074/jbc.M908162199. [DOI] [PubMed] [Google Scholar]
- 28.Cho KA, Ryu SJ, Oh YS, Park JH, Lee JW, Kim HP, Kim KT, Jang IS, Park SC. Morphological adjustment of senescent cells by modulating caveolin-1 status. J Biol Chem. 2004;279:42270–42278. doi: 10.1074/jbc.M402352200. [DOI] [PubMed] [Google Scholar]
- 29.Cho KA, Ryu SJ, Park JS, Jang IS, Ahn JS, Kim KT, Park SC. Senescent phenotype can be reversed by reduction of caveolin status. J Biol Chem. 2003;278:27789–27795. doi: 10.1074/jbc.M208105200. [DOI] [PubMed] [Google Scholar]
- 30.Dasari A, Bartholomew JN, Volonte D, Galbiati F. Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Res. 2006;66:10805–10814. doi: 10.1158/0008-5472.CAN-06-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Volonte D, Zhang K, Lisanti MP, Galbiati F. Expression of caveolin-1 induces premature cellular senescence in primary cultures of murine fibroblasts. Mol Biol Cell. 2002;13:2502–2517. doi: 10.1091/mbc.01-11-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kathuria H, Cao Y, Hinds A, Ramirez MI, Williams MC. ERM is expressed by alveolar epithelial cells in adult mouse lung and regulates caveolin-1 transcription in mouse lung epithelial cell lines. J Cell Biochem. 2007;102:13–27. doi: 10.1002/jcb.21270. [DOI] [PubMed] [Google Scholar]
- 33.van den Heuvel AP, Schulze A, Burgering BM. Direct control of caveolin-1 expression by FOXO transcription factors. Biochem J. 2005;385:795–802. doi: 10.1042/BJ20041449. [DOI] [PMC free article] [PubMed] [Google Scholar]