

Background: The TIP60 acetyltransferase mediates p53 acetylation at K120 and is required for p53-dependent growth arrest and apoptosis.
Results: UHRF1 suppresses TIP60 interaction with p53 and TIP60-mediated p53 acetylation. UHRF1 depletion significantly enhances p53 and Tip60-dependent transactivation of growth arrest and apoptotic targets.
Conclusion: UHRF1 negatively regulates p53 transactivity upstream of the TIP60-p53 interplay.
Significance: Inhibition of the TIP60-p53 interplay contributes to the oncogenic functions of UHRF1.
Keywords: Apoptosis, Oncogene, p53, Tumor Suppressor Gene, Ubiquitination, TIP60, UHRF1, Acetylation, Growth Arrest
Abstract
Numerous studies indicate the importance of acetylation in p53-mediated stress responses upon DNA damage. We and others previously showed that TIP60 (Tat-interacting protein of 60 kDa)-mediated acetylation of p53 at K120 is crucial for p53-dependent apoptotic responses. Nevertheless, it remains unclear how TIP60-mediated effects on p53 are dynamically regulated in vivo. Here, we report that UHRF1 (ubiquitin-like with PHD and RING finger domains 1) interacts with TIP60 both in vitro and in vivo and induces degradation-independent ubiquitination of TIP60. Moreover, UHRF1 expression markedly suppresses the ability of TIP60 to acetylate p53. In contrast, RNAi-mediated knockdown of UHRF1 increases the endogenous levels of p53 acetylation at K120 and p53-mediated apoptosis is significantly enhanced in UHRF1-depleted cells. To elucidate the mechanisms of this regulation, we found that the interaction between TIP60 and p53 is severely inhibited in the presence of UHRF1, suggesting that UHRF1 modulates TIP60-mediated functions in both K120 acetylation-dependent and -independent manners. Consistent with this notion, UHRF1 knockdown promotes activation of p21 and PUMA but not MDM2. These findings demonstrate that UHRF1 is a critical negative regulator of TIP60 and suggest that UHRF1-mediated effects on p53 may contribute, at least in part, to its role in tumorigenesis.
Introduction
The p53 tumor suppressor maintains cellular and genomic stability by monitoring and responding to a plethora of stress signals through transcription regulation of target genes that are involved in cell growth arrest, apoptosis, DNA repair, senescence, etc. (1). The key steps in p53 transactivation are derepression from its key inhibitor MDM2, acquirement of a combination of protein modifications, and promoter-specific recruitment of coactivators/coregulators (2). p53 can be modified post-translationally on a multitude of residues and accumulating evidence supports the indispensability of acetylation in p53 activation during stress response (3). p53-dependent growth arrest and apoptosis are entirely abolished upon loss of acetylation at all major p53 acetylation sites (4). Lys(K) 120, the acetylation of which is mediated by the TIP60 (also known as KAT5) acetyltransferase, is of particular interest because the tumor-derived non-acetylable arginine (R) mutant selectively diminishes induction of apoptosis (5, 6).
TIP60 is a ubiquitously expressed and evolutionarily conserved member of the MYST family lysine acetyltransferases with specificity for histones and nonhistone proteins including the androgen receptor and c-MYC transcription factors (7–10). TIP60 plays important roles in the DNA damage response pathway: activation of the ataxia telangiectasia-mutated (ATM)3 DNA damage sensor is dependent on the acetyltransferase activity of TIP60, and TIP60 is recruited to sites of DNA lesions in Drosophila to facilitate DNA repair (11, 12). A key study of large-scale inhibitory RNA (RNAi) screening identified TIP60 to be essential for p53-dependent cell growth arrest, thereby suggesting TIP60 as a component of the p53 pathway (13). This was soon confirmed by studies demonstrating TIP60 interaction with p53 and TIP60-mediated p53 acetylation at K120, which specifically favors expression of p53-dependent apoptotic targets (5, 6). Although TIP60 is a potent positive regulator of p53 activation, the dynamically regulated levels of TIP60-mediated K120 acetylation and the unstable interaction between TIP60 and p53 suggests additional players in regulating the TIP60-p53 interplay.
UHRF1 (also known as ICBP90 in humans and Np95 in mice) is a key component and critical coordinator of the epigenetic regulating complex that comprises the maintenance DNA methyltransferase DNMT1, the de novo DNMT3a/3b, HDAC1 (histone deacetylase 1), USP7(ubiquitin specific protease 7, also known as HAUSP), PCNA (proliferating cell nuclear antigen), and the more recently identified members G9a/EHMT2 (euchromatic histone-lysine N methyltransferase 2) and TIP60 (14–18). The implication of UHRF1 involvement in tumorigenesis stems from studies reporting high UHRF1 expression level in proliferating tissues and low expression in quiescent cells and tissues (19, 20). Indeed, UHRF1 was found up-regulated in numerous cancers, suggesting that it may serve as an attractive therapeutic target for cancer treatment (21–28). UHRF1 possesses multiple protein modalities (Fig. 1A) that facilitate coordination of other epigenetic regulators to collectively regulate propagation of genomic DNA methylation patterns, heterochromatin formation at certain tumor suppressor genes, and euchromatic gene expression (22, 29–34). The carboxyl (C-) terminal RING domain of UHRF1 confers intrinsic E3 ligase activity toward histones and non-histone proteins (25, 34–36). A recent study reported that UHRF1 ubiquitinates and targets DNMT1 for proteasomal degradation through coordinating other DNMT1-associated proteins including TIP60 (17).
FIGURE 1.

UHRF1 is a bona fide TIP60-interacting protein. A, schematic representation of UHRF1 domain structure. B, UHRF1 co-immunoprecipitates with TIP60 in an overexpression system. Whole cell extracts or immunoprecipitates with M2/FLAG antibody from H1299 cells transiently transfected with plasmid DNA expressing HA-UHRF1 or/and FLAG Tip60 were subjected to Western blot with α-FLAG and α-HA antibodies. C, TIP60 interacts with UHRF1 endogenously in U2OS cells. U2OS-derived nuclear extracts or immunoprecipitates with a control IgG or α-UHRF1 antibody were subjected to Western blot with α- TIP60 and α-UHRF1 antibodies. D, UHRF1 interacts with TIP60 directly in vitro. In vitro translated [35S]methionine-labeled 3×FLAG-UHRF1 protein was incubated with purified GST-TIP60 or GST alone. Complexes immobilized with GST resins and recovered using reduced glutathione were subjected to SDS-PAGE and analyzed by autoradiography. The levels of purified GST-TIP60 and GST are shown in the bottom panel stained by Coomassie Blue.
Although coexistence of TIP60 and UHRF1 in the same macro-molecular protein complex is indicated, direct interaction has not been reported (16, 17). Here we have identified UHRF1 as a direct interacting partner of TIP60 and a unique negative regulator of the TIP60-p53 interplay. UHRF1 expression induces TIP60 ubiquitination, which does not trigger proteolysis but partially contributes to marked suppression of p53 K120 acetylation mediated by TIP60. Ablation of UHRF1 promotes K120 acetylation and p53-mediated apoptosis. Through its SRA and RING domains UHRF1 binds to TIP60 and severely inhibits TIP60-p53 interaction, thereby modulating transcription of K120 acetylation-dependent and -independent p53 targets PUMA and p21. These data reveal that UHRF1 negatively regulates TIP60 and modulates TIP60 function in the p53 response pathway both dependent and independent of K120 acetylation.
EXPERIMENTAL PROCEDURES
Cell Culture
H1299 and U2OS cells were maintained in DMEM (Cellgro) and HCT116 cells in McCoy's 5A medium. All media were supplemented with 10% fetal bovine serum (Invitrogen). Transfections with plasmid DNA and siRNA were performed using Lipofectamine2000 (Invitrogen) according to the manufacturer's protocol.
Plasmid Construction
All UHRF1 expression vectors were constructed by PCR amplification from a pET28a-UHRF1 expression vector that was generously provided by Dr. Zhenghe Wang, and subcloned into either the pCMV-Myc-N or pCMV-HA expression vectors (Clontech). The 3×FLAG-UHRF1 construct used for in vitro translation is also a gift from Dr. Zhenghe Wang. Deletion mutants of UHRF1 were further constructed by PCR amplification from the full-length expression plasmids and subcloned into respective vectors. Point mutants of UHRF1 were introduced using the Quikchange site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol.
Antibodies
Antibodies used in this study include UHRF1 (H-65 and H-8), p53 (DO-1), p21 (SX118), PUMA (H-136), TIGAR (E-2), and Myc (9E10) from Santa Cruz Biotechnology, β-actin (AC-15) and Flag (M2) from Sigma, MDM2 (Ab-5) from EMD Biosciences, HA (3F10) from Roche Applied Science, GFP (JL-8) from Clontech, and α-Acp53K120 antibody (Tang et al., Ref. 5). α-TIP60 (CLHF) antibody was a generous gift from Dr. Bruno Amati.
Western Blot and Immunoprecipitation
For Western blot analysis, cells were lysed in cold RIPA buffer (20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Triton X-100, 1% DOC, 1 mm EDTA, 0.05% SDS, and freshly supplemented protease inhibitor mixture). Co-immunoprecipitation assays were performed as described previously (Dai et al., Ref. 48). In brief, cells were lysed in cold BC100 buffer (20 mm Tris-HCl, pH 7.9, 100 mm NaCl, 10% glycerol, 0.2 mm EDTA, 0.2% Triton X-100, and freshly supplemented protease inhibitor) with sonication. 5% of cell extracts were saved for input, and the rest was incubated with the antibody or control IgG at 4 °C overnight. A/G PLUS-agarose beads (Santa Cruz Biotechnology) were then added for 1.5 h of incubation at 4 °C. After five washes with the lysis buffer, the bound proteins were eluted by boiling with SDS sample buffer. For immunoprecipitation of ectopically expressed FLAG-tagged proteins, cells were lysed in Flag lysis buffer (50 mm Tris-HCl, pH 7.9, 137 mm NaCl, 10 mm NaF, 1 mm EDTA, 1% Triton X-100, 0.2% sarkosyl, 10% glycerol, and freshly supplemented protease inhibitor). When detection of acetylated proteins was desired, lysis buffer was freshly supplemented with 2 μm trichostatin A and 10 mm nicotinamide. Cell extracts were incubated with the monoclonal M2/Flag-agarose beads (Sigma) at 4 °C overnight. After five washes with the lysis buffer, the bound proteins were eluted with Flag-peptide (Sigma) in BC100 for 2 h at 4 °C.
siRNA-mediated Ablation of UHRF1, TIP60, and p53
Ablation of UHRF1 was performed by transfection of HCT116 cells or U2OS cells with siRNA duplex oligonucleotides (Silencer Select S26553, S26554, S26555) synthesized by Ambion. Ablation of TIP60 was performed by transfection with a siRNA duplex (5′-ACGGAAGGUGGAGGUGGUUdTdT-3′/5′-AACCACCUCCACCUUCCGUdTdT-3′); ablation of p53 was performed by transfection with siRNA duplex oligoset (On-Target-Plus Smartpool L00332900, Dharmacon). Control siRNA (On-Target-Plus siControl nontargeting pool D00181010, Dharmacon) was also used for transfection. RNAi transfections were performed 2 times in HCT116 cells and 3 times in U2OS cells with Lipofectamine2000 according to the manufacturer's protocol (Invitrogen).
Apoptosis
Apoptosis was measured by FACS analysis of DNA content. After treatment with DNA damage reagents for the indicated time, cells were collected by mild trypsinization, washed with PBS, fixed in cold 80% methanol, and stored at −20 °C until stained. After fixation, cells were washed twice with cold PBS and incubated 20 min at room temperature with 50 μg/ml of RNaseA in PBS and stained 5 min with 25 μg/ml of propidium iodide. Flow cytometry was performed with a FACScalibur flow cytometer (BD Biosciences).
Cell-based Ubiquitination Assay
Cell-based ubiquitination assays were performed essentially as described (58) with some modifications. H1299 cells were transfected with FLAG-Tip60, HA-UHRF1, and His-ubiquitin. 24 h post-transfection; 10% of the cells were lysed with FLAG lysis buffer, and extracts were saved as input. The rest of the cells were lysed with phosphate/guanidine buffer (6 m guanidine-HCl, 0.1 m Na2HPO4, 6.8 mm NaH2PO4, 10 mm Tris-HCl, pH 8.0, 0.2% Triton X-100, freshly supplemented with 10 mm β-mercaptoethanol and 5 mm imidazole) with mild sonication and subjected to Ni-NTA (Qiagen) pulldown for 3 h. Ni-NTA-captured fractions were then washed with phosphate/guanidine buffer and urea wash buffer (8 m urea, 0.1 m Na2HPO4, 6.8 mm NaH2PO4, 10 mm Tris-HCl, pH 8.0, 0.2% Triton X-100, freshly supplemented with 10 mm β-mercaptoethanol and 5 mm imidazole) once each, and further washed three times with buffer (8 m urea, 18 mm Na2HPO4, 80 mm NaH2PO4, 10 mm Tris-HCl, pH 6.3, 0.2% Triton X-100, freshly supplemented with 10 mm β-mercaptoethanol and 5 mm imidazole). Precipitates were eluted by 30 min of incubation with Elution buffer (0.5 m imidazole, 0.125 m DTT) and resolved by SDS-PAGE.
RNA Isolation and Quantitative RT-PCR
Total RNA was isolated from cells using TRIzol (Invitrogen) and treated with DNase I (Ambion). 2 μg of total RNA was reverse-transcribed using SuperScript III First-Strand Synthesis Supermix (Invitrogen) and random primers following the manufacturer's protocol. PCR was performed in triplicates using SYBR green mix (Applied Biosystems) with a 7500 Fast Real Time PCR System (Applied Biosystems). The relative amount of specific mRNA was first normalized to β-actin and then to control sample (ctl RNAi, 0 h). For the qRT-PCR analysis of human transcripts the following primers were used: MDM2 forward 5′-CGATGAATCTACAGGGACGCCATCG-3′, MDM2 reverse 5′-TCCTGATCCAACCAATCACCTG-3′, p21 forward 5′-CCATGTGGACCTGTCACTGTCTT-3′, p21 reverse 5′-CGGCCTCTTGGAGAAGATCAGCCG-3′, PUMA forward 5′-GGTCCTCAGCCCTCGCTCTC-3′, PUMA reverse 5′-GTACGACTTGTCTCCGCCGCTCGTAC-3′.
RESULTS
UHRF1 Interacts with TIP60 Both in Vitro and in Vivo
Recent studies demonstrated correlation of UHRF1 overexpression with tumor growth and aggressiveness and poor prognosis in prostate cancer and colorectal cancer (37, 38). The oncogenic role of UHRF1 has long been implicated through epigenetic regulation, however recent indication of coexistence of UHRF1 in the same multi-protein complex with the TIP60 acetyltransferase (16, 17) that modulates p53-dependent growth arrest and apoptosis leads to the attractive hypothesis that UHRF1 may be linked to the TIP60-p53 interplay and modulate the p53-dependent damage response pathway.
We first sought to confirm interaction between UHRF1 and TIP60 by performing co-immunoprecipitation experiments in H1299 cells transfected with FLAG-Tip60 and HA-UHRF1 expression vectors. Western blot analysis of M2/FLAG immunoprecipitates from transfected cell extracts revealed that UHRF1 was specifically detected in TIP60-associated immunoprecipitates (Fig. 1B). To further demonstrate endogenous interaction, we subjected extracts from U2OS cells to immunoprecipitation with the α-UHRF1 antibody or with the control IgG. As expected, endogenous UHRF1 was specifically immunoprecipitated with the α-UHRF1 antibody; more importantly, TIP60 readily coprecipitateed with UHRF1 (Fig. 1C). An in vitro GST-pulldown assay was performed to further assess direct interaction. Purified GST or GST-tagged TIP60 protein was incubated with in vitro translated [35S]methionine-labeled FLAG-UHRF1 protein. Following capture with GST resins and recovery of immobilized complexes, the eluted complexes were resolved by SDS-PAGE and analyzed by autoradiography. [35S]UHRF1 strongly bound immobilized GST-TIP60, but not GST alone (Fig. 1D), demonstrating direct UHRF1-TIP60 binding in vitro. Taken together, these data confirm that UHRF1 is a bone fide interacting partner of TIP60.
UHRF1 Induces Degradation-independent Ubiquitination of TIP60
The C-terminal RING domain endows UHRF1 with intrinsic E3 ubiquitin ligase activity. Recent studies have identified PML (promyelocytic leukemia protein) and DNMT1 to be substrates for UHRF1-mediated ubiquitin-dependent proteolysis (17, 36). In an effort to test the possibility of UHRF1-mediated TIP60 ubiquitination, a cell-based ubiquitination assay was performed where ubiquitinated proteins were captured with nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography from H1299 transfected with FLAG-Tip60, His-ubiquitin and HA-UHRF1. Coexpression of UHRF1 and TIP60 produced significant levels of poly-ubiquitinated TIP60 (Fig. 2A). To confirm the indispensability of UHRF1 E3 ubiquitin ligase activity, we further utilized a UHRF1 RING finger point mutant that retains affinity for TIP60 (Fig. 2B) but was previously reported deficient in in vitro autoubiquitination (25). Wild-type UHRF1 displayed robust in-cell autoubiquitination while the C724A mutant lacked E3 ligase activity (Fig. 2C, middle panel). More importantly, TIP60 was strongly ubiquitinated in the presence of wild-type UHRF1 but not C724A UHRF1 (Fig. 2C, upper panel), demonstrating that UHRF1 induces TIP60 ubiquitination directly via its E3 ligase activity conferred by the RING domain.
FIGURE 2.

UHRF1 promotes degradation-independent ubiquitination of TIP60. A, UHRF1 induces ubiquitination of TIP60 in vivo. H1299 cells were cotransfected with expression vectors encoding FLAG-Tip60 or/and HA-UHRF1 in combination with His6-ubiquitin. Whole cell extracts and Ni-NTA affinity-purified fractions were analyzed by Western blot with α-FLAG and α-HA antibodies. GFP was used as a control to confirm equal transfection. B, C724A UHRF1 mutant retains interaction with TIP60. H1299 were transiently transfected with expression vectors for FLAG-Tip60 in combination with Myc-tagged wild-type UHRF1 or C724A UHRF1. Whole cell extracts or immunoprecipitates with M2/FLAG antibody were analyzed by Western blot with α-Myc and α-FLAG antibodies. C, UHRF1 directly ubiquitinates TIP60 through its E3 ubiquitin ligase activity. H1299 cells were cotransfected with expression vectors encoding FLAG-Tip60 and His6-ubiquitin alone or together with either HA-UHRF1 or HA-C724A UHRF1 expression vectors. Whole cell extracts and Ni-NTA affinity-purified fractions were analyzed by Western blot with α-FLAG and α-HA antibodies. GFP was used as a control to confirm equal transfection. D, UHRF1 expression does not induce TIP60 degradation. H1299 cells were transfected with FLAG-HA-Tip60 in combination with increasing levels of HA-UHRF1. Whole cell extracts were analyzed by Western blot with α-HA antibody. E, UHRF1 depletion does not affect TIP60 protein level. HCT116 cells were treated with 2 rounds of knock-down with either control RNAi, UHRF1 RNAi, or TIP60 RNAi. Whole cell extracts were analyzed by Western blot with α-UHRF1 and α- TIP60 antibodies. F, UHRF1 depletion does not affect TIP60 mRNA level. Total RNA was extracted from control RNAi or UHRF1 RNAi-treated HCT116. Following reverse transcription, the abundance of TIP60 mRNA was assessed using quantitative real time PCR.
Because poly-ubiquitination most frequently targets its substrate for proteolysis, we then sought to test the hypothesis of UHRF1-mediated TIP60 degradation. A cotransfection experiment in H1299 revealed that exogenous TIP60 protein levels remain unchanged in the presence of increasing levels of UHRF1 (Fig. 2D). To confirm that UHRF1 does not regulate TIP60 stability, we further assessed endogenous TIP60 protein levels following RNAi-mediated UHRF1 ablation. Endogenous UHRF1 and TIP60 proteins in HCT116 cells were severely depleted after transfection with the respective siRNA; however TIP60 protein level was unaffected by UHRF1 ablation (Fig. 2E). Furthermore, quantitative real time PCR (qRT-PCR) analysis of TIP60 mRNA following UHRF1-RNAi or control-RNAi revealed that the level of TIP60 mRNA remains unaffected by UHRF1 ablation (Fig. 2F). These data collectively suggest that UHRF1 does not promote degradation of TIP60 or regulate TIP60 at the transcription level.
UHRF1 Depletion Increases TIP60-mediated p53 Acetylation at K120 and Enhances Apoptosis
Because TIP60 is a known regulator of p53 acetylation at the K120 residue, we first assessed whether UHRF1 modulates TIP60-dependent acetylation of p53. M2/FLAG immunoprecipitation of extracts from H1299 transfected with FLAG-p53, Tip60, and HA-UHRF1, revealed that p53 is easily acetylated by TIP60 and acetylation was markedly attenuated upon UHRF1 expression (Fig. 3A). We further ablated UHRF1 in U2OS via RNAi and assessed endogenous p53 acetylation by immunoprecipitating cell extracts with the α-Ac-p53K120 antibody. Prior to harvesting, we subjected cells to trichostatin A (for inhibiting HDAC1/HDAC2-mediated p53 deacetylation) and nicotinamide (for inhibiting SIRT1-mediated p53 deacetylation) treatment to enrich acetylated endogenous p53. As shown in Fig. 3B, p53 K120 acetylation was readily detected with treatment of deacetylase inhibitors, and significantly enhanced upon UHRF1 ablation, suggesting that UHRF1 is a potent suppressor of TIP60-mediated acetylation of p53 at K120.
FIGURE 3.

UHRF1 suppresses TIP60-mediated p53 acetylation at K120 and UHRF1 depletion augments damage-induced apoptosis. A, UHRF1 expression inhibits p53 acetylation by TIP60 at K120. H1299 cells were transiently transfected with plasmid DNA expressing FLAG-p53, Tip60, and HA-UHRF1. Total cell extracts and M2 immunoprecipitates were assayed by Western blot using antibodies against HA, p53, and p53-AcK120. B, UHRF1 inactivation significantly increases p53 acetylation at K120. U2OS cells were transiently transfected with either control siRNA or UHRF1 siRNA, and treated for 6 h with 1 μm trichostatin A (TSA) and 5 mm nicotinamde (NTA) prior to harvesting. Cell extracts and immunoprecipitates obtained with α-Acp53K120 or control IgG were analyzed with Western blot using α-UHRF1, α-p53, and α-Acp53K120 antibodies. C, FACS analysis of UHRF1-inactivated U2OS cells treated with etoposide. U2OS cells transiently transfected with either control siRNA or UHRF1 siRNA were treated with 20 μm etoposide for the indicated time. Cells were subsequently fixed in 80% cold methanol, stained with propidium iodide and subjected to DNA content analysis by flow cytometry. D, UHRF1 RNAi increases apoptosis. Apoptosis was assessed as in C, and percentages of apoptotic cells are presented as average values of three independent experiments. Error bars, ±1 S.D.
We and others previously demonstrated that p53 K120 acetylation is indispensable for apoptosis (5, 6), leading to our speculation that UHRF1 might modulate apoptosis in cells at risk of DNA damage. We therefore assessed apoptosis by flow cytometric analysis of DNA fragmentation in UHRF1-ablated U2OS cells treated with etoposide and stained with propidium iodide (PI). As shown in Fig. 3C, UHRF1 inactivation minimally affected basal level sub-G1 content but markedly increased apoptosis following etoposide challenge. Quantitative analysis revealed that following 36 or 44 h of etoposide treatment, an average of 10.21% and 21.58% of control RNAi-treated cells were apoptotic, whereas a dramatically elevated 26.91% and 34.96% of UHRF1 RNAi-treated cells underwent apoptosis (Fig. 3D). Collectively, these data demonstrate that UHRF1 negatively regulates damage-induced apoptosis through attenuating TIP60-mediated p53 K120 acetylation.
UHRF1 Inhibits TIP60-p53 Interaction
We further assessed UHRF1 regulation of TIP60-p53 interaction by transfecting H1299 with increasing amounts of Myc-UHRF1 in the presence of p53 and FLAG-Tip60 and subjecting cell extracts to M2/FLAG immunoprecipitation. p53 was readily detected in TIP60-associated immunoprecipitates; however, upon UHRF1 expression the amount of TIP60-bound p53 decreased drastically (Fig. 4A), indicating that UHRF1 inhibits TIP60 interaction with p53.
FIGURE 4.
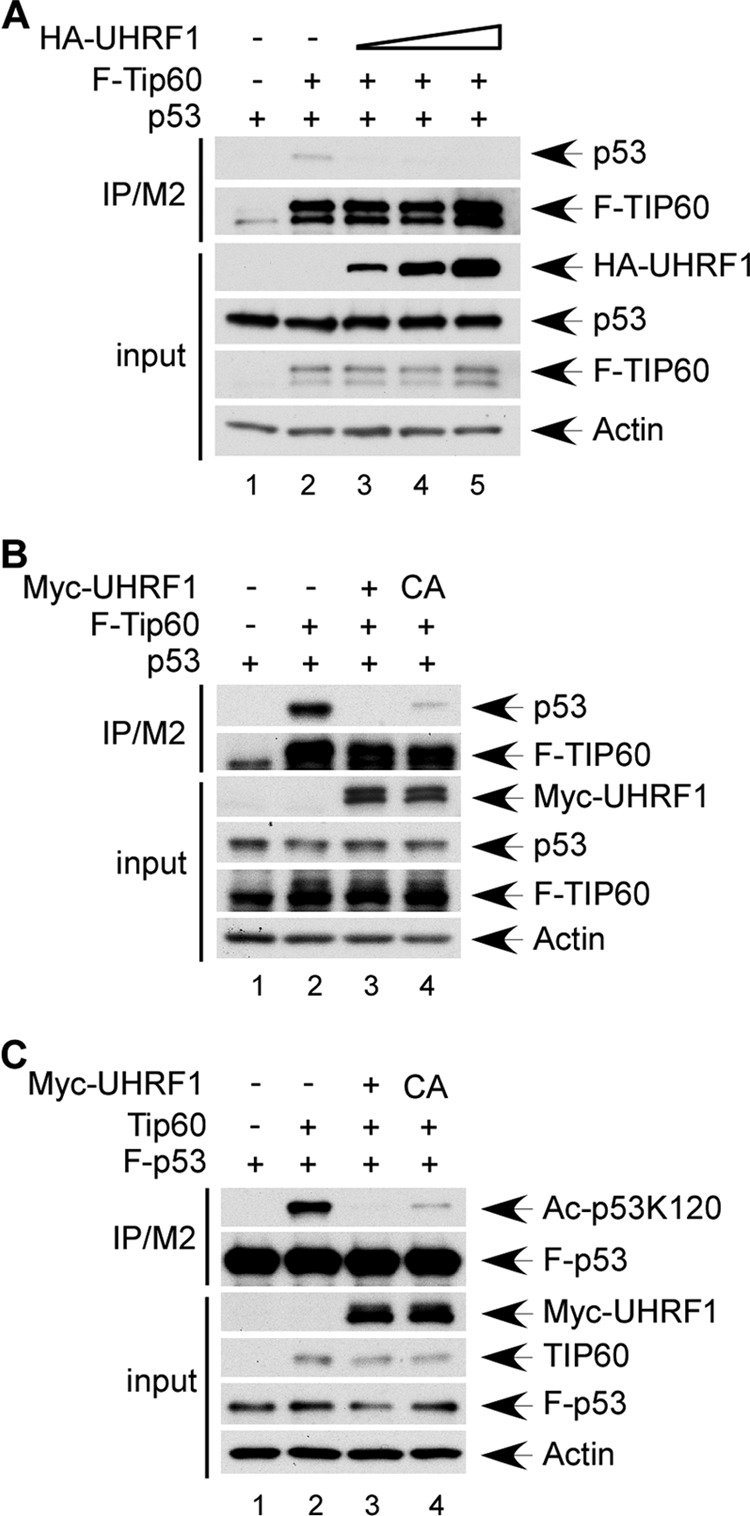
UHRF1 suppresses TIP60-p53 interaction partially through promoting TIP60 ubiquitination. A, UHRF1 expression inhibits TIP60 interaction with p53. H1299 cells were transiently transfected with expression vectors for FLAG-Tip60 and p53, in the absence of or in combination with HA-UHRF1 expression vector. Cell extracts and M2 immunoprecipitates were assayed by Western blot using α-HA, α-FLAG, and α-p53 antibodies. B, loss of UHRF1-mediated TIP60 ubiquitination partially suppresses TIP60 interaction with p53. H1299 cells were transiently transfected with expression vectors for FLAG-Tip60 and p53 in combination with Myc-tagged UHRF1 or C724A UHRF1. Total cell extracts and M2 immunoprecipitates were assayed by Western blot using antibodies against Myc, FLAG, and p53. C, loss of UHRF1-mediated TIP60 ubiquitination partially inhibits p53 acetylation by TIP60 at K120. H1299 cells were transiently transfected with expression vectors for FLAG-p53, Tip60, and Myc-tagged UHRF1 or C724A UHRF1. Total cell extracts and M2 immunoprecipitates were assayed by Western blot using antibodies against Myc, TIP60, p53, and p53-AcK120.
Having established that UHRF1 is capable of attenuating the TIP60-p53 interplay, we next sought to test whether this is achieved through TIP60 ubiquitination. Co-immunoprecipitation of p53 and TIP60 was performed in extracts of H1299 transfected with p53, FLAG-Tip60, and Myc-tagged UHRF1 or C724A UHRF1. As shown in Fig. 4B, expression of wild-type UHRF1 completely diminished the amount of p53 detected in TIP60-associated immunoprecipitates, whereas the ligase activity deficient C724A mutant only partially inhibited TIP60 interaction with p53. Furthermore, while wild-type UHRF1 completely abolished p53 K120 acetylation by TIP60, the C724A mutant mildly diminished p53 acetylation (Fig. 4C). Taken together, these results demonstrate that UHRF1 inhibits TIP60-p53 interaction and TIP60-mediated p53 acetylation, and that UHRF1-induced TIP60 ubiquitination may contribute partially to the suppression of the TIP60-p53 interplay.
SRA and RING Domains of UHRF1 Are Indispensable for UHRF1 Suppression of TIP60-p53 Interaction
The partial suppression of the TIP60-p53 interplay by the ligase activity-deficient UHRF1 mutant suggests that UHRF1 may elicit inhibitory effects through mechanisms including but not limited to TIP60 ubiquitination. It would therefore be interesting to elucidate what other functional domains might be responsible for exerting the inhibition. To this end, we generated a series of Myc-tagged UHRF1 truncation mutants as diagrammed in Fig. 5A with deletion of one or more functional domains. UHRF1-TIP60 interaction was determined by transfecting H1299 with F-Tip60 and Myc-tagged mutant UHRF1, followed by Western blot analysis of M2/FLAG immunoprecipitates. Mutant UHRF1 lacking the N-terminal ubiquitin-like domain (ΔNIRF), the Plant Homeo domain (ΔPHD), or the truncation mutant possessing only the SRA (Set and Ring Associated) and RING domains (S+R) bound strongly to TIP60 (Fig. 5B, lanes 3, 6, and 7); mutant UHRF1 that lacked either the SRA domain (ΔSRA) or the RING domain (ΔRING) showed weaker interaction with TIP60 (Fig. 5B, lanes 4 and 8); whereas loss of both SRA and RING domains (ΔS+R) completely abolished TIP60 interaction (Fig. 5B, lane 5), indicating that the SRA and RING domains are indispensable for UHRF1 interaction with TIP60.
FIGURE 5.

UHRF1 inhibits TIP60-p53 interaction through its SRA and RING domains. A, schematic representation of UHRF1 deletion mutants used in interaction domain mapping. Full-length UHRF1 and all deletion mutants were subcloned into pCMV-Myc expression vector. B, UHRF1 interacts with TIP60 through its SRA and RING domains. H1299 cells were transiently transfected with expression vectors for FLAG-Tip60 and Myc-tagged UHRF1 deletion constructs. Total cell extracts and M2 immunoprecipitates were assayed by Western blot using antibodies again Myc and FLAG. C, SRA and RING domains of UHRF1 are indispensable for inhibition of TIP60-p53 interaction. H1299 cells were transiently transfected with expression vectors for FLAG-Tip60 and p53, in combination with Myc-tagged full-length UHRF1 or ΔS+R UHRF1. Total cell extracts and M2 immunoprecipitates were analyzed by Western blot using antibodies against Myc, FLAG, and p53. D, UHRF1ΔSRA+RING mutant loses inhibition of TIP60-mediated p53 acetylation at K120. H1299 cells were transiently transfected with expression vectors for FLAG-p53 and Tip60, in combination with Myc-tagged full-length UHRF1 or ΔS+R UHRF1. Total cell extracts and M2 immunoprecipitates were analyzed by Western blot using α-Myc, α- TIP60, α-p53, and α-Acp53K120 antibodies. E, UHRF1ΔSRA+RING mutant retains nuclear localization. U2OS cells were transiently transfected with expression vectors for Myc-tagged full-length UHRF1 or ΔS+R UHRF1. 24 h post-transfection, cells were fixed with paraformaldehyde, immunostained with α-Myc antibody, and counterstained with 4,6-diamidino-2-phenylindole (DAPI). Subcellular localization was assessed by fluorescent microscopy.
To test whether the inhibition of TIP60-p53 interaction requires UHRF1 affinity for TIP60, we performed co-immunoprecipitation of p53 with TIP60 in extracts of H1299 transfected with p53, FLAG-Tip60, and Myc-UHRF1 or Myc-ΔS+R UHRF1. As expected, Western blot analysis of M2/FLAG eluates revealed that the level of TIP60-associated p53 was severely ablated upon UHRF1 expression (Fig. 5C, lane 3 versus lane 2). Strikingly, the ΔS+R mutant, which lacks binding affinity for TIP60, was incapable of suppressing TIP60-p53 interaction (Fig. 5C, lane 4 versus lane 2). In an effort to test whether p53 K120 acetylation is affected by loss of UHRF1-TIP60 interaction, we transfected H1299 with expression vectors for FLAG-p53, Tip60, and Myc-UHRF1 or Myc-ΔS+R UHRF1 and assessed the level of K120 acetylation in M2/FLAG immunoprecipitated total p53. Consistent with Fig. 4C, wild-type UHRF1 significantly diminished p53 acetylation by TIP60 (Fig. 5D, lane 2 and lane 3). However the ΔS+R mutant completely lost inhibition of p53 K120 acetylation (Fig. 5D, lane 4). We then sought to validate that the loss of TIP60 affinity is conferred through the deletion of SRA and RING domains rather than altered protein localization. We therefore assessed cellular localization of full-length and mutant UHRF1 by immunostaining Myc-UHRF1- or Myc-ΔS+R UHRF1-transfected U2OS cells. Co-staining with the α-Myc antibody and the nuclei labeling reagent 4,6-diamidino-2-phenylindole (DAPI) demonstrated that both the full-length UHRF1 protein and the ΔS+R mutant localize to the nucleus. These data collectively suggest that UHRF1 inhibits TIP60-p53 interaction through competitively binding to TIP60 via its SRA and RING domains, and ablation of UHRF1-TIP60 binding completely loses inhibition of the TIP60-p53 interplay.
UHRF1 Depletion Up-regulates Activation of PUMA and p21 but Not MDM2
Previous studies demonstrate that apart from promoting p53 acetylation at the K120 site, which is specifically required for the activation of apoptotic targets, TIP60 also regulates p53 transcription of p21 in the absence of K120 acetylation, through p53-dependent recruitment to the p21 promoter and modulation of histone H4 acetylation (5). In contrary, TIP60 is not recruited to the MDM2 promoter and activation of the p53 feedback target MDM2 is not dependent on TIP60 (5). Therefore our finding that UHRF1 inhibits not only p53 acetylation but also TIP60-p53 interaction predicts that UHRF1 ablation should result in increased activation of both PUMA and p21 in response to DNA damage because an increased amount of TIP60 now becomes available for promoter co-recruitment and/or co-activation of p53. Conversely, MDM2 induction by p53, which is independent of TIP60, should remain unaffected regardless of UHRF1 status.
To test this hypothesis, we performed RNAi-mediated inactivation of UHRF1 in HCT116 cells followed by treatment with the 5-fluorouracil (5-FU) antimetabolite that strongly activates p53 and induces p53-dependent growth arrest and apoptosis in HCT116 (39). As expected, upon 5-FU treatment p53 levels increased drastically; and notably UHRF1 inactivation affected neither basal p53 level (Fig. 6A, lane 6 versus lane 1) nor the accumulation of p53 protein. Furthermore, at all time points assessed, MDM2 induction was unaffected by UHRF1 depletion but PUMA and p21 activation was significantly increased in the UHRF1-depleted group (Fig. 6A, lanes 7–10 versus lanes 2–5). We further confirmed that UHRF1 ablation affected p53-dependent transcription of PUMA and p21 but not MDM2 by examining the mRNA levels of these targets using qRT-PCR (Fig. 6B). Indeed, basal MDM2, PUMA and p21 mRNA levels were unaffected by UHRF1 depletion. Upon 5-FU treatment, PUMA and p21 transcription was considerably augmented following UHRF1-RNAi, whereas MDM2 mRNA level increased upon 5-FU treatment and remained unaffected in the UHRF1-RNAi-treated group (Fig. 6B). To exclude off-target effects of RNAi and validate that our finding was not specific to HCT116 cells or the 5-FU drug, we ablated UHRF1 in U2OS using 3 siRNA oligos targeting different regions of the UHRF1 mRNA, and further treated these cells with another DNA damage reagent doxorubicin. UHRF1 was effectively ablated by all 3 siRNA oligos and doxorubicin-induced MDM2 induction remain unaffected by UHRF1 depletion, however PUMA and p21 activation was significantly increased by UHRF1 ablation using all 3 oligos (Fig. 6C). Together, these data validate UHRF1 ablation does not affect p53-mediated transcription of MDM2, but increases transcription of PUMA and p21.
FIGURE 6.

UHRF1 depletion up-regulates activation of PUMA and p21 but not MDM2 following DNA damage. A, UHRF1 RNAi in HCT116 cells up-regulates 5-FU induced PUMA and p21 activation but not MDM2 activation. HCT1116 cells were treated with 2 rounds of knock-down with either control RNAi or UHRF1 RNAi. Following treatment with 400 μm 5-FU for the indicated time, whole cell extracts were analyzed by Western blot with the indicated antibodies. B, UHRF1 RNAi in HCT116 cells up-regulates 5-FU induced PUMA and p21 but not MDM2 transcription. HCT116 were treated with 400 μm 5-FU for the indicated time following control RNAi or UHRF1 RNAi. Total RNA was extracted, and cDNA was prepared by reverse transcription. mRNA abundance for MDM2, p21, and PUMA was assessed using quantitative real time PCR. C, UHRF1 RNAi up-regulates doxorubicin induced PUMA and p21 but not MDM2 activation in U2OS cells. U2OS cells were transiently transfected with control siRNA or 3 U2OS-specifc siRNA oligos and treated with or without 0.5 μm doxorubicin for 16 h. Total cell extracts were analyzed by Western blot using the indicated antibodies. D, UHRF1 modulation of damage-induced PUMA and p21 activation is dependent on p53. UHRF1 is inactivated by RNAi in either p53+/+ or p53−/− HCT116 cells. Subsequently, cells were treated with or without 400 μm 5-FU for 8 h before extraction and Western blot analysis using the indicated antibodies. E, UHRF1 modulates damage-induced PUMA and p21 activation in a TIP60-dependent manner. UHRF1 or TIP60 alone, or both UHRF1 and TIP60 were inactivated in U2OS using RNAi. Subsequently, cells were subjected to 16 h of 0.5 μm doxorubicin treatment before extraction and Western blot analysis using the indicated antibodies.
To verify that the effect of UHRF1 on PUMA and p21 activation is p53 dependent, we ablated UHRF1 in the HCT116 p53+/+ and p53−/− pair prior to 5-FU treatment. Following damage, p53 accumulated in p53+/+ HCT116 and PUMA and p21 were activated, the extent to which was significantly increased by UHRF1 depletion (Fig. 6D, lanes 1–4). In a p53-deficient background 5-FU failed to activate PUMA and p21, and UHRF1 ablation did not increase PUMA and p21 level upon 5-FU treatment in the absence of p53 (Fig. 6D, lanes 5–8). 5-FU induced MDM2 only in the presence of p53, and no difference in MDM2 activation was observed upon UHRF1 RNAi.
To further validate that these effects are also dependent on TIP60, we inactivated UHRF1 or TIP60 alone, or together in U2OS cells using RNAi. p53 levels accumulated and MDM2 was activated normally following doxorubicin treatment in samples that were depleted of either UHRF1, TIP60 or both. However the induction of PUMA and p21 expression was severely diminished by TIP60 ablation (Fig. 6E, lanes 5 and 6); and more importantly, in the TIP60-deficient background UHRF1 depletion displayed no effect on PUMA and p21 levels before and after doxorubicin treatment (Fig. 6E, lanes 5–8). Together these data suggest that UHRF1 modulates p53 activity through negatively regulating TIP60-mediated functions in both K120 acetylation-dependent and -independent manners.
DISCUSSION
Our findings identify that UHRF1 is a direct interacting partner of TIP60 and a potent negative regulator of the TIP60-p53 interplay. UHRF1 expression induces degradation-independent ubiquitination of TIP60, which partially contributes to the marked suppression of TIP60-mediated p53 acetylation at K120. In contrast, UHRF1 ablation significantly increases p53 K120 acetylation, upon which p53-mediated apoptosis is dependent. Further elucidating the underlying mechanism, we found that UHRF1 severely inhibits TIP60 interaction with p53, leading to UHRF1 modulation of TIP60 function both dependent and independent of its ability to acetylate p53 at K120. Upon DNA damage, UHRF1 inactivation augments PUMA and p21 transcription, both of which rely on TIP60 but are differentially dependent on p53 K120 acetylation; in contrast, UHRF1 depletion does not affect stressed-induced MDM2 transcription by p53, which is independent of TIP60 status. Therefore, our findings suggest that UHRF1 acts as a critical negative regulator of TIP60 upstream of the p53 pathway, thereby negatively regulating TIP60-dependent transcription of key targets involved in growth arrest and apoptosis in cells at risk of DNA damage.
Based on our observation, we propose a model of tumorigenesis and/or tumor progression in the presence of high levels of cellular UHRF1 (Fig. 7). In normal cells, p53 is stabilized and activated upon DNA damage and induces transcription of the MDM2 feedback regulator in the absence of Tip60 recruitment and histone H4 acetylation in the vicinity of the MDM2 promoter. Tip60-p53 interaction is required for Tip60 recruitment to p53 target promoters and the induction of histone H4 acetylation, leading to p21 transactivation and growth arrest. Finally in cells that have undergone excessive damage, Tip60 acetylates p53 at K120, resulting in induction of PUMA and activation of the irreversible apoptotic pathway, thereby maintaining cellular and genomic stability and suppressing tumorigenesis (Fig. 7A). In contrast, UHRF1 overexpression leads to excessive UHRF1-Tip60 interaction and sequestration of Tip60 from associating with p53. This suppresses stress-induced Tip60 recruitment to p53 target promoters, acetylation of histone H4, and p53 actylation at K120. As a result, p53-dependent p21 and PUMA transactivation are abolished, and propagation of the damaged genome leads to tumorigenesis and/or tumor progression (Fig. 7B).
FIGURE 7.

A model for tumorigenesis/tumor progression in cells with UHRF1 overexpression. See text for details.
TIP60 is a haplo-insufficient tumor suppressor with well-documented functions in regulating transcription, DNA damage repair, and p53-mediated growth arrest and apoptosis (40). Therefore TIP60 function should require tight regulation, which to date has been shown achievable through post-translational modification or protein-protein interaction. Post-translationally, phosphorylation and autoacetylation of TIP60 upon DNA damage are required for TIP60 HAT activity and p53-mediated apoptosis (41, 42); whereas the E3 ligases MDM2 and CUL3 have been reported to target TIP60 directly for ubiquitin-dependent proteolysis (43, 44). In the respect of protein-protein interaction, a few studies have implicated that interaction with viral transforming proteins attenuates TIP60 HAT activity, de-stabilizes TIP60, or abrogates p53-dependent apoptosis (45–47). Recently, we have identified the p90 protein (also known as CCDC8) to specifically enhance p53-dependent apoptotic response through binding to TIP60 and promoting TIP60-mediated p53 acetylation (48). Our current study identifies UHRF1 to be another upstream regulator but functions to repress p53-dependent damage response through binding to TIP60, inhibiting TIP60-p53 interaction and TIP60-mediated p53 acetylation. Thus p90 and UHRF1, while both regulating upstream of TIP60 through protein-protein interaction, controls p53 function via distinct and opposing mechanisms.
At present, it is unclear how UHRF1 binding to TIP60 releases p53 from TIP60 interaction and renders TIP60 inactive in acetylating p53. TIP60 comprises an N-terminal chromodomain and a catalytic MYST domain. It is possible that UHRF1 and p53 compete for the same binding site within TIP60, or that UHRF1 binding induces a conformation change that makes TIP60 inaccessible for p53 binding. Further mapping of UHRF1-TIP60 and TIP60-p53 interaction domains and structural analysis of binding pockets may shed light in this respect. In addition to directly inhibiting TIP60-p53 binding, UHRF1 may change TIP60 conformation to compromise its HAT activity toward p53 and histone H4, the acetylation of which is required on p21 and PUMA promoters but not MDM2 promoter for transcription activation of respective gene targets (5).
Our results also reveal a previously unrecognized mechanism of TIP60 regulation through degradation-independent ubiquitination. Two previous studies report regulation of TIP60 stability by E3 ubiquitin ligases (43, 44), which likely contribute to maintenance of low TIP60 protein level in the absence of damage. Here we show that UHRF1 mediates TIP60 ubiquitination, which does not affect protein stability but rather negatively regulates TIP60-p53 interaction and TIP60 acetyltransferase activity. Although ectopic expression of E3 ligase-deficient mutant UHRF1 mildly suppresses the TIP60-p53 interplay, this could be owing to limited amount of cellular TIP60 being ubiquitinated by UHRF1. While physiological functions of degradation-independent ubiquitination are poorly understood, there have been a few studies implicating signal transduction, recruitment of interacting partners, and regulation of enzymatic activities (49). That ubiquitinated TIP60 loses acetyltransferase activity as suggested by our data is an interesting hypothesis and needs to be investigated further. It is also possible that UHRF1-mediated ubiquitination of TIP60 decreases its affinity for p53 or compromises recruitment to chromatin.
Overexpression of UHRF1 is found in a wide array of human tumors, including breast cancer, pancreatic cancer, brain tumor, lung cancer, bladder cancer, kidney cancer, cervical cancer, and colon cancer (21–27). Recently UHRF1 overexpression has been linked to tumor progression and poor prognosis in prostate cancer and colorectal cancer (37, 38). UHRF1 function in heterochromatin formation and inheritance of genomic DNA methylation patterns has long been implicated as its major oncogenic role. A number of anticancer drugs have been developed to target UHRF1 complex members such as HDAC1 and DNMT1 (26, 50), both of which are up-regulated in tumors (18, 51–56). However, the presence of multiple members of the HDAC family and the ubiquitous basal expression of HDACs and DNMT1 in normal cells create significant challenge for high specificity and low side effect (26, 56). In contrast to HDAC1 and DNMT1, the basal expression of UHRF1 in normal tissues is significantly lower and almost non-detectable in differentiated tissues (18, 57), making UHRF1 a very attractive therapeutic target and suggests that UHRF1 inhibitors, if available, could have fewer side effects than current drugs. Our study suggests that apart from epigenetic regulation, the oncogenic functions of UHRF1 may also be conferred through inhibition of the TIP60-p53 interplay and p53-dependent damage-induced apoptosis and growth arrest. It will be interesting to test whether UHRF1 overexpression and p53 mutation are mutually exclusive in human tumors, and in tumors with wild-type p53 whether down-regulation of UHRF1 or treatment with small molecule inhibitors targeting UHRF1-TIP60 interaction would de-repress the TIP60-p53 interplay and reactivate p53-dependent growth arrest and apoptosis, thereby inhibiting tumor growth.
Acknowledgments
We thank Dr. Zhenghe Wang from Case Western Reserve University for the UHRF1 plasmids, and Dr. Bruno Amati from European Institute of Oncology for the Tip60 antibody.
This work was supported, in whole or in part, by NCI, National Institutes of Health Grants 5R01 CA172023, 5R01 CA085533, and 2P01 CA080058 (to W. G.).
- ATM
- ataxia telangiectasia-mutated
- UHRF
- ubiquitin-like with PHD and RING finger domain
- RNAi
- inhibitory RNA
- SRA
- Set and Ring associated
- 5-FU
- 5-fluorouracil
- RING
- really interesting new gene domain
- TTD
- tandem tudor domain
- PHD
- plant homeo domain
- NIRF_N
- ubiquitin-like domain.
REFERENCES
- 1. Vousden K. H., Prives C. (2009) Blinded by the Light: The Growing Complexity of p53. Cell 137, 413–431 [DOI] [PubMed] [Google Scholar]
- 2. Kruse J. P., Gu W. (2009) Modes of p53 regulation. Cell 137, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dai C., Gu W. (2010) p53 post-translational modification: deregulated in tumorigenesis. Trends Mol. Med. 16, 528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tang Y., Zhao W., Chen Y., Zhao Y., Gu W. (2008) Acetylation is indispensable for p53 activation. Cell 133, 612–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tang Y., Luo J., Zhang W., Gu W. (2006) Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 24, 827–839 [DOI] [PubMed] [Google Scholar]
- 6. Sykes S. M., Mellert H. S., Holbert M. A., Li K., Marmorstein R., Lane W. S., McMahon S. B. (2006) Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 24, 841–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gaughan L., Logan I. R., Cook S., Neal D. E., Robson C. N. (2002) Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J. Biol. Chem. 277, 25904–25913 [DOI] [PubMed] [Google Scholar]
- 8. Frank S. R., Parisi T., Taubert S., Fernandez P., Fuchs M., Chan H. M., Livingston D. M., Amati B. (2003) MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep 4, 575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patel J. H., Du Y., Ard P. G., Phillips C., Carella B., Chen C. J., Rakowski C., Chatterjee C., Lieberman P. M., Lane W. S., Blobel G. A., McMahon S. B. (2004) The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell Biol. 24, 10826–10834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamamoto T., Horikoshi M. (1997) Novel substrate specificity of the histone acetyltransferase activity of HIV-1-Tat interactive protein Tip60. J. Biol. Chem. 272, 30595–30598 [DOI] [PubMed] [Google Scholar]
- 11. Sun Y., Jiang X., Chen S., Fernandes N., Price B. D. (2005) A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. U.S.A. 102, 13182–13187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kusch T., Florens L., Macdonald W. H., Swanson S. K., Glaser R. L., Yates J. R., 3rd, Abmayr S. M., Washburn M. P., Workman J. L. (2004) Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 306, 2084–2087 [DOI] [PubMed] [Google Scholar]
- 13. Berns K., Hijmans E. M., Mullenders J., Brummelkamp T. R., Velds A., Heimerikx M., Kerkhoven R. M., Madiredjo M., Nijkamp W., Weigelt B., Agami R., Ge W., Cavet G., Linsley P. S., Beijersbergen R. L., Bernards R. (2004) A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 428, 431–437 [DOI] [PubMed] [Google Scholar]
- 14. Ma H., Chen H., Guo X., Wang Z., Sowa M. E., Zheng L., Hu S., Zeng P., Guo R., Diao J., Lan F., Harper J. W., Shi Y. G., Xu Y., Shi Y. (2012) M phase phosphorylation of the epigenetic regulator UHRF1 regulates its physical association with the deubiquitylase USP7 and stability. Proc. Natl. Acad. Sci. U.S.A. 109, 4828–4833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim J. K., Estève P. O., Jacobsen S. E., Pradhan S. (2009) UHRF1 binds G9a and participates in p21 transcriptional regulation in mammalian cells. Nucleic Acids Res. 37, 493–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Achour M., Fuhrmann G., Alhosin M., Rondé P., Chataigneau T., Mousli M., Schini-Kerth V. B., Bronner C. (2009) UHRF1 recruits the histone acetyltransferase Tip60 and controls its expression and activity. Biochem. Biophys. Res. Commun. 390, 523–528 [DOI] [PubMed] [Google Scholar]
- 17. Du Z., Song J., Wang Y., Zhao Y., Guda K., Yang S., Kao H. Y., Xu Y., Willis J., Markowitz S. D., Sedwick D., Ewing R. M., Wang Z. (2010) DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci. Signal 3, ra80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Unoki M. (2011) Current and potential anticancer drugs targeting members of the UHRF1 complex including epigenetic modifiers. Recent Patents on Anti-cancer Drug Discovery 6, 116–130 [DOI] [PubMed] [Google Scholar]
- 19. Fujimori A., Matsuda Y., Takemoto Y., Hashimoto Y., Kubo E., Araki R., Fukumura R., Mita K., Tatsumi K., Muto M. (1998) Cloning and mapping of Np95 gene which encodes a novel nuclear protein associated with cell proliferation. Mamm. Genome 9, 1032–1035 [DOI] [PubMed] [Google Scholar]
- 20. Hopfner R., Mousli M., Jeltsch J. M., Voulgaris A., Lutz Y., Marin C., Bellocq J. P., Oudet P., Bronner C. (2000) ICBP90, a novel human CCAAT binding protein, involved in the regulation of topoisomerase IIα expression. Cancer Res. 60, 121–128 [PubMed] [Google Scholar]
- 21. Mousli M., Hopfner R., Abbady A. Q., Monté D., Jeanblanc M., Oudet P., Louis B., Bronner C. (2003) ICBP90 belongs to a new family of proteins with an expression that is deregulated in cancer cells. Br. J. Cancer 89, 120–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Unoki M., Nishidate T., Nakamura Y. (2004) ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG through its SRA domain. Oncogene 23, 7601–7610 [DOI] [PubMed] [Google Scholar]
- 23. Crnogorac-Jurcevic T., Gangeswaran R., Bhakta V., Capurso G., Lattimore S., Akada M., Sunamura M., Prime W., Campbell F., Brentnall T. A., Costello E., Neoptolemos J., Lemoine N. R. (2005) Proteomic analysis of chronic pancreatitis and pancreatic adenocarcinoma. Gastroenterology 129, 1454–1463 [DOI] [PubMed] [Google Scholar]
- 24. Oba-Shinjo S. M., Bengtson M. H., Winnischofer S. M., Colin C., Vedoy C. G., de Mendonça Z., Marie S. K., Sogayar M. C. (2005) Identification of novel differentially expressed genes in human astrocytomas by cDNA representational difference analysis. Brain Res. Mol. Brain Res. 140, 25–33 [DOI] [PubMed] [Google Scholar]
- 25. Jenkins Y., Markovtsov V., Lang W., Sharma P., Pearsall D., Warner J., Franci C., Huang B., Huang J., Yam G. C., Vistan J. P., Pali E., Vialard J., Janicot M., Lorens J. B., Payan D. G., Hitoshi Y. (2005) Critical role of the ubiquitin ligase activity of UHRF1, a nuclear RING finger protein, in tumor cell growth. Mol. Biol. Cell 16, 5621–5629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Unoki M., Kelly J. D., Neal D. E., Ponder B. A., Nakamura Y., Hamamoto R. (2009) UHRF1 is a novel molecular marker for diagnosis and the prognosis of bladder cancer. Br. J. Cancer 101, 98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lorenzato M., Caudroy S., Bronner C., Evrard G., Simon M., Durlach A., Birembaut P., Clavel C. (2005) Cell cycle and/or proliferation markers: what is the best method to discriminate cervical high-grade lesions? Hum. Pathol. 36, 1101–1107 [DOI] [PubMed] [Google Scholar]
- 28. Kofunato Y., Kumamoto K., Saitou K., Hayase S., Okayama H., Miyamoto K., Sato Y., Katakura K., Nakamura I., Ohki S., Koyama Y., Unoki M., Takenoshita S. (2012) UHRF1 expression is upregulated and associated with cellular proliferation in colorectal cancer. Oncol. Rep 28, 1997–2002 [DOI] [PubMed] [Google Scholar]
- 29. Rajakumara E., Wang Z., Ma H., Hu L., Chen H., Lin Y., Guo R., Wu F., Li H., Lan F., Shi Y. G., Xu Y., Patel D. J., Shi Y. (2011) PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol. Cell 43, 275–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arita K., Ariyoshi M., Tochio H., Nakamura Y., Shirakawa M. (2008) Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature 455, 818–821 [DOI] [PubMed] [Google Scholar]
- 31. Hashimoto H., Horton J. R., Zhang X., Bostick M., Jacobsen S. E., Cheng X. (2008) The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature 455, 826–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Avvakumov G. V., Walker J. R., Xue S., Li Y., Duan S., Bronner C., Arrowsmith C. H., Dhe-Paganon S. (2008) Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature 455, 822–825 [DOI] [PubMed] [Google Scholar]
- 33. Sharif J., Muto M., Takebayashi S., Suetake I., Iwamatsu A., Endo T. A., Shinga J., Mizutani-Koseki Y., Toyoda T., Okamura K., Tajima S., Mitsuya K., Okano M., Koseki H. (2007) The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450, 908–912 [DOI] [PubMed] [Google Scholar]
- 34. Karagianni P., Amazit L., Qin J., Wong J. (2008) ICBP90, a novel methyl K9 H3 binding protein linking protein ubiquitination with heterochromatin formation. Mol. Cell. Biol. 28, 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Citterio E., Papait R., Nicassio F., Vecchi M., Gomiero P., Mantovani R., Di Fiore P. P., Bonapace I. M. (2004) Np95 is a histone-binding protein endowed with ubiquitin ligase activity. Mol. Cell. Biol. 24, 2526–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guan D., Factor D., Liu Y., Wang Z., Kao H. Y. (2012) The epigenetic regulator UHRF1 promotes ubiquitination-mediated degradation of the tumor-suppressor protein promyelocytic leukemia protein. Oncogene [DOI] [PubMed] [Google Scholar]
- 37. Babbio F., Pistore C., Curti L., Castiglioni I., Kunderfranco P., Brino L., Oudet P., Seiler R., Thalman G. N., Roggero E., Sarti M., Pinton S., Mello-Grand M., Chiorino G., Catapano C. V., Carbone G. M., Bonapace I. M. (2012) The SRA protein UHRF1 promotes epigenetic crosstalks and is involved in prostate cancer progression. Oncogene 31, 4878–4887 [DOI] [PubMed] [Google Scholar]
- 38. Sabatino L., Fucci A., Pancione M., Carafa V., Nebbioso A., Pistore C., Babbio F., Votino C., Laudanna C., Ceccarelli M., Altucci L., Bonapace I. M., Colantuoni V. (2012) UHRF1 coordinates peroxisome proliferator activated receptor γ (PPARG) epigenetic silencing and mediates colorectal cancer progression. Oncogene 31, 5061–5072 [DOI] [PubMed] [Google Scholar]
- 39. Bunz F., Hwang P. M., Torrance C., Waldman T., Zhang Y., Dillehay L., Williams J., Lengauer C., Kinzler K. W., Vogelstein B. (1999) Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J. Clin. Invest. 104, 263–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gorrini C., Squatrito M., Luise C., Syed N., Perna D., Wark L., Martinato F., Sardella D., Verrecchia A., Bennett S., Confalonieri S., Cesaroni M., Marchesi F., Gasco M., Scanziani E., Capra M., Mai S., Nuciforo P., Crook T., Lough J., Amati B. (2007) Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 448, 1063–1067 [DOI] [PubMed] [Google Scholar]
- 41. Charvet C., Wissler M., Brauns-Schubert P., Wang S. J., Tang Y., Sigloch F. C., Mellert H., Brandenburg M., Lindner S. E., Breit B., Green D. R., McMahon S. B., Borner C., Gu W., Maurer U. (2011) Phosphorylation of Tip60 by GSK-3 determines the induction of PUMA and apoptosis by p53. Mol. Cell 42, 584–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang J., Chen J. (2010) SIRT1 regulates autoacetylation and histone acetyltransferase activity of TIP60. J. Biol. Chem. 285, 11458–11464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Legube G., Linares L. K., Lemercier C., Scheffner M., Khochbin S., Trouche D. (2002) Tip60 is targeted to proteasome-mediated degradation by Mdm2 and accumulates after UV irradiation. EMBO J. 21, 1704–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bhoumik A., Singha N., O'Connell M. J., Ronai Z. A. (2008) Regulation of TIP60 by ATF2 modulates ATM activation. J. Biol. Chem. 283, 17605–17614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Creaven M., Hans F., Mutskov V., Col E., Caron C., Dimitrov S., Khochbin S. (1999) Control of the histone-acetyltransferase activity of Tip60 by the HIV-1 transactivator protein, Tat. Biochemistry 38, 8826–8830 [DOI] [PubMed] [Google Scholar]
- 46. Jha S., Vande Pol S., Banerjee N. S., Dutta A. B., Chow L. T., Dutta A. (2010) Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol Cell 38, 700–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gupta A., Jha S., Engel D. A., Ornelles D. A., Dutta A. (2012) Tip60 degradation by adenovirus relieves transcriptional repression of viral transcriptional activator EIA. Oncogene, DOI 10.1038/onc.2012.534 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dai C., Tang Y., Jung S. Y., Qin J., Aaronson S. A., Gu W. (2011) Differential effects on p53-mediated cell cycle arrest vs. apoptosis by p90. Proc. Natl. Acad. Sci. U.S.A. 108, 18937–18942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ikeda F., Dikic I. (2008) Atypical ubiquitin chains: new molecular signals. 'Protein Modifications: Beyond the Usual Suspects' review series. EMBO Rep 9, 536–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Batty N., Malouf G. G., Issa J. P. (2009) Histone deacetylase inhibitors as anti-neoplastic agents. Cancer Lett. 280, 192–200 [DOI] [PubMed] [Google Scholar]
- 51. Witt O., Lindemann R. (2009) HDAC inhibitors: magic bullets, dirty drugs or just another targeted therapy. Cancer Lett. 280, 123–124 [DOI] [PubMed] [Google Scholar]
- 52. Weichert W. (2009) HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 280, 168–176 [DOI] [PubMed] [Google Scholar]
- 53. Etoh T., Kanai Y., Ushijima S., Nakagawa T., Nakanishi Y., Sasako M., Kitano S., Hirohashi S. (2004) Increased DNA methyltransferase 1 (DNMT1) protein expression correlates significantly with poorer tumor differentiation and frequent DNA hypermethylation of multiple CpG islands in gastric cancers. Am. J. Pathol. 164, 689–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Peng D. F., Kanai Y., Sawada M., Ushijima S., Hiraoka N., Kosuge T., Hirohashi S. (2005) Increased DNA methyltransferase 1 (DNMT1) protein expression in precancerous conditions and ductal carcinomas of the pancreas. Cancer Sci. 96, 403–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sawada M., Kanai Y., Arai E., Ushijima S., Ojima H., Hirohashi S. (2007) Increased expression of DNA methyltransferase 1 (DNMT1) protein in uterine cervix squamous cell carcinoma and its precursor lesion. Cancer Lett. 251, 211–219 [DOI] [PubMed] [Google Scholar]
- 56. Witt O., Deubzer H. E., Milde T., Oehme I. (2009) HDAC family: What are the cancer relevant targets? Cancer Lett. 277, 8–21 [DOI] [PubMed] [Google Scholar]
- 57. Bronner C., Achour M., Arima Y., Chataigneau T., Saya H., Schini-Kerth V. B. (2007) The UHRF family: oncogenes that are drugable targets for cancer therapy in the near future? Pharmacol. Ther. 115, 419–434 [DOI] [PubMed] [Google Scholar]
- 58. Li M., Brooks C. L., Wu-Baer F., Chen D., Baer R., Gu W. (2003) Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science 302, 1972–1975 [DOI] [PubMed] [Google Scholar]