

Summary
The macrophage migration inhibitory factor (MIF) receptor (CD74) was cloned recently, but the signaling mechanism is not evident. We hypothesized that signaling requires an additional molecule such as CD44, which activates nonreceptor tyrosine kinases. We utilized the CD74- and CD44-deficient COS-7/M6 cell to create stable transfectants expressing CD74, CD44, and a truncated CD44 lacking its intracytoplasmic signaling domain. CD74 alone mediated MIF binding; however, MIF-induced ERK1 and ERK2 kinase phosphorylation required the coexpression of full-length CD44. MIF binding was associated with the serine phosphorylation of CD74 and CD44. Investigations that used siRNA or kinase inhibitors indicate that MIF-induced ERK1 and ERK2 activation through CD44 required the Src tyrosine kinase. Studies of CD74, CD44, and CD74-CD44 transformants and corresponding mutant cells showed that CD74 and CD44 were necessary for MIF protection from apoptosis. These data establish CD44 as an integral member of the CD74 receptor complex leading to MIF signal transduction.
Introduction
Macrophage migration inhibitory factor (MIF) is an upstream activator of innate immunity that regulates subsequent adaptive responses (Bacher et al., 1996; Calandra and Roger, 2003). MIF antagonizes the action of glucocorticoids (Calandra et al., 1995; Calandra and Roger, 2003), upregulates Toll-like receptor 4 (TLR-4) expression (Roger et al., 2001), controls Jab1 transcriptional effects (Kleemann et al., 2000), and suppresses activation-induced, p53-dependent apoptosis (Hudson et al., 1999; Mitchell et al., 2002; Nguyen et al., 2003). This latter action may sustain inflammatory responses in the face of activation-induced apoptosis, and it may underlie MIF’s broad inflammatory and proproliferative effects on diverse cell types (Hudson et al., 1999; Mitchell et al., 2002; Fingerle-Rowson et al., 2003; Leech et al., 2003).
Interest in the biology of MIF has been heightened by the protein’s role in septic shock (Bernhagen et al., 1993; Calandra et al., 2000), by the description of functional polymorphisms in the gene that are associated with inflammatory disease (Gregersen and Bucala, 2003), and by an emerging role for MIF in tumorigenesis (Meyer-Siegler and Hudson, 1996; Hudson et al., 1999; Fingerle-Rowson et al., 2003). A cell-surface receptor for MIF was cloned in 2003 and identified as the widely expressed Type II transmembrane protein, CD74 (Leng et al., 2003). Known features of MIF signal transduction include the phosphorylation of the ERK1 and ERK2 MAP kinases, which may be sustained in certain circumstances (Mitchell et al., 1999). In addition, MIF activates the ERK effectors cytoplasmic phospholipase A2, which initiates arachidonic metabolism and has a role in p53 suppression (Mitchell et al., 2002), and the Elk-1 and Ets transcription factors, which regulate TLR4 expression (Roger et al., 2001). MIF-dependent ERK activation also promotes maximal expression of cyclin D1 leading to cyclin-dependent kinase activation, RB phosphorylation, and adhesion and/or growth factor stimulation of mesenchymal cells (Liao et al., 2003; Swant et al., 2005).
In an initial report, evidence was provided for a high-affinity binding interaction between MIF and the CD74 ectodomain (Kd ~9 × 10−9) (Leng et al., 2003). Like MIF, CD74 is expressed as a homotrimer, but the precise mechanism by which signal transduction proceeds by MIF engagement of CD74 is unknown. The CD74 intracellular domain is only 46 amino acids long and it lacks homology with tyrosine or serine/threonine kinases, or with the interaction domains for nonreceptor kinases or nucleotide binding proteins. The intracytoplasmic tail of CD74 nevertheless may undergo phosphorylation (Anderson et al., 1999), and there are data supporting a pathway for this protein’s regulated, intramembrane cleavage (Matza et al., 2002). Two studies also have reported a functional, cell-surface association between CD74 and CD44 (Naujokas et al., 1993, 1995), which has known tyrosine kinase activation properties (Turley et al., 2002). In the present study, we explored the possibility that MIF signaling through CD74 requires the simultaneous expression and activation of CD44. We performed studies in cell lines engineered to stably express CD74 or CD44, their combination, or CD74 together with a truncated CD44 lacking its cytoplasmic signaling domain (CD44Δ67). We also investigated the responses of primary cells genetically deficient in CD74 or CD44.
Results
Creation and Characterization of Stably Expressing CD74 and CD44 Transformants
Mammalian COS-7 cells do not bind MIF unless engineered to express CD74 (Leng et al., 2003), and the COS-7/M6 subline additionally is CD44 deficient (Jiang et al., 2002). The absence of CD74 and CD44 was confirmed in COS-7/M6 cells by immunoblotting, and the cells then were used as hosts for the stable transfection of plasmid DNA encoding full-length human CD74 (1–232 aa), full-length CD44 (1–361 aa of the hematopoietic “H” isoform of CD44), or a truncated CD44 lacking its cytoplasmic domain (CD44Δ67) (Figure 1). Cell lines expressing the corresponding cDNAs were propagated, subcloned, and selected for further study based on the stable expression of these proteins (Figure 2A). We confirmed the cell-surface expression of the transfected proteins by flow cytometry. We also verified equivalent surface expression of the full-length and truncated forms of CD44, which is important for functional analyses, and confirmed that expression of the MIF binding receptor, CD74, was not influenced by the presence of CD44 or CD44Δ67 (Figure 2B).
Figure 1.
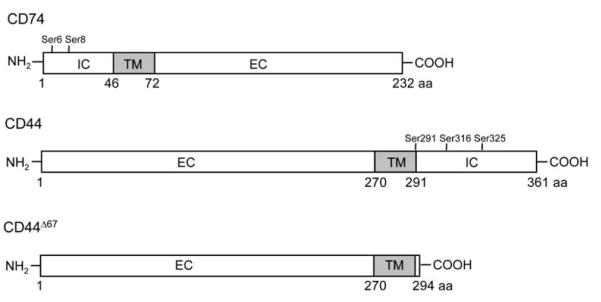
Schematic Diagram of the Structures of the Human CD74 and CD44 Proteins Used to Create Stable Cell Lines
IC, TM, and EC are the intracellular, transmembrane, and extracellular domains, respectively. The locations of the known intracytoplasmic serine phosphorylation sites are indicated.
Figure 2.

Analysis of COS-7/M6 Cells Stably Transfected with CD44, CD74, CD74+CD44, or CD74+CD44Δ67
(A) Cell lysates were immunoblotted with human CD44 and human CD74 antibodies. CD44 cannot be distinguished from CD44Δ67 by molecular weight because of posttranslational glycosylation (Jiang et al., 2002).
(B) Cell-surface expression of COS-7/M6 cell controls or COS-7/M6 cells expressing CD44, CD74, CD74+CD44, or CD74+CD44Δ67. Flow cytometry was performed after incubation of 5 × 104 cells at 0°C with FITC-labeled anti-CD44 or anti-CD74. Each analysis included an isotypic control.
(C) Flow cytometry analysis of Alexa-MIF binding to COS-7/M6 cells and COS-7/M6 cells stably transfected with CD44, CD74, or CD74+CD44. Each panel shows the fluorescence profile of 5 × 104 cells incubated at 0°C for 30 min with Alexa-MIF. Control studies showed specific competition of Alexa-MIF binding by unlabeled MIF (25× excess) or by anti-CD74 (clone LN2, 50 μg/ml) (Leng et al., 2003, and data not shown). In all figure labels, COS-7 cells refers to the CD74- and CD44-deficient COS-7/M6 line. Numerical values show the MFI ± SD (p values by unpaired t test).
We first utilized these stable cell lines to assess the potential contribution of CD44 to the MIF binding interaction with CD74. We prepared an Alexa 488, fluorescently labeled MIF that was used previously in the expression cloning of CD74 (Leng et al., 2003), and we tested its binding activity with the different transfected cell lines (Figure 2C). These data supported the conclusion that CD74 alone is sufficient to mediate MIF binding to cells. CD44 alone did not bind MIF, nor did CD44 confer additional binding above that provided by CD74 alone.
CD44 Is Required for MIF-Mediated ERK Phosphorylation
The phosphorylation of the ERK1 and ERK2 subfamily of MAP kinases is an established feature of MIF signal transduction (Mitchell et al., 1999; Lacey et al., 2003; Amin et al., 2003). Initial studies showed that the COS-7–CD74+CD44 cells responded to MIF stimulation, and in accord with prior work (Lue et al., 2006), the response was “bell shaped” and dose dependent (Figure 3A). We then examined ERK phosphorylation in the different transfected cell lines over time in response to a maximal stimulatory concentration of MIF and showed MIF induced ERK phosphorylation only in those cells that expressed both CD74 and full-length CD44 (Figure 3B). Although MIF could induce a sustained pattern (>90 min) of ERK1 and ERK2 phosphorylation in some cell types (Mitchell et al., 1999; Liao et al., 2003), this was not observed in the stable COS-7/M6 expression system. ERK phosphorylation decayed at 30 min, which may be due to the absence of adhesion signals that mediate the sustained phase of activation (Liao et al., 2003; Swant et al., 2005). Stable transfection of CD74 and CD44 did not affect the baseline sensitivity of COS-7/M6 cells to non-MIF stimuli, as assessed by an experiment in which no differences in ERK phosphorylation in untransfected versus transfected cells was observed after 10% serum stimulation (see Figure S1 in the Supplemental Data available online).
Figure 3.

MIF-Induced ERK Phosphorylation Requires CD74 and Full-Length, Intact CD44
(A) Dose-dependent phosphorylation of ERK1 and ERK2 (ERK1/2) by MIF stimulation of the COS-7, CD74+CD44-expressing cell line.
(B) COS-7/M6 cells stably transfected with CD74, CD44, CD74+CD44, or CD74+CD44Δ67 were stimulated with MIF (100 ng/ml) for the indicated times.
(C) Murine embryonic fibroblasts (MEFs) from wild-type mice, but not CD74-KO or CD44-KO mice, respond to MIF by ERK phosphorylation.
(D) Preformed MIF/CD74 complexes do not stimulate ERK phosphorylation in CD44-expressing COS-7/M6 cells. Recombinant sCD74 was incubated with MIF overnight in a 3:1 molar ratio prior to addition to cells for 10 min. Epidermal growth factor (EGF) was used as a positive control for ERK phosphorylation.
(E) COS-7 cells stably transfected with CD74+CD44, CD74, or CD44 were cocultured with the indicated cell line partner (1 × 106 of each cell type) and MIF-induced ERK phosphorylation measured. The numerical ratio between phosphorylated to the total kinase protein was determined by densitometric scanning of three experiments and expressed as a fold-change below each lane. The p values were calculated for each of the comparisons (100, 50, and 10 versus 0 ng/ml MIF in [A], 100 versus 0 ng/ml MIF in [B] and [C] by the Student’s t test. ***p < 0.01, **p < 0.02, *p < 0.05. The p values for comparisons with p > 0.05 are not displayed). KO, knockout.
We verified the requirement for CD44 in MIF signal transduction by examining primary cells prepared from wild-type, CD74-deficient, and CD44-deficient mice. Wild-type murine embryonic fibroblasts (MEFs) express both CD74 and CD44 (Figure S2) and responded to MIF by ERK phosphorylation (Figure 3C). MIF induced ERK phosphorylation only in wild-type cells, and not in cells genetically deficient in CD74 or CD44. Similar results were obtained in peritoneal macrophages (Figure S3).
We did not obtain evidence for a cell-surface interaction between CD44 and MIF (Figure 2C). However, CD44 might mediate signaling of MIF by binding to a MIF that has undergone conformational modification as a result of binding to CD74. As a test of this possibility, we prepared soluble, recombinant CD74 ectodomain (sCD74) that binds MIF with a Kd ~9 × 10−9 (Leng et al., 2003) and added increasing concentrations of preformed MIF-sCD74 complexes to cells expressing CD44 alone (Figure 3D). No increase in ERK phosphorylation was observed under these conditions, supporting the requirement for an interaction between membrane-expressed CD74 and CD44. We also tested the possibility that CD44 may be activated in trans by a MIF-CD74 complex by examining MIF-induced ERK phosphorylation in 1:1 mixtures of the CD74- or CD44-expressing COS-7/M6-derived cell lines. An MIF-induced increase in ERK phosphorylation was not detected in these mixing experiments (Figure 3E). These data, taken together, are consistent with the interpretation that MIF-induced ERK phosphorylation requires the cellular expression of two integral membrane proteins: a binding receptor (CD74) and a signaling protein (CD44).
MIF Modulates the Serine Phosphorylation of CD74 and CD44
Two serine residues within the CD74 intracytoplasmic domain (Ser6 and Ser8) undergo phosphorylation in a protein kinase-dependent manner (Anderson et al., 1999). We quantified the content of phospho-serine residues in CD74 by a specific sandwich ELISA (Perez et al., 2003) and found the phosphorylation of CD74 in the COS-7–CD74+CD44 cell line and in wild-type MEFs increased in response to MIF, but it did not change in the other COS-7/M6 transformants and genetically deficient, primary cell lines (Figure 4A). The observation that the phosphoserine content of CD74 increased only in cells that expressed CD74 and full-length CD44 is consistent with the hypothesis that the MIF-mediated phosphorylation of CD74 is dependent on the activity of the CD44 intracytoplasmic domain.
Figure 4.

Phosphoserine Content of CD74 and CD44 Measured by ELISA
(A) COS-7/M6-derived cell lines and MEFs were treated with MIF (100 ng/ml) for 10 min and the cell lysates analyzed for phospho-serine by a CD74-specific sandwich ELISA.
(B) The COS-7/CD74+CD44 cell line was pretreated with the protein kinase A (PKA) inhibitor, H-89 (20 μM), or the protein kinase C (PKC) inhibitor, RO-31-28801 (10 μM), for 30 and 60 min prior to MIF (100 ng/ml) stimulation. Cell lysates then were analyzed for phospho-serine content.
(C) COS-7/M6-derived cell lines and MEFs were treated with MIF (100 ng/ml) for 10 min and the cell lysates analyzed for phospho-serine by a CD44-specific sandwich ELISA.
(D) Analysis of CD44 phospho-serine content in control and MIF-stimulated, COS-7 cell lines after preincubation with the PKA inhibitor, H-89, or the PKC inhibitor, RO-31-2880, for 30 min. Phosphoserine contents are expressed as relative absorbance values for ELISA of cell lysates for cell treatment versus nontreatment. *p < 0.02 versus corresponding control.
(E) Western analysis of COS-7–CD74+CD44 cells after stimulation with MIF for 10 min. Cell lysates were probed for PKA and PKC with phospho-specific and total PKA and PKC antibodies, *p < 0.01. KO, knockout.
Error bars denote the mean ± SD.
Protein kinase A (PKA) has been implicated in the MIF-dependent phosphorylation of ERK leading to downstream activation events (Mitchell et al., 1999). We tested the potential role of PKA and PKC, which may phosphorylate CD74 in vitro (Anderson et al., 1999), by analyzing MIF-induced CD74 phosphorylation in the presence of the PKA inhibitor, H-89, or the PKC inhibitor, RO-31-2880. MIF stimulation of CD74 phosphorylation decreased markedly upon PKA inhibition. No effect was seen in the presence of the PKC inhibitor RO-31-2880 at the effective and PKC-selective concentration of 10 μM (Figure 4B; Padfield and Panesar, 1998; Hill et al., 2003).
We also analyzed the effect of MIF stimulation on the phosphorylation of CD44, which may occur on three intracytoplasmic serine residues (Ser291, Ser316, and Ser325) (Ponta et al., 2003; Thorne et al., 2004). Ser325 is constitutively phosphorylated in resting cells, but undergoes dephosphorylation in response to PKC-activating stimuli. Conversely, Ser291 and Ser316 are unphosphorylated in resting cells, but then may be phosphorylated by the activation of PKC (Ser291) and PKA (Ser316) (Legg et al., 2002; Ponta et al., 2003; Thorne et al., 2004). We analyzed the effect of MIF stimulation CD44 phosphorylation by sandwich ELISA with a CD44 capture antibody to quantify CD44 phospho-serine residues (Perez et al., 2003). In contrast to the phosphoserine content of CD74, which increased in response to MIF (Figure 4A), the net content of CD44 serine phosphorylation did not change after MIF stimulation (Figure 4C). We interpreted this result to be due either to the lack of effect of MIF on CD44 serine phosphorylation or, alternatively, to a mixed effect on CD44 phosphorylation and dephosphorylation such that net phosphoserine content did not change. We obtained evidence for this second possibility by analyzing CD44 phosphoserine content after stimulation in the presence of the protein kinase inhibitors. Under these conditions, the serine phosphorylation of CD44 was reduced by PKA inhibition (H-89, 20 μM) but not by PKC inhibition (RO-31-2880, 10 μM) (Figure 4D). A role for MIF activation of PKA was confirmed by immunoblotting of the COS-7–CD74+CD44-expressing cell line, which showed MIF-dependent phosphorylation of PKA but not PKC (Figure 4E). These results suggest that MIF stimulation is indeed associated with a PKA-dependent alteration in the serine phosphorylation of CD44, but because the net phosphoserine content is unaltered, there is a reciprocal decrease in the amount of constitutively phosphorylated serines.
The Protein Tyrosine Kinase Src Is Activated by MIF Engagement of the CD74-CD44 Complex
Members of the nonreceptor protein tyrosine kinase family, c-Src (Src), may physically associate with the intracytoplasmic domain of CD44 (Taher et al., 1996; Bourguignon et al., 2001), leading to downstream ERK phosphorylation via a Ras-Raf-MEK-dependent pathway (Ishida et al., 1998; Migliaccio et al., 2000; Mahabeleshwar and Kundu, 2003). There is also recent evidence for the activation of Src kinase family members by MIF, although the receptor proteins that may be involved in the activation are unknown (Onodera et al., 2002; Amin et al., 2006; Lue et al., 2006). We analyzed the phosphorylation state of Src in the different COS-7/M6 transformants and MEFs after MIF stimulation by immunoblotting with a phospho antibody specific for Src (Tyr416). Src kinase was phosphorylated by MIF treatment, but only in the cells expressing both CD74 and full-length CD44 (Figures 5A and 5B).
Figure 5.

Src Tyrosine Kinase Mediates MIF-Induced ERK Phosphorylation
(A and B) COS-7/M6 derived cell lines (A) or MEFs (B) were stimulated with MIF for 10 min and the cell lysates analyzed for phospho-Src (p-Src, Tyr416), GAPDH, phospho-ERK1 and ERK2 (p-ERK1/2), and total ERK1 and ERK2 (ERK1/2) by specific antibodies.
(C) COS-7/M6 cells expressing CD74+CD44 treated with siRNA directed against c-Src or a control siRNA prior to stimulation with MIF. Lysates were analyzed for p-Src, total Src, GAPDH, and ERK by western blotting.
(D and E) COS-7/M6 cells expressing CD74+CD44 (D) or primary macrophages from wild-type mice (E) were treated with the kinase inhibitor, PP2, for 60 min prior to MIF stimulation (10 min) and western blotting. Densitometric values below lanes refer to the ratio of phospho-protein to total, reference protein, *p < 0.05, **p < 0.04, ***p < 0.01.
To confirm a role for Src tyrosine kinase in MIF signal transduction leading to ERK phosphorylation, we prepared a short interfering RNA (siRNA) directed against Src (Coluccia et al., 2006), and we tested its ability to prevent the MIF-stimulated phosphorylation of Src and ERK1 and ERK2. Treatment of COS-7–CD74+CD44-expressing cells with Src-specific siRNA decreased both Src protein expression and MIF-induced Src phosphorylation (Figure 5C). We also tested the potential effect of the Src kinase inhibitor PP2 (Salazar and Rozengurt, 1999). PP2 addition to the COS-7–CD74+CD44 cell line (Figure 5D) or to primary macrophages (Figure 5E) resulted in an inhibition of the phosphorylation of Src and ERK. These data are consistent with a role for a CD44-associated Src kinase in MIF signal transduction leading to ERK1/2 phosphorylation.
CD74 and CD44 Are Required for MIF-Mediated Protection from p53-Dependent Apoptosis
An important biologic action of MIF is to sustain proinflammatory responses by inhibiting activation-induced, p53-dependent apoptosis (Hudson et al., 1999; Mitchell et al., 2002; Nguyen et al., 2003). We next asked whether CD74 and CD44 were necessary for this action of MIF by assessing the apoptotic response of the different COS-7/M6 cell lines and primary macrophages genetically deficient in CD74 or CD44. The COS-7/M6 cell lines showed a brisk response to apoptotic induction, but only in the case of the COS-7–CD74+CD44-expressing cell line did MIF exert a significant protective effect (Figure 6A). The antiapoptotic action of MIF was associated with a reduction in the intracytoplasmic content of a Ser15-phosphorylated, p53 species (Figure 6B). MIF protection from apoptosis of primary macrophages also was found to be dependent on CD74 and CD44 (Figure 6C), and in these cells the protective effect of MIF was almost complete, as in prior reports (Hudson et al., 1999; Mitchell et al., 2002). MIF treatment of macrophages during apoptosis induction also was associated with a diminution in the cellular content of Ser15-phosphorylated p53 (Figure 6D). In contrast to reports in a macrophage cell line (Mitchell et al., 2002) or in cultured human synovial fibroblasts and mouse synovium (Leech et al., 2003), however, we observed no effect of MIF on total cellular p53 protein content.
Figure 6.

MIF-Mediated Protection from Apoptosis Requires CD74 and CD44
(A) COS-7/M6 cell lines were stimulated for apoptosis induction (AI) in the presence of MIF (100 ng/ml) for 24 hr, and the apoptotic response was measured by caspase-3 activity (*p < 0.02, Student’s t test, two-tailed).
(B) Western blotting analysis of p53 in cytosolic fractions from the different COS-7 cell lines showing reduced phospho-p53 (p-p53) content in the CD74+CD44 expressing cells after stimulation with MIF, *p < 0.02.
(C) Caspase-3 activity in primary macrophages after apoptosis induction in the presence of MIF (*p < 0.02, Student’s t test, two-tailed).
(D) Western blotting analysis of cytosolic fractions from primary mouse macrophages after apoptosis induction in the presence and absence of MIF. Note that the murine (total) p53 antibody detects an immunoreactive, p53 doublet, which is consistent with previous observations (Mitchell et al., 2002). Densitometric values below lanes refer to the ratio of phospho-protein to total p53 protein, *p < 0.02.
(E) Enhanced activation-induced apoptosis in macrophages from CD74-KO and CD44-KO mice. Wild-type, MIF-KO, CD74-KO, and CD44-KO mice were injected i.p. with LPS, and macrophages were isolated from the peritoneal exudates 24 hr later. Representative high-power fields of fluorescent-annexin stained macrophages are shown. The control image shows wild-type macrophages obtained from saline-treated mice. For quantification, macrophages (5 × 104 cells per mouse, n = mice per experimental group) were examined in multiple high-power fields and the positively stained cells (intense, punctate fluorescence) enumerated. No differences were observed between saline treatment of wild-type, MIF-KO, CD74-KO, or CD44-KO (data not shown). *p < 0.01 versus saline-treated control (Student’s t test, two-tailed).
(F) Macrophage apoptosis quantified by oligonucleosome ELISA. The values shown are mean 6 SD of triplicate wells and are representative of two experiments. *p < 0.02 versus LPS-treated, wild-type mice, **p < 0.01 versus no LPS treatment (Student’s t test, two-tailed).
Error bars denote the mean ± SD.
Finally, we sought to examine the impact of the MIF–CD74-CD44 signal transduction pathway in vivo by examining macrophage apoptosis in endotoxemic mice. Wild-type, MIF-KO, CD74-KO, and CD44-KO mice were primed with endotoxin (LPS), and their macrophages were harvested 1 day later by peritoneal lavage. Initial assessment of macrophage viability by fluorescent-annexin staining showed that endotoxemic, wild-type mice had a several-fold increase in apoptotic macrophage numbers when compared to saline-treated controls (Figure 6E). The apoptotic response was enhanced in the MIF-KO mice when compared to the wild-type controls, which is in agreement with a prior report (Mitchell et al., 2002), and apoptosis also increased in the CD74-deficient and the CD44-deficient mouse strains. Oligonucleosome ELISA analysis showed an equivalent amount of LPS-induced apoptosis in macrophages isolated from the MIF-deficient, CD74-deficient, and CD44-deficient strains that in turn was enhanced when compared to wild-type controls (Figure 6F). These data support a role for CD74 and CD44 in the MIF-mediated protection of macrophage apoptosis in vivo.
Discussion
We provide support for an important role for CD44 in MIF-mediated signal transduction through CD74. Prior to the identification of MIF as an extracellular ligand for CD74, there was evidence for an accessory function for CD74 in immune stimulation that was mediated by an interaction with CD44 (Naujokas et al., 1993). Given CD74’s short intracytoplasmic domain and the absence of motifs for second messenger activation, we hypothesized that CD44 might be necessary for many, if not all, of MIF’s signal transduction properties. By using COS-7/M6 (deficient in both CD74 and CD44) cell lines engineered to stably express combinations of CD74, CD44, and CD44Δ67, we found that CD74 alone imparts MIF binding to cells and that the presence of CD44 did not confer additional binding activity over that of CD74 alone. Of note, evidence in support of a molecular complex between MIF, CD74, and CD44 was provided by Meyer-Siegler et al. (2004), who utilized an immunoprecipitation approach in cultured carcinoma cells. Our data indicate that MIF induces the serine phosphorylation of the CD74 intracytoplasmic domain in a CD44-dependent manner. This implies that although CD74 functions to bind MIF, it also undergoes ligand-induced activation events that may be necessary for the CD44-dependent signaling pathways described herein or for additional signaling mechanisms yet to be elucidated.
We did not obtain evidence for a specific interaction between MIF and CD44; however, MIF-mediated ERK1 and ERK2 activation requires the expression of a full-length CD44. MIF stimulation was associated with the PKA-dependent serine phosphorylation of CD74 and CD44. Investigations with siRNA and the kinase inhibitor PP2 indicate that MIF-induced ERK activation proceeds via the Src tyrosine kinase, which previously has been shown to associate physically with the CD44 intracytoplasmic domain (Taher et al., 1996).
An important feature of MIF’s biologic action is its ability to sustain monocyte/macrophage activation in the face of activation-induced, p53-dependent apoptosis (Hudson et al., 1999; Mitchell et al., 2002). Apoptosis and p53 studies of MIF-stimulated CD74, CD44, and CD74/CD44 transformants and genetically deficient primary cells are consistent with the conclusion that CD74 and CD44 are necessary for MIF protection from apoptosis. Endotoxemic MIF-deficient mice showed a higher amount of macrophage apoptosis than wild-type controls, which is in accord with prior work (Mitchell et al., 2002), and the level of enhanced macrophage apoptosis was recapitulated in mice lacking the MIF binding (CD74) or signaling (CD44) receptor.
From the above data, we propose an MIF signal transduction pathway based on evidence for the proximal involvement of CD74 and CD44 (Figure 7). The respective binding and signaling roles for CD74 and CD44 are reminiscent of the signaling mechanism established for the IL-6 family of cytokines, whereby a binding receptor (i.e., IL-6Rα) associates with a transmembrane glycoprotein (i.e., gp130) leading to kinase activation (Hibi and Hirano, 2001). Several of the biologic activities of MIF have been identified to proceed via ERK activation. These include arachidonic acid metabolism and prostaglandin production (via cytoplasmic phospholipase A2 and cyclooxygenase-2) (Mitchell et al., 1999; Sampey et al., 2001), regulation of p53 activity (Mitchell et al., 2002), and the activation of additional ERK effectors, such as the Ets family of transcription factors that regulate Toll-like receptor expression (Roger et al., 2001). One question of interest is the precise role of CD74 and/or CD44 in additional features of MIF action, such as its ability to bind to and regulate the intracytoplasmic action of c-Jun activation domain binding protein-1 (JAB1). (Kleemann et al., 2000). Whether CD74 mediates MIF internalization and/or facilitates interaction with JAB1 will be important to consider. Recent information suggests that JAB1 regulates the sustained phase of MIF-induced ERK phosphorylation via activation of a Src family kinase member (Lue et al., 2006). Sustained ERK phosphorylation by MIF also exhibits adhesion dependence (Liao et al., 2003), which may now be better understood given CD44’s known role in matrix interaction (Bourguignon et al., 2001; Ponta et al., 2003). Mitchell et al. also have established that cell adhesion results in an autocrine MIF release response and to a pathway for ERK activation involving the sequence of Rho GTPase, myosin light chain kinase, and focal adhesion kinase (Liao et al., 2003; Swant et al., 2005).
Figure 7.

Model of MIF Signal Transduction Involving MIF Binding to CD74 and Activation of the CD44 Coreceptor
The pathway leading from the CD44 intracytoplasmic domain through c-Src and toward the activation of guanine nucleotide exchange factors (GEFs) has been described (Bourguignon et al., 2001; Taher et al., 1996). The pathway downstream of Ras has been described (Mitchell et al., 1999; Lue et al., 2006), and Swant et al. (2005) recently reported a role for the Ras effector, RhoGTPase, acting via myosin light chain kinase (MLCK) and focal adhesion kinase (FAK), in the induction of sustained phase, MIF-mediated ERK activation.
The present data also add to the emerging cellular biology of CD74 and CD44. CD74 has been shown to undergo regulated intramembrane cleavage in B lymphocytes, leading to the activation of transcription by the p65-RelA homodimer (Matza et al., 2001, 2002; Starlets et al., 2006). Whether MIF participates as a ligand in these events is presently under examination (I. Shachar, personal communication). Notably, CD74-KO and MIF-KO mice share phenotypic features, such as an impairment in T helper 2 cell responses that may be attributable, in part, to deficiencies in maturation of an adaptive immune response (Topilski et al., 2002; Mizue et al., 2005). CD44 is a genetically polymorphic molecule with an important role in cell-extracellular matrix interaction (Ponta et al., 2003). A potential overlapping function for CD44 and MIF, in addition to survival and antiapoptotic functions (Allouche et al., 2000; Chen et al., 2001), include proinflammatory actions for CD44 when engaged by an activating antibody or by hyaluronan fragments. In studies reminiscent of MIF biologic function, however, many of these observations emphasize the regulatory nature of CD44 (Bradbury, 2002; Teder et al., 2002; Webb et al., 1990; Sun et al., 2001). Finally, pharmacologic strategies directed at MIF are in preclinical development (Lolis and Bucala, 2003). Closer consideration of antireceptor strategies that target the CD74/CD44 interaction may offer a basis for enhanced therapeutic specificity.
Experimental Procedures
Mice and Primary Cells
Wild-type (BALB/c or C57/Bl6), CD74-KO (Shachar and Flavell, 1996), CD44-KO (Teder et al., 2002), and MIF-KO (Bozza et al., 1999) mice were bred at the Yale Animal Resource Center. The KO strains were in BALB/c or C57/Bl6 backgrounds (each at generation N10). Mouse embryonic fibroblasts (MEFs) were prepared as described (Fingerle-Rowson et al., 2003). Initial studies revealed no differences in MIF signaling responses of cells derived from mice of the BALB/c or C57/Bl6 backgrounds. Primary macrophages from peritoneal exudate fluid were obtained as described previously (Calandra et al., 1994). Mouse studies were approved by the Yale Institutional Animal Use and Care Committee.
Plasmid DNA Vectors
The pcDNA3.1-CD74 plasmid was constructed by inserting a full-length human CD74 cDNA fragment (1–232 aa) into the pcDNA3.1/V5-His-TOPO vector (Invitrogen) (Leng et al., 2003). The pTracer-CD44 and pTracer-CD44Δ67 plasmids, which encode the human hematopoietic form of CD44 (CD44H, 1–361 aa) and a truncated CD44 lacking the cytoplasmic domain (CD44Δ67, 1–294 aa), respectively, were created by subcloning into the pTracer-SV40 vector (Invitrogen) a human full-length CD44 cDNA or a CD44 cDNA created by substitution of a stop codon for cysteine 295 via site-directed mutagenesis (Jiang et al., 2002). Structural fidelity was confirmed by DNA sequencing. Figure 1 illustrates the CD74 and CD44 cDNA constructs.
Creation of Stable Transformants
COS-7/M6 cells, which express neither CD74 nor CD44 (Figure 2; Jiang et al., 2002), were stably transfected with pcDNA3.1-CD74, pTracer-CD44, pcDNA3.1-CD74 plus pTracer-CD44, or pcDNA3.1-CD74 plus pTracer-CD44Δ67 with the LipofectAMINE PLUS Kit (Invitrogen). Stable transformants were selected by culture in G418 (1.5 mg/ml, Sigma). The different transformants were subcloned and stable expression confirmed periodically by immunoblotting with human CD74 antibody (BD Pharmingen) and human CD44 antibody (sc-7297, Santa Cruz). Clones stably expressing immunoequivalent levels of CD74 and CD44 were used for functional studies. COS-7/M6 cells were used as negative controls, and HeLa cells (Bouchard and Racaniello, 1997) expressing CD44 were used as positive controls.
Flow Cytometry
Washed cells (~4.0 × 105) were resuspended in PBS/2% FBS and stained on ice for 30 min with isotypic controls, human CD74, human CD44, or mouse CD44 (BD Pharmingen) antibodies conjugated with FITC or Alexa 488-labeled MIF (Leng et al., 2003). The labeled cells were studied with a FACS-Calibur (BD Pharmingen) and the data were analyzed with CellQuest software (BD Pharmingen).
Signal Transduction Assays
Cells were stimulated with native sequence, recombinant human MIF (for COS-7 transformants) or mouse MIF (for MEFs and macrophages) produced in our laboratory (Bernhagen et al., 1994). The human CD74 ectodomain (CD7473-232) was prepared recombinantly in soluble form (sCD74) as described previously (Leng et al., 2003). In a typical experiment, 2 × 106 cells were rendered quiescent by incubation in low serum for 16 hr prior to stimulation with MIF for 5–30 min (Mitchell et al., 1999). Cells then were harvested and lysed in buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1% Glycerol, and 1% Triton X-100, and freshly added phosphatase and proteinase inhibitors (1 mM orthovanandate, 10 mM NaF, 10 μg/ml leupeptin, 25 μg/ml aprotinin, and 50 μg/ml PMSF). For immunoblotting, 15–20 μg of cell lysate proteins were separated by 10% SDS-PAGE and transferred to PVDF Immobilon-P transfer membranes (Millipore). The blots were developed with specific primary antibodies that included a pair of phospho-ERK1and ERK2 (sc-7383, Santa Cruz) and (total) ERK1 and ERK2 (sc-94, Santa Cruz) antibodies, phospho-Src (Tyr416, Cell Signaling) and Src (GD11, Upstate) antibodies, and GAPDH antibodies (Cell Signaling). The ECL detection reagents (Amersham) were used to visualize bands. The blots displayed are representative of stimulation studies that were performed at least three times. For quantitation of signal intensity, western blots were scanned and analyzed as recently described (McDevitt et al., 2006). The ratio between phosphorylated to the total kinase protein was determined and expressed as a fold-change for each lane. The p values were calculated for each of the comparisons shown from multiple experiments (Student’s t test).
The serine phosphorylation of CD74 and CD44 was quantified by specific sandwich ELISA (Perez et al., 2003). 96-well microtiter plates were coated with 1–2 μg/ml of CD74 or CD44 antibody at 4°C overnight. Lysates from MIF-stimulated or control cultures were added to antibody-coated wells in triplicate and incubated at 4°C overnight. After several washes (0.5% Tween-20 in PBS), 0.2 μg/ml phospho-serine antibody (Hypromatrix) conjugated with horseradish peroxidase was added, and immunoreactivity was measured by the TMB reagent (DakoCytomation) and the data were analyzed with SpectraMax Plus (Molecular Devices). Phospho-serine values were expressed as relative ELISA absorbance for cell treatment versus nontreatment.
Protein kinase inhibition was performed by preincubating cells for 30–120 min with 20 μM of the PKA inhibitor H-89, 10 μM of the PKC inhibitor RO-31-2880, or 10 μM of the Src inhibitor PP2 (Calbiochem) (Faltynek et al., 1995). Cell lysates were collected and analyzed by immunoblots with a pair of phospho-PKA and (total)-PKA antibodies, a pair of phospho-PKC (all from Cell Signaling) and (total)-PKC (Santa Cruz) antibodies, or phospho-Src and GAPDH antibodies.
siRNA Studies
An siRNA specific for Src and a nontargeting siRNA control were obtained from Dharmacon (Iversen et al., 2004). Cells were transfected with siRNA with RNAiFect (Qiagen) for 24 hr. The cells then were incubated in DMEM/0.3% FBS for 19 hr prior to stimulation with MIF (100 ng/ml) for 30 min. Cell lysates were analyzed with immunoblots with phospho-Src, Src (total), GAPDH, phospho-ERK1 and ERK2, and total ERK1 and ERK2 antibodies.
Apoptosis Studies
Cells (2 × 106) cultured in 10 cm plates were treated for 24 hr with the apoptosis inducers 1 mM sodium nitroprusside (SNP, Sigma) and 2 μg/ml camptothecin (Sigma) (Hudson et al., 1999; Mitchell et al., 2002). Caspase-3 activity was analyzed with a colorimetric assay kit (R&D Systems). Cytoplasmic p53 content was analyzed as described previously (Mitchell et al., 2002) by immunoblotting of cytosolic fractions with a pair of phospho-p53 (Ser15) (R&D Systems) and total p53 antibodies (Cell Signaling). In vivo apoptosis studies followed the procedures of Mitchell et al. (2002) and were conducted after i.p. injection of 15 mg/kg lipopolysaccharide (LPS) (E. coli 0111:B4; Sigma). Mice were sacrificed 24 hr after priming, and the peritoneal macrophages were collected and immediately analyzed for apoptosis by an oligonucleosome ELISA (Roche Diagnostics). Primary macrophages also were immunostained by Alexa 488-conjugated, Annexin V (Invitrogen), and apoptotic cells were enumerated by fluorescence microscopy.
Supplementary Material
Acknowledgments
We thank B. Ma for assistance with fluorescence microscopy and J. Griffith for helpful discussions. Supported by NIH grants to R.B. (AR049610, AI42310, AR050498), W.K. (AR43384), P.N. (HL060539), and the Alliance for Lupus Research (R.B., E.L.).
Footnotes
Supplemental Data Three Supplemental Figures can be found with this article online at http://www.immunity.com/cgi/content/full/25/4/595/DC1/.
The authors declare that they have no competing financial interests.
References
- Allouche M, Charrad RS, Bettaieb A, Greenland C, Grignon C, Smadja-Joffe F. Ligation of the CD44 adhesion molecule inhibits drug-induced apoptosis in human myeloid leukemia cells. Blood. 2000;96:1187–1190. [PubMed] [Google Scholar]
- Amin MA, Volpert OV, Woods JM, Kumar P, Harlow LA, Koch AE. Migration inhibitory factor mediates angiogenesis via mitogen-activated protein kinase and phosphatidylinositol kinase. Circ. Res. 2003;93:321–329. doi: 10.1161/01.RES.0000087641.56024.DA. [DOI] [PubMed] [Google Scholar]
- Amin MA, Haas CS, Zhu K, Mansfield PJ, Kim MJ, Lackowski NP, Koch AE. Migration inhibitory factor up-regulates vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 via Src, PI3 kinase, and NF kappa B. Blood. 2006;107:2252–2261. doi: 10.1182/blood-2005-05-2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson HA, Bergstralh DT, Kawamura T, Blauvelt A, Roche PA. Phosphorylation of the invariant chain by protein kinase C regulates MHC class II trafficking to antigen-processing compartments. J. Immunol. 1999;163:5435–5443. [PubMed] [Google Scholar]
- Bacher M, Metz CN, Calandra T, Mayer K, Chesney J, Lohoff M, Gemsa D, Donnelly T, Bucala R. An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc. Natl. Acad. Sci. USA. 1996;93:7849–7854. doi: 10.1073/pnas.93.15.7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, Manogue KR, Cerami A, Bucala R. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365:756–759. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- Bernhagen J, Mitchell RA, Calandra T, Voelter W, Cerami A, Bucala R. Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF) Biochemistry. 1994;33:14144–14155. doi: 10.1021/bi00251a025. [DOI] [PubMed] [Google Scholar]
- Bouchard MJ, Racaniello VR. CD44 is not required for poliovirus replication. J. Virol. 1997;71:2793–2798. doi: 10.1128/jvi.71.4.2793-2798.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourguignon LY, Zhu H, Shao L, Chen YW. CD44 interaction with c-Src kinase promotes cortactin-mediated cytoskeleton function and hyaluronic acid-dependent ovarian tumor cell migration. J. Biol. Chem. 2001;276:7327–7336. doi: 10.1074/jbc.M006498200. [DOI] [PubMed] [Google Scholar]
- Bozza M, Satoskar AR, Lin G, Lu B, Humbles AA, Gerard C, David JR. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J. Exp. Med. 1999;189:341–346. doi: 10.1084/jem.189.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury J. A therapeutic future for CD44 in inflammation? Lancet. 2002;359:2008. doi: 10.1016/S0140-6736(02)08854-2. [DOI] [PubMed] [Google Scholar]
- Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat. Rev. Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J. Exp. Med. 1994;179:1895–1902. doi: 10.1084/jem.179.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calandra T, Bernhagen J, Metz CN, Spiegel LA, Bacher M, Donnelly T, Cerami A, Bucala R. MIF as a glucocorticoid-induced modulator of cytokine production. Nature. 1995;377:68–71. doi: 10.1038/377068a0. [DOI] [PubMed] [Google Scholar]
- Calandra T, Echtenacher B, Le Roy D, Pugin J, Metz CN, Hültner L, Heumann DMD, Bucala R, Glauser MP. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat. Med. 2000;6:164–169. doi: 10.1038/72262. [DOI] [PubMed] [Google Scholar]
- Chen D, McKallip RJ, Zeytun A, Do YLC, Robertson JLMTW, Nagarkatti PS, Nagarkatti M. CD44-deficient mice exhibit enhanced hepatitis after concanavalin A injection: evidence for involvement of CD44 in activation-induced cell death. J. Immunol. 2001;166:5889–5897. doi: 10.4049/jimmunol.166.10.5889. [DOI] [PubMed] [Google Scholar]
- Coluccia AML, Benati D, Dekhil H, De Filippo A, Lan C, Gambacorti-Passerini C. SKI-606 decreases growth and motility of colorectal cancer cells by preventing pp60(c-Src)-dependent tyrosine phosphorylation of beta-catenin and its nuclear signaling. Cancer Res. 2006;66:2279–2286. doi: 10.1158/0008-5472.CAN-05-2057. [DOI] [PubMed] [Google Scholar]
- Faltynek CR, Schroeder J, Mauvais P, Miller D, Wang S, Murphy D, Lehr R, Kelley M, Maycock A, Michne W, et al. Damnacanthal is a highly potent, selective inhibitor of P56(Lck) tyrosine kinase activity. Biochemistry. 1995;34:12404–12410. doi: 10.1021/bi00038a038. [DOI] [PubMed] [Google Scholar]
- Fingerle-Rowson G, Petrenko O, Netz CN, Forsthuber TG, Mitchell R, Huss R, Moll U, Müller W, Bucala R. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc. Natl. Acad. Sci. USA. 2003;100:9354–9359. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregersen PK, Bucala R. Macrophage migration inhibitory factor, MIF alleles, and the genetics of inflammatory disorders: incorporating disease outcome into the definition of phenotype. Arthritis Rheum. 2003;48:1171–1176. doi: 10.1002/art.10880. [DOI] [PubMed] [Google Scholar]
- Hibi M, Hirano T. IL-6 receptor. In: Oppenheim JJ, Feldmann M, editors. Cytokine Reference. Vol 2. Academic Press; San Diego: 2001. pp. 1761–1778. [Google Scholar]
- Hill KJ, Webber AC, Hill SJ. A role of protein kinase C mu in signalling from the human adenosine A(1) receptor to the nucleus. Br. J. Pharmacol. 2003;139:721–732. doi: 10.1038/sj.bjp.0705294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J. Exp. Med. 1999;190:1375–1382. doi: 10.1084/jem.190.10.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida M, Ishida T, Thomas SM, Berk BC. Activation of extracellular signal-regulated kinases (ERK1/2) by angiotensin II is dependent on c-Src in vascular smooth muscle cells. Circ. Res. 1998;82:7–12. doi: 10.1161/01.res.82.1.7. [DOI] [PubMed] [Google Scholar]
- Iversen TM, Vang T, Amarzguioui M, Tasken K. Knockdown of Csk, Lck and Fyn(T) by siRNA-mediated RNA interference in T cells. FASEB J. 2004;18:C258. [Google Scholar]
- Jiang H, Peterson RS, Wang W, Bartnik E, Knudson CB, Knudson W. A requirement for the CD44 cytoplasmic domain for the hyaluronan binding, pericellular matrix assembly, and receptor-mediated endocytosis in COS-7 cells. J. Biol. Chem. 2002;277:10531–10538. doi: 10.1074/jbc.M108654200. [DOI] [PubMed] [Google Scholar]
- Kleemann R, Hausser A, Geiger G, Mischke R, Burger-Kentischer A, Flieger O, Johannes FJ, Roger T, Calandra T, Kapurniotu A, et al. Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature. 2000;408:211–216. doi: 10.1038/35041591. [DOI] [PubMed] [Google Scholar]
- Lacey D, Sampey A, Mitchell R, Bucala R, Santos L, Leech M, Morand E. Control of fibroblast-like synoviocyte proliferation by macrophage migration inhibitory factor. Arthritis Rheum. 2003;48:103–109. doi: 10.1002/art.10733. [DOI] [PubMed] [Google Scholar]
- Leech M, Lacey D, Xue JR, Santos L, Hutchinson P, Wolvetang E, David JR, Bucala R, Morand EF. Regulation of p53 by macrophage migration inhibitory factor in inflammatory arthritis. Arthritis Rheum. 2003;48:1881–1889. doi: 10.1002/art.11165. [DOI] [PubMed] [Google Scholar]
- Legg JW, Lewis CA, Parsons M, Ng T, Isacke CM. A novel PKC-regulated mechanism controls CD44-ezrin association and directional cell motility. Nat. Cell Biol. 2002;4:399–407. doi: 10.1038/ncb797. [DOI] [PubMed] [Google Scholar]
- Leng L, Metz C, Fang Y, Xu J, Donnelly S, Baugh J, Delonery T, Chen Y, Mitchell RA, Bucala R. MIF signal transduction initiated by binding to CD74. J. Exp. Med. 2003;197:1467–1476. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H, Bucala R, Mitchell RA. Adhesion-dependent signaling by MIF. J. Biol. Chem. 2003;278:76–81. doi: 10.1074/jbc.M208820200. [DOI] [PubMed] [Google Scholar]
- Lolis E, Bucala R. Macrophage migration inhibitory factor. Expert Opin. Ther. Targets. 2003;7:153–164. doi: 10.1517/14728222.7.2.153. [DOI] [PubMed] [Google Scholar]
- Lue HQ, Kapurniotu A, Fingerle-Rowson G, Roger T, Leng L, Thiele M, Calandra T, Bucala R, Bernhagen J. Rapid and transient activation of the ERK MAPK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on JAB1/CSN5 and Src kinase activity. Cell. Signal. 2006;18:688–703. doi: 10.1016/j.cellsig.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Mahabeleshwar GH, Kundu GC. Tyrosine kinase p56(lck) regulates cell motility and nuclear factor kappa B-mediated secretion of urokinase type plasminogen activator through tyrosine phosphorylation of I kappa B alpha following hypoxia/reoxygenation. J. Biol. Chem. 2003;278:52598–52612. doi: 10.1074/jbc.M308941200. [DOI] [PubMed] [Google Scholar]
- Matza D, Wolstein O, Dikstein R, Shachar I. Invariant chain induces B cell maturation by activating a TAFII105-NF-κB-dependent transcription program. J. Biol. Chem. 2001;276:27203–27206. doi: 10.1074/jbc.M104684200. [DOI] [PubMed] [Google Scholar]
- Matza D, Kerem A, Medvedovsky H, Lantner F, Shachar I. Invariant chain-induced B cell differentiation requires intramembrane proteolytic release of the cytosolic domain. Immunity. 2002;17:549–560. doi: 10.1016/s1074-7613(02)00455-7. [DOI] [PubMed] [Google Scholar]
- McDevitt MA, Xie J, Shanmugasundaram G, Griffith J, Liu A, McDonald C, Thuma P, Gordeuk VR, Metz CN, Mitchell R, et al. A critical role for the host mediator macrophage migration inhibitory factor in the pathogenesis of malarial anemia. J. Exp. Med. 2006;203:1185–1196. doi: 10.1084/jem.20052398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Siegler K, Hudson PB. Enhanced expression of macrophage migration inhibitory factor in prostatic adenocarcinoma metastases. Urology. 1996;48:448–452. doi: 10.1016/S0090-4295(96)00207-5. [DOI] [PubMed] [Google Scholar]
- Meyer-Siegler KL, Leifheit EC, Vera PL. Inhibition of macrophage migration inhibitory factor decreases proliferation and cytokine expression in bladder cancer cells. BMC Cancer. 2004;4:34. doi: 10.1186/1471-2407-4-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Lombardi M, Barone MV, Ametrano D, Zannini MS, Abbondanza C, Auricchio F. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000;19:5406–5417. doi: 10.1093/emboj/19.20.5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell RA, Metz CN, Peng T, Bucala R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J. Biol. Chem. 1999;274:18100–18106. doi: 10.1074/jbc.274.25.18100. [DOI] [PubMed] [Google Scholar]
- Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, Bucala R. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc. Natl. Acad. Sci. USA. 2002;99:345–350. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizue Y, Ghani S, Leng L, McDonald C, Kong P, Baugh J, Lane SJ, Craft J, Nishihira J, Donnelly SC, et al. Role for macrophage migration inhibitory factor (MIF) in asthma. Proc. Natl. Acad. Sci. USA. 2005;102:14410–14415. doi: 10.1073/pnas.0507189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naujokas MF, Morin M, Anderson MS, Peterson M, Miller J. The chondroitin sulfate form of invariant chain can enhance stimulation of T cell responses through interaction with CD44. Cell. 1993;74:257–268. doi: 10.1016/0092-8674(93)90417-o. [DOI] [PubMed] [Google Scholar]
- Naujokas MF, Arneson LS, Fineschi B, Peterson ME, Sitterding S, Hammond AT, Reilly C, Lo D, Miller J. Potent effects of low levels of MHC class II associated invariant chain on CD4+ T cell development. Immunity. 1995;3:359–372. doi: 10.1016/1074-7613(95)90120-5. [DOI] [PubMed] [Google Scholar]
- Nguyen MT, Lue HQ, Kleemann R, Thiele M, Tolle G, Finkelmeier D, Wagner E, Braun A, Bernhagen J. The cytokine macrophage migration inhibitory factor reduces pro-oxidative stress-induced apoptosis. J. Immunol. 2003;170:3337–3347. doi: 10.4049/jimmunol.170.6.3337. [DOI] [PubMed] [Google Scholar]
- Onodera S, Nishihira J, Iwabuchi K, Koyama Y, Yoshida K, Tanaka S, Minami A. Macrophage migration inhibitory factor up-regulates matrix metalloproteinase-9 and -13 in rat osteoblasts. Relevance to intracellular signaling pathways. J. Biol. Chem. 2002;277:7865–7874. doi: 10.1074/jbc.M106020200. [DOI] [PubMed] [Google Scholar]
- Padfield PJ, Panesar N. Cholecystokinin octapeptide inhibits Ca2+-dependent amylase secretion from permeabilized pancreatic acini by blocking the MgATP-dependent priming of exocytosis. Biochem. J. 1998;330:329–334. doi: 10.1042/bj3300329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez OD, Mitchell D, Jager GC, South S, Murriel C, McBride J, Herzenberg LA, Kinoshita S, Nolan GP. Leukocyte functional antigen 1 lowers T cell activation thresholds and signaling through cytohesin-1 and Jun-activating binding protein 1. Nat. Immunol. 2003;4:1083–1092. doi: 10.1038/ni984. [DOI] [PubMed] [Google Scholar]
- Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003;4:33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- Roger T, David J, Glauser MP, Calandra T. MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature. 2001;414:920–924. doi: 10.1038/414920a. [DOI] [PubMed] [Google Scholar]
- Salazar EP, Rozengurt E. Bombesin and platelet-derived growth factor induce association of endogenous focal adhesion kinase with Src in intact Swiss 3T3 cells. J. Biol. Chem. 1999;274:28371–28378. doi: 10.1074/jbc.274.40.28371. [DOI] [PubMed] [Google Scholar]
- Sampey AV, Hall PH, Mitchell RA, Metz CN, Morand EF. Regulation of synoviocyte phospholipase A2 and cyclooxygenase 2 by macrophage migration inhibitory factor. Arthritis Rheum. 2001;44:1273–1280. doi: 10.1002/1529-0131(200106)44:6<1273::AID-ART219>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Shachar I, Flavell RA. Requirement for invariant chain in B cell maturation and function. Science. 1996;274:106–108. doi: 10.1126/science.274.5284.106. [DOI] [PubMed] [Google Scholar]
- Starlets D, Gore Y, Binsky I, Haran M, Harpaz N, Shvidel L, Becker-Herman S, Berrebi A, Shachar I. Cell-surface CD74 initiates a signaling cascade leading to cell proliferation and surviva. Blood. 2006;107:4807–4816. doi: 10.1182/blood-2005-11-4334. [DOI] [PubMed] [Google Scholar]
- Sun LK, Wahl P, Bilic G, Wuthrich RP. CD44-mediated cyclooxygenase-2 expression and thromboxane A(2) production in RAW 264.7 macrophages. Inflamm. Res. 2001;50:496–499. doi: 10.1007/PL00000224. [DOI] [PubMed] [Google Scholar]
- Swant JD, Rendon BE, Symons M, Mitchell RA. Rho GTPase-dependent signaling is required for macrophage migration inhibitory factor-mediated expression of cyclin D1. J. Biol. Chem. 2005;280:23066–23072. doi: 10.1074/jbc.M500636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taher TE, Smit L, Griffioen AW, Schilder-Tol EJ, Borst J, Pals ST. Signaling through CD44 is mediated by tyrosine kinases. J. Biol. Chem. 1996;271:2863–2867. doi: 10.1074/jbc.271.5.2863. [DOI] [PubMed] [Google Scholar]
- Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Puré E, Henson PM, Noble PW. Resolution of lung inflammation by CD44. Science. 2002;296:155–158. doi: 10.1126/science.1069659. [DOI] [PubMed] [Google Scholar]
- Thorne RF, Legg JW, Isacke CM. The role of the CD44 transmembrane and cytoplasmic domains in co-ordinating adhesive and signalling events. J. Cell Sci. 2004;117:373–380. doi: 10.1242/jcs.00954. [DOI] [PubMed] [Google Scholar]
- Topilski I, Harmelin A, Flavell RA, Levo Y, Shachar I. Preferential Th1 immune response in invariant chain-deficient mice. J. Immunol. 2002;168:1610–1617. doi: 10.4049/jimmunol.168.4.1610. [DOI] [PubMed] [Google Scholar]
- Turley EA, Noble PW, Bourguignon LYW. Signaling properties of hyaluronan receptors. J. Biol. Chem. 2002;277:4589–4592. doi: 10.1074/jbc.R100038200. [DOI] [PubMed] [Google Scholar]
- Webb DSA, Shimizu Y, Van Seventer GA, Shaw S, Gerrard TL. LFA-3, CD44, and CD45: physiologic triggers of human monocyte TNF and IL-1 release. Science. 1990;249:1295–1297. doi: 10.1126/science.1697984. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.