

Background: Modified heptoses are important for bacterial virulence.
Results: We characterized novel l-gluco-heptose synthesis enzymes from Campylobacter jejuni and compared their specificity with d-altro-heptose synthesis enzymes.
Conclusion: Significant differences of activities and specificities were observed despite high sequence similarities.
Significance: The versatility of the enzymes can be exploited to synthesize novel carbohydrates for technological applications and to develop therapeutic inhibitors.
Keywords: Bacterial Pathogenesis, Campylobacter, Carbohydrate Biosynthesis, Enzyme Catalysis, Glycobiology, Capsule, Epimerases, Heptose Modification
Abstract
Uniquely modified heptoses found in surface carbohydrates of bacterial pathogens are potential therapeutic targets against such pathogens. Our recent biochemical characterization of the GDP-6-deoxy-d-manno- and GDP-6-deoxy-d-altro-heptose biosynthesis pathways has provided the foundation for elucidation of the more complex l-gluco-heptose synthesis pathway of Campylobacter jejuni strain NCTC 11168. In this work we use GDP-4-keto,6-deoxy-d-lyxo-heptose as a surrogate substrate to characterize three enzymes predicted to be involved in this pathway: WcaGNCTC (also known as Cj1427), MlghB (Cj1430), and MlghC (Cj1428). We compare them with homologues involved in d-altro-heptose production: WcaG81176 (formerly WcaG), DdahB (Cjj1430), and DdahC (Cjj1427). We show that despite high levels of similarity, the enzymes have pathway-specific catalytic activities and substrate specificities. MlghB forms three products via C3 and C5 epimerization activities, whereas its DdahB homologue only had C3 epimerase activity along its cognate pathway. MlghC is specific for the double C3/C5 epimer generated by MlghB and produces l-gluco-heptose via stereospecific C4 reductase activity. In contrast, its homologue DdahC only uses the C3 epimer to yield d-altro-heptose via C4 reduction. Finally, we show that WcaGNCTC is not necessary for l-gluco-heptose synthesis and does not affect its production by MlghB and MlghC, in contrast to its homologue WcaG81176, that has regulatory activity on d-altro-heptose synthesis. These studies expand our fundamental understanding of heptose modification, provide new glycobiology tools to synthesize novel heptose derivatives with biomedical applications, and provide a foundation for the structure function analysis of these enzymes.
Introduction
C3/C5 epimerases and C3/C5 epimerases/C4 reductases have attracted much attention as antibacterial drug targets due to their essential role in the biosynthesis of bacterial surface carbohydrates involved in virulence. For example, the putative C3/C5 epimerase WbmF is important for O-antigen synthesis in Bordetella species (1), and the well characterized C3/C5 epimerases RmlC from Escherichia coli and Pseudomonas aeruginosa and RfbC from Salmonella enterica are involved in the production of dTDP-l-rhamnose for O-antigen synthesis (2–5). Likewise, GDP-4-keto-6-deoxy-α-d-mannose C3/C5 epimerases/C4 reductases such as GFS and GMER involved in making GDP-l-fucose from GDP-mannose have been studied extensively as the process of synthesis of GDP-l-fucose is well conserved throughout evolution (6–8) and l-fucose is important in a variety of cellular processes. The GFS from E. coli and Helicobacter pylori play a role in virulence (6, 9), and in the case of the H. pylori, the l-fucose generated by GFS is displayed on the cell surface where it plays a role in host mimicry (10, 11). Eukaryotic GDP-mannose epimerases/reductases have also been extensively studied due to the importance of l-fucose as a determinant of blood group antigens as a cell surface ligand involved in inflammatory responses and in Notch signaling (12, 13). Plant homologues involved in the synthesis of vitamin C have also been characterized in depth, such as the GME GDP-mannose C3/C5 epimerase/C4 reductase of Arabidopsis thaliana (14).
All the enzymes described above are involved in the synthesis of hexose derivatives. Heptose derivatives also play an important role in bacterial virulence, as demonstrated previously in Yersinia pseudotuberculosis (15). Heptose derivatives are also present in the capsule of the human gastro-intestinal pathogen Campylobacter jejuni (16, 17). Because this bacterium is a commensal in poultry, most cases of human campylobacteriosis result directly from ingestion of contaminated poultry meat in developed countries (18–20). Thus, elimination of Campylobacter colonization at the source, during chicken rearing, is an appealing option. In light of developing antibiotic resistance in Campylobacter (19, 21), this requires novel intervention options. The capsule is an important virulence factor of C. jejuni (22, 23). Therefore, like their hexose-modifying counterparts, the heptose-modifying enzymes responsible for making the heptose derivatives that are found within the capsule are potential anti-Campylobacter targets applicable to decrease commensal colonization of broiler chicken by C. jejuni or to treat infected patients. In the absence of human homologous pathways, it may be possible to identify safe inhibitors of bacterial heptose modification pathways for therapeutic applications. To explore this possibility, the bacterial heptose modification pathways must be elucidated. Furthermore, the comparative analysis of similar Campylobacter enzymes involved in the formation of related but not identical heptose derivatives will provide important clues as to the type of inhibitors that could ultimately be designed against such enzymes; that is, highly specific inhibitors versus broad spectrum inhibitors acting against various Campylobacter strains that produce different heptose derivatives.
We recently reported the first characterization of two RmlC and GFS homologues involved in heptose modification, namely DdahB (previously known as Cjj1430) and DdahC (formerly known as Cjj1427), which are encoded by the capsular cluster of C. jejuni strain 81-176 and are responsible for the synthesis of GDP-6-deoxy-d-altro-heptose along with the GDP-manno-heptose C6 dehydratase DdahA (also known as WcbK; Fig. 1A) (24, 25). The new Ddah* names assigned to the enzymes in this study were chosen to reflect the involvement of these enzymes in GDP-6-deoxy-d-altro-heptose synthesis (Tables 1 and 2) and are introduced to facilitate distinguishing them from other enzymes that are involved in 6-O-methyl-l-glucoheptose and are named Mlgh*. Although DdahB was anticipated to have C3/C5 epimerase activity based on sequence homologies to RmlC and RfbC, and DdahC was anticipated to have C3/C5 epimerase and C4 reductase activities similarly to GFS and GMER, we showed that DdahB only exerted its C3 epimerase activity and that DdahC only served as a reductase in the GDP-6-deoxy-d-altro-heptose synthesis pathway (25).
FIGURE 1.

Comparative GDP-6-deoxy-d-altro-heptose and GDP-6-O-Me-l-gluco-heptose synthesis pathways of C. jejuni. The 6-deoxy-d-altro-heptose synthesis pathway shown in panel A is as established previously (24, 25). The 6-O-Me-l-gluco-heptose synthesis pathway shown in panel B is hypothetical. The C4 oxidase MlghA necessary to form the 4-keto derivative necessary for MlghB activity has not been identified. A putative methyltransferase MlghD has been identified but not characterized so that it is not currently known when the 6-O methyl group is introduced along this pathway, as indicated by the question marks. R refers to the fact that in vivo the enzymes from C. jejuni strain NCTC 11168 may use 6-O-methyl or non-methylated derivatives.
TABLE 1.
Correspondence between enzymes involved in heptose modification in strains NCTC 11168 and 81-176
| Strain NCTC 11168 |
Identity | Similarity | Strain 81–176 |
||
|---|---|---|---|---|---|
| Name in this studya | ORFb | Name in this studya | ORF/former nameb | ||
| % | % | ||||
| MlghA | X | NAc | NAc | DdahA | Cjj1426 /WcbK |
| MlghB | Cj1430 | 81 | 98 | DdahB | Cjj1430 |
| MlghC | Cj1428 | 57 | 90 | DdahC | Cjj1427 |
| MlghD | Cj1426 | NAc | NAc | NA. c | NA. c |
| WcaGNCTC | Cj1427 | 98 | 100 | WcaG81176 | Cjj1425/WcaG |
a The enzymes directly involved in making 6-deoxy-d-altro-heptose in strain 81-176 are named using the Ddah* nomenclature, whereas the enzymes directly involved in making 6-O-methyl-l-glucoheptose in strain NCTC 11168 are named using the Mlgh* nomenclature.
b The ORFs and names were as indicated in the genome databases (see Ref. 26) for strain NCTC 11168 and www.ncbi.nlm.nih.gov for strain 81-176. Former names were as previously used in Refs. 24 and 25.
c NA stands for not applicable as the oxidase MlghA of strain NCTC 11168 has not been identified to date, and there is no methyltransferase involved in the pathway for strain 81-176.
TABLE 2.
Summary of names, substrates and functions of all enzymes used in this study
| Name in this study | ORFa Prior name | Predicted function | Observed function | Substrate | Product |
|---|---|---|---|---|---|
| Strain 81–176 for 6-deoxy-d-altro-heptose pathway | |||||
| DdahA | Cjj1426 | C4, C6 dehydratase | C4, C6 dehydratase | GDP-manno-heptose | GDP-6-deoxy-4-keto-d-lyxo-heptose |
| DdahB | Cjj1430 | C3, C5 epimerase/C4 reductase | C3 epimerase | GDP-6-deoxy-4-keto-d-lyxo-heptose | GDP-6-deoxy-4-keto-d-arabino heptose |
| DdahC | Cjj1427 | C3, C5 epimerase/C4 reductase | C4 reductase | GDP-6-deoxy-4-keto-d-arabino heptose | GDP-6-deoxy-4-keto-d-altro heptose |
| WcaG81176 | Cjj1425 WcaG | C4 reductase | C4 reductase | GDP-6-deoxy-4-keto-d-lyxo-heptose | GDP-6-deoxy-d-manno-heptose |
| Strain NCTC 11168 for 6-OMe-l-gluco-heptose pathwayb | |||||
| MlghA | X | C4 oxidase | Not identified | GDP-manno-heptose | GDP-4-keto-d-lyxo-heptose |
| MlghD | Cj1426 | 6-O Methyl Transferase | Not determined | GDP-4-keto-d-lyxo-heptose | GDP-6-OMe-4-keto-d-lyxo-heptose |
| MlghB | Cj1430 | C3, C5 epimerase/C4 reductase | C3, C5 epimerase | GDP-6-OMe-4-keto-d-lyxo-heptose | GDP-6-OMe-4-keto-l-xylo-he ptose |
| MlghC | Cj1428 | C3, C5 epimerase/C4 reductase | C4 reductase | GDP-6-OMe-4-keto-l-xylo-heptose | GDP-6-OMe-4-keto-l-gluco-heptose |
| WcaGNCTC | Cj1427 | C4 reductase | C4 reductase | GDP-6-OMe-4-keto-d-lyxo-heptose | GDP-6-OMe-d-manno-heptose |
a The ORFs and names were as indicated in the genome databases (see Ref. 26) for strain NCTC 11168 and www.ncbi.nlm.nih.gov for strain 81-176. Former names were as previously used in Refs. 24 and 25.
b The enzymes are listed in the anticipated order of participation in the pathway. The assignment of the methyltransferase in the early steps of the pathway after the oxidation step is likely but speculative and awaits biochemical confirmation. Based on this assignment, the natural substrates for MlghB, MlghC, and WcaGNCTC are anticipated to be 6-OMe-4-keto derivatives. The 6-deoxy-4-keto derivatives used as substrates in this study are surrogate substrates obtained by the initial activity of DdahA.
Interestingly, similar enzymes (MlghB (also known as Cj1430) and MlghC (also known as Cj1428) are encoded by the capsular cluster of C. jejuni strain NCTC 11168 (26), which produces 6-O-methyl-l-gluco-heptose (16). MlghB is 81% identical and 98% similar to DdahB, and MlghC is 57% identical and 90% similar to DdahC (Table 1). These degrees of similarity are high enough to anticipate similar functions in both strains but low enough to allow for strain-specific activity. The homologous enzymes may perform strain-specific epimerizations/reductions to generate strain-specific modified heptoses: d-altro in strain 81-176 versus l-gluco in strain NCTC 11168 (Table 2, Fig. 1).
Although d-altro heptose synthesis only involves C3 epimerization of the d-manno-heptose precursor followed by its reduction at C4 (25), the synthesis of l-gluco-heptose is anticipated to require epimerizations both on C3 and on C5 followed by reduction at C4 (Fig. 1B). This pathway, therefore, potentially involves the predicted reductase MlghC as a stereospecific reductase. However, this l-gluco-heptose synthesis pathway presents the conundrum that two enzymes (MlghB and MlghC) can potentially perform the C3/C5 epimerase activities (Table 2), whereas only one such enzyme would suffice. Both enzymes nevertheless seem to be involved in heptose modification based on mutagenesis studies (16, 27), but their specific function remains unknown. Based on the sequence homologies indicated above and based on our biochemical analysis of the d-altro-heptose synthesis pathway, one could anticipate that MlghB will only have C3 epimerase activity, whereas MlghC will have C5 epimerase and C4 reductase activities. Alternatively, MlghB may have both C3 and C5 epimerase activities, whereas MlghC would only serve as a C4 reductase. Therefore, the biochemical pathway for l-gluco-heptose synthesis needs investigating at the biochemical level to resolve the issue.
The capsular cluster of strain NCTC 11168 also encodes a predicted C4 reductase (WcaGNCTC) that is 98% identical to the C4 reductase WcaG from strain 81-176 (now called WcaG81176, Table 1). The strikingly high identity suggests identical functions in both strains. We showed previously that WcaG81176 is a reductase as predicted (24) but that, contrary to expectations, WcaG81176 is not part of the mainstream GDP-6-deoxy-d-altro-heptose synthesis pathway (Fig. 1A). However, it affected the outcome of this pathway via modulation of DdahC activity (25). The role of WcaGNCTC on the l-gluco heptose synthesis pathway remains to be elucidated.
We hypothesized that DdahB, MlghB, DdahC, and MlghC would perform strain-specific epimerizations and/or reductions to generate either the d-altro or l-gluco form of the capsule-linked heptose in C. jejuni and that WcaGNCTC may exert regulatory effects on the l-gluco heptose synthesis pathway. To evaluate this hypothesis, we cloned, overexpressed, and purified the yet uncharacterized WcaGNCTC, MlghB, and MlghC from C. jejuni strain NCTC 11168. Using a combination of capillary electrophoresis, NMR spectroscopy, and mass spectrometry analyses and using GDP-6-deoxy-4-keto-d-lyxo-heptose as a substrate, we compared the activities of these three enzymes with the activities of the previously characterized WcaG81176, DdahB, and DdahC homologues from the GDP-6-deoxy-d-altro-heptose modification pathway. We show that despite high levels of similarities, the MlghB and DdahB enzymes have different activities, and we demonstrate a high level of substrate specificity of MlghC and DdahC for different heptose epimers. We also show that WcaG81176 and WcaGNCTC have the same biochemical activity as suggested by their quasi identity, but their impact on their cognate heptose modification pathway is different.
MATERIALS AND METHODS
Cloning of cj1427c, cj1428c, and cj1430c into the pET Vector and Protein Expression
The cj1427c, cj1428c, and cj1430c genes from C. jejuni strain NCTC 11168 coding for WcaGNCTC, MlghC, and MlghB, respectively, were PCR-amplified from genomic DNA using primers CJ1427 P2/P3, CJ1428 P2/P3, and CJ1430 P2/P3, respectively (Table 3), and cloned in the pET23 derivative (28) using standard procedures as done before for WcaG81176, DdahB, and DdahC from strain 81-176 (24, 25). All constructs were sequenced at the Robarts Institute Sequencing Facility (London, Canada). Expression was performed in Luria-Bertani broth (LB) using E. coli ER2566 for DdahC, WcaGNCTC, and MlghC and E. coli BL21(DE3)pLysS for all other enzymes. Protein expression was induced by the addition of 0.1 mm isopropyl β-d-1-thiogalactopyranoside, and expression was carried out at 37 °C, except for DdahB and WcaGNCTC (25 °C). The proteins were purified by nickel chelation using standard methods reported previously (24, 25). The proteins were analyzed by SDS-PAGE and Coomassie Blue staining or by anti-histidine-tag Western blotting as described previously (24, 25).
TABLE 3.
Sequences of primers used in these studies
NcoI and BamHI restriction sites used for cloning purposes are highlighted in bold.
| Primer name | Primer sequence |
|---|---|
| CJ1427P2 | AGGGTCCATGGGCATGTCAAAAAAAGTTTTAATTAC |
| CJ1427P3 | GCGTCGGATCCTTAATTAAAATTTGCAAAGCGA |
| CJ1428P2 | AGGTACCATGGGCATGCAAACAAATTCAAAAATATA |
| Cj1428P3 | GCTGGATCCTCAATTTTGTGTTTTATACCA |
| CJ1430P2 | AGGGTCCATGGCAATAGAATTTGATATA |
| CJ1430P3 | GCGTCGGATCCTTATCCTTTATTTTTAGTTGCAA |
Capillary Electrophoresis (CE) of Sugar Nucleotides
CE3 analyses were performed using a bare silica capillary with 200 mm borax buffer pH 9 as reported before (24, 25).
Preparation of GDP-d-glycero-d-manno-heptose Substrate
The enzymatic preparation of GDP-d-glycero-d-manno-heptose from sedoheptulose 7-phosphate and its purification by anion exchange chromatography were performed as reported previously (29).
Enzyme Assays
Reactions usually contained ∼0.10 mm GDP-d-glycero-d-manno-heptose substrate (from above), 0.12 mm NADPH/NADP+ mix (70/30%/%) in 200 mm Tris-HCl, pH 8.0, and were incubated at 37 °C. The amounts of enzymes added and incubation times and any other variations from the standard reaction composition indicated above are specified in the legends to the figures. Large-scale reactions were prepared by direct proportional increase of all components for anion-exchange purification followed by mass spectrometry or NMR spectroscopy analyses.
Mass Spectroscopy (MS) Analyses of Reaction Products
Reactions containing ∼3 nmol of the product were prepared in ammonium bicarbonate buffer and were lyophilized before MS analysis. MS analyses were performed in the negative ion mode at the Dr. Don Rix Protein Identification Facility of the University of Western Ontario as described before (25).
MALDI MS Analyses of Purified Enzymes
MALDI MS analyses were performed in the linear positive ion mode at the MALDI mass spectrometry facility of the University of Western Ontario (London, ON, Canada) as described before (29).
NMR Spectroscopy
A large scale reaction containing 0.75 μmol of GDP-manno-heptose, 0.87 μmol of NADH, 0.20 μg of DdahA, 0.26 μg of MlghB, and 0.24 μg of MlghC in 6.25 ml of 0.2 m triethylammonium bicarbonate, pH 8.5, was incubated for 1 h at 37 °C. It was filtered through a 3-kDa cutoff ultrafiltration membrane (Pall Filtron), and the reaction product was purified by anion exchange chromatography as described above. The purity of the fractions was monitored by CE. The purified product (P5γ) was lyophilized repeatedly after resuspension in Milli Q water (twice) and in D2O (four times) before NMR analysis.
All 1H NMR data were collected with a Varian Inova 600 MHz NMR spectrometer at 25 °C. One-dimensional 1H NMR spectra were collected using a 2-s presaturation pulse centered on the residual HDO resonance. 1H and 13C assignments were determined from two-dimensional 1H total correlation spectroscopy experiments (30) using a 6-kHz spinlock for 256 complex increments and natural abundance 13C,1H heteronuclear single quantum correlation experiment (31, 32). All spectra were processed using VnmrJ 2.1B and 1H and 13C chemical shifts referenced to 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt standard at 0.00 ppm. Assignments were also aided by selective one-dimensional 1H total correlation spectroscopy and NOESY experiments. All spectra were processed using unshifted Gaussian weighting in VnmrJ 3.2A. 1H and 13C chemical shifts referenced directly to 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt standard at 0.00 ppm.
RESULTS
Protein Expression and Purification
MlghB, MlghC, and WcaGNCTC were overexpressed from the pET23 plasmid with a N-terminal His6 tag and purified by nickel affinity chromatography. All proteins could be obtained in high yields in a soluble and pure form as determined by SDS-PAGE analysis with Coomassie Blue staining and anti-histidine tag Western blotting (supplemental Fig. S1). As reported previously for DdahB (25), MlghB migrated anomalously at a slightly higher molecular weight (25 kDa) than expected (22.2 kDa), but MALDI MS analysis confirmed its proper size (data not shown).
Identifying a Substrate for MlghB
It is likely that a C4 oxidase generates a 4-keto derivative that serves as a substrate for epimerization by MlghB (Fig. 1B). Although no gene coding for a putative C4 oxidase could be identified in proximity to heptose modifying genes (Tables 1 and 2), the MlghB enzyme did not have any catalytic activity on GDP-d-glycero-d-manno-heptose (data not shown), which was consistent with the need for prior oxidation of the substrate. Also, it is likely that MlghB uses a 6-O-methylated substrate based on knock-out mutagenesis data obtained with the candidate methyltransferase MlghD (also known as Cj1426 (27)) (Fig. 1B, Tables 1 and 2), but this enzyme has not been characterized at the biochemical level. Therefore, the 4-keto-6-O-methyl derivative that likely represents the natural substrate for MlghB is to this day not available for biochemical studies. In light of the high levels of similarities of MlghB with the DdahB enzyme that uses GDP-6-deoxy-4-keto-d-lyxo-heptose (called P1) as a substrate, we tested whether MlghB could also use P1 as a surrogate substrate. P1 was generated by incubating the C4/C6 dehydratase DdahA with GDP-d-glycero-d-manno-heptose (abbreviated as GDP-manno-heptose thereafter) (Fig. 2). Upon the addition of MlghB to the DdahA/GDP-manno-heptose reaction mix, a reduced amount of peak P1 was observed, whereas new product peaks appeared, clearly indicating that MlghB could use P1 as a surrogate substrate (Fig. 3A, trace b). This finding that MlghB can efficiently process P1 allowed us to perform a direct comparison of the activities of MlghB versus DdahB using an identical substrate. This also allowed the comparative analysis of the enzymes that serve downstream in the l-gluco- and d-altro-heptose modification pathways: MlghC and DdahC.
FIGURE 2.

Experimental GDP-6-deoxy-l-gluco-heptose synthesis pathway. This experimental pathway was obtained using the surrogate 6-deoxy derivative generated by the C4/C6 dehydratase from the 6-deoxy-d-altro-heptose synthesis pathway DdahA. The pathway results from the combination of the CE, MS, and NMR spectroscopy analyses described in these studies.
FIGURE 3.

Comparative analysis of strain-specific reactions catalyzed by MlghB versus DdahB and by MlghC versus DdahC. For both panels, a base reaction containing ∼0.10 mm concentrations of freshly preformed P1 was prepared by incubating GDP-d-manno-heptose with 3 μm DdahA (which was still present in the reaction), 0.10 mm NADPH, 0.03 mm NADP+, and 200 mm Tris-HCl, pH 8.0. All reactions were incubated at 37 °C for 30 min. For panel A, the base reaction was further incubated as such or supplemented by 7 μm MlghB and 3 μm MlghC or DdahC as indicated on the figure. For panel B, the reaction composition was the same as in panel A except that MlghB was replaced by DdahB. Note that DdahA is still present and active under these conditions and replenishes the stock of P1 substrate as long as heptose is available. P1, DdahA product and substrate of MlghB and DdahB. P4α, -β, and -γ, MlghB products. P4α is also produced by DdahB. P5α: DdahC product. P5γ, MlghC product. * denotes a small impurity present in the heptose preparation, and + denotes a small amount of NADP+ that was present in the NADPH stock. Integration data are provided in supplemental Table S1.
MlghB Generates Several Products, Indicating Both C3 and C5 Epimerase Activities
The reaction of MlghB with P1 yielded three products: P4α, P4β, and P4γ (Fig. 3A, trace b, supplemental Table S1). This was in contrast to reactions of DdahB with P1, which only yielded one product (Fig. 3B, trace b, supplemental Table S1). Co-injection experiments showed that the P4α product obtained with MlghB was the same as the single reaction product obtained upon reaction of DdahB with P1 (data not shown). The fact that the MlghB product P4α was identical to the DdahB product, which serves as the normal substrate for DdahC, was further supported by the fact that DdahC could use either the DdahB product or the MlghB P4α product as a substrate to generate the same P5α product (Fig. 3, A and B, traces d). This was further demonstrated by reacting DdahC with a P1/MlghB reaction mix containing the P4α, -β, and -γ products but from which MlghB had been removed to prevent the continuous replacement of P4α. Under these conditions, the appearance of P5α was clearly accompanied by exclusive disappearance of P4α (Fig. 4A, traces a and c, integration data in supplemental Table S2).
FIGURE 4.

CE electrophoregrams highlighting the differences between the reductases WcaGNCTC and WcaG81176. The identity of all peaks is as described in Fig. 3. In addition, P3 is reduced GDP-6-deoxy-manno-heptose. Panel A, ∼0.10 mm preformed P1, 7 μm MlghB, 0.10 mm NADPH, and 200 mm Tris-HCl, pH 8.0 were incubated at 37 °C for 15 min, and the reaction was filtered through a 10-kDa membrane to remove MlghB. The filtrate obtained comprised products P1, P4α, P4β, and P4γ in fixed proportions. It was incubated for 30 min at 37 °C as such (trace a) or supplemented with 3 μm concentrations each of the indicated enzymes. The data show that WcaGNCTC and WcaG81176 have the same activity and cannot replace the reductase of their cognate pathway, MlghC or DdahC. Panel B, 6 μl of ∼0.07 mm heptose and 3 μm DdahA (trace a, base line) were supplemented with 3 μm concentrations of each of the indicated enzymes (without elimination of DdahA). The activity was recorded after 2.5 h incubation at 37 °C. The data show that the activity of MlghC is not affected by WcaGNCTC and vice versa. Integration data are provided in supplemental Table S2.
P4α produced by DdahB was previously identified as GDP-6-deoxy-4-keto-d-arabino-heptose (25). MS analysis of the DdahA/MlghB reaction mix showed the appearance of a single ion at m/z 616. The MS/MS spectrum was identical to that of GDP-6-deoxy-4-keto-d-arabino-heptose (25), with a fragment that comprised the heptose ring detected at m/z 333 (Fig. 5A). This mass is consistent with the predicted epimerization of P1, and the presence of a single ion despite the presence of three distinct reaction products (P4α, P4β, and P4γ, as per CE analysis) indicates that P4α, P4β, and P4γ are epimers of one another. Because P4α was previously shown to be the C3 epimer (which corresponds to a d-arabino form (25)) and because MlghB is a predicted C3,C5 epimerase, P4β and P4γ represent the C5 epimer (corresponding to a l-ribo form) and the C3/C5 epimer (corresponding to a l-xylo form), but it is not possible at this stage to tell which of these two products corresponds to which epimer. The products were too unstable to withstand purification for identification by NMR spectroscopy. Further CE, MS, and NMR spectroscopy analyses of reactions performed with the downstream enzyme and that yield a stable product are presented below that allowed us to assign the CE peak labeled P4γ to the double epimer and the peak labeled P4β to the single C5 epimer as indicated on Fig. 2.
FIGURE 5.
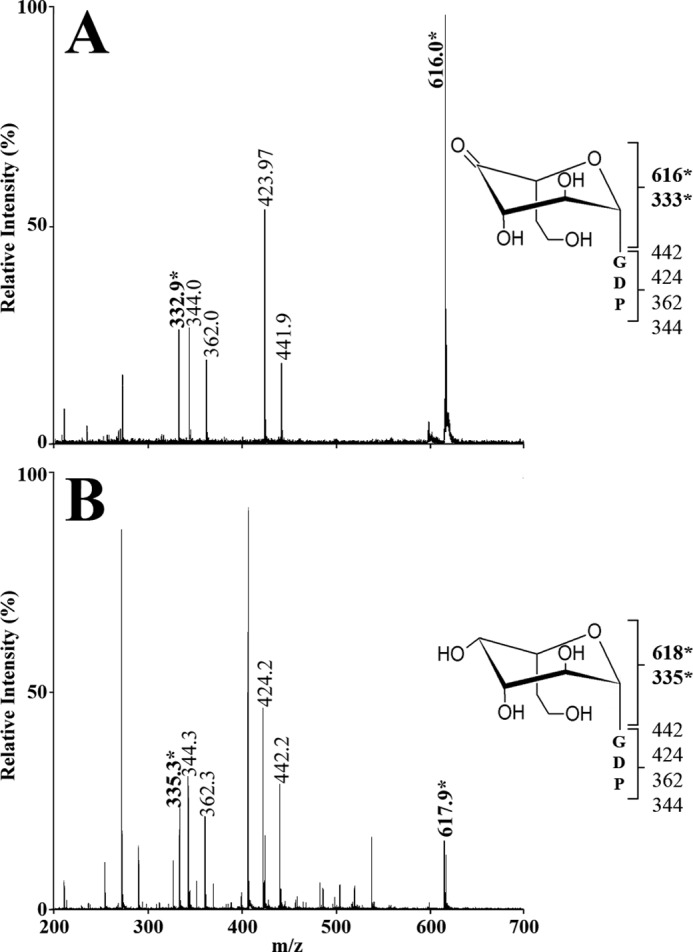
Mass spectrometry analysis of the MlghB and MlghC reaction products. The MS/MS spectra of the reaction products were obtained from full reactions comprising DdahA and GDP-d-glycero-d-manno-heptose plus MlghB (panel A) or MlghB and MlghC (panel B, product P5γ). For both panels, the structure of the expected sugar is depicted along with the fragment sizes that pertain to the GDP moiety only (lower bracket) and those that include the heptose ring (upper bracket). The latter are the full sugar nucleotide (largest m/z value) and a fragment lacking the guanosine. They are highlighted in bold and with an asterisk.
The data so far also indicate that there is no preferred order for the C3 and C5 epimerizations carried out by MlghB. Reaction sequentiality would be expected to yield only two products at equilibrium, one of the two single epimers (either C3 or C5 but not both) and the double epimer, instead of three products. Time course experiments performed with short reaction times (5 min) and very diluted enzyme consistently revealed the appearance of the three products concomitantly in roughly equal proportions (data not shown). This also argues against the sequentiality of appearance of the products.
These data demonstrate that, although MlghB is able to use the same P1 substrate as DdahB, it differs drastically from DdahB in that it readily exerts its predicted C5 epimerase activity in addition to its C3 epimerase activity. Moreover, both activities can occur on the P1 substrate with no preferential order, yielding a mixture of C3, C5, and C3/C5 epimers in equilibrium with P1.
Comparison of the Catalytic Efficiencies of MlghB and DdahB on GDP-6-deoxy-4-keto-d-lyxo-heptose
Comparison of reactions performed in parallel with equal amounts of DdahB and MlghB indicated that P1 conversion was more efficient with MlghB than with DdahB. Indeed, in reactions performed with MlghB, leftover P1 represented only about 32% of the total sugar nucleotides present in the reaction at equilibrium, whereas the new products represented ∼60% of all sugar nucleotides. The remaining components were the degradation product P2 (which corresponds to GDP) and a small impurity comprised in the heptose preparation. In contrast, as high as ∼75–80% P1 remained, and only 15–20% product were formed when reactions were performed with DdahB (Fig. 3, A and B, traces a and b, supplemental Table S1 for integration data). It was not possible to establish Km and Vmax values due to the lack of stability and limited availability of P1.
MlghC Generates a Single Product of the Three MlghB Epimers
No reactivity of MlghC was observed on the P1 product in the absence of MlghB (data not shown) even in the presence of NAD(P)H cofactor. However, when MlghC was incubated with P1 and NADPH along with MlghB, a single new reaction product appeared (called P5γ), with the disappearance in P1, P4α, P4β, and P4γ peaks (Fig. 3A, trace c, supplemental Table S1). This indicates that P5γ originated from at least one of the MlghB reaction products P4α, P4β, and/or P4γ. P5γ migrated well upstream of P1 by CE and was, therefore, a product distinct from P5α obtained through DdahB/DdahC reactions, which migrated downstream of P1 (Fig. 3B, trace d, supplemental Table S1).
MS analysis of the MlghB/MlghC reaction mixture revealed a peak at m/z 618, indicative of the reduction of the substrate(s) provided. The MS/MS pattern (Fig. 5B) was identical to that of reduced product P5α produced by DdahC (25), indicating that it is likely an epimer of P5α. Altogether, these data suggest that MlghC bound and catalyzed at least one of the MlghB products to generate a reduced product P5γ. The formation of a single product indicates that the reduction step is stereospecific.
Which of the Three MlghB Products Does MlghC Catalyze?
MlghC may catalyze any of the three products generated by MlghB. Its ability to use the single epimers (C3 or C5) would imply that it can complete the epimerization steps not carried out by MlghB due to its predicted epimerase activities. Although epimerization reactions result in equilibrium between at least two products, the stereospecific reduction activity of MlghC would irreversibly pull the equilibrium toward formation of a single product, the reduced C3/C5 epimer. Alternatively, MlghC may only serve as a reductase and reduce only one of the three MlghB products, but assignment of this product a priori from the data presented on Fig. 3 is not possible because the active MlghB present in the reaction supplies replacement P4α, -β, and -γ products to MlghC as long as enough P1 is available.
To help discriminate which of the P4α, P4β, and P4γ was a substrate for MlghC, hybrid reactions were performed between MlghC and DdahB that can only produce P4α. MlghC was unable to use P4α as a substrate under the conditions tested (Fig. 3B, trace c, supplemental Table S1).
To determine which of the two remaining products, P4β and P4γ, was the preferred substrate for MlghC, a mixture of P4α, -β, and -γ products was generated by incubation of GDP-manno-heptose with DdahA and MlghB, and the enzymes were removed by ultrafiltration so that the proportions of P4α, -β, and -γ products could not change any further. The addition of MlghC resulted in the expected production of P5γ, with exclusive consumption of P4γ (Fig. 4A, traces a and e, integration data in supplemental Table S2). Thus, of the three reaction products generated by MlghB, the P4γ product serves as the preferred substrate for MlghC. Therefore, P5γ stems directly from P4γ.
Identity of the P5γ Reaction Product
Because no other enzyme with predicted C3/C5 epimerase activity than MlghC and MlghB is encoded by the capsular cluster of C. jejuni strain NCTC 11168, we predicted that the reaction product P5γ obtained from catalysis of GDP-manno-heptose by DdahA, MlghC, and MlghB would be in the l-gluco configuration that corresponds to the capsular l-gluco-heptose. NMR spectroscopy was performed on pure P5γ to ascertain its identity. Several unique features of the NMR spectroscopy data clearly indicated that the sugar was in the gluco configuration. First, the chemical shift of H1 (4.97 ppm) was >0.3 ppm upfield of that found in GDP-6-deoxy-d-altro-heptose (5.32 ppm, Table 4). Furthermore, chemical shift assignment of the P5γ product showed that H2, H3, and H4 were all shifted upfield between 3.2 and 3.5 ppm. A comparison of these chemical shifts to pyranose derivative lacking the GDP moiety revealed these positions are particularly indicative of the gluco configuration (33). In addition, coupling constant analysis showed that H1 possessed a larger coupling to H2 (3J1,2 = 7.8 Hz), giving rise to an apparent triplet pattern indicative of a trans arrangement of H1 and H2 in the ring (Fig. 6). This was in contrast to the familiar doublet-of-doublets observed for GDP-6-deoxy-d-altro-heptose resulting from the syn arrangement and small couplings between H1 and H2 (3J1,2 = 2.3 Hz). Moreover, NMR spectroscopy data clearly indicated a β configuration, i.e. that both substituents at C5 and C1 were located on the same side of the sugar ring. The β configuration was identified from the series of large 1H-1H coupling constants around the sugar ring (7.8–9.4 Hz) that indicated H1, H3, and H5 were on the same side of the ring. In addition, one-dimensional selective NOE experiments showed an NOE between H1 and H3/H5, as expected for the β configuration.
TABLE 4.
1H and 13C NMR data for the P5γ and P5α products of the GDP-6-deoxy-l-gluco and d-altro-heptose synthesis pathways
| Compound |
1H and 13C chemical shifts and coupling constantsa |
|||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
| P5αb | ||||||||
| 1H, ppm | 5.32 | 3.93 | 3.94 | 3.70 | 4.18 | 1.99, 1.74 | 3.77, 3.71 | |
| 3JHH (3JHP), Hz | 2.3 (7.7) | 3.7 | 3.1 | 8.2 | ||||
| 13C, ppm | 98.5 | 72.6 | 73.0 | 71.0 | 70.5 | 35.5 | 60.7 | |
| P5γc | ||||||||
| 1H, ppm | 4.97 | 3.34 | 3.48 | 3.23 | 3.50 | 2.08, 1.62 | 3.76, 3.71 | |
| 3JHH (3JHP), Hz | 7.8 (7.8) | 9.2 | 9.4 | 9.4 | ||||
| 13C, ppm | 100.6 | 76.5 | 77.8 | 76.9 | 75.7 | 36.0 | 60.3 | |
a Chemical shifts referenced to internal 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt standard; 1H = 0.00 ppm. Only chemical shifts from the sugar portions of the compounds are reported.
b Data were previously reported in Ref. 25.
c Chemical shifts for GDP portion of P5γ in ppm; H1′ (5.92, 89.4), H2′ (4.79, 76.2), H3′ (4.52, 73.1), H4′ (4.34, 86.5), H5′ (4.20, 68.0).
FIGURE 6.

NMR spectroscopic identification of the MlghC product P5γ. Selected regions of the 600 MHz 1H NMR spectra for GDP-6-deoxy-d-altro-heptose P5α, obtained via the activity of DdahA, DdahB and DdahC (A) and for GDP-6-deoxy-l-gluco-heptose P5γ (B), obtained by activity of DdahA, MlghB and MlghC on GDP-d-glycero-d-manno-heptose. The spectrum (C) shows regions from the selective one-dimensional total correlation spectroscopy experiment obtained from excitation of H1 at 4.97 ppm. The spectrum shows that protons H1, H2, H3, and H4 all have large coupling constants to neighboring protons. Each spectrum shows the 1H assignments for the pyranose (1–7) and ribose (1′-5′) portions of the molecules. Peaks labeled (*) arise from buffer impurities.
NMR spectroscopy was not able to distinguish between l and d conformations because of their mirror image symmetry. The fact that the P5γ product has a β-gluco configuration nevertheless supports the hypothesis that it has an l conformation. Indeed, formation of the d-gluco form from the original d-manno substrate would only require a C2 epimerization and no C3 or C5 epimerization, but none of the enzymes used in this study has predicted C2 epimerization, and MlghB can perform both C3 and C5 epimerizations as per CE and MS data shown above. Therefore, the gluco configuration implies a l conformation. Moreover, the β configuration observed by NMR spectroscopy is consistent with the d to l switch that essentially amounts to swapping the C5 substituent across the sugar plane in the absence of any further modification at C1, as expected for C3/C5 epimerases. In contrast, a β d configuration would imply a C1 configuration switch that is not consistent with the nature of the enzymes used in these assays. Collectively, the CE, MS, and NMR spectroscopy data indicate that P5γ is GDP-6-deoxy-β-l-gluco-heptose.
MlghC Has No Epimerase Activity and Has Stereospecific C4 Reductase Activity
The CE data above showed that MlghB had both C3 and C5 epimerase activities, and MlghC used the P4γ MlghB product. Because P4α is the C3 epimer based on our prior work (25), P4γ is either the C5 epimer or the double C3/C5 epimer. NMR spectroscopy data on P5γ imply that both C3 and C5 epimerizations have been completed upon combined activities of MlghB and MlghC (Fig. 6). If P4γ was the single C5 epimer (and, therefore, P4β would be the double epimer), MlghC would need to perform the C3 epimerization on this substrate to generate the double epimer before reduction to yield l-gluco-heptose. Therefore, it would have the ability to reduce the double C3/C5 epimer that it just formed. This double epimer would be the same as the MlghB P4β product, but CE data showed that MlghC cannot use P4β as a reduction substrate (Fig. 4A, trace e). Altogether, the CE, NMR, and MS data argue (i) that MlghC used P4γ as a substrate for C4 reductase activity, (ii) that P4γ is the double C3/C5 epimer, (iii) that MlghC did not exert any of its predicted C3 or C5 epimerase activities in the context of this pathway, and (iv) that the C4 reductase activity of MlghC resulted in stereospecific inversion of the configuration at C4 (which is necessary to generate l-gluco). Finally, the data show that under conditions where MlghC did not use the P4α and P4β products, these products nevertheless disappeared totally from MlghC reaction mixtures when MlghB was still present (Fig. 3A, trace c). This indicates that MlghB is able to interconvert P4α and P4β continuously into P4γ upon consumption of P4γ by MlghC. The complete reaction scheme accounting for all these findings is presented in Fig. 2.
DdahC and MlghC Have Non-overlapping Substrate Specificity
The experiments above indicate that DdahC and MlghC have at least one difference in terms of substrate specificity as MlghC did not use P4α, which is the normal substrate for DdahC. Conversely, because DdahB only produces the C3 epimer P4α, it was not known if DdahC could also potentially use the single C5 epimer P4β or the double C3/C5 epimer P4γ that serves as substrate for MlghC. To determine this, DdahC and MlghC were incubated for short amounts of time with identical DdahA/MlghB reaction mixes in which MlghB was still active and could replenish the stocks of the various P4α, P4β, and P4γ as long as P1 was still available in excess. This led to entirely different outcomes: almost complete conversion of the original substrates into P5γ by MlghC (Fig. 3A, trace c) versus exclusive and limited formation of P5α by DdahC (Fig. 3A, trace d, supplemental Table S1). The amount of catalysis obtained with DdahC under these conditions was not affected by the presence of the P4β and P4γ products as it was comparable with the conversion observed in the presence of P4α only (Fig. 3B, trace d, supplemental Table S1). If DdahC was able to use P4β and/or P4γ in addition to P4α, the production of peak P5γ would also be expected. Therefore, these experiments demonstrate that DdahC and MlghC have non-overlapping substrate and product specificities. This was further demonstrated using an enzyme-free mixture of P4α, P4β, and P4γ (Fig. 4A, traces c and e, supplemental table S2).
The C4 Reductase WcaGNCTC Is Not Part of the Mainstream l-Gluco-heptose Synthesis Pathway and Has No Detectable Regulatory Function
The predicted C4 reductase WcaGNCTC did not have any catalytic activity on GDP-d-glycero-d-manno-heptose (data not shown). However, upon incubation of WcaGNCTC with GDP-manno-heptose and DdahA, a new product was formed (product P3, Fig. 4B, trace b, supplemental Table S2). Product P3 co-migrated with product P3 obtained with WcaG81176 (Fig. 4A, traces b and d). This suggested that both enzymes use the DdahA reaction product P1 and have the same C4 reductase catalytic activity, as expected from their 98% identity.
WcaGNCTC could not replace MlghC for the reduction of the MlghB reaction products into P5γ (Fig. 4A, traces d and e). Likewise, WcaG81176 could not replace DdahC for reduction of P4α into P5α (Fig. 4A, traces b and c). These data are consistent with our previous finding that WcaG81176 was not part of the mainstream d-altro-heptose synthesis pathway (25) and suggest that WcaGNCTC is also not part of the l-gluco-heptose synthesis pathway. However, although WcaG81176 could inhibit the activity of the reductase DdahC of the d-altro-heptose pathway (25), WcaGNCTC did not exert much inhibitory activity on the reductase MlghC of the l-gluco-heptose pathway even when present in 2-fold excess compared with MlghC (Fig. 4B, traces e and g, supplemental Table S2). Both MlghC and WcaGNCTC enzymes could perform their own catalysis in the presence of one another, resulting in formation of product P3 upon catalysis of P1 by WcaGNCTC even in the presence of MlghC (traces b and c) and of product P5γ upon catalysis of P4γ by MlghC (traces d–g). Therefore, contrary to WcaG81176, WcaGNCTC did not have any apparent regulatory function on l-gluco-heptose synthesis in our in vitro conditions.
DISCUSSION
Heptose-modifying Enzymes as New Glycobiology Tools
This work provides a detailed comparative analysis of C3/C5 epimerases and C3/C5 epimerases/C4 reductases involved in heptose modification. The enzymes studied are unique as they use heptose-based substrates, whereas all other C3/C5 epimerases and C3/C5 epimerases/C4 reductases studied to date use hexose-based substrates (2, 6, 7, 14, 34). Also the ring configuration and the nucleotide portion of the substrates used by these enzymes differ widely compared with the substrates of previously studied enzymes. This may prove useful for the production of novel sugar nucleotides whose limited availability hampers progress in glycobiology. In addition, this study reveals that sequence-based prediction of enzymatic activities and of associated substrates is a very risky enterprise in the absence of sound biochemical data. For example, GFS, GMER, and GME use the epimerization substrate GDP-6-deoxy-4-keto-d-lyxo-mannose that is closely related to the GDP-6-deoxy-4-keto-d-lyxo-heptose used by MlghB and DdahB in this study. However, these enzymes differ from MlghB and DdahB in that they all have additional C4 reductase activity, and GME even generates its own 4-keto from GDP-mannose via additional C4 oxidase activity (6, 7, 14). Sequence-wise, MlghB and DdahB are less related to these GFS, GMER, and GME enzymes, although they use related substrates, than to the RmlC and RfbC enzymes that only have C3/C5 epimerase activities like MlghB and DdahB but use a very different substrate: dTDP-6-deoxy-d-xylo-4-hexulose (=dTDP-4-keto-6-deoxy-glucose) (2, 34). Likewise, based on sequence features, both DdahC and MlghC are related to the GME, GFS, and GMER enzymes, which traditionally has C3/C5 epimerization and C4 reductase activities and use GDP-linked substrates (35). DdahC and MlghC have actually been annotated as a GDP-fucose synthase (gene name fcl) (26, 36). However, this study demonstrates their lack of epimerase activity. Knock-out mutagenesis and capsular structural studies have also led to global assignments of MlghC and WcaGNCTC as GDP-heptose epimerases (16, 27). In light of our biochemical data that showed no C3/C5 epimerization activities and utilization of heptose-based substrates by all these enzymes, these annotations may need revisiting to heptulose C4 reductase for DdahC, MlghC, and WcaGNCTC with the understanding that they use substrates presenting different configurations.
Benefits of the Surrogate GDP-6-deoxy-4-keto-d-lyxo-heptose Substrate for the Direct Comparison of Homologous Enzymes from the d-altro-Heptose and l-Gluco-heptose Synthesis Pathways
As observed previously for DdahB, DdahC, and WcaG81176 (24, 25), the MlghB, MlghC, and WcaGNCTC enzymes did not have any catalytic activity on GDP-d-glycero-d-manno-heptose (data not shown). This was consistent with the fact that C3/C5 epimerases and C3/C5 epimerases/C4 reductases use a 4-keto derivative as a substrate for epimerization (6, 34, 37). Apart from the GDP-mannose epimerase GME from A. thaliana which generates its own 4-keto via C4 oxidase activity (14), this 4-keto intermediate is usually generated by prior activity of a C4/C6 dehydratase. Examples are RfbB in S. enterica, RmlB in E. coli, HP0044 in H. pylori, or DdahA in C. jejuni strain 81-176 (2, 9, 24, 34, 38). No such dehydratase is encoded in the capsular cluster of C. jejuni strain NCTC 11168 (26), which is consistent with the fact that the final product incorporated in the capsule is 6-O-Me-l-gluco-heptose, which is not C6-dehydrated (16). But no putative C4 oxidase could be identified in the capsular cluster (Tables 1 and 2). Therefore, the natural substrate for MlghB and MlghC is unknown and not available for biochemical studies.
Although the quest for this substrate continues, the fact that the enzymes from the l-gluco-heptose pathway could use the C6 dehydrated product of DdahA as a substrate, allowed their biochemical characterization. This allowed defining their role in l-gluco-heptose synthesis based on NMR spectroscopy analyses of the P5γ product that was obtained via combined activities of DdahA, MlghB, and MlghC. The NMR spectroscopy and CE data collectively indicated that P5γ is in a β l-gluco configuration, whereas the heptose found within the capsule is in the α l-gluco configuration. The transferase responsible for release of the GDP moiety and attachment of the modified heptose to the capsular backbone is likely responsible for the inversion of the C1 configuration (resulting in the β to α switch). Inverting transferases that make glycosidic bonds with a stereochemistry opposite that of the sugar donor are widespread in all biological kingdoms, including among enzymes involved in bacterial polysaccharide synthesis (39–41). To date, the heptosyltransferase is predicted to be Cj1431 (26, 36), but its activity has not been determined for lack of substrate. The possibility to synthesize the substrate enzymatically based on the work presented herein will now allow testing the activity of this enzyme.
Moreover, the use of the surrogate 6-deoxy substrate also allowed the direct comparison of MlghB and MlghC with enzymes of the 6-deoxy-d-altro-heptose pathway. This revealed several significant differences despite the overall high degree of sequence conservation of the sets of enzymes between both pathways.
The Highly Similar MlghB and DdahB Have Different Activities
Both MlghB and DdahB were predicted to be C3/C5 epimerases, but only MlghB performed both epimerizations, whereas DdahB only exerted its C3 epimerization activity along its cognate heptose modification pathway. Prolonged incubations showed that the C5 epimerase functionality of DdahB was nevertheless preserved in this enzyme (data not shown) but yielded minimal catalytic activity compared with the C3 epimerization activity. On-going structural modeling and site-directed mutagenesis may allow unraveling the reasons for such selectivity of activity. It is likely that steric hindrance in the binding cavity of DdahB (but not of MlghB) may not favor accommodation of the C5 epimer, whose structure is drastically different from those of the C3 epimer and of the original substrate.
Order of C3 and C5 Epimerizations and Release of Multiple Products by MlghB
MlghB proceeds with the C3 and C5 epimerizations without any sequentiality, which is rather unusual compared with other C3/C5 epimerases. For example, RmlC (34) and GME (14) start with C5 epimerization, whereas GFS starts with C3 epimerization (7). As a result, although most other C3/C5 epimerases only lead to the formation of two products, a mixture of three products was obtained with MlghB. Whether the three different products would be released by MlghB in vivo is up for debate. As represented in Fig. 1, the biological l-gluco-heptose synthesis pathway is anticipated to involve mostly the double epimer, and the observation of the two single epimers in our in vitro data could be due to drastically different enzyme/substrate stoichiometries in vitro versus in vivo. It is also possible that the enzymes interact in vivo, which may prevent the release of reaction intermediates. Finally, the release of the three different products may also be due to the fact that the experiments were performed with a surrogate substrate.
Similar Enzymes Lead to Different Pathway Outcomes
Overall, our data indicated that MlghB and DdahB are key to lead the heptose modification pathway toward the desired product in each strain by performing strain-specific epimerization reactions despite their very high levels of identity (81%) and similarity (98%). This was surprising as, for example, their E. coli RmlC and S. enterica RfbC homologues are only 51.4% identical (75% similar) to one another but have the same function on the same substrate (2, 34). The differences of activity observed between MlghB and DdahB would nevertheless not be sufficient to lock each pathway in the desired direction if the downstream enzymes MlghC and DdahC also expressed their predicted C3/C5 epimerization capacity and had relaxed substrate specificity. Our data show that DdahC and MlghC, both, only have C4 reductase activity along their cognate pathway but have distinct substrate specificities: GDP-6-deoxy-4-keto-d-arabino- heptose (P4α) for DdahC versus l-xylo-heptose (double epimer, P4γ) for MlghC. Such strong substrate specificity is in contrast to the reported ability of GME to reduce both the single C5 epimer and the double C3/C5 epimer (14) but is similar to the strong specificity for the double epimer exhibited by GFS (7). The 57% identity and 90% similarity between MlghC and DdahC are low enough to allow different substrate specificity so that each of these enzymes only accommodates the epimer that leads to the desired product by simple reduction. Specifically, the lack of C5 epimerization activity of DdahB combined with lack of epimerization activity of DdahC locks the pathway toward formation of the d-altro heptose derivative. Likewise, despite the formation of three products by MlghB, the pathway is refocused into the formation of a single l-gluco product thanks to the lack of epimerization activity of MlghC combined with its specificity for the C3/C5 epimer. By consuming a single product, MlghC pulls the MlghB epimerization equilibrium between the three epimers toward replenishment of its favored C3/C5 double epimer substrate, thereby resulting in complete conversion of the original substrate into the final l-gluco-heptose derivative.
WcaGNCTC Does Not Affect GDP-6-deoxy-l-gluco-heptose Synthesis
It is puzzling that both C. jejuni strains 81-176 and NCTC 11168 strains would encode almost identical WcaG81176 and WcaGNCTC enzymes in their heptose modification clusters, whereas they produce very different modified heptoses. Moreover, as observed before for the production of 6-deoxy-d-altro-heptose, the production of 6-deoxy-l-gluco-heptose could be obtained in the absence of WcaGNCTC. Based on the high sequence identity levels of WcaG81176 and WcaGNCTC, it came as no surprise that WcaGNCTC could also use the DdahA reaction product P1 as a substrate and that its reaction product was identical to the P3 reaction product obtained with WcaG81176, but the physiological significance of this result is elusive as no obvious C4/C6 heptose dehydratase is available in strain NCTC 11168. Based on our prior findings with WcaG81176, it also came as no surprise that WcaGNCTC could not use any of the MlghB reaction products. However, in contrast to the complete inhibition of DdahC observed in the presence of WcaG81176 (25), the presence of WcaGNCTC had no inhibitory effect on MlghC and did not prevent the production of l-gluco-heptose under the experimental conditions used in these studies. Only a slight decrease in product yield was observed due to competition for the P1 intermediate. Whether such competition would affect the production of l-gluco-heptose in vivo is unknown and whether similar effects would be observed in the presence of the original 6-O-methyl substrate instead of the 6-deoxy surrogate substrate is unknown. The biological role of WcaGNCTC, therefore, remains to be elucidated. Because WcaGNCTC and WcaG81176 are quasi identical, the differences observed in terms of inhibition of downstream reductases MlghC and DdahC likely reflect differences between the reductases, which allow inhibitory protein/protein interactions between WcaG81176 and DdahC but not between WcaGNCTC and MlghC.
Conclusion
This detailed comparative analysis of heptose-modifying enzymes from C. jejuni is contributing to a better understanding of the intricacies of heptose modification and is important to fully harness the potential of these enzymes for biotechnological and biomedical applications. To decipher the activities of these enzymes and their substrate specificities, numerous practical hurdles had to be overcome in light of their usage and production of numerous closely related and unstable sugars. This work establishes a solid foundation for extensive site-directed mutagenesis and structural studies to fully understand the mechanism of action of these enzymes and guide the rationale design of therapeutic inhibitors.
In terms of biological significance, the global conservation of the heptose modification pathways and the presence of strain-specific features both suggest that the modified heptoses are likely important functional components of the capsule. Although methylation and the addition of phosphoramidate moieties on the heptose seem to be dispensable based on the predicted phase variability of the genes involved, the changes of configurations of the sugar ring brought about by the enzymes described in this manuscript seem to be essential for incorporation of the heptose into the capsule (16, 27). Having identified the key enzymes involved in these configuration changes and the enzymes involved in regulating the process will help further delineate the biological role of modified heptoses in vivo via knock-out mutagenesis studies.
Supplementary Material
Acknowledgments
We thank M. Wong, J. Griffith, and A. Merkx-Jacques for cloning the cj1427, cj1428, and cj1430 genes in the pET vector at the onset of this project. We thank Dr. J. S. Lam (University of Guelph) for advice on nomenclature.
This work was supported through operating grants from the Natural Sciences and Engineering Research Council (NSERC) of Canada (RGPIN 240762-2010]; to C. C.) and the Canadian Institutes of Health Research (MOP 93520) and the Canada Research Chairs program (to G. S.).

This article contains supplemental Tables S1 and S2 and Fig. S1.
- CE
- capillary electrophoresis.
REFERENCES
- 1. King J. D., Harmer N. J., Preston A., Palmer C. M., Rejzek M., Field R. A., Blundell T. L., Maskell D. J. (2007) Predicting protein function from structure. The roles of short-chain dehydrogenase/reductase enzymes in Bordetella O-antigen biosynthesis. J. Mol. Biol. 374, 749–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stern R. J., Lee T. Y., Lee T. J., Yan W., Scherman M. S., Vissa V. D., Kim S. K., Wanner B. L., McNeil M. R. (1999) Conversion of dTDP-4-keto-6-deoxyglucose to free dTDP-4-keto-rhamnose by the rmlC gene products of Escherichia coli and Mycobacterium tuberculosis. Microbiology 145, 663–671 [DOI] [PubMed] [Google Scholar]
- 3. Xiang S. H., Haase A. M., Reeves P. R. (1993) Variation of the rfb gene clusters in Salmonella enterica. J. Bacteriol. 175, 4877–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yao Z., Valvano M. A. (1994) Genetic analysis of the O-specific lipopolysaccharide biosynthesis region (rfb) of Escherichia coli K-12 W3110. Identification of genes that confer group 6 specificity to Shigella flexneri serotypes Y and 4a. J. Bacteriol. 176, 4133–4143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rahim R., Burrows L. L., Monteiro M. A., Perry M. B., Lam J. S. (2000) Involvement of the rml locus in core oligosaccharide and O polysaccharide assembly in Pseudomonas aeruginosa. Microbiology 146, 2803–2814 [DOI] [PubMed] [Google Scholar]
- 6. Rizzi M., Tonetti M., Vigevani P., Sturla L., Bisso A., Flora A. D., Bordo D., Bolognesi M. (1998) GDP-4-keto-6-deoxy-d-mannose epimerase/reductase from Escherichia coli, a key enzyme in the biosynthesis of GDP-L-fucose, displays the structural characteristics of the RED protein homology superfamily. Structure 6, 1453–1465 [DOI] [PubMed] [Google Scholar]
- 7. Lau S. T., Tanner M. E. (2008) Mechanism and active site residues of GDP-fucose synthase. J. Am. Chem. Soc. 130, 17593–17602 [DOI] [PubMed] [Google Scholar]
- 8. Menon S., Stahl M., Kumar R., Xu G. Y., Sullivan F. (1999) Stereochemical course and steady state mechanism of the reaction catalyzed by the GDP-fucose synthetase from. Escherichia coli. J. Biol. Chem. 274, 26743–26750 [DOI] [PubMed] [Google Scholar]
- 9. Järvinen N., Mäki M., Räbinä J., Roos C., Mattila P., Renkonen R. (2001) Cloning and expression of Helicobacter pylori GDP-l-fucose synthesizing enzymes (GMD and GMER) in Saccharomyces cerevisiae. Eur. J. Biochem. 268, 6458–6464 [DOI] [PubMed] [Google Scholar]
- 10. Moran A. P., Prendergast M. M. (2001) Molecular mimicry in Campylobacter jejuni and Helicobacter pylori lipopolysaccharides. Contribution of gastrointestinal infections to autoimmunity. J. Autoimmun. 16, 241–256 [DOI] [PubMed] [Google Scholar]
- 11. Rasko D. A., Keelan M., Wilson T. J., Taylor D. E. (2001) Lewis antigen expression by Helicobacter pylori. J. Infect. Dis. 184, 315–321 [DOI] [PubMed] [Google Scholar]
- 12. Brückner K., Perez L., Clausen H., Cohen S. (2000) Glycosyltransferase activity of Fringe modulates Notch-Delta interactions. Nature 406, 411–415 [DOI] [PubMed] [Google Scholar]
- 13. Ayukawa T., Matsumoto K., Ishikawa H. O., Ishio A., Yamakawa T., Aoyama N., Suzuki T., Matsuno K. (2012) Rescue of Notch signaling in cells incapable of GDP-l-fucose synthesis by gap junction transfer of GDP-l-fucose in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 109, 15318–15323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Major L. L., Wolucka B. A., Naismith J. H. (2005) Structure and function of GDP-mannose-3′,5′-epimerase, an enzyme that performs three chemical reactions at the same active site. J. Am. Chem. Soc. 127, 18309–18320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ho N., Kondakova A. N., Knirel Y. A., Creuzenet C. (2008) The biosynthesis and biological role of 6-deoxyheptose in the lipopolysaccharide O-antigen of Yersinia pseudotuberculosis. Mol. Microbiol. 68, 424–447 [DOI] [PubMed] [Google Scholar]
- 16. St Michael F., Szymanski C. M., Li J., Chan K. H., Khieu N. H., Larocque S., Wakarchuk W. W., Brisson J. R., Monteiro M. A. (2002) The structures of the lipooligosaccharide and capsule polysaccharide of Campylobacter jejuni genome sequenced strain NCTC 11168. Eur. J. Biochem. 269, 5119–5136 [DOI] [PubMed] [Google Scholar]
- 17. Aspinall G. O., McDonald A. G., Pang H. (1992) Structures of the O chains from lipopolysaccharides of Campylobacter jejuni serotypes O:23 and O:36. Carbohydr. Res. 231, 13–30 [DOI] [PubMed] [Google Scholar]
- 18. Kirkpatrick B. D., Tribble D. R. (2011) Update on human Campylobacter jejuni infections. Curr. Opin. Gastroenterol. 27, 1–7 [DOI] [PubMed] [Google Scholar]
- 19. Deckert A., Valdivieso-Garcia A., Reid-Smith R., Tamblyn S., Seliske P., Irwin R., Dewey C., Boerlin P., McEwen S. A. (2010) Prevalence and antimicrobial resistance in Campylobacter spp. isolated from retail chicken in two health units in Ontario. J. Food Prot. 73, 1317–1324 [DOI] [PubMed] [Google Scholar]
- 20. Bohaychuk V. M., Checkley S. L., Gensler G. E., Barrios P. R. (2009) Microbiological baseline study of poultry slaughtered in provincially inspected abattoirs in Alberta, Canada. Can. Vet. J. 50, 173–178 [PMC free article] [PubMed] [Google Scholar]
- 21. Angulo F. J., Baker N. L., Olsen S. J., Anderson A., Barrett T. J. (2004) Antimicrobial use in agriculture: controlling the transfer of antimicrobial resistance to humans. Semin. Pediatr. Infect. Dis. 15, 78–85 [DOI] [PubMed] [Google Scholar]
- 22. Maue A. C., Mohawk K. L., Giles D. K., Poly F., Ewing C. P., Jiao Y., Lee G., Ma Z., Monteiro M. A., Hill C. L., Ferderber J. S., Porter C. K., Trent M. S., Guerry P. (2013) The polysaccharide capsule of Campylobacter jejuni modulates the host immune response. Infect. Immun. 81, 665–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guerry P., Poly F., Riddle M., Maue A. C., Chen Y. H., Monteiro M. A. (2012) Campylobacter polysaccharide capsules. Virulence and vaccines. Front Cell Infect. Microbiol. 2, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCallum M., Shaw G. S., Creuzenet C. (2011) Characterization of the dehydratase WcbK and the reductase WcaG involved in GDP-6-deoxy-manno-heptose biosynthesis in Campylobacter jejuni. Biochem. J. 439, 235–248 [DOI] [PubMed] [Google Scholar]
- 25. McCallum M., Shaw S. D., Shaw G. S., Creuzenet C. (2012) Complete 6-deoxy-d-altro-heptose biosynthesis pathway from Campylobacter jejuni. More complex than anticipated. J. Biol. Chem. 287, 29776–29788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parkhill J., Wren B. W., Mungall K., Ketley J. M., Churcher C., Basham D., Chillingworth T., Davies R. M., Feltwell T., Holroyd S., Jagels K., Karlyshev A. V., Moule S., Pallen M. J., Penn C. W., Quail M. A., Rajandream M. A., Rutherford K. M., van Vliet A. H., Whitehead S., Barrell B. G. (2000) The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403, 665–668 [DOI] [PubMed] [Google Scholar]
- 27. Sternberg M. J., Tamaddoni-Nezhad A., Lesk V. I., Kay E., Hitchen P. G., Cootes A., van Alphen L. B., Lamoureux M. P., Jarrell H. C., Rawlings C. J., Soo E. C., Szymanski C. M., Dell A., Wren B. W., Muggleton S. H. (2013) Gene function hypotheses for the Campylobacter jejuni glycome generated by a logic-based approach. J. Mol. Biol. 425, 186–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Newton D. T., Mangroo D. (1999) Mapping the active site of the Haemophilus influenzae methionyl-tRNA formyltransferase. Residues important for catalysis and tRNA binding. Biochem. J. 339, 63–69 [PMC free article] [PubMed] [Google Scholar]
- 29. Butty F. D., Aucoin M., Morrison L., Ho N., Shaw G., Creuzenet C. (2009) Elucidating the formation of 6-deoxyheptose. Biochemical characterization of the GDP-d-glycero-d-manno-heptose C6 dehydratase, DmhA and its associated C4 reductase, DmhB. Biochemistry 48, 7764–7775 [DOI] [PubMed] [Google Scholar]
- 30. Bax A., Davis D. G. (1985) MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 65, 355–360 [Google Scholar]
- 31. Kay L. E., Keifer P., Saarinen T. (1992) Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 114, 10663–10665 [Google Scholar]
- 32. John B. K., Plant D., Hurd R. E. (1992) Improved proton-detected heteronuclear correlation using gradient-enhanced z and zz filters. J. Magn. Reson. 101, 113–117 [Google Scholar]
- 33. Shashkov A. S., Pakulski Z., Grzeszczyk B., Zamojski A. (2001) Distribution of pyranose and furanose forms of 6-deoxyheptoses in water solution. Carbohydr. Res. 330, 289–294 [DOI] [PubMed] [Google Scholar]
- 34. Dong C., Major L. L., Srikannathasan V., Errey J. C., Giraud M. F., Lam J. S., Graninger M., Messner P., McNeil M. R., Field R. A., Whitfield C., Naismith J. H. (2007) RmlC, a C3′ and C5′ carbohydrate epimerase, appears to operate via an intermediate with an unusual twist boat conformation. J. Mol. Biol. 365, 146–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reeves P. R., Hobbs M., Valvano M. A., Skurnik M., Whitfield C., Coplin D., Kido N., Klena J., Maskell D., Raetz C. R., Rick P. D. (1996) Bacterial polysaccharide synthesis and gene nomenclature. Trends Microbiol. 4, 495–503 [DOI] [PubMed] [Google Scholar]
- 36. Karlyshev A. V., Champion O. L., Churcher C., Brisson J. R., Jarrell H. C., Gilbert M., Brochu D., St Michael F., Li J., Wakarchuk W. W., Goodhead I., Sanders M., Stevens K., White B., Parkhill J., Wren B. W., Szymanski C. M. (2005) Analysis of Campylobacter jejuni capsular loci reveals multiple mechanisms for the generation of structural diversity and the ability to form complex heptoses. Mol. Microbiol. 55, 90–103 [DOI] [PubMed] [Google Scholar]
- 37. Rosano C., Bisso A., Izzo G., Tonetti M., Sturla L., De Flora A., Bolognesi M. (2000) Probing the catalytic mechanism of GDP-4-keto-6-deoxy-d-mannose epimerase/reductase by kinetic and crystallographic characterization of site-specific mutants. J. Mol. Biol. 303, 77–91 [DOI] [PubMed] [Google Scholar]
- 38. Wu B., Zhang Y., Wang P. G. (2001) Identification and characterization of GDP-d-mannose 4,6-dehydratase and GDP-l-fucose synthetase in a GDP-l-fucose biosynthetic gene cluster from Helicobacter pylori. Biochem. Biophys. Res. Commun. 285, 364–371 [DOI] [PubMed] [Google Scholar]
- 39. Logan S. M., Altman E., Mykytczuk O., Brisson J. R., Chandan V., Schur M. J., St Michael F., Masson A., Leclerc S., Hiratsuka K., Smirnova N., Li J., Wu Y., Wakarchuk W. W. (2005) Novel biosynthetic functions of lipopolysaccharide rfaJ homologs from Helicobacter pylori. Glycobiology 15, 721–733 [DOI] [PubMed] [Google Scholar]
- 40. Llull D., Garcia E., Lopez R. (2001) Tts, a processive β-glucosyltransferase of Streptococcus pneumoniae, directs the synthesis of the branched type 37 capsular polysaccharide in Pneumococcus and other gram-positive species. J. Biol. Chem. 276, 21053–21061 [DOI] [PubMed] [Google Scholar]
- 41. Coutinho P. M., Deleury E., Davies G. J., Henrissat B. (2003) An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 328, 307–317 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.