

Background: p38α MAP kinase has two recognition sites, the CD domain and DEF pocket.
Results: DEF pocket mutations decreased phosphorylation levels of several substrates and autophosphorylation capabilities.
Conclusion: The DEF pocket directs selective substrate activation and also mediates autophosphorylation.
Significance: Understanding how substrates interact selectively with the two p38α docking sites could become the basis for the design of inhibitors.
Keywords: MAP Kinases (MAPKs), Molecular Docking, p38, Protein Phosphorylation, Threonine-serine Protein Kinase, Docking Region, MAP Kinase, Autophosphorylation, p38, Substrate Selectivity
Abstract
Signaling processes are primarily promoted by molecular recognition and corresponding protein-protein interactions. One of the key eukaryotic signaling pathways is the MAP kinase cascade involved in vital cellular processes such as cell proliferation, differentiation, apoptosis, and stress response. The principle recognition site of MAP kinases, the common docking (CD) region, forms selective interactions with substrates, upstream activators, and phosphatases. A second docking site, defined as the DEF site interaction pocket (DEF pocket), is formed subsequent to ERK2 and p38α activation. Both crystal structures of p38α in its dually phosphorylated form and of intrinsically active mutants showed the DEF pocket, giving motivation for studying its role in substrate activation and selectivity. Mutating selected DEF pocket residues significantly decreased the phosphorylation levels of three p38α substrates (ATFII, Elk-1, and MBP) with no apparent effect on the phosphorylation of MK2 kinase. Conversely, mutating the CD region gave the opposite effect, suggesting p38α substrates can be classified into DEF-dependent and DEF-independent substrates. In addition, mutating DEF pocket residues decreased the autophosphorylation capability of intrinsically active p38α mutants, suggesting DEF-mediated trans-autophosphorylation in p38α. These results could contribute to understanding substrate selectivity of p38α and serve as a platform for designing p38α-selective DEF site blockers, which partially inhibit p38α binding DEF-dependent substrates, whereas maintaining its other functions intact. In this context, preliminary results using synthetic peptides reveal significant inhibition of substrate phosphorylation by activated p38α.
Introduction
Controlling vital cellular processes such as proliferation, differentiation, apoptosis, and stress response are primarily mediated by protein kinases. Abnormal activities of kinases are often associated with human diseases (1). Thus, understanding kinase mechanisms of activation and regulation is a pivotal goal in biological research. There are several manners by which cells regulate kinase activity such as timing of activation, specificity, and down-regulation. All these processes involve protein-protein interactions with partner proteins such as scaffold proteins, upstream activators, substrates and phosphatases, which are generally mediated through recognition sites on both interacting molecules.
One of the central signaling cascades in eukaryotic cells is the mitogen-activated protein kinase (MAPK) pathway, which is involved in most essential cellular processes. The MAPKs are activated via dual phosphorylation by their upstream activators, the MKKs. Consequently, MAPKs phosphorylate many substrates in the cytoplasm and nucleus including transcription factors, tumor suppressors, and other kinases (2). More than 200 substrates have been identified for MAPKs that are involved in numerous cellular processes. Phosphorylation of different substrates activates different signaling pathways and consequent phenotypes (3, 4). As with many enzymes, the interactions between MAPKs and substrates, upstream activators and down-regulators are mainly dictated by recognition (docking) regions in both interacting molecules. Two principle docking regions have been identified for substrates on MAPKs affecting substrate specificity and selectivity. The principle docking site identified in all MAP kinases is defined as a common docking (CD)2 region that selectively interacts with corresponding substrates, upstream activators, and phosphatases. This domain comprises a central cluster of 2–3 acidic residues with several hydrophobic residues in its vicinity (5–7). The CD site is located on the surface of the MAPK in the region connecting the C′ and N′ kinase lobes, distal from the catalytic site. Molecules that bind the CD region contain a complementary interacting site, termed the D-site (also defined as the DEJL domain and δ-domain), and are characterized by a consensus cluster of 2–3 positively charged residues with several proximate hydrophobic residues (7–11).
A second docking site, initially identified on ERK2 (6), is defined as the DEF site interaction pocket (DEF pocket). The site is formed by local conformational changes following activation (dual phosphorylation) and consists of hydrophobic residues (Fig. 1A). The DEF pocket is shaped as a hydrophobic cavity designed to accommodate a distinctive hydrophobic segment in substrates defined as the “docking site for ERK FXF” (F-site or DEF site). The F-site in substrates is generally characterized by two Phe residues separated by one amino acid (FXF motif) located 6–20 amino acids downstream to the substrate phosphoacceptor (12–15).
FIGURE 1.

a, sequence alignment of the segments contributing to the DEF pocket in p38α and ERK2. The individual residues that participate in the DEF pocket show high similarity between the two proteins and are shown in red. The pocket is defined by hydrophobic residues from the activation loop, a loop connecting the P+1 site and F-helix, the G-helix, and the MAP kinase insert region. b, surface presentation of ERK2wt (left) and activated ERK2wt (right) highlighting the residues contributing to the DEF pocket. The TEY motif is shown in red and the remaining residues in green. For clarity, only the C′-lobe of the molecular surface is displayed. The formation of the DEF pocket was initially characterized as part of the conformational changes occurring upon ERK2 activation. The DEF pocket, as an additional docking site, is considered to accommodate hydrophobic residues of substrates by directing them to the active site (shown by the arrow). c, surface presentation of p38αwt (left), activated p38αwt (middle), and p38αY323T active mutant (right) highlighting the residues contributing to the DEF pocket where the TGY motif is in red and the remaining residues in green. Activation by dually phosphorylated p38αwt and the intrinsic activity of the Y323T mutant result in conformational changes in the kinase interlobe orientation and the formation of the DEF pocket (residues labeled) in a similar contour as also observed for activated ERK2.
Mutational analysis of ERK2 docking regions emphasized their importance in substrate selectivity. Mutating selected residues in the DEF pocket in ERK2 resulted in decreased phosphorylation levels of Elk-1 and c-Fos transcription factors, whereas phosphorylation of RSK (kinase) was not affected (6, 16). In contrast, the D319N (“sevenmaker” (17, 18)) mutation in the CD region resulted in decreased RSK phosphorylation but did not affect Elk-1 and c-Fos phosphorylation levels, indicating different substrate preferences (6, 16).
Little is known about the availability and function of the DEF pocket in p38s, yet a F-site was identified in the p38α substrate, the transcription factor SAP-1 (19). It was then suggested, based on a substrate-derived peptide screen, that p38α and p38β also contain a DEF pocket consisting of residues homologous to ERK2 (20) (Fig. 1a), whereas p38γ and p38δ do not show an indication to its presence. The DEF pocket in p38α was recently identified by structural analysis of the dually phosphorylated form (21) and the intrinsically active mutants at the Tyr323 position (22), similar to that found for activated ERK2 (21, 23) (Fig. 1, b and c). In both ERK2 and p38α the DEF pocket is located in a similar region in the C-lobe and is made up of identical residues although it adopts a somewhat different contour.
Structural studies showed that the DEF pocket in p38α was available, leading us to investigate its role in substrate selectivity and autophosphorylation. Mutagenesis of DEF pocket residues resulted in a significant decrease of phosphorylation levels in three p38α substrates (ATFII, Elk-1, and MBP) with no notable change of the MK2 kinase. Conversely, mutagenesis in the CD region had the opposite effect on the substrates. The in vitro and in situ results suggest possible classification of p38α DEF-dependent and DEF-independent substrates. Autophosphorylation levels of intrinsically active mutants of p38α were also decreased by mutating the DEF pocket residues, indicating that it is mediated by the DEF pocket in trans. This study extends the understanding of how substrates interact selectively with the two docking sites of p38α. The DEF pocket could thus become a target for designing p38α inhibitors, which could prevent activation of DEF-dependent substrates, whereas maintaining its activity toward others. By using such an approach we have shown that a 15-amino acid peptide derived from the Elk-1 F-site displays inhibitory properties on ATFII phosphorylation.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis of the p38α Mutants and Structural Analysis
Site-directed mutagenesis was performed by polymerase chain reaction according to the recommendations of the manufacturer. Mutagenesis was performed on the human p38αwt cDNA subcloned into a pET-28b (Novagen) vector downstream and in-frame with the hexahistidine coding sequence. All mutated cDNAs were verified by sequencing the entire p38α cDNA.
Expression, purification, and crystallization protocols of the p38αY258A+ML194–5AA+HI228–9AA penta-mutant were conducted as previously described for the p38α (22, 24). Crystallographic data were collected at the European Synchrotron Radiation Facility (ESRF) (see Table 1), integrated and scaled using the HKL suite (25). The structure of the penta-mutant was solved via molecular replacement using p38αwt as the search model and further refined at the resolution range of 50–1.66 Å using Phenix (26) (Table 1).
TABLE 1.
Data collection and refinement statistics
| PDB entry 4GEO | p38αY258A+ ML194–5AA+HI228–9AA |
|---|---|
| ESRF beamline | ID23–1 |
| Wavelength (Å) | 0.97 |
| Space group | P212121 |
| Unit cell parameters (Å) | a = 66.9, b = 74.6, c = 74.8 |
| Resolution range (Å) (last resolution shell) | 52.0–1.66 (1.69–1.66) |
| Unique reflections | 44,163 |
| Redundancy | 4.3 |
| Rsym(I)a | 5.1% (44.9%) |
| Completeness | 98.2 (99.1) |
| I/σ | 40 |
| Number of protein atoms | 2622 |
| Number of ligand atoms | 40 |
| Number of solvent atoms | 326 |
| R-factor | 0.217 |
| R-freeb | 0.255 |
| Average B-factor (Å2) | |
| Protein | 27.1 |
| Solvent | 35.5 |
| Root mean square deviation from ideality | |
| Bond length | 0.015 Å |
| Bond angle | 1.49° |
| Ramachandran plot (PROCHECK) | |
| Favored | 92.8% |
| Allowed | 6.8% |
| Generously allowed | 0.3% |
| Disallowed | 0.0% |
a Rsym(I) = Σ|I−〈I〉|/ΣI.
b Test set consists of 5% for all data.
Protein Expression and Purification for in Vitro Kinase Assay
Protein expression and purification of p38α proteins and GST-ATF2 were conducted as previously described (27). The GST-Elk1 (amino acid 310–428), GST-Elk1ΔD (amino acid 329–428), and GST-MK2 (pGEX-MK2-K76R) were expressed in Escherichia coli as with the p38α proteins. The cell cultures of GST-Elk1 and GST-MK2 were grown at 30 °C for 20 and 5 h, respectively. Cells were collected by centrifugation and washed in phosphate-buffered saline (PBS) and the pellet was then stored at −20 °C. The frozen pellet was gently thawed on ice, and suspended in PBS and protease inhibitors mixture (Sigma p8849). After mechanical disruption of the cells using a microfluidizer (model M-110 EHIS, Microrofluidics Corp., Newton, MA) the lysate was centrifuged at 20,000 × g for 30 min at 4 °C. The supernatant, containing the soluble proteins was loaded on a glutathione-Sepharose column (Amersham Biosciences), washed in PBS, and eluted using 50 mm Tris buffer, pH 8, and 20 mm glutathione. The protein solution was then dialyzed overnight against 12.5 mm Hepes buffer, pH 7.5, 100 mm KCl, 6.25% glycerol, and 1 mm dithiothreitol (DTT). After dialysis, the protein concentration was determined using the Bradford method and the purified protein was then divided into aliquots, flash-frozen in liquid nitrogen, and stored at −80 °C. MBP (Sigma M-1891) was dissolved with 50 mm Tris buffer, pH 8.
In Vitro Kinase Assay
The paper-spotted kinase reactions were performed as previously described (27). In parallel, a quality assay was done in which samples from the paper-spotted kinase reactions were mixed with Laemmli sample buffer and boiling at 100 °C for 5 min. The assay samples were run on SDS-PAGE stained with Coomassie staining and then expose to x-ray film. The kinetic kinase assays were carried out for 10 min with substrate concentrations ranging between 0 and 140 μg (0–75 μm for GST-ATFII or 0–85 μm for GST-Elk-1). The peptide competitive kinase assay was performed using increasing concentrations (0.1 μm to 1 mm) of peptides derived from the Elk-1 F-site (APRSPAKLSFQFPSS) or a mutated F-site (APRSPAKLSAQAPSS) as a negative control.
The autophosphorylation kinase assay was performed in a similar buffer as the paper-spotted kinase assay with no substrate where each reaction contained 1.25 μg of purified protein in a final volume of 25 μl. Reactions were carried out for durations of 0, 15, 30, and 60 min at 30 °C and terminated by cooling to 4 °C and adding 6 μl of 5× Laemmli sample buffer and then heating to 100 °C for 5 min. The assay samples were run on SDS-PAGE with Coomassie staining and then exposed to x-ray film. For quantifying the autophosphorylation levels the relevant bands for the dried SDS-PAGE were counted using a scintillation counter running a 32P Cherenkov program.
Western Blot Analysis
For the Western blot analysis, 0.2 μg of purified recombinant protein were heated at 100 °C for 5 min, separated by SDS-PAGE, and then transferred to a nitrocellulose membrane. After incubating the membrane with the appropriate antibodies, specific proteins were visualized using an enhanced chemiluminescence detection reagent and then monitored by exposing the membranes to x-ray film. The antibodies used in the assays were as follows: goat anti-p38 from Santa Cruz Biotechnology, rabbit anti-phosphor-p38, rabbit anti-MK2 (3042S), rabbit anti-phosphor-MK2 (3007S), and rabbit anti-Elk-1 from Cell Signaling (9182), rabbit anti-phosphor-Elk-1 from Santa Cruz Biotechnology (SC8406), anti-HA tag from 12CA5 hybridomas and mouse anti-phosphothreonine from Cell Signaling (9386S); and anti-phosphotyrosine from 4G10 hybridomas.
Cell Culture and Luciferase Assay
The transfected recombinant p38α cDNAs containing an HA tag were cloned into pCEFL vectors (Invitrogen). The active MKK6 double mutant (MKK6-EE) with an HA tag was cloned into the pBabe plasmid. The transfected recombinant MK2 and Elk-1 cDNAs were cloned into the pΔCR and pEXV3 plasmids (28). HEK293 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, penicillin, and streptomycin (Biological Industries, BeitHa'emek, Israel) and incubated at 37 °C in 5% CO2. Mouse embryonic fibroblasts cells, lacking p38α (MEFα−/−), were grown in a similar medium solution as described above, supplemented with nonessential amino acids, sodium pyruvate, and β-mercaptoethanol (Invitrogen). The MEF cells were transfected using the TurboFectTM transfection reagent (Fermentas) according to the manufacturer's instructions and HEK293 cells were transfected using the calcium-phosphate method. Cells were washed with phosphate-buffered saline 48 h post-transfection, harvested by Laemmli's buffer, and then scraped using rubber policeman.
For the luciferase assay, MEF cells were plated on 12-well plates (0.1 × 106 cells/well). The cells were transfected with 2 μg of pCEFL containing wild type p38α or the different mutants, 0.1 μg of 6× AP-1-luc, 0.2 μg of pBabe-MKK6-EE, and 30 ng of Renilla luciferase (pRL-TK) as a control for transfection efficiency. The cells were harvested 48 h post-transfection and the luciferase activity was measured using the dual luciferase reporter assay system (Promega).
RESULTS
Selection of DEF Pocket Mutants
The conformational changes shown for the Tyr323 intrinsically active mutants resulted in the formation of the DEF site interaction pocket in the C-lobe (22, 24). This pocket acquires a similar contour to that observed in the dually phosphorylated ERK2 and p38α using homologous amino acids (21, 23) (Fig. 1).
The DEF pocket in p38α, as in ERK2, consists of 13 residues from several segments, including the activation loop (residues 180–182), the loop connecting P+1 and F-helix (residues 190–203), the G-helix (residues 227–237), and the MAP kinase insert region (residues 240–262) (Fig. 1). Site-directed mutagenesis was done on selected residues to examine the role of the DEF pocket residues for p38α substrate recognition and selectivity. Of the 13 residues forming the DEF pocket, three are the Thr-Gly-Tyr phosphorylation motif (180–182) and two (Arg189 and Trp197) stabilize the active form. Mutating these residues drastically decrease p38α activity altogether (29).3 Two additional residues (Leu195 and Leu232) are buried in the internal part of the pocket and are probably considered to play a minor role in substrate binding. Based on the structural and biochemical analyses we have elected to mutate 5 amino acid residues obtaining three distinct variants (p38αY258A, p38αML194–5AA, and p38αHI228–9AA).
The Role of the DEF Pocket in p38α Substrate Selectivity
The mutants in the DEF pocket were first examined for their capability to be phosphorylated and activated in vitro by MKK6EE. The results indicate that all mutants were phosphorylated similarly to p38αwt (not shown). Additionally, the crystal structure of the DEF pocket penta-mutant (inactive p38αY258A+ML194–5AA+HI228–9AA), determined at a resolution of 1.66 Å (Table 1), shows a highly similar overall fold compared with the nonactivated p38αwt (not shown).
The activities of the DEF pocket mutants were assayed for their ability to phosphorylate in vitro four p38α substrates, GST-ATF-II, GST-Elk-1, GST-MAPKAPK2 (GST-MK2), and MBP. The results show that some of the DEF pocket mutants display differences in their ability to phosphorylate different substrates. In this context, p38αY258A displayed a reduced capability in phosphorylating ATF-II, Elk-1, and MBP (∼25% of p38αwt activity), but it phosphorylates MK2 at levels almost identical to those of p38αwt (Fig. 2). The p38αML194–5AA double mutant has shown very low activity toward all four substrates although still capable of phosphorylating MK2 to some extent (13% of p38αwt) (Fig. 2). The p38αHI228–9AA double mutant showed somewhat decreased activity toward Elk-1 and MBP (to about ∼72% of the activated wild type) but had no activity-decreasing effect on ATFII and MK-2 (Fig. 2). In summary, ATFII, Elk-1, and MBP were more affected by the DEF pocket mutations than MK2. Also, the ML194–5AA double mutation resulted in a notable decrease in activity toward all substrates. Met194 and Leu195 are located in the αEF helix near the αEF/αF loop, a conserved functional region in all kinases that contribute to stabilization of the active form of the kinase, and has also been shown to be critical for p38α activity (30). One could thus assume that the double mutant affects the overall catalytic properties of p38α rather than substrate selectivity.
FIGURE 2.

a, bar graphs showing the results of the in vitro paper-spotted kinase assay of the phosphorylation levels of four p38α substrates (ATF-II, Elk-1, MK2, and MBP) by the DEF pocket mutants. The p38αwt and DEF pocket mutants (Y258A, ML194–5AA, and HI228–9AA) were initially activated in vitro by MKK6EE. For each substrate the activity of activated p38αwt was normalized to 100% and nonactivated wild type was used as a negative control. Mutagenesis of Tyr258 and ML194–5 into Ala decreased the phosphorylation level of ATFII to approximately ∼25 and 3%, respectively. In contrast, the double mutant HI228–9AA had no significant influence on ATFII phosphorylation levels. For MK2 only, mutagenesis of ML194–5AA dramatically decreased the phosphorylation levels. Both Elk-1 and MBP have a similar phosphorylation profile, whereas all three mutants show decreased phosphorylation levels to approximately ∼25, 4, and 72% for Y258A, ML194–5AA, and HI228–9AA, respectively. The results show the average of two independent experiments (each in triplicates) and error bars are shown. b, qualitative results of the kinase assay for each substrate are shown by SDS-PAGE emphasizing the differences in the phosphorylation levels of selected DEF pocket mutants. Radioactivity was monitored by exposing the gels to x-ray film (upper images) and Coomassie staining of the gels verified equal amounts of substrate (lower images).
We further determined the catalytic parameters of p38αY258A and p38αHI228–9AA against ATFII and Elk1 (Table 2). The mutants display differences in the catalytic parameters in comparison to p38αwt with a high correlation to the kinase assay results (Table 2). In this context, the p38αY258A mutant displays a decrease of the specificity constant (Km/kact) to ∼23 and 40% compare with the wild type (for the ATFII and Elk1 substrates, respectively). The Km/kact values of the p38αHI228–9AA mutant are 107 and 83% compared with the wild type (for the ATFII and Elk1 substrates, respectively).
TABLE 2.
Catalytic parameters of activation of ATFII and Elk-1 by p38α DEF site mutants
| GST-ATFll |
GST-Elk-1 |
|||||
|---|---|---|---|---|---|---|
| Km | Kcat | Kcat/Km | Km | Kcat | Kcat/Km | |
| μm | min−1 | min−1* μm−1 | μm | min−1 | min−1* μm−1 | |
| wt | 119 | 100 | 0.84 | 472 | 6 | 0.013 |
| Y258A | 297 | 57 | 0.19 | 244 | 1 | 0.005 |
| HI228–9AA | 445 | 400 | 0.89 | 201 | 2 | 0.011 |
| D361N | 114 | 100 | 0.87 | 483 | 7 | 0.014 |
Substrate Preferences of the DEF Pocket and the CD Region
The specificity of substrate to the CD docking region in p38α was examined by mutating Asp316 to Asn as in ERK2D319N (17, 18). Western blot analysis showed that the phosphorylation level of MKK-activated p38αD316N was similar to that of p38αwt indicating that Asp316 is not critical for the interaction of p38α with its upstream activator MKK6 in vitro (Fig. 3a). The ability of the activated mutants to phosphorylate each of the four substrates was determined by the kinase assay against the different substrates that show that the activated p38αD316N phosphorylates MK2 at a lower level (29%) than p38αwt. Conversely, p38αD316N activity toward ATF-II, Elk-1, and MBP was increased by 11–18% (Fig. 3b). Catalytic studies of the p38αD316N mutant revealed a similar kinetic profile to that of p38αwt (Table 2). These results indicated that substrates that are less influenced by mutation in the CD domain are more dependent on interactions with the DEF pocket and vice versa. A truncated fragment of the GST-fused Elk-1 substrate (GST-Elk1ΔD) was used to corroborate this result. This substrate lacks the D-domain region of Elk-1 but includes the F-site region. GST-Elk1ΔD was phosphorylated by p38α to a higher level than Elk-1, which had both docking domains (Fig. 3c). In addition, the degree of phosphorylation of GST-Elk1ΔD by p38α DEF pocket mutants was similar to that previously observed for GST-Elk1 (Fig. 3d).
FIGURE 3.

a, Western blot analysis of the MKK6EE-activated p38αD316N mutant using the anti-p-p38 antibody (upper image). The analysis reveals similar phosphorylation levels to those of the p38α wild type indicating that Asp316 is not critical for the interaction of p38α with its upstream activator MKK6. Anti-p38 antibody verified the amount of proteins assayed (lower image). b, in vitro paper-spotted kinase assay of the activated D316N mutant against the four substrates in comparison to activated p38αwt, which was set to 100% for each substrate. The results show a significant decrease in the phosphorylation level of only MK2 to ∼30%, whereas the levels for the remaining substrates are increased. The results show the average of two independent experiments (each in triplicates) and error bars are shown. c, qualitative results of the kinase assay comparing the phosphorylation levels of Elk-1 (left) and Elk-1ΔD (right) lacking the D-domain region (amino acid 310–328) by activated p38αwt. Elk-1ΔD is phosphorylated to a higher degree than Elk-1 (upper image). Coomassie staining verifies equal amounts of each substrate (lower image). d, quantitative results of the kinase assay of the DEF pocket mutants and the D316N mutant against Elk1ΔD reveals similar phosphorylation levels as observed for the Elk1, which might imply that the CD docking region is less essential for the Elk1 interaction with p38α. The results show the average of two independent experiments (each in triplicates) and error bars are shown.
The Role of the DEF Pocket in p38α Autophosphorylation
The alternative activation modes of p38α and the intrinsically active mutants have been shown to induce autophosphorylation and subsequent activation (22, 24, 31, 32). Autophosphorylation of the intrinsically active mutants previously identified in p38α (Asp176, Phe327, and Tyr323 sites) occurs in trans and may involve interactions with the DEF pocket (22, 24). This assumption was examined by combining and assaying the intrinsically active mutations and the DEF pocket mutations of p38α. The autophosphorylation activity of each intrinsically active mutant was normalized to 100% activity and the wild type molecule was used as a negative control for basal (low) autophosphorylation activity. A notable decrease in the autophosphorylation capability was found for all the combined mutants, probably resulting from mutations at the DEF pocket (Fig. 4a). More specifically, the Y258A and H228A/I229A mutations resulted in decreased autophosphorylation levels (after 60 min) to 16–60% compared with the autophosphorylation level of the intrinsically active mutants. A more dramatic effect was found for the M194A/L195A mutants that exhibited only a 4–5% autophosphorylation level (Fig. 4A). Western blot analyses revealed that the decreased phosphorylation levels of both Thr and Tyr residues resulted from mutations in the DEF pocket (Fig. 4B).
FIGURE 4.

a, in vitro autophosphorylation kinase assay of p38α intrinsically active mutants (p38αD176A, p38αF327S, and p38αD176A+F327S) combined with the DEF pocket mutants (p38αY258A, p38αML194–5AA, and p38α HI228–9AA) at 30 °C as a function of time. Coomassie staining (lower images) verified the amount of p38α in each lane. The radiographs (upper images) show the decreased autophosphorylation capability of the intrinsically active mutants as a result of the different DEF pocket mutations. Qualitative analysis of the autophosphorylation levels of p38α mutants (by counting the bands from the SDS-PAGE using a scintillation counter) is shown in the tables on the right. The autophosphorylation activity of each intrinsically active mutant was normalized to 100%. The results reveal a significant decrease to 16–47% for the Y258A mutant and 38–60% for the HI228–9AA mutant compared with the autophosphorylation level of the intrinsically active mutants. The p38αD176A+ML194–5AA triple mutant shows a decreased autophosphorylation level by 96% compared with p38αD176A. b, Western blot analysis of the p38αD176A mutant combined with the DEF mutants using the anti-p-p38α, anti p-Thr and anti p-Tyr antibodies shows a significant decrease in the intrinsic phosphorylation levels of the DEF mutants compared with those of p38αD176A showing a significant effect of the DEF pocket mutagenesis on the potential autophosphorylation capability of the p38αD176A active mutant. Anti-p38α verified the amount of protein assayed (lower image).
The F-site of MAPK substrates is considered to accommodate aromatic amino acids 6–20 residues downstream to the substrate phosphoacceptor (12–15). The activation loop of p38α contains two aromatic residues (Trp187 and Tyr188) 7 and 8 residues downstream to the Thr-180 phosphoacceptor (Fig. 5a). Because autophosphorylation of p38α was shown to occur in trans, it is plausible that the aromatic region may interact with a DEF pocket of another p38α molecule directing the phosphoacceptors toward the active site thus facilitating autophosphorylation. The p38αW187A mutant was constructed to examine this assumption. The p38αW187A mutant exhibits highly similar characteristics as the p38αwt in terms of upstream activation and activity.
FIGURE 5.

a, sequence alignment of the segments including the Thr phosphoacceptor in p38α and the Ser/Thr phosphoacceptor of the three selected substrates (Elk1, ATF-II, and MBP). Elk-1 contains the canonical DEF site motif and the FXF motif six amino acids downstream to the Ser phosphoacceptor. Analysis of ATFII, MBP, and p38α sequences reveal segments of hydrophobic/aromatic residues (shown in red) 6–12 amino acids downstream to Thr180 phosphoacceptor. b, in vitro autophosphorylation kinase assay of p38α intrinsically active mutants p38αD176A and p38αF327S and the combined p38αD176A/W187A and p38αF327S/W187A mutants. Recombinant p38α mutants were incubated with kinase assay buffer without substrate for increasing time intervals at 30 °C. The radiographs (upper image) reveal a decreased autophosphorylation capability of the intrinsically active mutants when combined with the W187A mutation, which could indicate that autophosphorylation is obtained by Trp187 interactions in the DEF pocket. Coomassie staining (lower image) verified the amounts of enzymes in each lane. The autophosphorylation results are part of the same experiment shown in Fig. 4a for comparison on the same scale. c, paper-spotted kinase assay of p38α intrinsically active mutants p38αD176A and p38αF327S and the combined p38αD176A/W187A and p38αF327S/W187A mutants with ATFII substrate. The result reveals that mutagenesis of Trp187 significantly decreased the phosphorylation levels of ATFII, apparently due to the decrease in the autophosphorylation capability of the intrinsically active mutants. The results show the average of two independent experiments (each in triplicates) and error bars are shown.
The autophosphorylation kinase assay of the intrinsically active mutants combined with the W187A mutation displayed a significant decrease in their autophosphorylation capabilities (Figs. 4c and 5b). An in vitro kinase assay was performed using ATFII as a substrate to examine the influence of the W187A mutation on the intrinsic activity of the active mutants. The ATFII phosphorylation assay showed a dramatic decrease in phosphorylation levels. The activation capability of the p38αD176A/W187A mutant was decreased by a factor of nine, whereas the p38αF327S/W187A showed no activity (Fig. 5C).
Cell Culture Assay of p38 DEF Pocket Mutants
To examine whether the DEF pocket analyzed in this study plays a role in living cells, the relevant mutants were transiently expressed in p38α−/− MEF cells with co-expression of MKK6EE. Western blot analysis showed that the DEF mutants were phosphorylated in situ by MKK to a similar level as the wild type protein (Fig. 6a). The effect of the DEF pocket mutants on phosphorylation of MK2 and Elk-1, which were co-transfected, was examined, showing that the phosphorylation levels of MK2 were not affected by the mutagenesis of Tyr258 or His228-Ile229 but the phosphorylation level of Elk-1 was significantly decreased by mutagenesis of Tyr258. For the p38αML194–5AA mutant, no phosphorylation was observed for either of the substrates. Taken together, these observations are of high correlation with the in vitro results.
FIGURE 6.
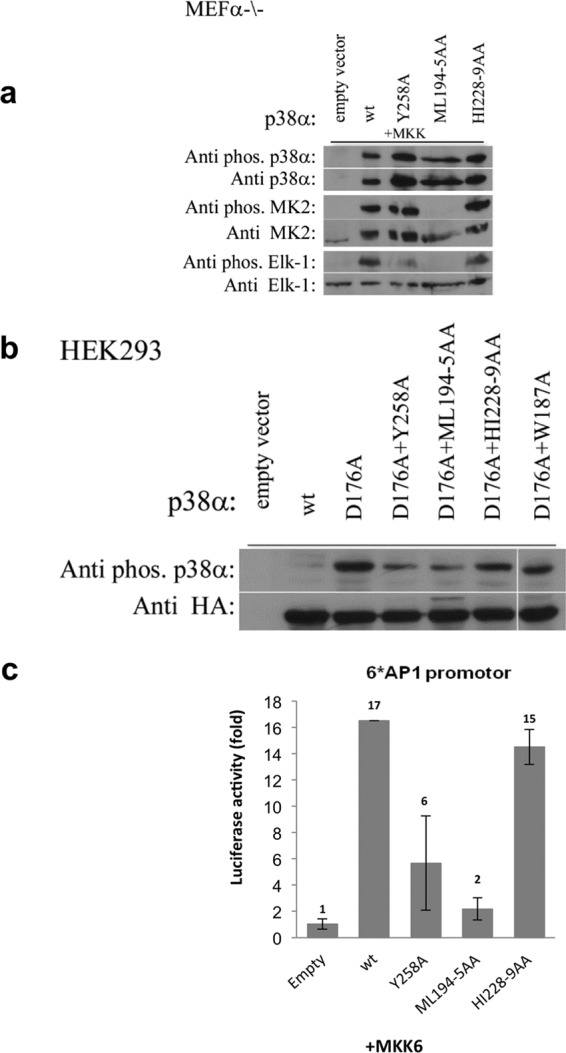
a, MEFα−/− cells were co-transfected with genes of the specified p38α DEF pocket mutants, MKK6EE, and the substrates, Elk-1 and MK2. The cells were harvested after 48 h for Western blot analysis using antibodies that specifically recognize the phosphorylated forms of p38α, Elk-1, and MK2 (upper panels). The blots were stripped and re-incubated with antibodies against p38α, Elk-1, and MK2 (lower panels). Analysis reveals that the Y258A mutation resulted in a notable decrease in phosphorylation levels of Elk-1 but not of MK2. The HI228–9AA mutant shows a slight decrease in the phosphorylation levels of Elk-1 but no effect on the MK2. Conversely, for the ML194–5AA mutant, no phosphorylation was observed for both substrates. These results show high coloration to the in vitro kinase assay. b, the autophosphorylation capabilities of p38αD176A combined with the DEF pocket mutants were examined in HEK293 cells. Cells were transfected with the relevant HA-tagged p38α mutant genes and 48 h post-transfection the cells were harvested for Western blot analysis. The results reveal that p38αD176A is spontaneously phosphorylated in cells but levels decrease by mutating DEF pocket residues or with W187A, verifying the involvement of these residues in autophosphorylation. c, to examine if the DEF pocket is required for natural activation of the p38α cascade, MEFα−/− cells were co-transfected with the genes of the specified p38α DEF pocket mutants, MKK6EE, the AP-1-luciferase reporter gene, and a Renilla luciferase gene. 48 h post-transfection, the cells were harvested and dual luciferase activity was measured. The results clearly indicate a significant decrease in luciferase activity of p38αY258A but not the p38αHI228–9AA mutant compared with p38αwt, indicating that the DEF pocket in p38α is required for its transcriptional activity. The results are the average of two independent experiments and normalized to the activity of empty vector-transfected cells (left bar).
p38α intrinsically active mutants have been shown to be spontaneously phosphorylated in cell culture (33). HEK293 cells were transfected with the D176A intrinsically active mutant or combined with DEF pocket mutants to determine whether autophosphorylation is mediated by the DEF pocket. The p38αD176A/W187A mutant was also examined. Western blot analysis showed a decrease in the spontaneous autophosphorylation of D176A, probably resulting from mutations in the DEF pocket or W187A mutation (Fig. 6B).
We then examined if formation of the DEF pocket in p38α is required for the natural activation of the p38α cascade by determining the ability of the DEF pocket mutants to induce transcription of a reporter gene driven by an AP-1-responsive cis element. These elements serve as binding sites for transcription activators of the AP-1 family (34, 35) and p38α was shown to stimulate AP-1 activity via several of its components (e.g. ATF2 and cAMP-response element-binding protein). HEK293 were co-transfected with the different p38α mutants, MKK6EE, and the AP1-luciferase constructs. There was a significant decrease in luciferase activity for the p38αY258A but not the p38αHI228–9AA mutant. As expected, the p38αML194–5AA mutant decreased luciferase activity to the basal activity similar to the activity measured for an empty vector (Fig. 6C).
DISCUSSION
The functionality of living cells requires tight regulation of the proteins mediating the different signals. One of the key signaling pathways in eukaryotic cells is the MAPKs cascade. Two main regulation modes of signaling are mediated by protein-protein interactions in the MAPK signaling cascade. One involves direct interactions between proteins at specific recognition sites, whereas the other is directed by scaffold proteins.
For MAPKs, it has been shown that the specificity for substrates is exclusively derived from docking site interactions rather than interactions in the active site (36, 37). Of the two characterized MAP kinase docking sites, the CD region is the principal region accountable for MAP kinase specificity toward upstream activators, substrates, and phosphatases. The DEF pocket, the second docking site, is distinctive for substrates formed mainly in the active dually phosphorylated state of the ERK and p38, and located in the C-lobe proximal to the catalytic site.
Here, the role of the DEF pocket in substrate selectivity in p38α was examined. Activation of the four substrates by p38α is influenced differently by DEF pocket mutations and could be classified into two main groups. The first includes the MK2 whose phosphorylation was not affected by DEF pocket mutants (p38αY258A and p38αHI228–9AA) and did not contain putative F-site residues near its phosphorylation site. The second group including ATFII, Elk-1, and MBP, can be defined as substrates whose phosphorylation was mediated by interactions with the DEF pocket of p38α. This classification into two groups was also supported by experiments where Asp316, one of the essential acidic residues of the CD region, was mutated into Asn. D316N significantly decreased the phosphorylation of MK2 but not the other three substrates. The notion that these two spatially segregated docking regions interact differently with substrates has already been suggested for ERK2 (16, 38). In this context, it was already shown in vivo and in vitro that phosphorylation of transcription factors c-Fos and Elk-1 is principally mediated by DEF pocket interactions, whereas phosphorylation of RSK (90-kDa ribosomal S6 kinase, a member of the MAPKAPK family) is mediated by the CD domain (16). In addition, it was also shown that in ERK2, the induction of epithelial to mesenchymal transformation is mediated by DEF pocket-dependent signaling events (38). The results for ERK2 showed that the phosphorylation of transcription factors is DEF-dependent, whereas the activation of kinase substrates is DEF-independent, as we now also show for p38α. The results of the luciferase assay indicate that the DEF pocket is also essential for activating the p38α signaling cascade. Thus interactions through any of the two docking regions in p38α can mediate different signaling events. In addition, we have shown that a short peptide derived from Elk-1 containing the FQF motif shows an inhibitory effect in ATFII phosphorylation, whereas the mutated peptide lacking the FQF (AQA) motif did not (Fig. 7). These results suggest that the DEF site binders could be optimized to become selective p38α inhibitors.
FIGURE 7.
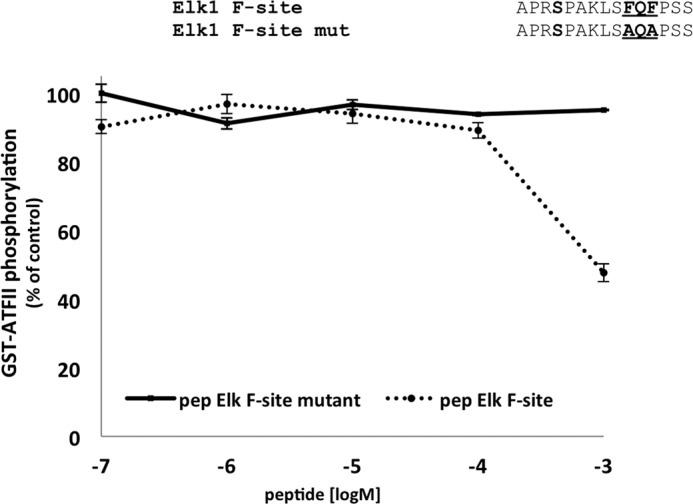
Inhibition of the ATFII phosphorylation activity of dually phosphorylated p38αwt by Elk-1 F-site derived peptides. p38αwt was initially activated in vitro by MKK6EE and the phosphorylation levels of ATFII in the presence of increasing concentrations of the peptides (between 0.1 μm to 1 mm) were monitored by a paper-spotted kinase assay. The Elk-1 F-site-mutated peptide, where the FQF motif was mutated into AQA, was used as a negative control (the full sequence is shown in the upper part of the figure). The phosphorylation levels of ATFII in the presence of 0.1 μm Elk-1 F-site mutant was normalized to 100%. These results clearly show a significant decrease in ATFII phosphorylation to ∼50% by the F-site peptide with no apparent effect by the negative control peptide.
Within the DEF-dependent subgroup of the three substrates, there are differences in their respective phosphorylation levels, which could result from variations in their F-sites. Only Elk-1 contains the characterized FXF canonical motif (defined as the F-site) downstream to its phosphorylation sites (12, 19). Hydrophobic/aromatic residues (Phe-Leu and Phe-Ser-Trp, which may serve as F-sites, were found in ATFII and MBP sequences, respectively, downstream to their phosphorylation site (Fig. 5a). Although they do not contain the canonical FXF motif, these hydrophobic segments may bind the DEF pocket although they would be accommodated differently in the hydrophobic DEF pocket of p38α. Presumably each substrate interacts somewhat differently with the DEF pocket, thus contributing to the specificity of the each substrate. The kinetic experiments for substrates Elk-1 and ATFII also support this assumption because the kinetic parameters of the DEF pocket mutants differ. The variation in the binding regions in the substrates that participate in binding the DEF pocket could also indicate a certain plasticity of the latter, which could also provide indications of how autophosphorylation in p38α occurs.
Activation by autophosphorylation is one of the main self-regulating mechanisms of kinases occurring either in cis (intramolecular) or trans (intermolecular). For many kinases autophosphorylation occurs upon stimulation or inhibition (ligand binding, phosphorylation, etc.) (39). Although it was previously thought that throughout evolution MAPKs lost their autophosphorylation capabilities, recent results show that p38 and ERK2 can also be autophosphorylated. The alternative activation pathways in p38α are probably the best indication for the involvement of autophosphorylation in their activation (31, 32, 40). In addition, intrinsically active mutants of p38s and ERKs are also shown to be activated by autophosphorylation (24, 27, 41–43). For p38α it was shown that autophosphorylation of intrinsically active mutants and TCR-induced activation occur in trans (24, 44). Two aromatic residues, Trp187 and Tyr188, downstream to the Thr180 phosphorylation site of p38α form a putative p38α F-site (Fig. 5a). The autophosphorylation kinase assays reveal that mutating either the DEF pocket residues or Trp187 to Ala significantly decreases the autophosphorylation capability of the intrinsically active mutants, as was also shown in cell culture assays. In addition, the W187A mutation combined with the intrinsically active mutants resulted in a dramatic decrease in the intrinsic activity probably due to low autophosphorylation levels. The autophosphorylation results of the DEF pocket mutants and W187A experimentally validate our previously proposed mechanism of trans-autophosphorylation in p38α (22, 24). Trp187 (and maybe Tyr188) may accommodate the DEF pocket thus orienting the activation loop in a conformation in which the phosphoacceptors are positioned in the active site of a neighboring molecule thereby promoting trans-autophosphorylation.
Although structural data of ERK2 and p38α have been available for almost two decades, contributing to the understanding of the canonical activation mechanisms of these molecules, the DEF pocket was only characterized at a later stage via structural analysis of the dually phosphorylated ERK2 and p38α as well as active Tyr323 mutants of p38α. The DEF pockets in ERK2 and p38α are composed of homologous residues forming a hydrophobic cavity in the C-lobe with somewhat different topological outlines. The DEF pocket, considered to be a secondary docking site for several MAP kinases, serves as a recognition region for substrates containing the complementary F-site, whereas not affecting other substrates lacking the F-site. Our study on p38α shows that the DEF pocket is responsible for regulating the phosphorylation of selected substrates (DEF pocket dependent; mainly transcription factors), whereas other substrates are DEF pocket-independent. The DEF pocket may provide a basis for designing specific inhibitors that block the pocket and preclude transcription activity, as was shown here for the Elk-1-derived peptide, without completely incapacitating the catalytic capacities of p38α MAP kinase.
Acknowledgments
We thank the staff of ESRF, Grenoble, France, for their outstanding help, by maintaining and upgrading the facility. We thank Dr. Deborah E. Shalev for insightful discussions. The pGEX-MK2-K76R plasmid was a generous gift from Prof. M. Gaestel, Institute of Biochemistry, Medical School Hannover, Hannover, Germany.
This work was supported by ISF Research Center of Excellence 180/09 awarded (to O. L. and D. E.).
The atomic coordinates and structure factors (code 4GEO) have been deposited in the Protein Data Bank (http://wwpdb.org/).
N. Tzarum, N. Komornik, D. Ben Chetrit, D. Engelberg, and O. Livnah, unpublished data.
- CD
- common docking
- MBP
- myelin basic protein.
REFERENCES
- 1. Blume-Jensen P., Hunter T. (2001) Oncogenic kinase signaling. Nature 411, 355–365 [DOI] [PubMed] [Google Scholar]
- 2. Zarubin T., Han J. (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res. 15, 11–18 [DOI] [PubMed] [Google Scholar]
- 3. Carlson S. M., Chouinard C. R., Labadorf A., Lam C. J., Schmelzle K., Fraenkel E., White F. M. (2011) Large-scale discovery of ERK2 substrates identifies ERK-mediated transcriptional regulation by ETV3. Sci. Signal. 4, rs11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yoon S., Seger R. (2006) The extracellular signal-regulated kinase. Multiple substrates regulate diverse cellular functions. Growth Factors 24, 21–44 [DOI] [PubMed] [Google Scholar]
- 5. Chang C. I., Xu B. E., Akella R., Cobb M. H., Goldsmith E. J. (2002) Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol. Cell 9, 1241–1249 [DOI] [PubMed] [Google Scholar]
- 6. Lee T., Hoofnagle A. N., Kabuyama Y., Stroud J., Min X., Goldsmith E. J., Chen L., Resing K. A., Ahn N. G. (2004) Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol. Cell 14, 43–55 [DOI] [PubMed] [Google Scholar]
- 7. Tanoue T., Adachi M., Moriguchi T., Nishida E. (2000) A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell. Biol. 2, 110–116 [DOI] [PubMed] [Google Scholar]
- 8. Bardwell A. J., Flatauer L. J., Matsukuma K., Thorner J., Bardwell L. (2001) A conserved docking site in MEKs mediates high-affinity binding to MAP kinases and cooperates with a scaffold protein to enhance signal transmission. J. Biol. Chem. 276, 10374–10386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bardwell L., Thorner J. (1996) A conserved motif at the amino termini of MEKs might mediate high-affinity interaction with the cognate MAPKs. Trends Biochem. Sci. 21, 373–374 [PubMed] [Google Scholar]
- 10. Enslen H., Brancho D. M., Davis R. J. (2000) Molecular determinants that mediate selective activation of p38 MAP kinase isoforms. EMBO J. 19, 1301–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kallunki T., Su B., Tsigelny I., Sluss H. K., Dérijard B., Moore G., Davis R., Karin M. (1994) JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 8, 2996–3007 [DOI] [PubMed] [Google Scholar]
- 12. Jacobs D., Glossip D., Xing H., Muslin A. J., Kornfeld K. (1999) Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 13, 163–175 [PMC free article] [PubMed] [Google Scholar]
- 13. Murphy L. O., MacKeigan J. P., Blenis J. (2004) A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol. Cell Biol. 24, 144–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Murphy L. O., Smith S., Chen R. H., Fingar D. C., Blenis J. (2002) Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 4, 556–564 [DOI] [PubMed] [Google Scholar]
- 15. Vinciguerra M., Vivacqua A., Fasanella G., Gallo A., Cuozzo C., Morano A., Maggiolini M., Musti A. M. (2004) Differential phosphorylation of c-Jun and JunD in response to the epidermal growth factor is determined by the structure of MAPK targeting sequences. J. Biol. Chem. 279, 9634–9641 [DOI] [PubMed] [Google Scholar]
- 16. Dimitri C. A., Dowdle W., MacKeigan J. P., Blenis J., Murphy L. O. (2005) Spatially separate docking sites on ERK2 regulate distinct signaling events in vivo. Curr. Biol. 15, 1319–1324 [DOI] [PubMed] [Google Scholar]
- 17. Bott C. M., Thorneycroft S. G., Marshall C. J. (1994) The sevenmaker gain-of-function mutation in p42 MAP kinase leads to enhanced signalling and reduced sensitivity to dual specificity phosphatase action. FEBS Lett. 352, 201–205 [DOI] [PubMed] [Google Scholar]
- 18. Chu Y., Solski P. A., Khosravi-Far R., Der C. J., Kelly K. (1996) The mitogen-activated protein kinase phosphatases PAC1, MKP-1, and MKP-2 have unique substrate specificities and reduced activity in vivo toward the ERK2 sevenmaker mutation. J. Biol. Chem. 271, 6497–6501 [DOI] [PubMed] [Google Scholar]
- 19. Galanis A., Yang S. H., Sharrocks A. D. (2001) Selective targeting of MAPKs to the ETS domain transcription factor SAP-1. J. Biol. Chem. 276, 965–973 [DOI] [PubMed] [Google Scholar]
- 20. Sheridan D. L., Kong Y., Parker S. A., Dalby K. N., Turk B. E. (2008) Substrate discrimination among mitogen-activated protein kinases through distinct docking sequence motifs. J. Biol. Chem. 283, 19511–19520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y. Y., Wu J. W., Wang Z. X. (2011) Mitogen-activated protein kinase (MAPK) phosphatase 3-mediated cross-talk between MAPKs ERK2 and p38α. J. Biol. Chem. 286, 16150–16162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tzarum N., Diskin R., Engelberg D., Livnah O. (2011) Active mutants of the TCR-mediated p38α alternative activation site show changes in the phosphorylation lip and DEF site formation. J. Mol. Biol. 405, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 23. Canagarajah B. J., Khokhlatchev A., Cobb M. H., Goldsmith E. J. (1997) Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell 90, 859–869 [DOI] [PubMed] [Google Scholar]
- 24. Diskin R., Lebendiker M., Engelberg D., Livnah O. (2007) Structures of p38α active mutants reveal conformational changes in L16 loop that induce autophosphorylation and activation. J. Mol. Biol. 365, 66–76 [DOI] [PubMed] [Google Scholar]
- 25. Otwinowski Z., Minor W. (1997) in Methods in Enzymology, pp. 307–326, Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- 26. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Diskin R., Askari N., Capone R., Engelberg D., Livnah O. (2004) Active mutants of the human p38α mitogen-activated protein kinase. J. Biol. Chem. 279, 47040–47049 [DOI] [PubMed] [Google Scholar]
- 28. Ben-Levy R., Leighton I. A., Doza Y. N., Attwood P., Morrice N., Marshall C. J., Cohen P. (1995) Identification of novel phosphorylation sites required for activation of MAPKAP kinase-2. EMBO J. 14, 5920–5930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tzarum N., Eisenberg-Domovich Y., Gills J. J., Dennis P. A., Livnah O. (2012) Lipid molecules induce p38α activation via a novel molecular switch. J. Mol. Biol. 424, 339–353 [DOI] [PubMed] [Google Scholar]
- 30. Nolen B., Taylor S., Ghosh G. (2004) Regulation of protein kinases. Controlling activity through activation segment conformation. Mol. Cell 15, 661–675 [DOI] [PubMed] [Google Scholar]
- 31. Ge B., Gram H., Di Padova F., Huang B., New L., Ulevitch R. J., Luo Y., Han J. (2002) MAPKK-independent activation of p38α mediated by TAB1-dependent autophosphorylation of p38α. Science 295, 1291–1294 [DOI] [PubMed] [Google Scholar]
- 32. Salvador J. M., Mittelstadt P. R., Guszczynski T., Copeland T. D., Yamaguchi H., Appella E., Fornace A. J., Jr., Ashwell J. D. (2005) Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat. Immunol. 6, 390–395 [DOI] [PubMed] [Google Scholar]
- 33. Askari N., Diskin R., Avitzour M., Capone R., Livnah O., Engelberg D. (2007) Hyperactive variants of p38α induce, whereas hyperactive variants of p38γ suppress, activating protein 1-mediated transcription. J. Biol. Chem. 282, 91–99 [DOI] [PubMed] [Google Scholar]
- 34. Karin M. (1995) The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 270, 16483–16486 [DOI] [PubMed] [Google Scholar]
- 35. Shaulian E., Karin M. (2001) AP-1 in cell proliferation and survival. Oncogene 20, 2390–2400 [DOI] [PubMed] [Google Scholar]
- 36. Callaway K. A., Rainey M. A., Riggs A. F., Abramczyk O., Dalby K. N. (2006) Properties and regulation of a transiently assembled ERK2·Ets-1 signaling complex. Biochemistry 45, 13719–13733 [DOI] [PubMed] [Google Scholar]
- 37. Rainey M. A., Callaway K., Barnes R., Wilson B., Dalby K. N. (2005) Proximity-induced catalysis by the protein kinase ERK2. J. Am. Chem. Soc. 127, 10494–10495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shin S., Dimitri C. A., Yoon S. O., Dowdle W., Blenis J. (2010) ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol. Cell 38, 114–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smith J. A., Francis S. H., Corbin J. D. (1993) Autophosphorylation. A salient feature of protein kinases. Mol. Cell. Biochem. 127, 51–70 [DOI] [PubMed] [Google Scholar]
- 40. Gills J. J., Castillo S. S., Zhang C., Petukhov P. A., Memmott R. M., Hollingshead M., Warfel N., Han J., Kozikowski A. P., Dennis P. A. (2007) Phosphatidylinositol ether lipid analogues that inhibit AKT also independently activate the stress kinase, p38α, through MKK3/6-independent and -dependent mechanisms. J. Biol. Chem. 282, 27020–27029 [DOI] [PubMed] [Google Scholar]
- 41. Avitzour M., Diskin R., Raboy B., Askari N., Engelberg D., Livnah O. (2007) Intrinsically active variants of all human p38 isoforms. FEBS J. 274, 963–975 [DOI] [PubMed] [Google Scholar]
- 42. Bell M., Capone R., Pashtan I., Levitzki A., Engelberg D. (2001) Isolation of hyperactive mutants of the MAPK p38/Hog1 that are independent of MAPK kinase activation. J. Biol. Chem. 276, 25351–25358 [DOI] [PubMed] [Google Scholar]
- 43. Levin-Salomon V., Kogan K., Ahn N. G., Livnah O., Engelberg D. (2008) Isolation of intrinsically active (MEK-independent) variants of the ERK family of mitogen-activated protein (MAP) kinases. J. Biol. Chem. 283, 34500–34510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mittelstadt P. R., Yamaguchi H., Appella E., Ashwell J. D. (2009) T cell receptor-mediated activation of p38{alpha} by monophosphorylation of the activation loop results in altered substrate specificity. J. Biol. Chem. 284, 15469–15474 [DOI] [PMC free article] [PubMed] [Google Scholar]