

GoldenBraid 2.0 is a comprehensive technological framework that facilitates the construction of increasingly complex multigene structures and exchange of genetic building blocks.
Abstract
Plant synthetic biology aims to apply engineering principles to plant genetic design. One strategic requirement of plant synthetic biology is the adoption of common standardized technologies that facilitate the construction of increasingly complex multigene structures at the DNA level while enabling the exchange of genetic building blocks among plant bioengineers. Here, we describe GoldenBraid 2.0 (GB2.0), a comprehensive technological framework that aims to foster the exchange of standard DNA parts for plant synthetic biology. GB2.0 relies on the use of type IIS restriction enzymes for DNA assembly and proposes a modular cloning schema with positional notation that resembles the grammar of natural languages. Apart from providing an optimized cloning strategy that generates fully exchangeable genetic elements for multigene engineering, the GB2.0 toolkit offers an ever-growing open collection of DNA parts, including a group of functionally tested, premade genetic modules to build frequently used modules like constitutive and inducible expression cassettes, endogenous gene silencing and protein-protein interaction tools, etc. Use of the GB2.0 framework is facilitated by a number of Web resources that include a publicly available database, tutorials, and a software package that provides in silico simulations and laboratory protocols for GB2.0 part domestication and multigene engineering. In short, GB2.0 provides a framework to exchange both information and physical DNA elements among bioengineers to help implement plant synthetic biology projects.
Synthetic biology is producing a paradigm shift in biotechnology based on the introduction of engineering principles in the design of new organisms by genetic modification (Check, 2005; Haseloff and Ajioka, 2009). Whereas synthetic biology has rapidly permeated microbial biotechnology, the engineering of multicelled organisms following synthetic biology principles is now emerging and is mainly driven by the so-called top-down approaches, where newly engineered genetic circuits are embedded into naturally existing organisms used as a “chassis.” The plant chassis offers an extraordinarily fertile ground for synthetic biology-like engineering. However, technology still faces the huge challenge of performing engineering-driven genetic designs. One of the main technological challenges of plant synthetic biology requires the construction and transfer of multigene structures to the plant genome. This is putting pressure on developing DNA assembly and transformation technologies adapted to plants. One main trend is the use of modular cloning, an engineering-inspired strategy consisting of the fabrication of new devices by combining prefabricated standard modules. In a modular strategy, predefined categories, the so-called “parts,” are assembled together following a number of rules known as the “assembly standard.” Modular DNA building has been enthusiastically adopted by microbial synthetic biologists because it offers a number of advantages such as speed, versatility, laboratory autonomy, combinatorial potential, and often lower cost (Ellis et al., 2011). Modular methods acquire full potential when parts are easily interchangeable and when one or a few assembly standards are shared by many manufacturers.
A number of features define the value of a modular cloning method. Speed and efficiency are important characteristics, as are also its simplicity and the ability to produce scarless or scar-benign assemblies. Moreover, any cloning strategy for synthetic biology should enable endless reusability; that is, it should ensure that new composite parts themselves can take part in new assemblies, therefore allowing unlimited growth. Several modular cloning strategies have been proposed in the literature, and each presents advantages and shortcomings. For instance, the original BioBrick standard widely used in microbial synthetic biology scores a maximum for simplicity because a single rule governs all the assemblies (a property known as idempotency). However, it is not scar benign and is only relatively efficient (Knight, 2003). Ligation-Independent Cloning (Aslanidis and de Jong, 1990), Uracil-Specific Excision Reagent Fusion (Geu-Flores et al., 2007), and especially Gibson Assembly (Gibson et al., 2009) are highly efficient DNA assembly methods, although they are neither strictly modular nor widely adopted by plant biotechnologists. In sharp contrast, Gateway Cloning (Hartley et al., 2000) is of widespread use in plant laboratories (Karimi et al., 2002, 2007; Estornell et al., 2009). Recently, MultiRound Gateway technologies opened Gateway capabilities to the sequential delivery of multiple transgenes by multiple rounds of recombination reactions (Chen et al., 2006; Vemanna et al., 2013). In general, Gateway-based technologies are highly efficient. Unfortunately, they are not always scar benign, as they leave 21-bp scars between building blocks. Other technologies involving rare cutters or homing endonuclease-based strategies have also been developed and adapted to plant transformation (Lin et al., 2003; Dafny-Yelin and Tzfira, 2007; Fujisawa et al., 2009), including combinations of homing endonucleases and engineered zinc finger nucleases (Zeevi et al., 2012) and iterative in vivo assembly rounds of Cre recombinase and phage1 site-specific recombination (Chen et al., 2010). Many of these techniques can serve as efficient assembly methods for multigene engineering. Nonetheless, a prerequisite to become a standard for plant synthetic biology is the development of a set of rules and tools based on those technologies that can be shared by as many laboratories as possible.
Recently, a very powerful DNA assembly method named Golden Gate was described (Engler et al., 2008, 2009). Golden Gate uses type IIS restriction enzymes to generate four-nucleotide sticky ends flanking each DNA piece, which can be subsequently joined together efficiently by T4 ligase. The assembly reaction is multipartite and is performed in a single-tube reaction to yield highly efficient scarless or scar-benign assemblies. This is because type IIS recognition sites are eliminated upon ligation, leaving only four-nucleotide seams, which can be user defined. These features make the Golden Gate technology an excellent candidate to set up a standardized modular cloning system. However, as originally conceived, Golden Gate is not a reusable system and, therefore, cannot be used efficiently for multigene engineering.
Most recently, two strategies were described to enable the reusability of the Golden Gate cloning scheme: MoClo (Weber et al., 2011a) and GoldenBraid (Sarrion-Perdigones et al., 2011). Both methods use the multipartite Golden Gate property to build transcriptional units (TUs) starting from basic standard building blocks, and both create specially designed destination vectors to enable Golden Gate-built TUs to be assembled among them. Whereas the GoldenBraid minimalist cloning strategy allows multigene growth by enabling binary assemblies between TUs, the MoClo destination vectors offer the interesting possibility of performing multipartite assembly at the TU level, albeit at the cost of the higher complexity of its vector toolkit.
The Golden Gate-based strategies MoClo and GoldenBraid are ideal to serve as modular assembly systems in plant synthetic biology, as they are efficient, reusable, and scar benign. To realize their full potential, it is very important to (1) advance in adopting common standards, so building blocks can be shared by as many users as possible; (2) further optimize the design of cloning strategies to improve speed and efficiency; and (3) improve the user experience by generating new hardware (building blocks and modules) and software (databases and assembly programs) tools that simplify and facilitate the engineering process.
To facilitate the implementation of plant synthetic biology approaches, we present GoldenBraid 2.0 (GB2.0), a new version of the GoldenBraid cloning strategy. In this new version, we defined, in concert with MoClo developers, a common assembly standard by establishing arbitrary, yet scar-benign, assembly seams within a TU that facilitate part exchangeability. In addition, we optimized the versatility of the GoldenBraid strategy by enhancing its minimalist design, creating a universal part entry vector and simplifying the cloning setup. Finally, we generated a collection of premade genetic modules and new software tools for the purpose of facilitating the building of frequently used genetic structures. In short, we present a new grammar for plant synthetic biology and introduce a comprehensive toolkit to facilitate the use of GB2.0 in composing new genetic designs.
RESULTS
The GB2.0 Cloning Strategy
To describe the GB2.0 assembly strategy, we follow an analogy with a natural language because we believe this comparison closely describes the GB2.0 cloning strategy structure and facilitates its understanding. This is because the hierarchical manner in which the different building blocks in GB2.0 are combined to form a multigenic structure become analogous to the way grammar elements (morphemes, words, phrases, and sentences) are combined hierarchically to create a composition. Figure 1A provides an equivalence table between the elements of English grammar and the elements of the GB2.0 system.
Figure 1.

Analogies between GB2.0 and English grammar. A, GB2.0 elements can be compared with those of a natural language. In English grammar (left), morphemes are joined together to make words; words are combined together to make phrases and sentences, which are further joined to make a composition. In GB2.0 (right), the simplest units are GBpatches, used to build any of the 11 standard GBparts. GBpatches can be also combined in GBSparts to facilitate cloning (e.g. a whole promoter). GBparts and GBSparts are combined in a multipartite reaction to build TUs, which can be used for plant transformation, or can be reused and combined with other TUs to build multigene modules. B, Flow chart of the GoldenBraid assembly steps. It starts with the GoldenBraid domestication of GBpatches into GBparts or GBSparts; GBparts are multipartitely combined to build up TUs; finally, TUs are binarily assembled to build modules and multigene constructs. [See online article for color version of this figure.]
GBparts: Words and Phrases. Definition of the GB2.0 Grammar
The first task in upgrading GoldenBraid was to define the minimal standard building blocks in GB2.0, the so-called GBparts, which can be considered the “words” of the GoldenBraid grammar. GBparts are fragments of DNA flanked by four-nucleotide overhangs. They are stored as inserts within a specially designed entry vector (pUPD), from where they are released by cleavage with BsaI or BtgZI restriction enzymes to generate the corresponding flanking overhangs. GBparts are classified into different classes or categories according to their specific functions. Each GoldenBraid class is defined by its flanking four nucleotides, which will overhang upon enzyme digestion and will determine its position within the TU. We defined 11 standard classes (Fig. 2A) that correspond to the basic functional categories in a typical TU. The first three categories (01, 02, and 03) were orderly set in the 5′ nontranscribed region and correspond to operators and promoter regions. Next, we defined seven categories in the transcribed region: one corresponded to the 5′ untranslated region (UTR; 11); one related to the 3′ UTR (17); four were reserved to the coding region (13–16); and an additional class was set as a buffer zone to facilitate, among other designs, the construction of noncoding TUs intended for gene silencing. Lastly, we set a final class (21) for standard 3′ untranscribed GBparts.
Figure 2.

The complete GB2.0 grammar and its most frequently used structures. A, Schematic overview of a TU structure where the 11 standard GoldenBraid classes are depicted: 01, 02, and 03 GBparts form the 5′ nontranscribed region (5′NT); position 11 is the 5′ UTR of mRNA; 12 is a linker region; 13 to 16 (TL1–TL4) are four divisions of the translated region; 17 is the 3′ UTR of mRNA; and 21 is the 3′ nontranscribed region of the TU (3′NT). B, Frequently used structures for the protein-coding TUs. The elements forming each frequently used structure and the class that they belong to are depicted. C, Frequently used structures for RNA silencing, including amiRNA, hpRNA, and tasiRNA. 5′NT and 3′NT as well as 5′ UTR and 3′ UTR are defined above; LINK represents a region between the 5′ UTR and the coding sequence where tags or fused proteins can be placed; PROM is a promoter; CDS is the coding DNA sequence; TER represents the terminator; SP is signal peptide; NT and CT are N- and C-terminal tags or fusion proteins; OP is a promoter operator; minPROM is a minimal promoter; 5′FS and 3′FS indicate the flanking sequences of the amiRNA precursor sequences; Target represents the region of the amiRNA structure comprising the loop and the complementary target sequences; GOI and IOG are the fragments of the gene of interest in an inverted orientation; INT is the intron for hpRNA processing; mir173 represents the mir173 target site for tasiRNA processing; fGOI indicates the fragment gene of interest to be silenced. [See online article for color version of this figure.]
Besides the basic classes, GB2.0 also employs “superclasses.” For practical purposes, it is convenient to group several contiguous basic GBparts that, together, perform a defined function (e.g. a complete promoter or a full coding region) in a single DNA element (a GoldenBraid superpart, abbreviated to GBSpart) instead of splitting it into its basic standard parts. This is analogous to an English phrase, which comprises a group of words that function as a single unit within the hierarchical structure of the sentence syntax (e.g. a subject or a direct complement). As with GBparts, GBSparts are ultimately DNA fragments stored within the pUPD vector. Upon digestion with BsaI or BtgZI, the whole phrase is released as a solid indivisible unit flanked by four-nucleotide “barcodes.” In practice, GBSparts are very convenient, as they reduce the number of elements that need to be assembled to produce a TU; therefore, they enhance efficiency. Frequently used superclasses are depicted in Figure 2, B and C. For example, the promoter regions normally employed in traditional cloning correspond to the superclass (01–12). GBparts and GBSparts are components of the GoldenBraid collection, and their sequence information is stored in the GBdatabase.
GBpart Domestication: Creating Words and Phrases
The process of adapting a DNA building block (GBpart or GBSpart) to the GoldenBraid grammar is referred to as domestication. GoldenBraid domestication usually involves PCR amplification of the target DNA (word or phrase) using GB-adapted primers (for details, see Fig. 3) and the subsequent cloning of the resulting PCR fragment into the pUPD vector using a BsmBI restriction-ligation reaction. Occasionally, domestication may involve the removal of internal BsaI, BsmBI, or BtgZI restriction sites. In order to facilitate an eventual automation of the cloning process, the GB2.0 system includes a standard procedure for internal site removal. This procedure, described in detail in Supplemental Figure S1, involves the amplification of the target DNA in separated fragments (named GBpatches) using GB-adapted primers, which incorporate single mismatches to disrupt the enzyme target sites. Once amplified, GBpatches are reassembled together in a single-tube BsmBI restriction-ligation reaction into pUPD to yield a domesticated GBpart or GBSpart.
Figure 3.

Standardized domestication of GBparts. GBparts are domesticated by amplifying the desired sequence with standard GBprimers (GB.F and GB.R). GBprimers include approximately 20 nucleotides of the gene-specific primer (GSP) and a tail region that includes a BsmBI recognition site, the cleavage site for cloning into pUPD, and the four-nucleotide barcode (1234 and 5678). The amplified DNA part is cloned into pUPD in a restriction-ligation reaction, with BsmBI as the restriction enzyme. The resulting GBpart is cleavable by BsaI and BtgZI to produce 1234 and 5678 flanking overhangs. BsmBI recognition sequences are depicted in orange in the DNA sequence; BsaI and BtgZI are labeled in red and blue, respectively. Enzyme cleavage sites are boxed. [See online article for color version of this figure.]
The GB2.0 Destination Plasmids Kit
GoldenBraid destination vectors (pDGBs) are binary vectors that function as recipients of new assemblies. Each pDGB contains a GBcassette (the selection LacZ gene flanked by two restriction/recognition sites corresponding to two different type IIS enzymes; Fig. 4A). In addition, GB2.0 plasmids include a watermark (i.e. a distinctive restriction site flanking the GBcassette) to help plasmid identification. Detailed information about the sequences of the different GBcassettes is also provided in Figure 4A. The special orientation and arrangement of the restriction enzymes define two levels of pDGBs, the α-level and Ω-level plasmids, which are used for the BsaI- and BsmBI-GoldenBraid reactions, respectively. Plasmids also differ in the resistance marker that is associated with each level (kanamycin for level α and spectinomycin for level Ω, allowing counterselection). To ensure an endless cloning design, a minimum set of four pDGBs is required (pDGBΩ1, pDGBΩ2, pDGBα1, and pDGBα2). Additionally, this set can be expanded to eight plasmids to enable assemblies in different orientations (pDGBΩ1R, pDGBΩ2R, pDGBα1R, and pDGBα2R). For GB2.0, we constructed two complete sets of pDGBs, one based on the pGreenII backbone and another set based on the pCAMBIA backbone. The sequence information of all 16 pDGBs in GB2.0 is uploaded in the GBdatabase.
Figure 4.

GB2.0 cassettes and assembly rules. A, GB2.0 cassettes and their comparison with the previous GoldenBraid version. GBcassettes comprise a LacZ selection cassette flanked by four type IIS restriction sites (BsaI and BsmBI) positioned in inverse orientation. The previous GoldenBraid plasmid kit comprised four destination plasmids, two in each assembly level. GB2.0 incorporates four additional plasmids that permit the assembly of transcriptional units in reverse orientation using the same GBparts. Additionally, the six four-nucleotide barcodes of GoldenBraid (A, B, C, 1, 2, and 3) collapsed in only three GB2.0 barcodes, where A ≡ 1, B ≡ 2, and C ≡ 3. This special design feature permits GBparts to be directly assembled in both level plasmids. Finally, GB2.0 plasmids incorporate distinctive restriction sites flanking the GBcassette as watermarks for plasmid identification. BsaI cleavage sequences are boxed in red, BsmBI cleavage sequences are boxed in orange, and sites where both enzymes can digest are boxed in green. The watermark restriction sites are underlined. B, Rules for multipartite assembly. The pUPD elements represent each GBpart and GBSpart that conforms to a grammatically correct TU, pDGBΩi is any level Ω destination vector, pDGBαi is any level α destination vector, and pEGBΩi (X) and pEGBαi (X) are the resulting expression plasmids harboring a well-constructed transcriptional unit X. C, Rules for binary assembly. (Xi) and (Xj) are composite parts assembled using the multipartite assembly option; (Xi+Xj) is a composite part of (Xi) and (Xj) that follows the same assembly rules as (Xi) and (Xj); pEGBα1(X), pEGBα2(X), pEGBΩ1(X), and pEGBΩ2(X) are expression plasmids hosting a composite part X; and pDGBΩ1, pDGBΩ2, pDGBα1, and pDGBα2 are destination plasmids hosting a LacZ cassette. [See online article for color version of this figure.]
The Composing Strategy: from Single Words to Full Compositions
The GB2.0 cloning strategy comprises two types of assemblies (see the GB2.0 chart in Fig. 1B): multipartite assemblies and binary assemblies. Multipartite assemblies are performed to create single TUs. The different GBparts and GBSparts required to produce a well-constructed TU are mixed together in a single tube in the presence of a pDGB, the corresponding type IIS restriction enzyme(s), and the T4 ligase, and they are incubated in cyclic restriction-ligation reactions. If all the elements are correctly set in the reaction, they orderly assemble within the destination vector and generate a so-called expression vector, which harbors the assembled composite part. Our pDGBs are binary vectors; therefore, the resulting expression clone is ready to be used directly for Agrobacterium tumefaciens-mediated plant transformation.
After building a new TU using a multipartite assembly, the resulting new expression clone can be binarily combined with another expression clone to produce increasingly complex multigene structures analogous to how sentences are combined to create a written composition. The solution provided by GoldenBraid cloning relies on the special design of GoldenBraid destination vectors, which introduces a double loop (braid) into the cloning strategy. A composite part (a TU or a group of TUs) cloned in a given entry vector can be combined only with a second composite part cloned in the complementary entry vectors at the same level. This is done in the presence of a destination vector of the opposite level and generates a new expression vector at the opposite level. A formal notation describing the rules for multipartite and binary assemblies is shown in Figure 4, B and C.
By choosing appropriate combinations of expression and destination vectors, it is possible to create increasingly complex structures, and the only limits are the capacity of the vector backbone or the biological restrictions imposed by bacteria. Moreover, all the new composite parts are fully reusable (they can be used directly for part transformation or can be employed in new assemblies) and exchangeable (they can be combined with the GoldenBraid modules that are produced separately in different laboratories by following the same assembly rules).
Innovative Features in the GB2.0 Cloning Strategy
Besides a proposal for a grammar, GB2.0 introduces a number of new elements that modify the original GoldenBraid cloning design to make it simpler and more versatile. Many of the new GB2.0 features rely on the design of the plasmid that harbors GBparts and GBSparts, the Universal Domesticator (pUPD). The pUPD cassette is designed to serve as a polyvalent entry vector for all the different GBparts and GBSparts, regardless of their category. This is because the four-nucleotide barcodes are incorporated into the GBpart by PCR instead of being imprinted in the plasmid itself. Such a universal plasmid enables us to establish a single standard protocol for all the domestication parts based solely on its sequence information and category specification.
Another innovative feature of pUPD is the incorporation of both BtgZI and BsaI sites flanking the GoldenBraid cassette. The enzyme target sites are arranged in such a way that both BsaI and BtgZI digestions release exactly the same piece of DNA, which contains the same four-nucleotide overhangs, regardless of the enzyme used. This opens up the possibility of GBparts being assembled into the α- and Ω-level vectors indistinctly by using either BsaI reactions and BtgZI/BsmBI reactions, respectively. To enable this option, the GoldenBraid cassettes in the pDGBs have also been redesigned and simplified. In the previous version, the sequences of the restriction sites for BsaI (named A, B, and C) differed from the restriction sites for BsmBI (named 1, 2, and 3). In GB2.0, we made A ≡ 1, B ≡ 2, and C ≡ 3 (for details, see Fig. 4A). In this way, and by making full use of the dual BsaI/BtgZI release from pUPD, any pDGB can be used as a recipient of a multipartite assembly, which, therefore, makes entry in the GoldenBraid loop fully symmetric. Thus, BsaI reactions are performed to build TUs in α-vectors, and BsmBI/BtgZI reactions (BsmBI to open pDGB and BtgZI to release the GBpart) are performed to build TUs in Ω-pDGBs. Furthermore, by choosing any of the reverse pDGB plasmids as recipients, TU orientation can be inverted. This opens up the possibility of creating new binary assemblies in all the possible relative orientations.
The pUPD design provides yet another interesting new feature to GB2.0, as it enables the use of a nonstandard assembly level operating below the standard GBpart level (referred to as the GBpatch level). This feature can be most convenient for a number of applications, including the generation of seamless junctures, introducing combinatorial arrangements into protein engineering, or promoter tinkering using nonstandard positions. The process is similar to the above-described domestication procedure. An example of the use of the GBpatch level for combinatorial antibody engineering is depicted in Supplemental Figure S2.
Frequently Used Structures
There are a limited number of structural types for the majority of synthetic transcriptional units and genetic modules. For instance, many protein-encoding TUs can be constitutively expressed, whereas others are regulated by 5′ (or 3′) operators. The resulting proteins can be preceded by a signal peptide or may contain C-terminal and N-terminal fusions. Besides, noncoding TUs can be used for silencing purposes. To cope with this functional diversity while simplifying the user toolbox, we defined a group of “frequently used structures,” for which specific prearranged GBparts and GBSparts were developed (Fig. 2, B and C). We now go on to describe some of the frequently used structures that are currently included in the GoldenBraid system and their associated tools.
Basic Expression Cassettes for Multigene Engineering
Multigene engineering may require the use of different regulatory regions to avoid the silencing associated with the repeated use of a DNA sequence in the same construct. To meet this requirement, we incorporated several regulatory 5′ and 3′ regions into the GB2.0 collection. Most 5′ regulatory regions are (01–12) GBSparts comprising a promoter and 5′ UTR, whereas 3′ regulatory regions are (17–21) GBSparts comprising 3′ UTR and terminator regions. According to this basic setup, full (13–16) open reading frames can be easily incorporated into tripartite reactions to obtain constitutively expressed TUs. In order to undertake synthetic biology projects, it is very important to have a range of regulatory regions available and that the expression strength provided by each promoter/terminator combination is properly characterized so that the multigene expression can be adjusted accordingly. As a first approach toward the characterization of a set of basic expression cassettes, we finely characterized the relative promoter/terminator strength of a number of cassettes using the Renilla/luciferase system (ratio determined by the Dual Glo Assay System [Promega]) in transiently transformed Nicotiana benthamiana leaves. The characterization of (01–12) and (17–21) regions as individual entities is a relatively straightforward procedure using GB2.0 cloning. However, as the collection grows, the individual characterization of all the possible combinations becomes an intractable task. Therefore, we decided to investigate to what extend the transcriptional strength provided by each “promoter/terminator” (i.e. 01–12_17–21) combination can be inferred from the separated contribution of each region. For this purpose, all the (01–12) promoter regions in the collection were tested by the Renilla/luciferase system in combination with a common (17–21) terminator region (TNos). In parallel, all the (17–21) terminator regions in the collection were tested in combination with a common (01–12) promoter region (PNos). The “experimental transcriptional activity” (ETA) of each region was calculated as being relative to the Renilla/luciferase values of a (01–12_17–21) reference combination (PNos_TNos), which was arbitrarily set as 1 (for construct details, see Fig. 5A). The ETA (01–12) values ranged between 0.47 ± 0.01 and 15.03 ± 1.44 relative luminescence units, whereas the ETA (17–21) values ranged between 0.77 ± 0.18 and 2.61 ± 0.54 (Fig. 5, B and C). Using these data, “theoretical transcriptional activity” (TTA) was calculated for each cassette combination (Fig. 5D) as the product of the individual ETAs of the two regulatory regions. Finally, the Renilla/luciferase ratio of a number of cassette combinations (covering 65% of total possibilities) was also tested experimentally. As we can see in Figure 5E and Supplemental Figure S3, there is good agreement between the theoretical and experimental activity values. Of the 34 experimental combinations assayed in the evaluation test, 31 showed deviation in relation to the theoretical values below 2-fold (±0.3 in logarithmic values; for detailed information, see Supplemental Fig. S3C).
Figure 5.

Characterization of regulatory regions for basic expression cassettes. A, Constructs for ETA quantification. The promoter (01–12)i_(17–21)TNos constructs comprise a first TU with the (01–12) promoter of interest, the firefly luciferase and the nopaline synthase terminator, followed by the Renilla reference module (top row). For the (01–12) TNos _(17–21)j terminator constructs, the first TU comprises the (17–21) terminator of interest and the firefly luciferase and the nopaline synthase promoter (middle row). For activity normalization, the PNos:luciferase:TNos construct combined with the Renilla reference module was used (bottom row). B, The ETA of the promoter regions in (01–12)i_(17–21)TNos constructs was determined as the firefly (FLuc)/Renilla (RLuc) luciferase activity ratio of each construct normalized with the equivalent ratio of the PNos:luciferase:TNos construct. Error bars represent the sd of at least three replicates. C, The ETA of terminator regions in the (01–12)PNos_(17–21)j constructs was estimated as described in B. D, Scheme of the combinatorial promoter/terminator constructs comprising a first TU with a (01–12) promoter, the firefly luciferase, and a (17–21) terminator and combined with the Renilla reference module. E, Correspondence between the ETA and TTA data in the combinatorial constructs. The logarithm of the ratios between the ETA and TTA values for 34 experimental promoter/terminator combinations is plotted. Bars represent average values ± sd. PNos, nopaline synthase promoter; TNos, nopaline synthase terminator; P35s, CaMV 35S promoter; p19, Tomato Bushy Stunt Virus silencing suppressor. [See online article for color version of this figure.]
Regulated Expression Cassettes
The GoldenBraid grammar contains several standard positions for the insertion of regulatory regions. In the 5′ untranscribed region, we defined three standard GBparts to allow combinatorial promoter tinkering and to facilitate the insertion of synthetic operators. As a functionality proof, we assembled and tested the premade cassettes for heat shock and dexamethasone-regulated expression; the latter is based on the “operated promoter A” scheme shown in Figure 2B. The Renilla/luciferase/p19 reporter cassettes constructed with promoters pHSP70 and pHSP18.2 showed clear induction after incubation at 37°C (Supplemental Fig. S4). The potential of the GoldenBraid modular assembly was further demonstrated with the construction of two regulated systems based on the fusion of the glucocorticoid receptor with the DNA-binding domains of LacI or Gal4 and the activation domain of Gal4. In this transactivation example, up to 15 premade modules comprising coding and noncoding regulatory regions were efficiently assembled de novo to produce two operated luciferase TUs, which clearly responded to the presence of dexamethasone (Supplemental Fig. S5).
Protein-Protein Interaction Tools
Reporter fusion partners are powerful analytical tools utilized in the study of protein-protein interactions. However, the use of unlinked cotransformation for the delivery of the interaction partners often compromises the extraction of reliable qualitative data, based on the poorly supported assumption that cotransformation efficiency in each cell is the same for all fusion partners. We reasoned that the linked cotransformation of fusion partners can help improve the sensitivity and accuracy of the protein-protein interaction analysis. Bearing this use in mind, we designed premade modules for the bifluorescence complementation (BiFC) assays. For this purpose, BiFC adaptors with a (01–12) structure were constructed containing the full 35S promoter and the corresponding yellow fluorescent protein fusion partners. Based on this setup, bait and prey with a canonical (13–16) structure can be easily assembled in multipartite reactions to form the required fusion proteins. The prearranged BiFC tools were functionally tested using transcription factors Akin10/Akinβ2 as positive interaction partners and a spermidine synthase as a negative partner (Belda-Palazón et al., 2012). As observed in Supplemental Figure S6, the number of cells showing positive interactions with the GB-assisted linked cotransformation setup outnumbers those of the unlinked cotransformation approach.
Silencing Tools
The negative regulation of endogenous genes often proves an engineering requirement. For this reason, special frequently used structures were defined for three RNA-silencing strategies: transacting small interfering RNA (tasiRNA), artificial microRNA (amiRNA), and hairpin RNA (hpRNA; Supplemental Fig. S7A). Details of all the elements used in the RNA interference designs are provided in Supplemental Table S1.
For the generation of tasiRNA constructs, special (01–11) GBSparts containing the mir173 trigger sequence are required. A cauliflower mosaic virus (CaMV) 35S-based GBSpart for constitutive tasiRNA expression is currently available in the GoldenBraid collection. Regulated or tissue-specific tasiRNA expression can be designed using the GBpatch special feature of GB2.0. For the functional characterization of the tasiRNA structure, a 410-bp fragment of Arabidopsis (Arabidopsis thaliana) phyotoene desaturase (PDS; Felippes et al., 2012) was incorporated as a (12–16) GBSpart and was transformed into Arabidopsis to yield approximately 0.1% seedlings with the albino phenotype (Supplemental Fig. S7C). The tasiRNA constructs require the coexpression of miR173 for effective silencing in plant species other than Arabidopsis (Felippes et al., 2012). To extend the species range of the tasiRNA tool, a new TU with a constitutively expressed miR173 was constructed and incorporated into the collection. The functionality of the dual construct was tested transiently in N. benthamiana using PDS as the silencing target, which resulted in the bleaching of the infiltrated area (Supplemental Fig. S7D).
An amiRNA silencing tool was also enabled with the creation of two special GBSparts, namely 5′FS and 3′FS. These GBSparts require noncanonical barcodes to allow the seamless assembly of 5′FS and 3′FS in the amiRNA precursor. The special categories are denoted as (12–13B) and (16B), respectively, where B indicates the four noncanonical flanking nucleotides (GTGA and TCTC, respectively). The standard (01–11) promoters without ATG and the (17–21) terminators were used in the amiRNA design. The central region (14B–15B), containing a fragment of the gene target sequence, was constructed using gene-specific oligonucleotides, as described in Supplemental Figure S7B. In order to validate the proposed structure, Arabidopsis PDS silencing was assayed using a gene target fragment that was formerly described by Yan et al. (2011; Supplemental Fig. S7E). The resulting amiRNA construct was transformed into Arabidopsis, yielding seedlings with the albino phenotype.
Finally, in the hpRNA structure, the regulatory regions lacking ATG are inserted as (01–11) parts. An intron from tomato (Solanum lycopersicum; SGN-U324070) was incorporated into the collection to serve as a (14–15) intron GBpart. The inverted fragments of the target gene of interest can be cloned at positions (12–13) and at position (16).
GW-GB Adapter Tool
A GW-GB adapter tool was incorporated into the GB2.0 collection in order to facilitate the transition between the Gateway and GB2.0 assembly methods (Supplemental Fig. S8). GW-GB adapters are GBparts or GBSparts [e.g. a (12–16) GBSpart to adapt coding regions] made of a Gateway cassette flanked by attR1-attR2 sites and embedded inside the pUPD plasmid. As such, adapter vectors can be used directly as destination plasmids for Gateway entry clones flanked by attL1-attL2 sites. In this way, Gateway entry clones can be transferred individually or in bulk to the pUPD plasmid and become ready-to-use GBSparts. Alternatively, the GW-GB adapter can be employed as an ordinary GBSpart to create a new multigene construct in a binary vector. Consequently, the resulting multigene construct becomes a Gateway destination vector containing an attR1-attR2 Gateway cassette, where Gateway entry clones can be inserted individually or in bulk. It should be noted that direct Gateway-to-GB2.0 adaptation does not remove internal enzyme target sites; therefore, the efficiency of subsequent assembly reactions can lower.
GoldenBraid Collection and Software Tools
When this paper was being written, our in-house GoldenBraid collection contained more than 400 entries. As the collection grows, engineering is becoming increasingly easy and fast, because, on occasion, the required GBparts, GBSparts, and TUs are already domesticated and/or constructed. To efficiently handle this collection, we developed a Web framework that hosts a GBdatabase and offers software tools to facilitate the assembly process. The GB2.0 Web site was implemented using Django, a Python Web framework that supports rapid design and the development of Web-based applications (version 1.5; Django Software Foundation; http://djangoproject.com). The object-relational database management system PostgreSQL was chosen to host our schema, which allowed incorporation of the sequences of all the elements included in the collection. Additional relevant information on part identity, functionality, and indexing is also provided.
Given the simplicity of the GoldenBraid assembly rules, it was relatively straightforward to develop software tools that assist in GB2.0 assembly. Therefore, we developed a software package comprising three programs, each program corresponding to one of the three basic processes in GB2.0 assembly. The first program, named GBDomesticator, assists the part adaptation process to the GoldenBraid standard. It takes an input DNA sequence provided by the user and offers the best PCR strategy to remove internal enzyme target sites and to add flanking nucleotides to it according to the specified category. A second program, known as the TUassembler, takes GBparts and GBSparts from the database and simulates a multipartite assembly in silico. The TUassembler includes shortcuts to frequently used structures assembly as well as a free-hand option. Finally, a third program, BinaryAssembler, performs in silico binary assemblies between the composite parts stored in the GoldenBraid database. BinaryAssembler offers the possibility of choosing the relative orientation of each member of the assembly. All three programs generate a detailed laboratory protocol to perform the domestication/assembly and to return a GenBank-formatted file containing the final domesticated/assembled sequence. The GoldenBraid database and software tools are available at http:/www.gbcloning.org.
DISCUSSION
The aim of this work is to provide a standard framework for DNA assembly in plant synthetic biology. We, and others, realized that the modularity of the multipartite assembly based on type IIS enzymes offers a great opportunity for standardization by following a positional information scheme that resembles the grammar of a sentence in many natural languages. Indeed, it is illustrative to conceive the transcriptional unit as a similar structure to a sentence, which is made up of hierarchically assembled elements like morphemes, words, and phrases. It is also interesting to envision the whole engineering process as a way to imprint instructions using DNA strings. Therefore, we, in concert with MoClo developers, propose a common grammar where the four-nucleotide overhangs are predefined for each position within the transcriptional unit. Overhang assignation is mainly arbitrary, but some decisions are made to make them scar benign. For instance, the 12-13 boundary defining the beginning of the coding DNA sequence was designed to include the start codon, whereas the 13-14 boundary was made compatible with signal-peptide cleavage sites.
In our view, this new GB2.0 cloning scheme has a number of features that make it a good candidate for a plant assembly standard. Many of those features are consubstantial to the Golden Gate system: very high efficiency, modularity, and the ability to produce scare-benign assemblies. GB2.0 also incorporates the reusability and modularity of the GoldenBraid and MoClo systems and goes beyond them in that it provides a standardized framework, goes deep into the versatility and the minimalist design of the GoldenBraid loop, and incorporates new tools to assist cloning.
A major drawback of defining a standard is loss in versatility, since no standard can cope with all custom design requirements. To deal with this problem, we incorporated an underlying nonstandard assembly level that makes full use of the newly designed pUPD vector. At this level, nonstandard GBpatches can be custom designed, for instance, for scarless assembly by choosing the appropriate four-nucleotide overhangs. GBpatches are assembled together into standard GBparts or GBSparts. We made full use of the GBpatch level for BiFC, amiRNA, and antibody engineering. Other possible uses include promoter tinkering or nonstandard combinatorial assemblies within the coding DNA sequence, as exemplified in the construction of customized Transcription Activator-Like effectors (Weber et al., 2011b; Li et al., 2012). Additionally, the GBpatch level is used for GBpart domestication (i.e. for the removal of internal enzyme recognition sites). This feature is also enabled by the special design of the new entry vector pUPD, which introduces inversely oriented BsmBI sites into the GoldenBraid cassette. This new design turns the pUPD plasmid into a universal entry vector, as the four nucleotides conferring part identity are not located in the entry vector, as they are in previous designs (Weber et al., 2011a). Instead, in the present setup, the four-nucleotide barcode is incorporated into the primers used during initial part/patch isolation. As a toll, this strategy involves the requirement of longer PCR primers during initial part isolation. This minor drawback is by far compensated by the simplicity introduced by the universal domesticator: in the absence of this solution, a minimum of 11 different entry vectors would be required to harbor the different categories in the GoldenBraid grammar, along with an unaffordable amount of additional vectors to allow the formation of all the possible “phrasal” combinations.
The underlying GoldenBraid cloning pipeline has been substantially simplified in the GB2.0 version to reduce redundancy and to achieve a minimalist design. Figure 6 depicts the comparison of GB2.0 and the previous GoldenBraid structure. Once again, most of the improvement achieved stems from the specific design of the new entry vector pUPD. First, the asymmetry of the cloning loop is corrected in GB2.0 with the introduction of a BtgZI site into the entry vector. BtgZI is a special enzyme that cuts 10 nucleotides away from its recognition site. This feature enables a dual-release option for each GBpart: BsaI release allows cloning in α-destination vectors, whereas BtgZI release allows cloning in Ω-destination vectors. We noted that BtgZI/BsmBI assemblies are less efficient than BsaI ones. Despite this drawback, the ability to create new TUs in both destination vectors can save one cloning step, which speeds up the construction of new multigene assemblies and opens up new possibilities for automation.
Figure 6.

GoldenBraid versus GB2.0. The previous GoldenBraid version had an asymmetric assembly flow (left). GBparts incorporated either the BsaI or the BsmBI releasable overhangs. BsaI-released GBparts were incorporated into the GoldenBraid cloning loop through level α vectors, whereas the BsmBI GBparts were used to build composite parts through the level Ω entry point. In the new GB2.0 symmetric design, the same GBparts can be incorporated into level α plasmids by a BsaI restriction/ligation reaction or into level Ω vectors by a mixed BsmBI/BtgZI reaction. Other differences between the previous GoldenBraid version and GB2.0 are also listed. TA Cloning, PCR bands cloned using adenosine/timine overgangs. [See online article for color version of this figure.]
We also developed a number of tools to assist users in their engineering projects. First, we anticipated genetic designer needs by prearranging a number of frequently used structures. Then, we populated our in-house collection with all the elements (GBparts, GBSparts, and software tools) required to enable the use of the frequently used structures. Finally, we assayed the functionality of newly developed elements using in planta assays. In certain cases, this implied an initial step toward part characterization. One of the hallmarks of synthetic biology is its ability to predict the behavior of a system based on the characteristics of its constitutive parts. We show herein that it is possible to infer the activity provided by a “promoter + terminator” pair from the activities that each individual element display when separately assayed. The differences observed between the theoretical and experimental activity values fall within a narrow range that comes close to 0, with very few combinations showing deviations that are slightly above 2-fold (±0.3 in log values). This finding is important for engineering attempts that, as in complex metabolic engineering, require the combination of many different noncoding parts to create large metabolic pathways, while avoiding the introduction of unstable repetitive regions into the genetic design. The promoter parts assayed herein reveal a wider range of activities than terminators. Nevertheless, we confirm that the use of strong terminators like TAtHSP18.2 can promote the promoter’s transcriptional activity, as described previously (Nagaya et al., 2010). It is interesting that most of the observed positive deviations result from the combinations involving CaMV 35S-derived parts, suggesting a nonlinear behavior of the CaMV 35S regulatory elements. We employed N. benthamiana transient expression and the Renilla/luciferase reporter system (Grentzmann et al., 1998) as a first step toward the characterization of regulatory elements. This transient methodology is simple and accurate and, therefore, facilitates the analysis. A more detailed characterization may need to include the developmental and tissue-specific information obtained through stable plant transformation.
Both GB2.0 and the GoldenBraid collections come into being with an open-source vocation. We reinforced this point by developing a new set of GoldenBraid destination vectors based on open-source pCAMBIA binary vectors (Roberts et al., 1997; Chi-Ham et al., 2012). As we see it, the intellectual commons Intellectual Property model is that which best suits the requirements for the free exchange of parts and modules in plant synthetic biology (Oye and Wellhausen, 2010). Nevertheless, a number of issues, such as the Intellectual Property of individual parts and the ability to freely distribute them, need to be addressed in a concerted manner. Undoubtedly, community effort made to create publicly available collections of synthetic parts will have an impact on the progress of this discipline.
Plant synthetic biology has the potential of bringing about a significant impact on crop production. Engineering enhanced abiotic stress tolerance for growth in marginal lands, turning C3 plants into C4 plants (Caemmerer et al., 2012), constructing whole-organism biosensors or sentinels (Antunes et al., 2011), engineering highly challenging metabolic routes (Farre et al., 2012), and combinations of these are just some examples of high-impact goals within the reach of biotechnologists. Also, it has not escaped our notice that the proposed grammar can be easily adopted by other nonplant systems as well. We believe that technologies like GB2.0, which enable the standardization and facilitate the characterization and exchange of genetic parts and modules, are important contributions to the achievement of the challenging biotechnology goals ahead.
MATERIALS AND METHODS
Strains and Growth Conditions
Escherichia coli DH5α was used for cloning. Agrobacterium tumefaciens strain GV3101 was used for transient expression and transformation experiments. Both strains were grown in Luria-Bertani medium under agitation (200 rpm) at 37°C and 28°C, respectively. Ampicillin (50 µg mL−1), kanamycin (50 µg mL−1), and spectinomycin (100 µg mL−1) were used for E. coli selection. Rifampicin, tetracycline, and gentamicin were also used for A. tumefaciens selection at 50, 12.5, and 30 µg mL−1, respectively. 5-Bromo-4-chloro-3-indolyl-β-d-galactopyranoside acid (40 μg mL−1) and isopropylthio-β-galactoside (0.5 mm) were used on Luria-Bertani agar plates for the white/blue selection of clones.
Restriction-Ligation Assembly Reactions
Restriction-ligation reactions were set up as described elsewhere (Sarrion-Perdigones et al., 2011) using BsaI, BsmBI, BtgZI, or BbsI as restriction enzymes (New England Biolabs) and T4 Ligase (Promega). Reactions were set up in 25- or 50-cycle digestion/ligation reactions (2 min at 37°C, 5 min at 16°C), depending on assembly complexity. One microliter of the reaction was transformed into E. coli DH5α electrocompetent cells, and positive clones were selected in solid medium. Plasmid DNA was extracted using the E.Z.N.A. Plasmid Mini Kit I (Omega Bio-Tek). Assemblies were confirmed by restriction analysis and sequencing.
GBpart Domestication
GBparts and GBpatches were obtained by PCR amplification using suitable templates. The Phusion High-Fidelity DNA Polymerase (ThermoScientific) was used for amplification following the manufacturer’s protocols. Primers smaller than 60-mers were purchased from Sigma-Aldrich. 60-mer or longer oligonucleotides were synthesized by IDTDNA (Coralville) by the Ultramer technology. Amplified bands were purified using the QIAquick PCR Purification Kit (Qiagen) and were quantified in the Nano Drop Spectrophotometer 2000. Then, 40 ng of each amplicon and 75 ng of the domestication vector (pUPD) were mixed and incubated in a BsmBI restriction-ligation reaction. The pUPD sequence is deposited in the GBdatabase. Positive clones were selected on ampicillin-, 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside acid, and isopropylthio-β-galactoside-containing plates, and the correct assembly was confirmed by restriction analyses and sequencing. A description of the GBparts and GBSparts employed in this work is provided in Supplemental Table S1. The nucleotide sequences of all the GoldenBraid parts in the collection are deposited in the GoldenBraid database.
pDGB Construction
Two pDGB series, pDGB1 and pDGB2, were constructed. pDGB1 is based on the pGreenII backbone (Hellens et al., 2000), and pDGB2 is based on pCAMBIA (Roberts et al., 1997). For pDGB construction, the backbone of each binary vector was divided into fragments (vector modules). The pDGB1 backbone comprised two fragments, whereas the pDGB2 backbone was divided into four modules, given the presence of internal sites. To build vector modules, each fragment was amplified by PCR in a similar procedure to that described for GBparts and was cloned into a vector domestication plasmid (pVD) using a BsaI digestion-ligation reaction. The pVD vector was derived from pUPD; its sequence was deposited in GBdatabase. In addition to the backbone modules, a number of common modules were built: eight GBcassettes (α1, α1R, α2, α2R, Ω1, Ω1R, Ω2, and Ω2R) and two fragments encoding spectinomycin and kanamycin resistance. To assemble each pDGB, a BbsI restriction-ligation reaction was performed by combining the modules of the vector backbone, the desired GBcassette, and appropriate antibiotic resistance.
Nicotiana benthamiana Transient Transformation
For the transient expression experiments, plasmids were transferred to A. tumefaciens strain GV3101 by electroporation. Agroinfiltration was performed as described previously (Wieland et al., 2006). Overnight-grown bacterial cultures were pelleted and resuspended in agroinfiltration medium (10 mm MES, pH 5.6, 10 mm MgCl2, and 200 µm acetosyringone) to an optical density at 600 nm of 0.5. Infiltrations were carried out using a needle-free syringe in leaves 2, 3, and 4 of 4- to 5-week-old N. benthamiana plants (growing conditions: 24°C day/20°C night in a 16-h-light/8-h-dark cycle). Depending on the purpose of the experiments, leaves were harvested 3 to 5 d post infiltration and examined for transgene expression.
Arabidopsis Stable Transformation
Arabidopsis (Arabidopsis thaliana) accession Columbia plants were transformed by the floral dip method (Clough and Bent, 1998). Seeds were sterilized and plated on plates of Murashige and Skoog medium with 0.8% (w/v) agar and 1% (w/v) Suc (growing conditions: 24°C day/20°C night in a 16-h-light/8-h-dark cycle). Transgenic lines were selected without antibiotic resistance, as PDS silencing-transformed lines showed the albino phenotype.
Renilla/Luciferase Expression Assays
In order to measure the activity of Renilla/luciferase reporters (Grentzmann et al., 1998), three or four N. benthamiana leaves were agroinfiltrated following the above-described procedure. Leaves were harvested 3 d post infiltration. Firefly luciferase and Renilla luciferase were assayed from 100-mg leaf extracts following the Dual-Glo Luciferase Assay System (Promega) standard protocol and were quantified with a GloMax 96 Microplate Luminometer (Promega). The ETA of each region was calculated in relation to the Renilla/luciferase values of a (01–12_17–21) reference combination (PNos_TNos), which was arbitrarily set as 1, according to the formulae:
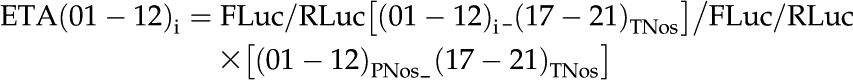 |
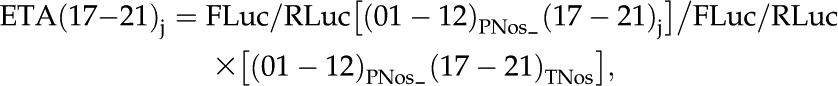 |
where FLuc/RLuc [(01–12)i_(17–21)j] refers to the ratio between the firefly luciferase activity (FLuc) of a (01–12)i:luciferase:(17–21)j construct and the Renilla luciferase activity (RLuc) of a 35S:Renilla:TNos internal standard construct. TTA was calculated for each cassette combination as the product of the individual ETA of the two regulatory regions, as follows:
Finally, the FLuc/RLuc of a number of cassette combinations was tested experimentally, and the ETA of each combination (ETAij) was calculated with the formula:
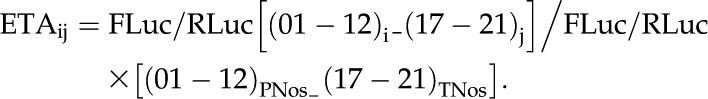 |
Glucocorticoid Receptor Induction and Heat-Shock Treatments
Discs of 1 cm2 from agroinfiltrated leaves were harvested at 3 d post infiltration, placed in a 350-μL solution containing 5 to 20 μm dexamethasone (Sigma-Aldrich) in 0.02% Tween 80, and incubated overnight in a growth chamber. Firefly luciferase and Renilla luciferase activities were measured after 24 h of treatment (6-h-light/8-h-dark, 24°C). For the heat-shock treatments, 1 cm2 of leaves at 3 d post infiltration was placed in 350 μL of water at 37°C for 2 h. Samples were collected at 3 and 14 h after treatment.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. GB Domestication with the removal of internal restriction sites.
Supplemental Figure S2. Combinatorial antibody engineering using the GBpatch assembly level.
Supplemental Figure S3. Theoretical and experimental transcriptional activity of different promoter/terminator combinations.
Supplemental Figure S4. Functional characterization of heat-shock promoters.
Supplemental Figure S5. Functional characterization of transactivation constructs.
Supplemental Figure S6. Frequently Used Structures (FUS) for the protein-protein interaction analysis.
Supplemental Figure S7. The Frequently Used Structures (FUS) for endogenous gene silencing.
Supplemental Figure S8. Adapting Gateway (GW) technology to GB2.0.
Supplemental Table S1. GBparts used in this work.
Supplementary Material
Acknowledgments
We particularly thank Dr. S. Marillonnet for his efforts in agreeing on a common standard for MoClo and GB2.0. Many people provided us with material and knowledge, which helped us to build our GBcollection. From our research center (Instituto de Biología Molecular y Celular de Plantas), Dr. M.A. Blázquez and Dr. D. Alabadí shared their knowledge on Arabidopsis promoters, Dr. A. Ferrando shared the BiFC vectors, Dr. C. Ferrandiz helped with the protein-protein interaction examples, Dr. J. Carbonell and Dr. C. Urbez helped with the amiRNA work, while Dr. J.A. Darós provided assistance with the fluorescent proteins and silencing suppressors. HSP70B was provided by Prof. A. Maule (John Innes Centre). Dr. F. Felippes and Dr. D. Wiegel (Max Planck Institute for Developmental Biology) shared detailed information on the microRNA-induced gene silencing design.
Glossary
- TU
transcriptional unit
- GB2.0
GoldenBraid 2.0
- UTR
untranslated region
- ETA
experimental transcriptional activity
- TTA
theoretical transcriptional activity
- BiFC
bifluorescence complementation
- tasiRNA
transacting small interfering RNA
- amiRNA
artificial microRNA
- hpRNA
hairpin RNA
- PDS
phyotoene desaturase
- CaMV
cauliflower mosaic virus
References
- Antunes MS, Morey KJ, Smith JJ, Albrecht KD, Bowen TA, Zdunek JK, Troupe JF, Cuneo MJ, Webb CT, Hellinga HW, et al. (2011) Programmable ligand detection system in plants through a synthetic signal transduction pathway. PLoS ONE 6: e16292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslanidis C, de Jong PJ. (1990) Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res 18: 6069–6074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belda-Palazón B, Ruiz L, Martí E, Tárraga S, Tiburcio AF, Culiáñez F, Farràs R, Carrasco P, Ferrando A. (2012) Aminopropyltransferases involved in polyamine biosynthesis localize preferentially in the nucleus of plant cells. PLoS ONE 7: e46907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caemmerer S, Quick W, Furbank R (2012) The development of C4 rice: current progress and future challenges. Science 336: 1671–1672 [DOI] [PubMed] [Google Scholar]
- Check E. (2005) Synthetic biology: designs on life. Nature 438: 417–418 [DOI] [PubMed] [Google Scholar]
- Chen QJ, Xie M, Ma XX, Dong L, Chen J, Wang XC. (2010) MISSA is a highly efficient in vivo DNA assembly method for plant multiple-gene transformation. Plant Physiol 153: 41–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QJ, Zhou HM, Chen J, Wang XC. (2006) A Gateway-based platform for multigene plant transformation. Plant Mol Biol 62: 927–936 [DOI] [PubMed] [Google Scholar]
- Chi-Ham CL, Boettiger S, Figueroa-Balderas R, Bird S, Geoola JN, Zamora P, Alandete-Saez M, Bennett AB. (2012) An intellectual property sharing initiative in agricultural biotechnology: development of broadly accessible technologies for plant transformation. Plant Biotechnol J 10: 501–510 [DOI] [PubMed] [Google Scholar]
- Clough SJ, Bent AF. (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16: 735–743 [DOI] [PubMed] [Google Scholar]
- Dafny-Yelin M, Tzfira T. (2007) Delivery of multiple transgenes to plant cells. Plant Physiol 145: 1118–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis T, Adie T, Baldwin GS. (2011) DNA assembly for synthetic biology: from parts to pathways and beyond. Integr Biol (Camb) 3: 109–118 [DOI] [PubMed] [Google Scholar]
- Engler C, Gruetzner R, Kandzia R, Marillonnet S. (2009) Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS ONE 4: e5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C, Kandzia R, Marillonnet S. (2008) A one pot, one step, precision cloning method with high throughput capability. PLoS ONE 3: e3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estornell LH, Orzáez D, López-Peña L, Pineda B, Antón MT, Moreno V, Granell A. (2009) A multisite Gateway-based toolkit for targeted gene expression and hairpin RNA silencing in tomato fruits. Plant Biotechnol J 7: 298–309 [DOI] [PubMed] [Google Scholar]
- Farre G, Naqvi S, Sanahuja G, Bai C, Zorrilla-López U, Rivera SM, Canela R, Sandman G, Twyman RM, Capell T, et al. (2012) Combinatorial genetic transformation of cereals and the creation of metabolic libraries for the carotenoid pathway. Methods Mol Biol 847: 419–435 [DOI] [PubMed] [Google Scholar]
- Felippes FF, Wang JW, Weigel D. (2012) MIGS: miRNA-induced gene silencing. Plant J 70: 541–547 [DOI] [PubMed] [Google Scholar]
- Fujisawa M, Takita E, Harada H, Sakurai N, Suzuki H, Ohyama K, Shibata D, Misawa N. (2009) Pathway engineering of Brassica napus seeds using multiple key enzyme genes involved in ketocarotenoid formation. J Exp Bot 60: 1319–1332 [DOI] [PubMed] [Google Scholar]
- Geu-Flores F, Nour-Eldin HH, Nielsen MT, Halkier BA. (2007) USER fusion: a rapid and efficient method for simultaneous fusion and cloning of multiple PCR products. Nucleic Acids Res 35: e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, III, Smith HO. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345 [DOI] [PubMed] [Google Scholar]
- Grentzmann G, Ingram JA, Kelly PJ, Gesteland RF, Atkins JF. (1998) A dual-luciferase reporter system for studying recoding signals. RNA 4: 479–486 [PMC free article] [PubMed] [Google Scholar]
- Hartley JL, Temple GF, Brasch MA. (2000) DNA cloning using in vitro site-specific recombination. Genome Res 10: 1788–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseloff J, Ajioka J. (2009) Synthetic biology: history, challenges and prospects. J R Soc Interface (Suppl 4) 6: S389–S391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellens RP, Edwards EA, Leyland NR, Bean S, Mullineaux PM. (2000) pGreen: a versatile and flexible binary Ti vector for Agrobacterium-mediated plant transformation. Plant Mol Biol 42: 819–832 [DOI] [PubMed] [Google Scholar]
- Karimi M, Bleys A, Vanderhaeghen R, Hilson P. (2007) Building blocks for plant gene assembly. Plant Physiol 145: 1183–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi M, Inzé D, Depicker A. (2002) GATEWAY vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci 7: 193–195 [DOI] [PubMed] [Google Scholar]
- Knight TF (2003) Idempotent Vector Design for Standard Assembly of BioBricks. MIT Synthetic Biology Working Group Technical Reports, Boston [Google Scholar]
- Li L, Piatek MJ, Atef A, Piatek A, Wibowo A, Fang X, Sabir JS, Zhu JK, Mahfouz MM. (2012) Rapid and highly efficient construction of TALE-based transcriptional regulators and nucleases for genome modification. Plant Mol Biol 78: 407–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Liu Y-G, Xu X, Li B. (2003) Efficient linking and transfer of multiple genes by a multigene assembly and transformation vector system. Proc Natl Acad Sci USA 100: 5962–5967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaya S, Kawamura K, Shinmyo A, Kato K. (2010) The HSP terminator of Arabidopsis thaliana increases gene expression in plant cells. Plant Cell Physiol 51: 328–332 [DOI] [PubMed] [Google Scholar]
- Oye KA, Wellhausen R (2010) The intellectual commons and property in synthetic biology. In M Schmidt, ed, Synthetic Biology: The Technoscience and Its Societal Consequences. Springer, Houten, The Netherlands, pp 121–140 [Google Scholar]
- Roberts C, Rajagopal S, Smith LM, Nguyen TA, Yang W, Nugrohu S, Ravi KS, Vijayachandra K, Harcourt RL, Dransfield L, et al (1997) A Comprehensive Set of Modular Vectors for advanced Manipulations and Efficient Transformation of Plants. pCAMBIA Vector Release Manual Rockefeller Foundation Meeting of the International Program on Rice Biotechnology, September 15-19, Malacca, Malaysia [Google Scholar]
- Sarrion-Perdigones A, Falconi EE, Zandalinas SI, Juárez P, Fernández-del-Carmen A, Granell A, Orzaez D. (2011) GoldenBraid: an iterative cloning system for standardized assembly of reusable genetic modules. PLoS ONE 6: e21622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemanna RS, Chandrashekar BK, Hanumantha Rao HM, Sathyanarayanagupta SK, Sarangi KS, Nataraja KN, Udayakumar M. (2013) A modified multisite Gateway cloning strategy for consolidation of genes in plants. Mol Biotechnol 53: 129–138 [DOI] [PubMed] [Google Scholar]
- Weber E, Engler C, Gruetzner R, Werner S, Marillonnet S. (2011a) A modular cloning system for standardized assembly of multigene constructs. PLoS ONE 6: e16765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber E, Gruetzner R, Werner S, Engler C, Marillonnet S. (2011b) Assembly of designer TAL effectors by Golden Gate cloning. PLoS ONE 6: e19722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland WH, Lammers A, Schots A, Orzáez DV. (2006) Plant expression of chicken secretory antibodies derived from combinatorial libraries. J Biotechnol 122: 382–391 [DOI] [PubMed] [Google Scholar]
- Yan H, Deng X, Cao Y, Huang J, Ma L, Zhao B. (2011) A novel approach for the construction of plant amiRNA expression vectors. J Biotechnol 151: 9–14 [DOI] [PubMed] [Google Scholar]
- Zeevi V, Liang Z, Arieli U, Tzfira T. (2012) Zinc finger nuclease and homing endonuclease-mediated assembly of multigene plant transformation vectors. Plant Physiol 158: 132–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.