

Comparison of the agronomic and metabolic trait variation between drought-sensitive and drought-tolerant wheat crosses uncovers novel correlations between agronomic traits and the levels of certain metabolites as well as important regions on the wheat genome that require further investigation.
Abstract
Drought is a major environmental constraint responsible for grain yield losses of bread wheat (Triticum aestivum) in many parts of the world. Progress in breeding to improve complex multigene traits, such as drought stress tolerance, has been limited by high sensitivity to environmental factors, low trait heritability, and the complexity and size of the hexaploid wheat genome. In order to obtain further insight into genetic factors that affect yield under drought, we measured the abundance of 205 metabolites in flag leaf tissue sampled from plants of 179 cv Excalibur/Kukri F1-derived doubled haploid lines of wheat grown in a field experiment that experienced terminal drought stress. Additionally, data on 29 agronomic traits that had been assessed in the same field experiment were used. A linear mixed model was used to partition and account for nongenetic and genetic sources of variation, and quantitative trait locus analysis was used to estimate the genomic positions and effects of individual quantitative trait loci. Comparison of the agronomic and metabolic trait variation uncovered novel correlations between some agronomic traits and the levels of certain primary metabolites, including metabolites with either positive or negative associations with plant maturity-related or grain yield-related traits. Our analyses demonstrate that specific regions of the wheat genome that affect agronomic traits also have distinct effects on specific combinations of metabolites. This approach proved valuable for identifying novel biomarkers for the performance of wheat under drought and could facilitate the identification of candidate genes involved in drought-related responses in bread wheat.
Bread wheat (Triticum aestivum) is one of the world’s most important food crops, contributing about one-fifth of human caloric intake (Shiferaw et al., 2013) and having an international trade volume greater than all other major food crops combined (Atchison and Head, 2010). Since arable land area will not increase much beyond present levels, increasing demands for wheat must be met through improved wheat yields. Furthermore, global environmental changes will intensify the need to develop crops with tolerance to abiotic stresses, especially water deficits. Worldwide, drought and other abiotic stresses significantly reduce agricultural productivity, with losses estimated at 50% or more (Bray et al., 2000). Wheat is an important crop in which to investigate tolerance to abiotic stress because it is grown in diverse environments and has great potential for adaptation to environmental conditions (Worland et al., 1994).
Plants are known to adjust morphologically, physiologically, and biochemically to water stress (Bohnert et al., 1995; Zhang et al., 2000; Vinocur and Altman, 2005). Epicuticular wax may accumulate, causing leaves to be glaucous, stomatal resistance may increase to reduce water loss, and root systems may adjust to increase water uptake either at depth or from intermittent rainfall events. Compatible solutes may accumulate, including amino acids (e.g. Pro), sugars (e.g. trehalose and fructans), sugar alcohols (e.g. mannitol), and amines (e.g. glycinebetaine) in plant cells.
Knowledge of the locations and the effects of genes that influence agronomic characters (traits) under drought stress could inform crop breeding and provide markers for selection. Many of these traits display quantitative (continuous) variation. Genetic loci affecting such traits are referred to as quantitative trait loci (QTLs). These loci may interact with each other (epistasis) and/or with environmental factors.
Despite considerable research to map QTLs and to quantify their effects and interactions, the biological and molecular basis of most quantitative trait variation remains poorly understood. With the application of metabolomic methods to tissue samples from mapping populations, it is possible to link metabolite profiles with genetic maps. Metabolomic phenotypes have been used for QTL mapping in mapping populations of several plant species, including Arabidopsis (Arabidopsis thaliana; Keurentjes et al., 2006; Lisec et al., 2008; Rowe et al., 2008), tomato (Solanum lycopersicum; Schauer et al., 2006), potato (Solanum tuberosum; Carreno-Quintero et al., 2012), and maize (Zea mays; Lisec et al., 2011). In some experiments, transcriptomic and/or proteomic data have been used in combination with metabolomic data and QTL analysis (Wentzell et al., 2007; Ferrara et al., 2008). Ultimately, the combination of transcriptomics, proteomics, metabolomics, and QTL analysis has the potential to reveal interactions and relationships among genes, transcripts, proteins, metabolites, and traits.
The parents of the mapping population used here, cv Excalibur and Kukri, were both developed for production in low-yielding environments in Australia that are subject to cyclic, Mediterranean-type drought (Izanloo et al., 2008). Under optimal growth conditions, either of these cultivars could yield over 10 tons ha−1, whereas under cyclic drought conditions, their yields can drop to about 2 tons ha−1, a loss of around 80% (Edwards, 2012). For wheat, this level of stress is common, with mean yields of only 3 tons ha−1 globally and 1.6 tons ha−1 in Australia (Shiferaw et al., 2013). The Excalibur cv is considered to be more drought tolerant than cv Kukri; under severe drought conditions, it outyields cv Kukri by between 10% and 40% (Izanloo et al., 2008; Fleury et al., 2010).
The Excalibur and Kukri cv differ for many traits, including plant height, grain number per spike, number of fertile tillers, osmotic adjustment, water use efficiency, stomatal conductance, chlorophyll content, and biomass (Izanloo et al., 2008). Recent studies have shown that metabolic and proteomic profiles of cv Excalibur differ from those of cv Kukri following cyclic drought (Ford et al., 2011; Bowne et al., 2012). Thus, it should be possible to use progeny from a cv Excalibur/Kukri cross to map QTLs affecting the levels of individual metabolites under drought stress conditions. To do this, we assessed metabolic profiles on tissue samples taken from a field experiment in which grain yield and other agronomic traits also had been measured. Since the effect of drought stress is usually perceived by the leaves, leading to stomatal closure, reduced transpiration rates, decreased leaf water potential, decrease in photosynthesis, and growth inhibition at the whole-plant level (Bartels and Sunkar, 2005), we chose leaf tissue for the metabolite analysis.
To obtain further insight into genetic factors that affect yield under drought conditions, we performed a parallel QTL analysis for yield-related phenotypic (agronomic) and metabolic traits. Because abiotic stress tolerance traits based upon glasshouse/laboratory-based experiments rarely translate into the field (Gaudin et al., 2013), we took a field-based approach to identify QTLs. The focus in this genetic study was to use the power of QTL analysis in a large population to examine the genetic control of grain yield, other agronomic traits, and metabolic traits for wheat grown under drought conditions, without explicitly examining drought tolerance per se, which would have required a well-watered control experiment for the entire population.
Our objectives were to assess pairwise genetic correlations among metabolic and agronomic traits, investigate the colocalization of QTLs that could be responsible for these correlations, and provide insights into the interconnected genetic and metabolic networks that underlie quantitative trait variation in bread wheat.
RESULTS
Genetic Correlations among Metabolic Traits Sharing Biochemical Pathways
In flag leaf tissue sampled from the cv Excalibur/Kukri doubled haploid (DH) lines, 205 compounds were detected, of which 112 could be identified. To distinguish between genetic and nongenetic sources of covariation, we estimated both genetic and phenotypic correlations for each possible pair of traits, including all 205 metabolites and 29 agronomic traits. Almost all correlations among metabolic traits (90% of all genetic and 97% of all phenotypic correlations) were positive (Fig. 1). For complete lists of genetic and phenotypic metabolite-metabolite correlation coefficients and their P values, see Supplemental Data Sets S1 and S2. Metabolites that are biochemically related were generally more strongly genetically correlated than biochemically unrelated metabolites. For example, metabolites of ascorbate metabolism (including gulose, ascorbate, glucarate, galactonate, threonate, and derivatives), Arg and Pro metabolism (including Glu, Gln, Orn, Arg, Pro, and 5-oxo-Pro), and glycolysis (including Glc and the hexose phosphates Glc-6-P and Fru-6-P) were strongly genetically correlated, indicating common genetic control. Similarly, most amino acids had strong genetic correlations with each other, as did sugars derived from Glc and raffinose. There were also some negative genetic correlations for metabolites that share common pathways. For example, the tricarboxylic acid (TCA) cycle intermediates cis-aconitate and isocitrate were negatively correlated with each other, indicating that the genetic regulation of biosynthetic routes drives the abundance of these intermediates in opposite directions.
Figure 1.

Heat map of genetic (top) and phenotypic (bottom) correlations between each of the measured metabolites of the DH wheat population. The color assigned to a point in the heat map grid indicates the strength of a particular correlation between two traits. The level of correlation is indicated by red for positive correlations and blue for negative correlations, as depicted in the color key. A fully annotated heat map is given in Supplemental Figure S1. The image was made with R (http://www.r-project.org/).
There were also some significant genetic correlations between metabolites belonging to distant pathways. For example, shikimate showed strong positive correlations with several metabolites of the TCA cycle, including succinate and cis-aconitate, and strong negative correlations with most amino acids, including Arg, Lys, and Tyr.
Several closely eluting unknowns (NA19, NA20, and NA21) showed strong negative genetic correlations with compounds related to the TCA cycle, including itaconate, aconitate, and 2-methyl maleate. This could reflect a “tradeoff” in which limited resources must be allocated between competing demands, such that increased allocation to NA19, NA20, and NA21 comes at the expense of reduced allocation to several TCA cycle intermediates.
Among 29 agronomic traits (Supplemental Table S2) that were measured, two traits related to phenological development, thermal time to heading and number of days to senescence, were strongly positively correlated with each other (Fig. 2). Similarly, grain yield exhibited strong positive correlation with one of its components, the number of grains per m2. For complete lists of phenotypic and genetic correlation coefficients among agronomic traits and their P values, see Supplemental Data Sets S3 and S4. Lines that headed early escaped drought stress and tended to have less glaucous leaves, less leaf rolling, longer peduncles and flag leaves, more spikes, more grains per spike, larger better-filled grains, higher yield, and higher harvest index than late-heading lines. By contrast, lines that matured slowly (and therefore scored higher in thermal time to heading and days to senescence) displayed a higher degree of rolling and glaucousness of their leaves and developed fewer spikes than lines that matured more quickly.
Figure 2.

Heat map of genetic (top) and phenotypic (bottom) correlations between each of the measured agronomic traits of the DH wheat population. The color assigned to a point in the heat map grid indicates the strength of a particular correlation between two traits. The level of correlation is indicated by red for positive correlations and blue for negative correlations, as depicted in the color key. Atl, Number of aborted tillers; CR, crown rot symptom severity; DS, days to senescence; Flth, flag leaf length; G2.8, G2.5, G2.2, the percentage of grains greater than 2.8, 2.5, and 2.2 mm, respectively; Gla, glaucousness; Gm2, grains m−2; Hi, harvest index; Hlth, head (spike) length; Hlwt, test weight; Ht, plant height; InfFL, number of infertile tillers; L2.2, screenings; Lfr_1, leaf rolling at time point 1; Lfr_2, leaf rolling at time point 2; Plth, peduncle length; SPAD_1, chlorophyll content at time point 1; SPAD_2, chlorophyll content at time point 2; SpM2, spikes m−2; TGW, thousand grain weight; TGW_5P, thousand grain weight of five plants; TTH, thermal time to heading; Yld, grain yield. For a detailed description of the agronomic traits, see Supplemental Table S1. The image was made with R (http://www.r-project.org/).
Correlations between Agronomic Traits and the Levels of Certain Primary Metabolites
Between 205 measured metabolites and 29 agronomic traits, we found 336 moderate to high genetic correlations (Fig. 3). For complete lists of genetic correlation coefficients and their P values, see Supplemental Data Sets S5 and S6. Consistent with the observations of Ferrara et al. (2008), we found that genetic correlations between traits belonging to different suites of characters (metabolic and agronomic) were generally not as strong as those among metabolites (Supplemental Data Set S1) or among agronomic traits (Supplemental Data Set S3). Not surprisingly, a substantial number of the metabolic traits showed strong positive or negative links (genetic correlation coefficient [rG] > +0.5 or rG < −0.5) with phenology-related traits. This indicates that certain metabolites were more abundant in the relatively young flag leaf tissue sampled from plants that headed and matured late, while others were more abundant in the older flag leaf tissue sampled from plants that headed and matured early.
Figure 3.

Clustered heat map of the genetic correlations between the agronomic traits and the measured metabolites of the DH wheat population. Clustering of the agronomic traits is depicted by the dendrogram at top, broken into six groups, labeled A through F. Clustering of the metabolic traits is depicted by the dendrogram at left, broken into five groups, labeled 1 through 5. The color assigned to a point in the heat map grid indicates the strength of a particular correlation between two traits. The level of correlation is indicated by red for positive correlation, blue for negative correlation, and white for no correlation, as depicted in the color key at top left. A fully annotated heat map is given in Supplemental Figure S2. The function “hclust” in the “stats” library of R (R Development Core Team, 2011) was used for hierarchical clustering, and Euclidean distances were used to calculate the distance matrix. The image was made with R (http://www.r-project.org/).
In a clustered heat map constructed based on genetic correlations between agronomic traits and metabolites (Fig. 3), there are several distinct clusters. The agronomic traits fall into six clusters: A, traits related to plant phenology; B, a mixed group of traits including tillering, head length, and crown rot symptom severity; C, chlorophyll content, number of spikes per plant, and grain size; D, plant biomass, number of infertile spikelets, and plant height; E, grain yield and number of grains per m2; and F, harvest index, number of spikes per m2, peduncle length, flag leaf length, test weight, and grain size (G2.5 and thousand grain weight).
The metabolites fall into five main clusters. The first two of these (1 and 2) consist of metabolites that exhibit negative genetic correlation with agronomic traits in cluster A or cluster B and positive genetic correlations with agronomic traits in clusters C, D, E, or F. Clusters 1 and 2 differ from each other in that the correlations with agronomic traits were stronger for the metabolites in cluster 1 than for cluster 2. The metabolites in these two clusters, which included sugars (e.g. Glc and Fru), amino acids (e.g. Lys and Tyr), phenolic alcohols and long-chain alcohols (e.g. stearyl alcohol), TCA cycle intermediates (e.g. citrate and isocitrate), and several distinct groups of closely eluting unknown metabolites, were less abundant in the relatively young leaf tissue sampled from lines that headed late and yielded poorly than in the older tissue sampled from lines that headed early and yielded more. The metabolites in clusters 4 and 5 exhibited opposite relationships to those in clusters 1 and 2, with strong (5) and less strong (4) positive genetic correlations with agronomic traits in clusters A and/or B and negative genetic correlations with agronomic traits in clusters C, D, E, and/or F. These metabolites include the amino acids Gln and β-Ala, metabolites related to the TCA cycle (e.g. cis-aconitate, dimethyl maleate, oxalate, and itaconate), metabolites of the phenylpropanoid pathway (e.g. quinate and caffeoyl quinate), the sugar alcohols inositol and galactinol, shikimate, and several fatty acids (e.g. stearate and linoleate). Finally, cluster 3, located in the center of the heat map, includes metabolites that were not strongly correlated with agronomic traits. Among these metabolites, some (e.g. Fru-6-P) exhibited weak positive genetic correlation with both phenological and grain traits, while others were positively correlated with either phenological traits (e.g. glycerol-3-phosphate and glycerol-2-phosphate) or grain traits (e.g. 3-amino-piperidin-2-one) but not both.
Detection of QTLs for 95 Metabolites
To investigate the genetic basis of relationships between agronomic traits and metabolic traits in the cv Excalibur/Kukri wheat mapping population grown under drought conditions in the field, we carried out parallel QTL analyses for 29 agronomic and 205 metabolic traits using the R package wgaim (Taylor and Verbyla, 2011). Prior to QTL mapping, we used ASReml/R to model variation from nongenetic sources of variation, including the positions of individual plots in the field and variation among and within batches of chromatographic runs.
To identify the proportion of metabolic variation that is genetically determined, we estimated broad-sense heritability for each metabolite. Over all metabolites, the mean of broad-sense heritability estimates was 0.50 (Supplemental Fig. S4). This relatively high value confirms the relevance of QTL analysis and the potential to alter metabolic traits via selection.
For each of the 95 metabolites, of which 43 were known compounds, between one and six metabolic quantitative trait loci (mQTLs) were detected (Fig. 4; Supplemental Fig. S3; Supplemental Table S3). For individual QTLs, the estimated broad-sense heritability ranged from close to 0 (glycerol-β-d-galactopyranoside) to 0.87 (methyl-O-β-d-glucopyranoside). The statistically significant mQTLs were distributed over 113 intervals on the genetic map. There were 12 mQTLs for amino acids, 45 for organic acids, 11 for sugars, six for sugar alcohols, 11 for sugar acids, six for phosphorylated compounds, 15 for fatty acids or fatty alcohols, five for phosphate, and one for an organic compound (2,4,6-tri-tert-butylbenzenethiol). Interestingly, most QTLs for amino acids, fatty acids, and organic acids were found on the A genome, whereas most QTLs for sugars, sugar acids, and sugar alcohols were mapped onto the B genome.
Figure 4.


QTL location on the cv Excalibur/Kukri genetic linkage map. Genetic distances are indicated as cM to the far left of the genetic map. QTLs for metabolites are shown on the left side, and QTLs for agronomic traits are shown on the right side, of each wheat chromosome (blue bars). For abbreviations of the metabolites and agronomic traits, see Supplemental Tables S1 and S4, respectively. Due to the larger number of mapped traits, chromosome 7A is shown on a larger scale to provide more detail.
For each of 36 metabolites, only one QTL was detected. The maximum number of QTLs detected per metabolite was six; this was for two unknown metabolites (NA13 and NA55; Supplemental Fig. S3). Some genomic regions (e.g. on chromosomes 4B, 4D, 5A, and 7A) affected multiple metabolites (Fig. 4; Supplemental Table S3). For example, mQTLs for the unknown compounds NA18 and NA55 each coincided with mQTLs for 26 other metabolites. In contrast, some genomic regions (e.g. on chromosomes 6B and 7B) had few mQTLs.
Pairs of metabolites sharing at least one mQTL usually had higher genetic correlations than pairs of metabolites sharing no mQTLs (Fig. 5). However, there were also cases in which metabolites were genetically highly correlated yet shared no mQTLs (e.g. maltose and NA40; rG = 0.84). Conversely, there were cases of metabolites sharing mQTLs yet exhibiting only weak genetic correlation with each other (e.g. Fru and isocitrate; rG = 0.28, despite two QTLs in common) or no significant correlations with each other (e.g. Arg and Asp; rG = −0.04, despite similar effects detected for these two amino acids in the 5-centimorgan [cM] interval between cwem0012 and PSP2999 on the short arm of chromosome 1A). Either of these situations could be due to the action of other QTLs whose effects were too small to detect.
Figure 5.

Box plot of the number of shared mQTLs versus the genetic correlation (absolute value) between any pair of two metabolites. The image was made with R (http://www.r-project.org/).
Detection of QTLs for 22 Agronomic Traits
The broad-sense heritability of individual agronomic traits varied from 0.05 (total biomass) and to 0.97 (thermal time to heading; Supplemental Table S4). For 22 of the 29 agronomic traits, we detected between two (chlorophyll 1, thousand grain weight, screenings L2.2) and 10 (crown rot symptom severity) QTLs. A complete list and description of QTLs for agronomic traits is provided in Supplemental Table S4, and the QTL positions are shown on the genetic linkage map in Figure 4. These QTLs are distributed over 53 intervals on the genetic map. In many cases, several traits were associated with the same marker loci, as on chromosomes 2B, 4A, and 7A, while other regions contain very few or no QTLs, as on chromosomes 1AS, 3B, 6B, 7B, and 7DS. On chromosome 7A, a single genomic interval between two adjacent markers affected 12 agronomic traits, including thermal time to heading, leaf rolling, flag leaf length, thousand grain weight, spikes per m2, test weight, and the grain size traits G2.5, G2.2, and L2.2. That interval, which explains 39% of the genetic variation for thermal time to heading (Supplemental Table S4), may contain a phenological locus with downstream effects on other traits. Another position on the same chromosome was found to affect grain yield, glaucousness, crown rot symptom severity, peduncle length, grains per m2, and harvest index.
Colocalization of Agronomic QTLs with mQTLs
The detection of coincident QTLs for either grain yield and grain yield-related traits or for grain yield and metabolic traits can provide information on traits that are associated with effects on grain yield under drought conditions. Each agronomic QTL coincided with at least one mQTL. In total, we found that mQTLs for 38 metabolites colocalized with QTLs for agronomic traits (Supplemental Tables S3 and S4, respectively). 2-Oxogulonate and the unknown compound NA57 shared the largest numbers of QTLs with agronomic traits (14 individual mQTLs sharing the same marker interval with 13 and 14 agronomic traits, respectively), closely followed by Fru and isocitrate, each of which shared QTLs with 12 agronomic traits.
Five genomic regions affected both metabolic and agronomic traits: one each on chromosomes 4B and 5A, two in close proximity on chromosome 7A, and one on chromosome 7DL. On chromosome 4B, a 29.1-cM interval flanked by wPt-7062 and wPt-8756 was associated with concentrations of phosphate and four unknown metabolites (NA13, NA45, NA55, and U22) and with the agronomic traits flag leaf length and leaf rolling. On chromosome 5A, a 3.8-cM interval flanked by wPt-0373 and wmc75b affected leaf rolling, cis-aconitate, itaconate, 2,4,-6-tri-tert-butylbenzenethiol, and four unknown metabolites (NA6, NA26, NA55, and NA60). On chromosome 7A, a 1.0-cM region between gwm60 and wmc28 affected several organic acids, including itaconate, dimethyl maleate, pipecolate, cis-aconitate, and caffeoyl quinate, the amino acid Gln, as well as a range of agronomic traits, including glaucousness, peduncle length, grains per m2, grain yield, and harvest index. Notably, this QTL is also close to one for grain yield and Glc-6-P (in the 1.1-cM interval between wmc83 and STM0511TCTG) and a known sodium-exclusion QTL (Edwards et al., 2008). Another genomic region on chromosome 7A, flanked by wmc283 and BS0001030, harbors QTLs for many traits: 12 agronomic traits (including thermal time to heading, days to senescence, and thousand grain weight) and the abundance of nine metabolites (including phosphate, Fru, 2-oxogulonate, and isocitrate). As noted above, this was the most important QTL for thermal time to heading (Supplemental Table S4). The colocalization of these metabolites with plant maturity is not unexpected; mQTLs for primary metabolites have been previously found to be colocalized with known plant maturity loci (Carreno-Quintero et al., 2012). Here, though, only nine out of 205 metabolites had QTLs mapped near the known vernalization locus Vrn-1A on chromosome 5A (Snape et al., 2001; Distelfeld et al., 2009), and there is no concentration of mQTLs on the homologous group 2 chromosomes, on which the photoperiod response (Ppd) genes are located (Fig. 4; Supplemental Tables S8 and S9).
Associations were also found between peduncle length and 1-kestose (chromosome 2B), between raffinose and harvest index (also on chromosome 2B), and between crown rot symptom severity and both phytol (chromosome 2D) and 1-kestose (chromosome 6A). However, all of these QTLs had low logarithm of odds (LOD) scores (LOD < 2.46), indicating that they may not be very important loci for these traits.
To compile information on connections between the metabolites, genetic correlations, and colocalization of metabolic with agronomic QTLs, we created an author-generated metabolite pathway map of primary metabolism (Fig. 6). This map shows significant correlations between pairs of metabolites that are adjacent in pathways and indicates whether mQTLs coincided with QTLs for agronomic traits. Most mQTLs coincided with QTLs for agronomic traits from clusters A, containing maturity-related traits, and F, containing grain yield-related traits. Metabolic QTLs for the sugars raffinose, 1-kestose, and Fru and the TCA cycle metabolite isocitrate overlapped with QTLs detected for grain yield-related traits, including thousand grain weight (cluster F). At the shared QTLs, cv Excalibur alleles increased both thousand grain weight and the levels of these metabolites, contributing to positive genetic correlation among these traits (Supplemental Tables S3 and S4). QTLs for most cis-aconitate metabolites and metabolites derived from the TCA cycle (itaconate and Gln) overlapped with QTLs detected for maturity-related traits (cluster A), including thermal time to heading, and with QTLs detected for grain yield-related traits (clusters E and F). At a shared QTL on 5A, the cv Excalibur allele delayed maturity and increased the levels of these metabolites (contributing to positive genetic correlations). In contrast, at a shared QTL on 7A, the cv Excalibur allele decreased the levels of these metabolites and increased grain yield (contributing to negative correlations). In several cases, mQTLs coincided with QTLs for both maturity and grain yield-related traits. With one exception (an mQTL for pipecolate), these overlaps had antagonistic effects on the two categories of agronomic traits. Among the agronomic traits from clusters C and D, only chlorophyll content had QTLs that coincided with mQTLs. Alleles that increased chlorophyll content decreased 2-oxogulonate (at a QTL on 6B) and increased monostearin (at a QTL on 1AL).
Figure 6.

Pathway map of the primary metabolism showing genetic correlations between metabolites detected in the DH population. Edges with positive genetic correlations are displayed in red, whereas edges with negative genetic correlations are displayed in blue. Corresponding correlation values are placed next to the edges, and significant correlations (P < 0.05) are marked with asterisks. Metabolites colored in pale gray were not measured in this study. Metabolites with mapped QTLs are displayed in boldface, and colocalizations with agronomic traits are visualized using boxes A to F; red boxes indicate a positive genetic correlation, and blue boxes indicate a negative genetic correlation, of the metabolite with the agronomic traits of this particular cluster (according to the clusters of agronomic traits in Fig. 3). Boxes are as follows: A, leaf rolling, screening L2.2, grain size G2.2, glaucousness, thermal time to heading, days to senescence; B, tiller number, number of aborted tillers, head length, crown rot symptom severity; C, chlorophyll content, fertile spikes per seed, grain size G2.8, total seeds per spike; E, grains per m2, grain yield; F, harvest index, peduncle length, flag leaf length, test weight, thousand grain weight, spikes per m2. The image was made using a modified author-created pathway map in VANTED (Junker et al., 2006).
Therefore, we were able to demonstrate that all agronomic QTLs overlap with at least one mQTL. Although we could not determine specific biochemically related groups of metabolites mapping to the aforementioned regions, several of the mapped metabolites remain unknown and are potentially under similar genetic control; however, at this stage, we cannot determine their definite relationships.
DISCUSSION
Drought stress, which has been identified as the most devastating abiotic constraint on crop productivity (Francia et al., 2005; Reynolds et al., 2005), causes substantial perturbations in plant metabolism (Bowne et al., 2011, 2012). Previous QTL studies of agronomic traits in wheat have lacked information on the biochemical pathways underlying those traits and their responses to drought. In this study, a drought-tolerant wheat cultivar (Excalibur), a drought-susceptible cultivar (Kukri), and a population of DH lines derived from a cross between cv Excalibur and Kukri were grown in the field under drought conditions and evaluated for agronomic traits, and leaves sampled from these materials were analyzed for metabolites of known and unknown identity.
Correlated Responses
The metabolic composition and agronomic traits of crop plants are expected to be influenced by genetics and by physiological and environmental conditions. When two or more characteristics have some kind of shared genetic basis, due to either pleiotropic loci or genetic linkage between loci (Gardner and Latta, 2007), they will tend to be coinherited and to exhibit correlated responses under selection. Estimates of genetic correlation between traits providing indicators of common genetic control can be used in the prediction of correlated responses to selection. Whether traits are genetically correlated, they may also respond to some of the same environmental factors, leading to positive or negative environmental correlations among traits. Phenotypic correlations among traits depend on genetic correlations, environmental correlations, and trait heritabilities (Cheverud, 1988; Falconer, 1989). Phenotypic correlation coefficients, therefore, are limited in their capacity to accurately reflect dependencies based on common genetic factors such as regulatory genes, especially under environmental conditions that affect multiple traits simultaneously. This may be particularly true under stress conditions, given that the genetic component of the overall stress response is usually small relative to the environmental component (Reynolds et al., 2009).
Here, we estimated phenotypic and genetic correlation coefficients (1) among agronomic traits, (2) among metabolites, and (3) between agronomic traits and metabolites. In most cases, phenotypic correlation estimates were lower than genetic correlations. Thus, examination of the genetic correlations made it possible to detect genetic relationships between metabolites and agronomic traits that would have been masked by environmental influences if only phenotypic correlations had been examined.
We found strong genetic correlations between plant maturity-related traits (thermal time to heading and days to senescence) and a range of agronomic traits, including grain yield under drought conditions. These strong correlations reflect the important influence of genetically determined differences in plant phenology on agronomic performance. They are consistent with previous reports on experimental populations with variable phenology, in which QTLs affecting flowering time also affected yield under water-limiting conditions (Reynolds and Tuberosa, 2008). Under conditions of either cyclic or terminal drought, lines that develop and mature at different rates would experience stress at different growth stages. In our experiment, lines that headed early may have achieved higher yields by either avoiding or escaping part or all of the drought stress.
Coregulation of metabolites may indicate that either a specific biological function controls different components or that a specific step in a biochemical pathway is affected. Here, almost all genetic correlations among metabolites were positive, indicating that there were hardly any notable tradeoffs involved in primary metabolism. Metabolites that are biochemically related had stronger genetic correlations compared with biochemically unrelated metabolites, indicating that genetically controlled changes in the content of one metabolite could modify the pools of other biochemically related metabolites (Fig. 2; Supplemental Data Sets S1 and S2). These findings, which are similar to those of Carreno-Quintero et al. (2012), may be due to genes encoding rate-limiting enzymes in the pathways and/or major regulators that influence the pathways.
Consistent with the fact that plant responses to drought stress are known to involve changes in the cellular antioxidative defense pathways (Reddy et al., 2004) and the synthesis of osmotically active compounds (Bartels and Sunkar, 2005), we found several clusters of highly genetically correlated metabolites belonging to both of these pathways. For example, metabolites of ascorbate metabolism (including glucarate, galactonate, threonate, ascorbate, and derivatives) were highly genetically correlated with each other and were positively genetically correlated with grain yield and grain yield-related agronomic traits (Supplemental Fig. S2; Supplemental Data Set S5). During drought stress, an imbalance between electron generation and consumption occurs due to a down-regulation of PSII activity. Diversion of excess light energy in the PSII core and antenna produces reactive oxygen species such as oxide ions, hydrogen peroxide, and hydroxide, which can cause substantial cellular damage (Peltzer et al., 2002). This damage can be mitigated by several types of nonenzymatic plant antioxidants, including ascorbic acid-like scavengers as well as pigments such as carotenoids, flavanones, and anthocyanins (Conklin, 2001; Reddy et al., 2004). Ascorbate (vitamin C) can act as an important water-soluble antioxidant by serving as a cofactor for a large number of enzymes and as a donor/acceptor in electron transport at the plasma membrane and in the chloroplasts (Conklin, 2001). Additionally, ascorbate has a role in either protecting or regenerating oxidized carotenoids and tocopherols (Imai et al., 1999). We found positive correlations between metabolites of ascorbate metabolism, ascorbate, α-tocopherol, and grain yield as well as several yield-related agronomic traits. This indicates that an effective system to scavenge reactive oxygen species may be needed in order to achieve high yield under detrimental environmental conditions.
Drought also induces osmotic stress in plants (Zhu, 2002). The biosynthesis and accumulation of compatible solutes can help plants cope with osmotic stress (Zhang et al., 2000; Ashraf and Foolad, 2007). These compounds are small, electrically neutral, nontoxic molecules that do not interfere with the normal metabolism of the plant. During osmotic stress, they are accumulated to high levels in the cytosol. They prevent water loss, stabilize membranes and proteins, help maintain cell turgor, and protect cells by scavenging reactive oxygen species (Pinhero et al., 1997; Valliyodan and Nguyen, 2006). During osmotic adjustment, nitrogen-containing compounds (including Pro, Asp, and Glu, polyamines, and quaternary ammonium compounds like glycinebetaine) accumulate, as do Suc, sugar alcohols, and sugars (Vinocur and Altman, 2005). Plants accumulate Pro by either activating Pro biosynthesis or by inactivating Pro degradation to Glu via D1 pyrroline-5-carboxylate (Bohnert and Sheveleva, 1998). Pro acts as a free radical scavenger, is involved in osmotolerance, and is known to protect membranes and proteins when relative water content decreases. Similarly, the accumulation and maintenance of a high sugar pool containing nonreducing sugars and oligosaccharides has been reported to act as a replacement for water by providing a hydration shell around proteins. Although there is evidence from some previous studies that enhanced accumulation of Pro (Hong et al., 2000) and sugars (Hoekstra et al., 2001; Bogdan and Zagdańska, 2006; Urano et al., 2009) improves the osmotic stress tolerance of plants, there have also been reports that the accumulation of organic compatible solutes makes only minor contributions to osmotic stress tolerance (Gagneul et al., 2007; Hill et al., 2013). Here, metabolites of Glu and Pro metabolism (including Gln, Orn, Arg, and 5-oxo-Pro) and sugar metabolism (raffinose, Fru, and Glc) were highly genetically correlated with grain yield and other yield-related agronomic traits. Further research on these metabolites could lead to opportunities to use engineered osmoprotectants to protect major crops from stress conditions (Supplemental Fig. S2; Supplemental Data Set S7).
We found higher levels of both Gln and Glu to be strongly genetically associated with late maturity and low grain yield. This is consistent with the role of these compounds in nitrogen metabolism. During periods of nitrogen availability, inorganic nitrogen is taken up from the soil and enzymatically converted into Glu and Gln, which can be incorporated into nitrogen-containing organic compounds, such as amino acids and nucleotides, providing nitrogen reserves for subsequent use in growth, defense, and reproductive processes (Forde and Lea, 2007). In wheat, Glu and Gln levels peak at flowering (Kichey et al., 2006). During grain filling, leaves transition from absorption and assimilation of organic and inorganic molecules to remobilization and translocation of assimilates to the grain. Our results for these metabolites, therefore, would be strongly influenced by phenological differences on the day of sampling, which was close to or during this transition period. Glu and Gln levels would have declined more in the leaves of earlier maturing (and ultimately higher yielding, probably due to avoidance of drought stress) lines than in the leaves of later maturing (and lower yielding) lines.
Mapping of Metabolic and Physiological Variation in Wheat Leaves
In the second part of this study, we combined QTL mapping with metabolomics. A genetic map of more than 430 molecular markers (Edwards, 2012) was used for mapping QTLs, and high heritabilities enabled the detection of QTLs for 22 agronomic and 205 metabolic traits (Fig. 4). As the two parents have some common ancestry, it is not surprising that some regions of the genome lack polymorphic markers, probably indicating that these regions are identical by descent.
Given that metabolites, particularly primary metabolites, are part of a densely interconnected metabolic network, we expect that many genetic effects on metabolites might be too small to detect as a metabolite QTL. For many metabolites, we detected just one QTL, in some cases with quite a large effect. In several distinct genomic regions, mQTLs were detected for numerous metabolites (Fig. 5; Supplemental Table S3). This is consistent with findings in several other mQTL studies (Keurentjes et al., 2006; Lisec et al., 2008; Rowe et al., 2008; Carreno-Quintero et al., 2012). It indicates that a large number of metabolites could be influenced by the manipulation of small genomic regions (Saito and Matsuda, 2010). However, it is currently not clear whether these distinct genomic regions exist due to only one gene that has pleiotropic effects or due to several closely linked genes in the same genomic region. The unknown compounds NA18 and NA55 both share QTLs with 26 other metabolites (Fig. 5; Supplemental Table S3), making those interesting targets for identification through analytical experiments.
Many QTLs for 20% of the metabolic traits colocalized with at least one agronomic QTL (Supplemental Tables S7 and S8). Such colocalization could be due to linkage or pleiotropy. Pleiotropy could involve direct effects of metabolite flux on agronomic traits or separate effects of regulatory genes on metabolic pathways and other mechanisms that influence agronomic traits.
Given the complexity of both metabolic pathways and agronomic performance, it is likely that there are epistatic interactions between genetic loci (Rowe et al., 2008) affecting the traits examined in this study. An exploration of these interactions would require an analysis of a larger population size than used here to give statistically meaningful results but would be worth exploring. Similarly, exploration of mQTL-environment interactions would be interesting, especially with metabolic profiling of the same population grown under a range of stress conditions.
CONCLUSION
Flag leaf samples were taken from a DH mapping population derived from a cross between a drought-tolerant and a drought-sensitive wheat cultivar grown in the field under terminal drought stress. Parallel analysis of relative metabolite abundance and an evaluation of agronomic traits, including grain yield as an indicator of drought tolerance, allowed us to estimate the genomic positions and effects of individual metabolite and agronomic trait QTLs. Our results confirm that metabolites can be mapped to distinct genetic regions, much like agronomic traits in conventional QTL mapping. The information from this research alone is not sufficient to determine whether a genetic locus and a phenotype are connected directly, for example, via a specific gene, or through multiple steps and pathways, such as transcription factors or other regulatory sequences. This is confounded due to the lack of a fully assembled and annotated wheat genomic sequence. With additional experimentation, it may be possible to determine whether coincident QTLs are due to linkage or pleiotropy and to more clearly define aspects of the biological basis of agronomic and metabolic traits. Although QTL mapping using a population of several hundred lines does not provide sufficient resolution to identify individual genes, it opens new opportunities to investigate the biochemical pathways underlying economically important traits.
High-resolution mapping of specific genomic regions would help clarify whether coincident QTLs are due to linkage or pleiotropy. Additional genetic resources, including 3,000 cv Excalibur/Kukri recombinant inbred lines (Fleury et al., 2010), are available for such studies, as are new marker platforms (Paux et al., 2012) and genotyping by sequencing (Poland et al., 2012). With increases in the availability of genomic sequence information for wheat, it should be possible to select genes or other sequences in the target regions to undergo functional analysis to investigate their roles in drought tolerance. This will provide a basis for future studies and validation of results obtained through the work proposed here. The information presented here represents a rich resource for further investigation and annotation of relevant genes as the wheat genome is sequenced and may pave the way to an improved understanding of the performance of cereal crops under drought. The metabolite QTL analysis provides useful information to support positional cloning and genetic analysis projects. In isolation, mQTL results do not support gene identification, but when combined with physiological and agronomic traits, as shown here, they support the development of testable hypotheses about possible mechanisms or processes underlying agronomic traits.
MATERIALS AND METHODS
Bread Wheat Cultivars and Mapping Population
The plant population consisted of 233 DH lines produced from a cross between ‘Excalibur’ (RAC177/‘Monoculm’//RAC311S; released by the University of Adelaide in 1991) and ‘Kukri’ (76ECN44/76ECN36//RAC549; MADDEN/6*RAC177; released by the University of Adelaide in 1999). Excalibur is a drought-adapted bread wheat (Triticum aestivum) cultivar that yields well in South Australian wheat regions but has low grain quality and is susceptible to rust. Kukri is a hard white wheat cultivar that has excellent grain quality and is rust resistant but has low drought and heat tolerance and produces lower grain yield compared with cv Excalibur when grown under water-limiting conditions (Izanloo et al., 2008).
Field Experimental Conditions
The field experiment was conducted in 2006 at the Roseworthy Campus at the University of Adelaide. The field experiment was randomized using a nearest-neighbor design with two replicates of each DH line, with additional plots of the parental lines and control varieties that included cv Axe, Carinya, Drysdale, Espada, Excalibur, Frame, Gladius, Kukri, Krichauff, RAC875, Stylet, Tincurren, Westonia, Wyalkatchem, and Yitpi. The plots were 1.25 m wide and 5 m long with six rows. Plots were reduced by herbicide application to 3.2 m long, just prior to anthesis. Seeds were sown on a volume basis, aiming for an average of 200 seeds m−2. The agronomic management regime followed local practice. The sampling of flag leaves was performed between 10 am and 3 pm on one day, when most lines were at late anthesis. Flag leaf samples were immediately stored at −80°C until extracted.
Gas Chromatography-Mass Spectrometry Metabolite Profiling
Chemicals for Metabolite Profiling
N-Methyl-N-(trimethylsilyl)-trifluoroacetamide (TMS) was purchased from Grace. HPLC-grade methanol was obtained from Scharlau. All other chemicals were purchased from Sigma-Aldrich.
Sample Extraction
A modified method for the preparation of plant extracts has been developed based on the method described by Jacobs et al. (2007). For each line, 30 mg of flag leaf tissue was weighted into cryo mill tubes (Precellys lysing kit; Bertin Technologies), and 0.5 mL of 100% methanol was added to the plant sample. Homogenization was performed for 30 s at 6,000 rpm using a cryo mill (Precellys 24; Bertin Technologies). After the addition of 20 μL of internal standard solution (20 μL per sample from a stock solution containing 1 mg mL−1 [13C]sorbitol in water), samples were extracted for 15 min at 70°C in a thermomixer at 750 rpm. Subsequently, the sample was mixed vigorously with 1 volume of water and then centrifuged for 10 min at 13,000 rpm. The supernatant was transferred into a new reaction tube, and 50-μL aliquots were transferred into glass vial inserts and dried in vacuo for further TMS derivatization.
Gas Chromatography-Mass Spectrometry Analysis
The gas chromatography (GC)-mass spectrometry (MS) system comprised a 7890A gas chromatograph and a 5975C Triple-Axis, quadrupole, mass selective detector (Agilent). A Gerstel MPS2XL GC-MS autosampler performed the derivatization procedure immediately prior to injection. The samples and the derivatization reagents were added to a glass vial and then placed in the autosampler tray. The autosampler then mixed sample with derivatization reagents automatically using the following program. Plant extracts were derivatized for 120 min at 37°C using 20 μL of methoxyamine hydrochloride (30 μL of 30 mg mL−1 in pyridine) per sample. This was followed by trimethylsilylation with 40 μL of TMS reagent per sample for 30 min. Finally, 2 μL of retention time standard mixture (0.029% [v/v] n-dodecane, n-pentadecane, n-nonadecane, n-docosane, n-octacosane, n-dotriacontane, and n-hexatriacontane dissolved in pyridine) was added per sample prior to injection onto the GC column.
One microliter of TMS-derivatized sample was injected onto the GC column using a hot-needle technique. The injector was operated in splitless mode isothermally at 250°C. Helium was used as the carrier gas with a flow rate of 0.8 mL min−1. Chromatographic separation was performed on a 30-m VF-5MS column (with 10-m Integra guard column, i.d. 0.25 mm, and 0.25-nm film thickness [Varian]). The MS transfer line to the quadrupole was fixed at 280°C, the electron impact ion source at 250°C, and the MS quadrupole at 150°C. The mass spectrometer was tuned according to the manufacturer’s protocols using Tris-(perfluorobutyl)-amine (CF43). The analysis was performed under the following oven temperature program. The injection temperature was set at 70°C, following by a 7°C min−1 oven temperature gradient to a final 325°C, and then held for 3.6 min at 325°C. The GC-MS system was then temperature equilibrated for 1 min at 70°C prior to injection of the next sample. Ions were generated by a 70-eV electron beam at an ionization current of 2.0 mA, and spectra were recorded at 2.91 scans per second with a mass-to-charge ratio of 50- to 550-atomic mass units scanning range. Retention time locking of the chromatographic peak of mannitol prior to the sample run ensured repeatable retention times across systems regardless of operator, detector type, or column maintenance.
Data Handling, Mining, and Statistics
Analytes were semiquantified after mass spectral deconvolution (AnalyzerPro; SpectralWorks). The chemical identification was manually supervised using the public domain mass spectra library of the Max-Planck-Institute for Molecular Plant Physiology (http://csbdb.mpimp-golm.mpg.de/csbdb/gmd/gmd.html) and the in-house Metabolomics Australia mass spectral library. All matching mass spectra were additionally verified by analysis of reference standard compounds. Compounds that were present in the library, denoted as “NA” (not assigned), as well as compounds that could not be further annotated, denoted as “U” (unknown), were included in the analysis to investigate the degree of associations with identified metabolites. Relative response ratios were calculated using the metabolite peak area divided by both the peak area of the internal standard sorbitol and the sample dry weight (g), as described by Roessner et al. (2001).
Suc is by far the most highly abundant metabolite in the tissue extracts. As we were aiming for a nonbiased metabolite approach that included the detection and identification of as many metabolites as possible, we deliberately “overloaded” the GC column with respect to Suc in order to detect and identify the majority of the tissue metabolites that were present at low abundance. Compounds that were detected in less than 50 lines were considered not informative enough for the analysis. Data represent two biological replicates per line measured in two replicate extractions (technical replicates).
Exploratory Analysis and Transformation
An initial exploratory analysis showed that nearly all phenotypic agronomic traits were normally distributed (data not shown). The traits grain size G2.8 and spikes per m−2 were the exception: these required log and square root transformation, respectively, prior to formal analysis. In contrast, the distributions of the metabolite traits were heavily skewed to the right (data not shown). For this reason, all metabolite traits were log transformed prior to formal analysis. For each metabolite, to help satisfy modeling assumptions, absolute measurements that deviated from mean values by more than 4 sd units were considered extreme outliers and set as missing values.
Linear Mixed-Model Analysis
Each of the agronomic and metabolic traits were initially analyzed using a linear mixed model that appropriately partitions and accounts for genetic and nongenetic information arising from the field and laboratory experiments. Let y be a vector of trait observations, then the linear mixed model has the form:
| (1) |
where β is a set of nongenetic fixed effects with associated design matrix X, u is a set of nongenetic random effects with indicator matrix Z, and g is a set of random genetic effects associated with the genetic lines involved in the experiment with indicator matrix Zg. In this model, the residual error, e, captures extraneous variation or correlation that may arise from known dependencies between the observations in the experiment. The set of effects (u, g, and e) are considered to be mutually independent.
For the agronomic traits, the fixed component of Equation 1 consisted of an indicator type to differentiate the DH or parental lines existing in the data. Other nongenetic fixed effects in this model included direction of seeding as well as linear row effects to appropriately model trends appearing across the rows of the field. Nongenetic random effects in the model include a field-block effect as well as separate row and range effects where required. Due to the experimental layout of the field trial, dependency between observations and, therefore, the residuals is expected. This is captured through the residual variance matrix using a separable AR1 × AR1 (where AR1 is an autoregressive process of order 1) correlation structure (Gilmour et al., 1997). The model also contains random genetic effects for the DH and parental lines.
For the metabolite traits, Equation 1 consisted of a fixed-type variable to genetically distinguish the DH or parental lines as well as extraneous nongenetic random effects stemming from the laboratory design, such as blocking or replication in the laboratory, sample batches, days in the sample batch, and day of extraction. As the GC-MS laboratory experiment was a multiphase designed experiment, the model also appropriately included information from the initial field trial, such as field replication and plot error. For this model, the residual error initially contained a simple correlation structure to model dependencies between observations across days. This structure was then simplified across all traits due to the nonsignificance of the correlation. Lastly, the model contained random genetic effects for the DH and parental lines used in the experiment.
All models were analyzed using the flexible linear mixed-modeling software ASReml-R (Butler et al., 2009) available in the open-source statistical software platform R (R Development Core Team, 2011). ASReml-R allows complex modeling of fixed effects, random effects, and residual correlation structures combined with the residual maximum likelihood approach of Patterson and Thompson (1971) to estimate model parameters. It has commonly been used in many modern plant-breeding experiments (Smith et al., 2002, 2004, 2005, 2006), and an academic Discovery version is freely available from http://www.vsni.co.uk.
Phenotypic and Genetic Correlations
Pairwise phenotypic correlations between agronomic traits and also between metabolic traits were calculated using Pearson’s correlation coefficient and were based on complete pairwise data. Before calculating the pairwise genetic correlations for the agronomic and metabolic traits, model-based predictions for the DH and parental lines were obtained for each trait from their associated linear mixed model described in the previous section. The collected set of predictions for the DH lines from each experiment were then used to calculate Pearson pairwise genetic correlations for the agronomic and metabolic traits. Similarly, pairwise genetic correlations between the agronomic and metabolic traits were based on the collated set of 177 mutual DH predictions extracted from each collected set of predictions obtained for each experiment.
Let r be any of the pairwise estimated correlation coefficients. Under a null hypothesis of zero correlation, the significance of r can be determined using the test statistic:
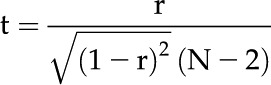 |
where N is the number of samples involved in estimating the correlation coefficient. Under the null hypothesis, this statistic is distributed as t with N − 2 degrees of freedom. If the null hypothesis is rejected at the α = 0.05 significance level, then the correlation coefficient is said to be significantly different from zero.
The discussion of the genetic and phenotypic correlations is based on the magnitude of the estimates as follows: (1) a correlation estimate of 0.5 or greater was considered strong; (2) a correlation estimate between 0.3 and 0.5 was considered moderate; and (3) a correlation estimate of less than 0.3 was considered weak.
Total phenotypic variance was partitioned into sources attributable to genotype and error. Components of variance were used to estimate broad-sense heritability according to the formula H2 = VG/(VG + VE), where VG is the among-genotype variance component and VE is the residual (error) variance component of the ANOVA.
Network Analysis
The genetic correlations between the metabolic and agronomic traits were displayed on an author-created metabolite network of the primary metabolism using the built-in graph editor in VANTED (Junker et al., 2006).
Linkage Map
Initially, a total of 233 lines of the cv Excalibur × Kukri double haploid population were genotyped with 438 polymorphic markers, consisting of 184 simple sequence repeats, 253 Diversity Arrays Technology markers, and the Vrn-1A marker. The linkage map was originally constructed at the Australian Centre for Plant Functional Genomics as described by Edwards (2012). After construction of the genetic map, an exploratory review of the map revealed 26 distinct groups of apparently genetically identical lines. From these groups, 43 genetic lines were omitted from the data set and the genetic map was constructed, with genetic distances, based on the Kosambi mapping function, reestimated using the hidden Markov algorithm of Lander and Green (1987) implemented in the R/qtl package (Broman et al., 2003). The final genetic linkage map for the population was based on data from 190 DH lines and had a total length of 3,281 cM (1,220, 1,228, and 833 cM for the A, B, and D genomes, respectively) and an average distance of 7.81 cM between markers. Missing genotypic data were imputed using the rules of Martinez and Curnow (1992).
QTL Analysis
The whole-genome average interval mapping (WGAIM) approach of Verbyla et al. (2007) was used for QTL analysis of the agronomic and metabolic traits. WGAIM uses extensions of the linear mixed model (Eq. 1) derived for each trait by incorporating a whole-genome approach for the detection and selection of QTLs. Initially, a working linear mixed model is proposed that includes a whole-genome contiguous block of QTL markers/intervals as random covariates. A simple likelihood ratio test of significance on the working model is then conducted to determine the requirement to search for QTLs on the genome. If it is significant, an outlier statistic is used to select the most likely putative QTL, which is then moved to the fixed part of the model. This process is repeated until no further QTLs are detected. Verbyla et al. (2007) showed that WGAIM is much more powerful than the more commonly used composite interval mapping approaches originally proposed by Zeng (1994). In addition, the simultaneous use of the whole genome in the analysis avoids repeated scans and the usual threshold calculations that are required for multiple testing problems. This considerably reduces computing time when a large number of traits require analysis. More importantly, during the WGAIM algorithm, nongenetic sources of variation arising from the environment or experimental design are estimated simultaneously with marker or interval QTLs, avoiding a two-stage modeling approach to QTL analysis. WGAIM has been implemented in the R package wgaim (Taylor et al., 2011), which uses ASReml-R as its core linear mixed-modeling algorithm. This package also includes QTL summary and diagnostic and plotting functions, including linkage map plots with highlighted QTLs. Further details of its use, including extended examples, can be found in Taylor and Verbyla (2011), and wgaim is freely downloadable from the Comprehensive R Archive Network at http://CRAN.R-project.org/package=wgaim.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. Heat map of genetic and phenotypic correlations between the measured metabolites.
Supplemental Figure S2. Heat map of the genetic correlations between the agronomic traits and the measured metabolites.
Supplemental Figure S3. Frequency distribution of the number of mQTLs detected for each metabolic trait.
Supplemental Figure S4. Frequency distribution of the broad-sense heritability of each detected metabolic trait.
Supplemental Figure S5. Frequency distribution of the number of QTLs detected for each yield or yield-related trait.
Supplemental Figure S6. Frequency distribution of the broad-sense heritability of each detected trait in the DH population.
Supplemental Table S1. List of metabolite traits.
Supplemental Table S2. List of yield and yield-related traits.
Supplemental Table S3. List of metabolic QTLs.
Supplemental Table S4. List of agronomic QTLs.
Supplemental Data Set S1. Genetic and phenotypic correlations between each of the measured metabolites.
Supplemental Data Set S2. P values of the genetic and phenotypic correlations between each of the measured metabolites.
Supplemental Data Set S3. Genetic and phenotypic correlations between each of the measured agronomic traits.
Supplemental Data Set S4. P values of the genetic and phenotypic correlations between each of the measured agronomic traits.
Supplemental Data Set S5. Genetic correlations between the measured agronomic and metabolic traits.
Supplemental Data Set S6. P-values of the genetic correlations between the measured agronomic and metabolic traits.
Supplementary Material
Acknowledgments
We thank the School of Botany (University of Melbourne) node of Metabolomics Australia for access to the GC-MS facility.
Glossary
- QTL
quantitative trait locus
- DH
doubled haploid
- TCA
tricarboxylic acid
- mQTL
metabolic quantitative trait locus
- cM
centimorgan
- LOD
logarithm of odds
- TMS
N-Methyl-N-(trimethylsilyl)-trifluoroacetamide
- GC
gas chromatography
- MS
mass spectrometry
- WGAIM
whole-genome average interval mapping
- rG
genetic correlation coefficient
References
- Ashraf M, Foolad MR. (2007) Roles of betaine and proline in improving plant abiotic stress resistance. Environ Exp Bot 59: 206–216 [Google Scholar]
- Atchison J, Head L. (2010) Wheat as food, wheat as industrial substance: comparative geographies of transformation and mobility. Geoforum 41: 236–246 [Google Scholar]
- Bartels D, Sunkar R. (2005) Drought and salt tolerance in plants. Crit Rev Plant Sci 24: 23–58 [Google Scholar]
- Bogdan J, Zagdańska B. (2006) Changes in the pool of soluble sugars induced by dehydration at the heterotrophic phase of growth of wheat seedlings. Plant Physiol Biochem 44: 787–794 [DOI] [PubMed] [Google Scholar]
- Bohnert HJ, Sheveleva E. (1998) Plant stress adaptations: making metabolism move. Curr Opin Plant Biol 1: 267–274 [DOI] [PubMed] [Google Scholar]
- Bohnert HJ, Nelson DE, Jensen RG. (1995) Adaptations to environmental stresses. Plant Cell 7: 1099–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowne J, Bacic A, Tester M, Roessner U. (2011) Abiotic stress and metabolomics. Annu Plant Rev 43: 61–85 [Google Scholar]
- Bowne JB, Erwin TA, Juttner J, Schnurbusch T, Langridge P, Bacic A, Roessner U. (2012) Drought responses of leaf tissues from wheat cultivars of differing drought tolerance at the metabolite level. Mol Plant 5: 418–429 [DOI] [PubMed] [Google Scholar]
- Bray EA, Bailey-Serres J, Weretilnyk E (2000) Responses to abiotic stresses. In W Gruissem, BB Buchannan, RL Jones, eds, Biochemistry and Molecular Biology of Plants. American Society of Plant Physiologists, Rockville, MD, pp 1158–1249 [Google Scholar]
- Broman KW, Wu H, Sen Ś, Churchill GA. (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19: 889–890 [DOI] [PubMed] [Google Scholar]
- Butler DG, Cullis BR, Gilmour AR, Gogel BJ (2009) ASReml-R Reference Manual. Technical report. Queensland Department of Primary Industries, Toowoomba, Australia. http://www.vsni.co.uk (May 31, 2013) [Google Scholar]
- Carreno-Quintero N, Acharjee A, Maliepaard C, Bachem CW, Mumm R, Bouwmeester H, Visser RG, Keurentjes JJ. (2012) Untargeted metabolic quantitative trait loci analyses reveal a relationship between primary metabolism and potato tuber quality. Plant Physiol 158: 1306–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheverud JM. (1988) A comparison of genetic and phenotypic correlations. Evolution 42: 958–968 [DOI] [PubMed] [Google Scholar]
- Conklin PL. (2001) Recent advances in the role and biosynthesis of ascorbic acid in plants. Plant Cell Environ 24: 383–394 [Google Scholar]
- Distelfeld A, Li C, Dubcovsky J. (2009) Regulation of flowering in temperate cereals. Curr Opin Plant Biol 12: 178–184 [DOI] [PubMed] [Google Scholar]
- Edwards J, Shavrukov Y, Ramsey C, Tester M, Langridge P, Schnurbusch T (2008) Identification of a QTL on chromosome 7AS for sodium exclusion in bread wheat. In R Appels, R Eastwood, E Lagudah, P Langridge, M Mackay, L McIntyre, eds, 11th International Wheat Genetics Symposium Proceedings. Sydney University Press, Brisbane, Australia. http://hdl.handle.net/2123/3263 (May 31, 2013) [Google Scholar]
- Edwards J (2012) A genetic analysis of drought related traits in hexaploid wheat. PhD thesis. University of Adelaide, Adelaide, Australia
- Falconer DS (1989) Introduction to Quantitative Genetics, Ed 3. Longman Science and Technology, London [Google Scholar]
- Ferrara CT, Wang P, Neto EC, Stevens RD, Bain JR, Wenner BR, Ilkayeva OR, Keller MP, Blasiole DA, Kendziorski C, et al. (2008) Genetic networks of liver metabolism revealed by integration of metabolic and transcriptional profiling. PLoS Genet 4: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleury D, Jefferies S, Kuchel H, Langridge P. (2010) Genetic and genomic tools to improve drought tolerance in wheat. J Exp Bot 61: 3211–3222 [DOI] [PubMed] [Google Scholar]
- Ford KL, Cassin A, Bacic A. (2011) Quantitative proteomic analysis of wheat cultivars with differing drought stress tolerance. Front Plant Sci 2: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forde BG, Lea PJ. (2007) Glutamate in plants: metabolism, regulation, and signalling. J Exp Bot 58: 2339–2358 [DOI] [PubMed] [Google Scholar]
- Francia E, Tacconi G, Crosatti C, Barabaschi D, Bulgarelli D, Dall’Aglio E, Vale G. (2005) Marker assisted selection in crop plants. Plant Cell Tissue Organ Cult 82: 317–342 [Google Scholar]
- Gagneul D, Aïnouche A, Duhazé C, Lugan R, Larher FR, Bouchereau A. (2007) A reassessment of the function of the so-called compatible solutes in the halophytic Plumbaginaceae Limonium latifolium. Plant Physiol 144: 1598–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner KM, Latta RG. (2007) Shared quantitative trait loci underlying the genetic correlation between continuous traits. Mol Ecol 16: 4195–4209 [DOI] [PubMed] [Google Scholar]
- Gaudin ACM, Henry A, Sparks AH, Slamet-Loedin IH. (2013) Taking transgenic rice drought screening to the field. J Exp Bot 64: 109–117 [DOI] [PubMed] [Google Scholar]
- Gilmour AR, Cullis BR, Verbyla AP. (1997) Accounting for natural and extraneous variation in the analysis of field experiments. J Biol Environ Stat 2: 269–293 [Google Scholar]
- Hill CB, Jha D, Bacic A, Tester M, Roessner U. (2013) Characterization of ion contents and metabolic responses to salt stress of different Arabidopsis AtHKT1;1 genotypes and their parental strains. Mol Plant 6: 350–368 [DOI] [PubMed] [Google Scholar]
- Hoekstra FA, Golovina EA, Buitink J. (2001) Mechanisms of plant desiccation tolerance. Trends Plant Sci 6: 431–438 [DOI] [PubMed] [Google Scholar]
- Hong Z, Lakkineni K, Zhang Z, Verma DP. (2000) Removal of feedback inhibition of delta(1)-pyrroline-5-carboxylate synthetase results in increased proline accumulation and protection of plants from osmotic stress. Plant Physiol 122: 1129–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T, Kingston-Smith AH, Foyer CH. (1999) Ascorbate metabolism in potato leaves supplied with exogenous ascorbate. Free Radic Res 31: 171–179 [DOI] [PubMed] [Google Scholar]
- Izanloo A, Condon AG, Langridge P, Tester M, Schnurbusch T. (2008) Different mechanisms of adaptation to cyclic water stress in two South Australian bread wheat cultivars. J Exp Bot 59: 3327–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs A, Lunde C, Bacic A, Tester M, Roessner U. (2007) The impact of constitutive expression of a moss Na+ transporter on the metabolomes of rice and barley. Metabolomics 3: 307–317 [Google Scholar]
- Junker BH, Klukas C, Schreiber F. (2006) VANTED: a system for advanced data analysis and visualization in the context of biological networks. BMC Bioinformatics 7: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keurentjes JJB, Fu J, de Vos CH, Lommen A, Hall RD, Bino RJ, van der Plas LHW, Jansen RC, Vreugdenhil D, Koornneef M. (2006) The genetics of plant metabolism. Nat Genet 38: 842–849 [DOI] [PubMed] [Google Scholar]
- Kichey T, Heumez E, Pocholle D, Pageau K, Vanacker H, Dubois F, Le Gouis J, Hirel B. (2006) Combined agronomic and physiological aspects of nitrogen management in wheat highlight a central role for glutamine synthetase. New Phytol 169: 265–278 [DOI] [PubMed] [Google Scholar]
- Lander ES, Green P. (1987) Construction of multilocus genetic linkage maps in humans. Proc Natl Acad Sci USA 84: 2363–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisec J, Meyer RC, Steinfath M, Redestig H, Becher M, Witucka-Wall H, Fiehn O, Törjék O, Selbig J, Altmann T, et al. (2008) Identification of metabolic and biomass QTL in Arabidopsis thaliana in a parallel analysis of RIL and IL populations. Plant J 53: 960–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisec J, Römisch-Margl L, Nikoloski Z, Piepho H-P, Giavalisco P, Selbig J, Gierl A, Willmitzer L. (2011) Corn hybrids display lower metabolite variability and complex metabolite inheritance patterns. Plant J 68: 326–336 [DOI] [PubMed] [Google Scholar]
- Martinez O, Curnow RN. (1992) Estimating the locations and the sizes of the effects of quantitative trait loci using flanking markers. Theor Appl Genet 85: 480–485 [DOI] [PubMed] [Google Scholar]
- Patterson HD, Thompson R. (1971) Recovery of inter-block information when block sizes are unequal. Biometrika 58: 545–554 [Google Scholar]
- Paux E, Sourdille P, Mackay I, Feuillet C. (2012) Sequence-based marker development in wheat: advances and applications to breeding. Biotechnol Adv 30: 1071–1088 [DOI] [PubMed] [Google Scholar]
- Peltzer D, Dreyer E, Polle A. (2002) Differential temperature dependencies of antioxidative enzymes in two contrasting species: Fagus sylvatica and Coleus blumei. Plant Physiol Biochem 40: 141–150 [Google Scholar]
- Pinhero RG, Rao MV, Paliyath G, Murr DP, Fletcher RA. (1997) Changes in the activities of antioxidant enzymes and their relationship to genetic and paclobutrazol-induced chilling tolerance of maize seedlings. Plant Physiol 114: 695–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland JA, Brown PJ, Sorrells ME, Jannink J-L. (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 7: e32253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2011) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna. http://www.R-project.org
- Reddy AR, Chaitanya KV, Vivekanandan M. (2004) Drought-induced responses of photosynthesis and antioxidant metabolism in higher plants. J Plant Physiol 161: 1189–1202 [DOI] [PubMed] [Google Scholar]
- Reynolds M, Manes Y, Izanloo A, Langridge P. (2009) Phenotyping approaches for physiological breeding and gene discovery in wheat. Ann Appl Bot 155: 309–320 [Google Scholar]
- Reynolds M, Tuberosa R. (2008) Translational research impacting on crop productivity in drought-prone environments. Curr Opin Plant Biol 11: 171–179 [DOI] [PubMed] [Google Scholar]
- Reynolds MP, Mujeeb-Kazi A, Sawkins M. (2005) Prospects for utilising plant adaptive mechanisms to improve wheat and other crops in drought- and salinity prone environments. Ann Appl Biol 146: 239–259 [Google Scholar]
- Roessner U, Luedemann A, Brust D, Fiehn O, Linke T, Willmitzer L, Fernie AR. (2001) Metabolic profiling allows comprehensive phenotyping of genetically or environmentally modified plant systems. Plant Cell 13: 11–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe HC, Hansen BG, Halkier BA, Kliebenstein DJ. (2008) Biochemical networks and epistasis shape the Arabidopsis thaliana metabolome. Plant Cell 20: 1199–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Matsuda F. (2010) Metabolomics for functional genomics, systems biology, and biotechnology. Annu Rev Plant Biol 61: 463–489 [DOI] [PubMed] [Google Scholar]
- Schauer N, Semel Y, Roessner U, Gur A, Balbo I, Carrari F, Pleban T, Perez-Melis A, Bruedigam C, Kopka J, et al. (2006) Comprehensive metabolic profiling and phenotyping of interspecific introgression lines for tomato improvement. Nat Biotechnol 24: 447–454 [DOI] [PubMed] [Google Scholar]
- Shiferaw B, Smale B, Braun H-J, Duveiller E, Reynolds M, Muricho G. (2013) Crops that feed the world 10. Past successes and future challenges to the role played by wheat in global food security. Food Sec (in press) [Google Scholar]
- Smith AB, Cullis BR, Thompson R. (2004) Analyzing variety by environment data using multiplicative mixed models and adjustments for spatial field trend. Biometrics 57: 1138–1147 [DOI] [PubMed] [Google Scholar]
- Smith AB, Cullis BR, Gilmour A. (2002) Applications: the analysis of crop evaluation data in Australia. Aust New Zeal J Stat 43: 129–145 [Google Scholar]
- Smith AB, Cullis BR, Thompson R. (2005) The analysis of crop cultivar breeding and evaluation trials: an overview of current mixed model approaches. J Agric Sci 143: 449–462 [Google Scholar]
- Smith AB, Lim P, Cullis BR. (2006) The design and analysis of multi-phase plant breeding experiments. J Agric Sci 43: 129–145 [Google Scholar]
- Snape JW, Butterworth K, Whitechurch E, Worland AJ. (2001) Waiting for fine times: genetics of flowering time in wheat. Euphytica 119: 185–190 [Google Scholar]
- Taylor JD, Diffey S, Verbyla AP, Cullis BR (2011) R/wgaim: Whole Genome Average Interval Mapping for QTL Detection Using Mixed Models. R package version 1.2. http://CRAN.R-project.org/package=wgaim
- Taylor JD, Verbyla AP. (2011) R package wgaim: QTL analysis in bi-parental populations using linear mixed models. J Stat Softw 40: 1–18 [Google Scholar]
- Urano K, Maruyama K, Ogata Y, Morishita Y, Takeda M, Sakurai N, Suzuki H, Saito K, Shibata D, Kobayashi M, et al. (2009) Characterization of the ABA-regulated global responses to dehydration in Arabidopsis by metabolomics. Plant J 57: 1065–1078 [DOI] [PubMed] [Google Scholar]
- Valliyodan B, Nguyen HT. (2006) Understanding regulatory networks and engineering for enhanced drought tolerance in plants. Curr Opin Plant Biol 9: 189–195 [DOI] [PubMed] [Google Scholar]
- Verbyla AP, Cullis BR, Thompson R. (2007) The analysis of QTL by simultaneous use of the full linkage map. Theor Appl Genet 116: 95–111 [DOI] [PubMed] [Google Scholar]
- Vinocur B, Altman A. (2005) Recent advances in engineering plant tolerance to abiotic stress: achievements and limitations. Curr Opin Biotechnol 16: 123–132 [DOI] [PubMed] [Google Scholar]
- Wentzell AM, Rowe HC, Hansen BG, Ticconi C, Halkier BA, Kliebenstein DJ. (2007) Linking metabolic QTLs with network and cis-eQTLs controlling biosynthetic pathways. PLoS Genet 3: 1687–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worland AJ, Appendino ML, Sayers EJ. (1994) The distribution, in European winter wheats, of genes that influence ecoclimatic adaptability whilst determining photoperiodic insensitivity and plant height. Euphytica 80: 219–228 [Google Scholar]
- Zeng ZB. (1994) Precision mapping of quantitative trait loci. Genetics 136: 1457–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Klueva NY, Wang Z, Wu R, Ho T-HD, Nguyen HT. (2000) Genetic engineering for abiotic stress resistance in crop plants. In Vitro Cell Dev Biol Plant 36: 108–114 [Google Scholar]
- Zhu JK. (2002) Salt and drought stress signal transduction in plants. Annu Rev Plant Biol 53: 247–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.