

Background: The integrin binding-defective mutant of IGF1 (R36E/R37E) is functionally defective and does not induce ternary complex formation (integrin-IGF1-IGF1R).
Results: R36E/R37E suppressed signaling induced by WT IGF1, the binding of WT IGF1 to cells, ternary complex formation, cell viability, and tumorigenesis.
Conclusion: R36E/R37E is a dominant-negative antagonist of IGF signaling.
Significance: R36E/R37E has potential as a therapeutic agent.
Keywords: Cancer, Drug Discovery, Extracellular Matrix, Growth Factors, Insulin-like Growth Factor (IGF), Integrins, Mutant, Signal Transduction
Abstract
Insulin-like growth factor-1 (IGF1) is a major therapeutic target for cancer. We recently reported that IGF1 directly binds to integrins (αvβ3 and α6β4) and induces ternary complex formation (integrin-IGF1-IGF1 receptor (IGF1R)) and that the integrin binding-defective mutant of IGF1 (R36E/R37E) is defective in signaling and ternary complex formation. These findings predict that R36E/R37E competes with WT IGF1 for binding to IGF1R and inhibits IGF signaling. Here, we described that excess R36E/R37E suppressed cell viability increased by WT IGF1 in vitro in non-transformed cells. We studied the effect of R36E/R37E on viability and tumorigenesis in cancer cell lines. We did not detect an effect of WT IGF1 or R36E/R37E in cancer cells under anchorage-dependent conditions. However, under anchorage-independent conditions, WT IGF1 enhanced cell viability and induced signals, whereas R36E/R37E did not. Notably, excess R36E/R37E suppressed cell viability and signaling induced by WT IGF1 under anchorage-independent conditions. Using cancer cells stably expressing WT IGF1 or R36E/R37E, we determined that R36E/R37E suppressed tumorigenesis in vivo, whereas WT IGF1 markedly enhanced it. R36E/R37E suppressed the binding of WT IGF1 to the cell surface and the subsequent ternary complex formation induced by WT IGF1. R36E/R37E suppressed activation of IGF1R by insulin. WT IGF1, but not R36E/R37E, induced ternary complex formation with the IGF1R/insulin receptor hybrid. These findings suggest that 1) IGF1 induces signals under anchorage-independent conditions and that 2) R36E/R37E acts as a dominant-negative inhibitor of IGF1R (IGF1 decoy). Our results are consistent with a model in which ternary complex formation is critical for IGF signaling.
Introduction
Insulin-like growth factor-1 (IGF1)2 is a polypeptide hormone that has a high degree of structural similarity to proinsulin (1). IGF1 has been implicated in cancer progression (2). Many cancer cells secrete abnormally high levels of IGF1 and IGF2 (3). Most IGF1 is synthesized in the liver (3). IGF1 acts through binding to the IGF1 receptor (IGF1R), a receptor tyrosine kinase. Ligand binding induces phosphorylation of specific tyrosine residues of IGF1R. These phosphotyrosines then bind to adaptor molecules such as Shc and insulin receptor substrate-1. Phosphorylation of these proteins leads to activation of PI3K and ERK1/2 signaling pathways, which release strongly anti-apoptotic signals (3). IGF1 thereby confers resistance to chemotherapy and radiation therapy. Several strategies to target IGF1 signaling have been developed, including siRNA and monoclonal antibodies for IGF1R and kinase inhibitors to inhibit the enzymatic activity of the receptor (4).
Integrins are a family of cell adhesion receptors that recognize extracellular matrix ligands and cell surface ligands (5, 6). It has been well established that integrin αvβ3 plays a critical role in regulating IGF1 signaling (2). It has been proposed that “ligand occupancy” of αvβ3 (i.e. the binding of extracellular matrix proteins such as vitronectin to αvβ3) enhances signaling induced by IGF1 binding to IGF1R (2). Indeed, antagonists to αvβ3 block IGF1 signaling. Anti-αvβ3 mAb and echistatin, a snake venom disintegrin that specifically inhibits αvβ3, block IGF1-induced cell migration (7). Also, echistatin blocks IGF1-stimulated DNA synthesis and insulin receptor substrate-1 phosphorylation and attenuates IGF1R-linked downstream signaling events such as activation of PI3K and ERK1/2 (8).
We have reported that IGF1 directly and specifically binds to αvβ3 and developed an integrin binding-defective mutant (R36E/R37E) of IGF1 (9). R36E/R37E is defective in enhancing cell viability and in inducing intracellular signals, although the mutant still binds to IGF1R (9). Also, WT IGF1 induces ternary complex formation (αvβ3-IGF1-IGF1R), but R36E/R37E does not. This suggests that the direct binding of integrins to IGF1 is critical for IGF signaling and a potential mechanism of integrin-IGF1R cross-talk. We have recently reported that IGF1 signals are more clearly detectable under anchorage-independent conditions (poly(2-hydroxyethyl methacrylate) (poly-HEMA)-coated plates) than under anchorage-dependent conditions (10). This suggests that IGF signaling is masked by signals from cell-matrix interaction under anchorage-dependent conditions. IGF signaling requires αvβ3 expression, and R36E/R37E is defective in inducing signals under anchorage-independent conditions. These results suggest that αvβ3-IGF1 interaction, but not αvβ3-extracellular matrix interaction, is essential for IGF signaling. Inhibitors of IGF1R, Src, AKT, and ERK1/2 does not suppress αvβ3-IGF-IGF1R ternary complex formation, suggesting that activation of these kinases is not required for ternary complex formation. Also, mutations of the β3 cytoplasmic tail (Y747F and Y759F) that block β3 tyrosine phosphorylation do not affect IGF1R phosphorylation or AKT activation. We propose a model in which IGF1 binding to IGF1R induces recruitment of integrin αvβ3 to the IGF-IGF1R complex and then β3 and IGF1R are phosphorylated. It is likely that αvβ3 is needed with the IGF1-IGF1R complex for triggering IGF signaling (10).
We recently reported that IGF1 binds to integrin α6β4, another integrin that is overexpressed in cancer cells. Using CHO cells expressing recombinant α6β4, we demonstrated that WT IGF1, but not R36E/R37E, induced intracellular signals in an α6β4-dependent manner under anchorage-independent conditions (in soft agar and in poly-HEMA-coated wells) (11). Thus, it is possible that α6β4-IGF interaction, rather than α6β4-extracellular matrix interaction, plays a role in α6β4-overexpressing cancer cell lines.
In this study, we determined that R36E/R37E suppressed cell viability increased by WT IGF1 (a dominant-negative effect by definition) in two non-transformed and five transformed cell lines. Using cancer cells stably expressing WT IGF1 or R36E/R37E, we demonstrated that WT IGF1 markedly enhanced tumorigenesis, but R36E/R37E suppressed it. Also, R36E/R37E suppressed the binding of WT IGF1 to the cell surface and subsequent ternary complex formation. These results suggest that R36E/R37E acts as a dominant-negative antagonist of IGF signaling and suggest a mechanism of the inhibitory action of R36E/R37E at the cell surface.
EXPERIMENTAL PROCEDURES
Materials
Recombinant WT IGF1 and R36E/R37E were synthesized as described (9). NIH3T3, C2C12, Colo205 human colon cancer, M21 human melanoma, MCF-7 human breast cancer, MBA-MB231 human breast cancer, and B16F10 mouse breast cancer cells were obtained from American Type Culture Collection. Anti-integrin β4, anti-phospho-AKT (Thr-308), anti-phospho-ERK1/2, anti-ERK1/2, anti-AKT, anti-phospho-IGF1Rβ (Tyr-1135/Tyr-1136), and anti-IGF1R antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Picropodophyllin (PPP) was purchased from Santa Cruz Biotechnology. Bovine insulin was obtained from Sigma.
Signaling Assays
IGF signaling in regular tissue culture was performed as described (10). We analyzed cell lysates by Western blotting using specific antibodies. Bound IgG was detected using HRP-conjugated second antibody and SuperSignal West Pico chemiluminescent substrate (Thermo Scientific). We analyzed images using a Fuji LAS 4000mini luminescent image analyzer and MultiGauge V3.0 software (Fujifilm, Tokyo, Japan). Under anchorage-independent conditions, poly-HEMA-coated plates were prepared as described (12), except that the final poly-HEMA concentration was 1.2 mg/cm2. We performed signaling assays as described above, except that the cells were serum-starved for 3 h in DMEM without FCS.
Transfection of Cancer Cells
We subcloned the cDNA fragment encoding WT or mutant IGF1 (9) into the BamHI/EcoRI sites of the pSecTagB secretion vector, which has been modified to have the His6 and S tags at the N terminus of the protein (13). We generated MCF-7 cells stably secreting WT or mutant IGF1 by transfecting WT or mutant IGF1 cDNA in the modified pSecTagB vector together with plasmid with a neomycin-resistant gene as described (9). After selection with G418, the cells were used without further enrichment. To detect the expression of IGF1 (His6-tagged) from cells stably expressing WT IGF1 or R36E/R37E, we inoculated 5 × 106 cells (DMEM + 10% FCS, 5 ml) and cultured them for 24 h and then concentrated the culture medium 5 times by ultrafiltration. We detected WT IGF1 and R36E/R37E in the medium by Western blotting using anti-His5 antibody.
In Vivo Tumorigenesis Assays
We injected MCF-7 and Met-1 cells expressing WT IGF1 or R36E/R37E or vector-only mock-transfected cells into the mammary fat pads of female severe combined immunodeficiency (SCID) or FVB mice (3 weeks old, two injections per mouse, 105 cells per injection), respectively. Tumor size was measured in two dimensions using calipers. Tumor size was calculated as described (14).
IGF1 Binding to the Cell Surface
Cells were cultured to near confluency in DMEM and 10% FCS, resuspended in DMEM and 0.1% BSA, and incubated for 30 min at room temperature to block nonspecific protein-binding sites. WT IGF1 was labeled with FITC. FITC-labeled WT IGF1 (20 μg/ml) was mixed with unlabeled WT IGF1, R36E/R37E, or FGF R50E (irrelevant control) and incubated for 15 min at room temperature. The cells were then incubated with the mixture for 5 min at room temperature. The cells were washed with PBS and 0.02% BSA, and bound FITC-labeled WT IGF1 was determined using a FACSCalibur (BD Biosciences).
Co-precipitation of Integrin β4, IGF1R/Insulin Receptor, and IGF
Cells were cultured to near confluency in DMEM and 10% FCS, plated in poly-HEMA-coated plates, and serum-starved in DMEM for 3 h. The cells were incubated with WT IGF1 and/or R36E/R37E for 30 min and lysed in lysis buffer (20 mm HEPES (pH 7.4), 100 mm NaCl, 10% glycerol, 1% Nonidet P-40, 1 mm MgCl2, 1 mm PMSF, and protease inhibitor mixture (Sigma-Aldrich)). Cell lysates were incubated overnight with anti-IGF1R or anti-insulin receptor (IR) antibody at 4 °C, and the immune complex was recovered by incubation with protein A-Sepharose (GE Healthcare) for 1 h at 4 °C and washed three times with wash buffer (20 mm HEPES (pH 7.4), 100 mm NaCl, 10% glycerol, 0.5% Nonidet P-40, 1 mm MgCl2, 1 mm PMSF, and protease inhibitors). The immunoprecipitated materials were analyzed by Western blotting with anti-integrin β4 antibody.
Other Methods
Cell viability was determined using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assays as described (15). Treatment differences were tested using analysis of variance (ANOVA) and Tukey's multiple-comparison test to control the global type I error.
RESULTS
Dominant-negative Effect of R36E/R37E on IGF Signaling
IGF1 binds directly to integrin αvβ3, and this interaction is critical for IGF signaling (9). The integrin binding-defective R36E/R37E IGF1 mutant is defective in inducing IGF1R phosphorylation, AKT activation, ERK1/2 activation, and enhancing cell viability in non-transformed NIH3T3 fibroblasts and C2C12 myoblasts, whereas R36E/R37E binds to IGF1R at a level comparable to WT IGF1 (9). WT IGF1 induces ternary complex formation (αvβ3-IGF1-IGF1R), but R36E/R37E is defective in this function (9). These previous results suggest that direct integrin-IGF interaction and subsequent ternary complex formation are critical for IGF signaling. These results predict that R36E/R37E, which is defective in inducing ternary complex formation, suppresses IGF signaling by competing with WT IGF1 for binding to IGF1R (a dominant-negative effect). To test this prediction, we studied the effect of excess R36E/R37E on cell viability increased by WT IGF1 in NIH3T3 and C2C12 cells. R36E/R37E by itself does not affect cell viability in these cell types (9). We found that excess R36E/R37E suppressed cell viability increase by WT IGF1 (Fig. 1, a and b). These results suggest that R36E/R37E acts as a dominant-negative mutant in these non-transformed cell types.
FIGURE 1.
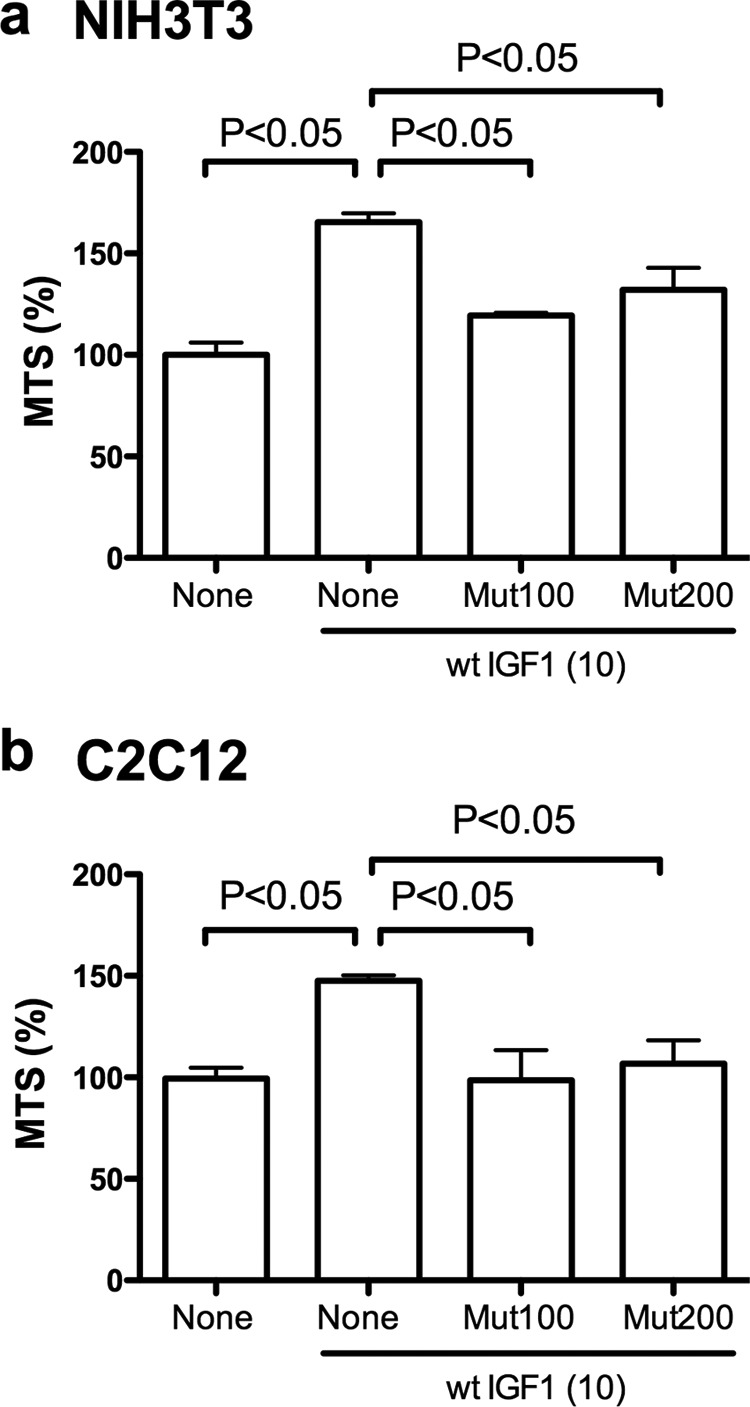
R36E/R37E suppresses cell viability increased by WT IGF1 in NIH3T3 and C2C12 cells in regular tissue culture. a and b, effect of R36E/R37E on cell viability. NIH3T3 (a) and C2C12 (b) cells were serum-starved overnight in DMEM and 0.4% FCS and cultured with 10 ng/ml WT IGF1 in the presence or absence of excess R36E/R37E (100 (Mut100) or 200 (Mut200) ng/ml) for 24 h. Cell viability was measured by MTS assays. Data are means ± S.E. (n = 4 for NIH3T3 cells and n = 12 for C2C12 cells). Statistical analysis was performed using ANOVA and Tukey's multiple-comparison test.
IGF Signaling in Transformed Cells under Anchorage-independent Conditions
We recently reported that WT IGF1 induces signals in transformed cells under anchorage-independent conditions (11). Under anchorage-dependent conditions, we did not clearly detect the effect of WT IGF1 or R36E/R37E on the cell viability of five transformed cell lines (MCF-7 human breast cancer, Met-1 mouse breast cancer, M21 human melanoma, Colo205 human colon cancer, and B16F10 mouse melanoma cells) (10) (Fig. 2). MCF-7 and Colo205 cells have been widely used to evaluate antagonists of IGF signaling (16–18). To determine the effect of WT IGF1 and R36E/R37E on transformed cells, we measured cell viability (using MTS assays) because we obtained essentially the same results for cell viability, colony formation in soft agar, and DNA synthesis (using BrdU incorporation assays) in our recent studies (10). We tested the effect of exogenous R36E/R37E on cell viability in these cancer cells under anchorage-independent conditions. WT IGF1 enhanced cell viability in these cells in a dose-dependent manner, whereas R36E/R37E was defective in this function (Fig. 3). We found that WT IGF1 induced phosphorylation of AKT, ERK1/2, and IGF1R in all five cell types under anchorage-independent conditions, but R36E/R37E was defective in these functions (Fig. 4a). These results suggest that WT IGF1 induces signaling, but R36E/R37E is defective in inducing signals under these conditions in five cancer cell lines.
FIGURE 2.

Transformed cells do not respond well to IGF1 in regular tissue culture. a, MCF-7 human breast cancer cells. b, Met-1 mouse breast cancer cells. c, Colo205 human colon cancer cells; d, M21 human melanoma cells. e, B16F10 mouse melanoma cells. Cells were plated in regular tissue culture plates and incubated with 0, 10, and 100 ng/ml WT IGF1 or R36E/R37E for 48 h. Cell viability was measured by MTS assays. Data are means ± S.E. (n = 6).
FIGURE 3.

R36E/R37E is defective in enhancing cell viability in cancer cell lines under anchorage-independent conditions. Cells were plated in poly-HEMA-coated plates and incubated with 0, 1, 10, and 100 ng/ml WT IGF1 or R36E/R37E for 48 h. Cell viability was measured by MTS assays. Data are means ± S.E. (n = 6). Statistical analysis was performed using ANOVA and Tukey's multiple-comparison test.
FIGURE 4.

R36E/R37E is defective in inducing IGF1 signaling in cancer cell lines under anchorage-independent conditions and suppresses IGF signaling induced by WT IGF1. a, R36E/R37E is defective in inducing IGF1 signaling in cancer cell lines under anchorage-independent conditions. Cells were plated in poly-HEMA-coated plates, serum-starved for 3 h, and stimulated with 100 ng/ml WT or R36E/R37E (R) IGF1 for 5 min. Cell lysates were analyzed by Western blotting. b, excess R36E/R37E suppresses IGF signaling induced by WT IGF1 in MCF-7 cells under anchorage-independent conditions. MCF-7 cells were stimulated for 5 min with WT IGF1 (10 ng/ml (wt IGF1 10)) alone and in the presence of R36E/R37E (50 (wt 10 + R 50) or 100 (wt 10 + R 100) ng/ml), and cell lysates were analyzed by Western blotting as described for a.
Notably, we found that excess R36E/R37E suppressed IGF1R phosphorylation and AKT phosphorylation induced by WT IGF1 (10 ng/ml) in MCF-7 cells under anchorage-independent conditions (Fig. 4b). We obtained essentially identical results with 5 ng/ml WT IGF1. These findings suggest that R36E/R37E may have potential as an antagonist of IGF1R. Consistent with the results described above, excess R36E/R37E suppressed cell viability increased by WT IGF1 in five different cancer cell lines under anchorage-independent conditions (Fig. 5). Taken together, our results suggest that R36E/R37E acts as a dominant-negative mutant in transformed cells as well.
FIGURE 5.

R36E/R37E suppresses cell viability increased by WT IGF1 in cancer cell lines under anchorage-independent conditions. Cells were plated in poly-HEMA-coated plates and cultured with 10 ng/ml WT IGF1 in the presence or absence of excess R36E/R37E mutant (Mut) for 48 h. Cell viability was measured by MTS assays. Data are means ± S.E. (n = 6). Statistical analysis was performed using ANOVA and Tukey's multiple-comparison test.
R36E/R37E Suppresses Tumorigenesis, whereas WT IGF1 Enhances It
We recently generated MCF-7 cells that stably express WT IGF-1 or R36E/R37E (11). The transfected cells secrete WT IGF1 or R36E/R37E at comparable levels (11). MCF-7 cells that express WT IGF1 grow much faster in soft agar than mock-transfected cells, but there is no difference in growth in soft agar between mock-transfected and R36E/R37E-expressing cells (11). In the present study, we found that WT IGF1-transfected MCF-7 cells showed enhanced viability under anchorage-independent conditions, as expected. There was only a small difference in cell viability between R36E/R37E-transfected MCF-7 and mock-transfected cells (Fig. 6a). To test the effect of R36E/R37E on tumorigenicity, we orthotopically injected WT IGF1-expressing and R36E/R37E-expressing MCF-7 cells into SCID mice and monitored tumorigenesis. We found that WT IGF1 markedly enhanced the tumorigenicity of MCF-7 cells compared with mock-transfected cells (Fig. 6b), whereas there was only a small difference between R36E/R37E-expressing and mock-transfected MCF-7 cells.
FIGURE 6.

R36E/R37E suppresses cell viability and tumorigenesis, whereas WT IGF1 enhances them in MCF-7 and Met-1 cells expressing WT IGF1 or R36E/R37E. MCF-7 (a) and Met-1 (c) transfectants were cultured in poly-HEMA-coated plates in DMEM for 48 h, and cell viability was measured by MTS assays. Data are means ± S.E. (n = 6). Statistical analysis was performed using ANOVA and Tukey's multiple-comparison test. Stably transfected MCF-7 (b) or Met-1 (d) cells were injected into the mammary fat pads of SCID or FVB mice, respectively (two injections per mouse, 105 cells per injection). Tumor growth was monitored by measuring tumor size using calipers. Statistical analysis was performed using Student's t test (n = 8 (day 48) for MCF-7 cells and n = 10 (days 25–31) for Met-1 cells).
It is possible that because MCF-7 cells do not grow very well in vivo, we did not detect an effect of R36E/R37E on tumorigenesis. Therefore, we decided to use Met-1 cells, which grow much faster in vivo. Met-1 cells contain the polyoma virus middle T transgene, form highly vascularized tumors, and metastasize effectively in nude mice (19, 20). We generated Met-1 cells that stably express WT IGF1 or R36E/R37E (11). The transfected cells secrete WT IGF1 or R36E/R37E at comparable levels (11). WT IGF1-transfected Met-1 cells grow much faster in soft agar compared with mock-transfected Met-1 cells. There is no difference between R36E/R37E-transfected and mock-transfected Met-1 cells in colony formation in soft agar (11). Consistently, in the present study, WT IGF1-transfected Met-1 cells showed enhanced viability under anchorage-independent conditions, as expected. There was only a small difference in cell viability between R36E/R37E-transfected and mock-transfected Met-1 cells (Fig. 6c). To test the effect of R36E/R37E on tumorigenesis in Met-1 cells, we orthotopically injected R36E/R37E-transfected and mock-transfected Met-1 cells into syngeneic FVB mice. As with MCF-7 cells, WT IGF1-transfected Met-1 cells grew much faster than mock-transfected cells in vivo. Notably, R36E/R37E-transfected cells grew significantly slower than mock-transfected cells (p < 0.05) (Fig. 6d). Our results suggest that R36E/R37E suppresses tumorigenesis in Met-1 cells, whereas WT IGF1 markedly enhances it.
R36E/R37E Suppresses the Binding of WT IGF1 to Cell Surface IGF1R and Ternary Complex Formation Induced by WT IGF1
Our results so far suggested that excess R36E/R37E suppressed the signaling functions of WT IGF1 in different cell lines, but it was still unclear how R36E/R37E suppressed WT IGF1. We measured the binding of FITC-labeled WT IGF1 to cell surface IGF1R in the presence of R36E/R37E by flow cytometry. Notably, excess R36E/R37E suppressed the binding of WT IGF1 to the cell surface in Met-1 and MCF-7 cells (Fig. 7, a–c) , suggesting that R36E/R37E inhibits the binding of WT IGF1 to IGF1R on the cell surface. We have reported that the binding of WT IGF1 to IGF1R recruits integrins αvβ3 and α6β4 to the IGF1-IGF1R complex, resulting in ternary complex formation (10, 11). Thus, R36E/R37E (integrin binding-defective) was expected to suppress recruitment of integrins and the resulting ternary complex formation induced by WT IGF1. To address this hypothesis, MCF-7 cells were stimulated with WT IGF1 in the presence of excess R36E/R37E, the complex was immunopurified with anti-IGF1R antibody, and the purified materials were analyzed by Western blotting using anti-β4 antibodies. We found that excess R36E/R37E reduced the levels of β4 in the complex (Fig. 7, d and e), suggesting that R36E/R37E effectively suppressed the ternary complex formation (α6β4-IGF1-IGF1R) in MCF-7 cells, as predicted. We propose a potential mechanism of inhibitory actions of R36E/R37E in which R36E/R37E competes with WT IGF1 for binding to IGF1R on the cell surface and R36E/R37E thereby suppresses subsequent recruitment of integrins and ternary complex formation.
FIGURE 7.

R36E/R37E competes with WT IGF1 for binding to IGF1R on the cell surface and ternary complex formation induced by WT IGF1. a–c, effect of R36E/R37E on the binding of WT IGF1 to the cell surface. Cells were incubated with FITC-labeled WT IGF1 (20 μg/ml) in the presence of unlabeled WT IGF1 (20, 50, and 200 μg/ml), R36E/R37E (20, 50, and 200 μg/ml), or FGF1 R50E (200 μg/ml) for 5 min at room temperature. Bound FITC-labeled WT IGF1 was determined using a FACSCalibur. The results suggest that R36E/R37E blocked the binding of WT IGF1 to the cell surface. d, effect of R36E/R37E on ternary complex formation (integrin β4-IGF1-IGF1R) induced by WT IGF1. MCF-7 cells were plated in poly-HEMA-coated plates and serum-starved in DMEM for 3 h. Cells were stimulated with WT IGF1 (20 ng/ml) in the presence of R36E/R37E (R; 50 and 200 ng/ml) for 30 min. IGF1R was immunopurified (IP) with anti-IGF1R antibodies from cell lysates. The immunoprecipitated materials were analyzed with anti-IGF1R or anti-β4 antibodies by Western blotting. The results suggest that R36E/R37E blocked the ternary complex formation (integrin β4-IGF1-IGF1R) induced by WT IGF1. IB, immunoblot. e, quantification of the density of β4 in the ternary complex in d. The experiments described for d were repeated three times, and the density of β4 was digitally quantified using a Fuji LAS 4000mini luminescent image analyzer. The density of β4 in the complex was normalized with that of IGF1R in the immunopurified materials.
R36E/R37E Suppresses Insulin-induced IGF1R Activation
Insulin binds to IGF1R at a much lower affinity compared with IGF1 (100-fold) and activates IGF1R (21, 22). If IGF1 competes with insulin for binding to IGF1R, we expected that R36E/R37E would suppress IGF1R activation by insulin. To address this hypothesis, we investigated whether R36E/R37E suppresses insulin-induced IGF1R activation. We stimulated MCF-7 cells with insulin (1 μg/ml = ∼100 nm) in the presence or absence of R36E/R37E (50 and 100 ng/ml) (Fig. 8a). R36E/R37E at these concentrations effectively suppressed IGF1R activation by insulin. This suggests that R36E/R37E suppresses IGF1R activation by insulin.
FIGURE 8.
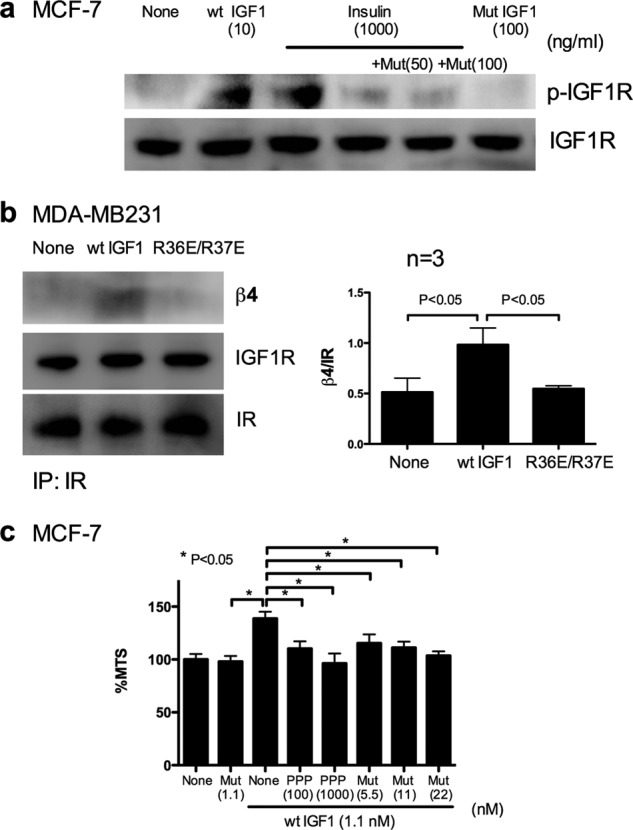
R36E/R37E suppresses IGF1R phosphorylation induced by insulin, does not induce ternary complex formation of the hybrid receptor, and suppresses cell viability increased by IGF1 at levels comparable to PPP. a, R36E/R37E suppresses IGF1R phosphorylation induced by insulin in MCF-7 cells. Cells were plated in poly-HEMA-coated plates and serum-starved in DMEM for 3 h. Cells were stimulated with a high concentration of bovine insulin (1 μg/ml) in the presence of R36E/R37E (50 (Mut(50)) and 100 (Mut(1000)) ng/ml) for 30 min. Cell lysates were analyzed by Western blotting. The results suggest that R36E/R37E blocked IGF1R phosphorylation induced by insulin. b, WT IGF1, but not R36E/R37E, induces ternary complex formation with the IGF1R/IR hybrid (integrin β4-IGF1-IGF1R/IR). MDA-MB231 cells were plated in poly-HEMA-coated plates and serum-starved in DMEM for 3 h. Cells were stimulated with WT IGF1 or R36E/R37E (100 ng/ml) for 30 min. IGF1R was immunopurified (IP) with anti-IR antibodies from cell lysates. The immunoprecipitated materials were analyzed with anti-IR, anti-IGF1R, or anti-β4 antibodies by Western blotting. The results suggest that WT IGF1, but not R36E/R37E, induced ternary complex formation with the hybrid receptor (integrin β4-IGF1-IGF1R/IR). c, PPP and R36E/R37E comparably suppress cell viability increased by WT IGF1 in MCF-7 cells under anchorage-independent conditions. Cells were plated in poly-HEMA-coated plates and cultured with 10 ng/ml (1.1 nm) WT IGF1 in the presence or absence of R36E/R37E (5.5–22 nm) or PPP (100 and 1000 nm) for 48 h. Cell viability was measured by MTS assays. Data are means ± S.E. (n = 6). Statistical analysis was performed using ANOVA and Tukey's multiple-comparison test.
Ternary Complex Formation with the IGF1R/IR Hybrid
Most human cancers overexpress both IGF1R and IR (21, 23, 24). Thus, it is reasonable to expect that they also overexpress the IGF1R/IR hybrid. It is unclear, however, if the hybrid receptor forms a ternary complex with integrins. We investigated whether IGF1 induces ternary complex formation with the hybrid receptor using MDA-MB231 cells expressing the hybrid receptor at high levels (21). We stimulated MDA-MB231 cells with IGF1 (100 ng/ml), immunopurified the ternary complex with anti-IR antibodies, and analyzed the immunopurified materials using anti-β4 antibodies. We detected β4 in the immunopurified complex when cells were stimulated with WT IGF1, but not with R36E/R37E (Fig. 8b). The immunopurified materials contained both IGF1R and IR. Because 1) IGF1 at 100 ng/ml did not bind to the IR homodimer and 2) MDA-MB231 cells express high levels of the hybrid receptor, it is highly likely that the immunopurified materials are predominantly hybrid receptors. This suggests that the hybrid receptor also forms a ternary complex with IGF1 and α6β4 through direct integrin binding to IGF1.
Comparison of the Ability of R36E/R37E and PPP, an Inhibitor of IGF1R Activation, to Suppress IGF1 Signaling
R36E/R37E is very similar to WT IGF1 and is expected to bind to IGF1R with an affinity and specificity comparable to those of WT IGF1. Thus, R36E/R37E has several potential advantages over currently tested antibodies or small compounds (see “Discussion”). We compared the ability of R36E/R37E to suppress IGF1 signaling with a currently available inhibitor of IGF1R phosphorylation, PPP, with an IC50 of 50 nm for blocking IGF1R activation (25). Under our experimental conditions, PPP at 100 nm and R36E/R37E at 5.5–20 nm effectively suppressed the IGF1-enhanced cell viability of MCF-7 cells (Fig. 8c). These results suggest that R36E/R37E is as effective as PPP in suppressing IGF1R activation.
DISCUSSION
In previous studies, we proposed a model in which direct binding of integrins to IGF1 and subsequent formation of a ternary complex (integrin-IGF1-IGF1R) play a role in IGF-IGF1R signaling (9, 11). We recently reported that IGF1-induced ternary complex formation does not require IGF1R or Src activation (10). We propose that ternary complex formation occurs before IGF1R or Src is activated. This model predicts that the integrin binding-defective R36E/R37E mutant is a dominant-negative mutant (IGF1 decoy) because R36E/R37E (functionally defective) is defective in inducing ternary complex formation and because it competes with WT IGF1 for binding to IGF1R. In the present study, we first demonstrated that excess R36E/R37E suppressed cell viability increased by WT IGF1 in two non-transformed cell lines under anchorage-dependent conditions. This is consistent with the prediction.
In transformed cells, however, we did not clearly detect effects of exogenous WT IGF1 or R36E/R37E on the cell viability of transformed cells under anchorage-dependent conditions. We suspect that cell-matrix interaction masks IGF signaling in the cancer cells. The potential effect of cell-matrix adhesion on another growth factor, heparin-binding EGF (HB-EGF), has been reported. Expression of HB-EGF in cancer cells enhances tumorigenesis in vivo, but expression of HB-EGF does not enhance the growth of cancer cells in vitro under anchorage-dependent conditions (26). This is probably because cell-matrix adhesion provides cells with sufficient proliferative signals. However, it is possible to detect the proliferative effect of HB-EGF on cancer cells in vitro in three- or two-dimensional culture in which cell-matrix interaction is reduced (26). It is likely that cell-matrix interaction masks signaling by exogenous growth factors. Notably, the results in an in vivo xenograft model correlate well with the results in three- or two-dimensional cultures in which cell-matrix interaction is reduced, but not with the results in regular tissue cultures (26). Thus, we studied the effect of exogenous IGF1 under anchorage-independent conditions.
We demonstrated that, under anchorage-independent conditions, WT IGF1 enhanced the viability of five cancer cell lines and that R36E/R37E was defective in this function. This suggests that 1) WT IGF1 induces signaling under anchorage-independent conditions and that 2) integrin-IGF1 interaction, rather than integrin-matrix interaction, is critical for IGF signaling in cancer cells under anchorage-independent conditions. IGF1 induced few or no signals in NIH3T3 or C2C12 cells under anchorage-independent conditions (data not shown), probably because these cells do not survive under anchorage-independent conditions.
We demonstrated that excess R36E/R37E suppressed signaling and cell viability increased by WT IGF1 in five cancer cell lines under anchorage-independent conditions. If the results under anchorage-independent conditions correlate with those in in vivo tumorigenesis as in HB-EGF (see above), it is expected that R36E/R37E would suppress in vivo tumorigenesis. Importantly, R36E/R37E suppressed tumorigenesis in vivo in rapidly growing Met-1 cells but with only a small difference in MCF-7 cells, whereas WT IGF1 markedly enhanced tumorigenesis in both cases. These findings suggest that R36E/R37E acts as a dominant-negative mutant in vitro and in vivo, as predicted. We also demonstrated that excess R36E/R37E suppressed the binding of WT IGF1 to the cell surface and WT IGF1-induced ternary complex formation (α6β4-IGF1-IGF1R) in MCF-7 cells, suggesting a mechanism of the inhibitory action of R36E/R37E at the cell surface as a decoy. Interestingly, R36E/R37E suppressed IGF1R activation by insulin, suggesting that insulin activates IGF1R in a manner similar to IGF1. Importantly, WT IGF1 induced ternary complex formation with the hybrid receptor, whereas R36E/R37E did not, suggesting that R36E/R37E can suppress activation of the hybrid receptor by IGF1. We demonstrated that R36E/R37E and PPP at comparable concentrations suppressed cell viability increased by IGF1, suggesting that R36E/R37E is as effective as PPP in suppressing IGF signaling. We propose that R36E/R37E is a novel antagonist of IGF1R.
We propose that R36E/R37E IGF1 may have potential as an IGF1R antagonist and an anticancer agent. R36E/R37E is highly specific to IGF1R, in contrast to kinase inhibitors, which are selective rather than specific. R36E/R37E also has high affinity for IGF1R. The currently used therapeutic agents for targeting therapy (antibodies and kinase inhibitors) almost always induce resistance after a while. This is partly due to point mutations in antibody epitopes or inhibitor-binding sites. Cancer cells obviously benefit from mutations that block the binding of antagonists. We expect that R36E/R37E may not induce such mutations in IGF1R because R36E/R37E and WT IGF1 bind to IGF1R exactly the same way, and blocking the binding of WT IGF1 to IGF1R by mutations in the IGF-binding site would not benefit cancer cells. Our discoveries are important because we can potentially exploit the property of R36E/R37E (IGF1 decoy) for targeting therapy. The potential of R36E/R37E as an inhibitor of IGF1R needs to be fully evaluated.
Using strategies similar to that used for IGF1, we have reported that FGF1 (27), neuregulin-1 (15), and fractalkine (28) directly bind to integrins. Direct binding of integrins to these growth factors/cytokines and ternary complex formation (e.g. integrin-FGF1-FGF1 receptor for FGF1) are critical for signaling. The integrin binding-defective mutants of these growth factors/cytokines are defective in signaling functions and act as antagonists in signaling in vitro and in vivo (28–30). Thus, integrin binding to growth factors/cytokines may be a common mechanism of integrin-growth factor/cytokine receptor cross-talk. The integrin binding-defective mutants of these growth factors/cytokines, including R36E/R37E IGF1, will be useful for studying the role of integrins in growth factor/cytokine signaling in future studies.
This work was supported, in whole or in part, by National Institutes of Health Grants CA13015 (to Y. T.) and HL096640 (to M. W.). This work also was supported by United States Department of Defense Grant W81XWH-10-1-0312 (to Y. T.).
- IGF1
- insulin-like growth factor-1
- IGF1R
- IGF1 receptor
- poly-HEMA
- poly(2-hydroxyethyl methacrylate)
- PPP
- picropodophyllin
- IR
- insulin receptor
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- ANOVA
- analysis of variance
- HB-EGF
- heparin-binding EGF
- SCID
- severe combined immunodeficiency.
REFERENCES
- 1. Stewart C. E., Rotwein P. (1996) Growth, differentiation, and survival: multiple physiological functions for insulin-like growth factors. Physiol. Rev. 76, 1005–1026 [DOI] [PubMed] [Google Scholar]
- 2. Clemmons D. R., Maile L. A., Ling Y., Yarber J., Busby W. H. (2007) Role of the integrin αVβ3 in mediating increased smooth muscle cell responsiveness to IGF-I in response to hyperglycemic stress. Growth Horm. IGF Res. 17, 265–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Clemmons D. R., Maile L. A. (2005) Interaction between insulin-like growth factor-I receptor and αVβ3 integrin linked signaling pathways: cellular responses to changes in multiple signaling inputs. Mol. Endocrinol. 19, 1–11 [DOI] [PubMed] [Google Scholar]
- 4. Chaves J., Saif M. W. (2011) IGF system in cancer: from bench to clinic. Anticancer Drugs 22, 206–212 [DOI] [PubMed] [Google Scholar]
- 5. Hynes R. O. (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 [DOI] [PubMed] [Google Scholar]
- 6. Takada Y., Ye X., Simon S. (2007) The integrins. Genome Biol. 8, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jones J. I., Doerr M. E., Clemmons D. R. (1995) Cell migration: interactions among integrins, IGFs and IGFBPs. Prog. Growth Factor Res. 6, 319–327 [DOI] [PubMed] [Google Scholar]
- 8. Zheng B., Clemmons D. R. (1998) Blocking ligand occupancy of the αVβ3 integrin inhibits insulin-like growth factor I signaling in vascular smooth muscle cells. Proc. Natl. Acad. Sci. U.S.A. 95, 11217–11222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saegusa J., Yamaji S., Ieguchi K., Wu C. Y., Lam K. S., Liu F. T., Takada Y. K., Takada Y. (2009) The direct binding of insulin-like growth factor-1 (IGF-1) to integrin αvβ3 is involved in IGF-1 signaling. J. Biol. Chem. 284, 24106–24114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fujita M., Takada Y. K., Takada Y. (2013) Insulin-like growth factor (IGF) signaling requires αvβ3-IGF1-IGF type 1 receptor (IGF1R) ternary complex formation in anchorage independence, and the complex formation does not require IGF1R and Src activation. J. Biol. Chem. 288, 3059–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fujita M., Ieguchi K., Davari P., Yamaji S., Taniguchi Y., Sekiguchi K., Takada Y. K., Takada Y. (2012) Cross-talk between integrin α6β4 and insulin-like growth factor-1 receptor (IGF1R) through direct α6β4 binding to IGF1 and subsequent α6β4-IGF1-IGF1R ternary complex formation in anchorage-independent conditions. J. Biol. Chem. 287, 12491–12500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fukazawa H., Mizuno S., Uehara Y. (1995) A microplate assay for quantitation of anchorage-independent growth of transformed cells. Anal. Biochem. 228, 83–90 [DOI] [PubMed] [Google Scholar]
- 13. Akakura N., Hoogland C., Takada Y. K., Saegusa J., Ye X., Liu F. T., Cheung A. T., Takada Y. (2006) The COOH-terminal globular domain of fibrinogen γ chain suppresses angiogenesis and tumor growth. Cancer Res. 66, 9691–9697 [DOI] [PubMed] [Google Scholar]
- 14. Feldman J. P., Goldwasser R., Mark S., Schwartz J., Orion I. (2009) A mathematical model for tumor volume evaluation using two-dimensions. J. Appl. Quant. Methods 4, 455–462 [Google Scholar]
- 15. Ieguchi K., Fujita M., Ma Z., Davari P., Taniguchi Y., Sekiguchi K., Wang B., Takada Y. K., Takada Y. (2010) Direct binding of the EGF-like domain of neuregulin-1 to integrins (αvβ3 and α6β4) is involved in neuregulin-1/ErbB signaling. J. Biol. Chem. 285, 31388–31398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mira E., Mañes S., Lacalle R. A., Márquez G., Martínez-A C. (1999) Insulin-like growth factor I-triggered cell migration and invasion are mediated by matrix metalloproteinase-9. Endocrinology 140, 1657–1664 [DOI] [PubMed] [Google Scholar]
- 17. Lu C., Shimaoka M., Ferzly M., Oxvig C., Takagi J., Springer T. A. (2001) An isolated, surface-expressed I domain of the integrin αLβ2 is sufficient for strong adhesive function when locked in the open conformation with a disulfide bond. Proc. Natl. Acad. Sci. U.S.A. 98, 2387–2392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perks C. M., Vernon E. G., Rosendahl A. H., Tonge D., Holly J. M. (2007) IGF-II and IGFBP-2 differentially regulate PTEN in human breast cancer cells. Oncogene 26, 5966–5972 [DOI] [PubMed] [Google Scholar]
- 19. Guy C. T., Cardiff R. D., Muller W. J. (1992) Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol. Cell. Biol. 12, 954–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheung A., Young L., Chen P. (1997) Microcirculation and metastasis in a new mouse mammary tumor model system. Int. J. Oncol. 11, 69–77 [PubMed] [Google Scholar]
- 21. Pandini G., Vigneri R., Costantino A., Frasca F., Ippolito A., Fujita-Yamaguchi Y., Siddle K., Goldfine I. D., Belfiore A. (1999) Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: evidence for a second mechanism of IGF-I signaling. Clin. Cancer Res. 5, 1935–1944 [PubMed] [Google Scholar]
- 22. Fox E. M., Miller T. W., Balko J. M., Kuba M. G., Sánchez V., Smith R. A., Liu S., González-Angulo A. M., Mills G. B., Ye F., Shyr Y., Manning H. C., Buck E., Arteaga C. L. (2011) A kinome-wide screen identifies the insulin/IGF-I receptor pathway as a mechanism of escape from hormone dependence in breast cancer. Cancer Res. 71, 6773–6784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Papa V., Gliozzo B., Clark G. M., McGuire W. L., Moore D., Fujita-Yamaguchi Y., Vigneri R., Goldfine I. D., Pezzino V. (1993) Insulin-like growth factor-I receptors are overexpressed and predict a low risk in human breast cancer. Cancer Res. 53, 3736–3740 [PubMed] [Google Scholar]
- 24. Vella V., Sciacca L., Pandini G., Mineo R., Squatrito S., Vigneri R., Belfiore A. (2001) The IGF system in thyroid cancer: new concepts. Mol. Pathol. 54, 121–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Girnita A., Girnita L., del Prete F., Bartolazzi A., Larsson O., Axelson M. (2004) Cyclolignans as inhibitors of the insulin-like growth factor-1 receptor and malignant cell growth. Cancer Res. 64, 236–242 [DOI] [PubMed] [Google Scholar]
- 26. Mizushima H., Wang X., Miyamoto S., Mekada E. (2009) Integrin signal masks growth-promotion activity of HB-EGF in monolayer cell cultures. J. Cell Sci. 122, 4277–4286 [DOI] [PubMed] [Google Scholar]
- 27. Mori S., Wu C. Y., Yamaji S., Saegusa J., Shi B., Ma Z., Kuwabara Y., Lam K. S., Isseroff R. R., Takada Y. K., Takada Y. (2008) Direct binding of integrin αvβ3 to FGF1 plays a role in FGF1 signaling. J. Biol. Chem. 283, 18066–18075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fujita M., Takada Y. K., Takada Y. (2012) Integrins αvβ3 and α4β1 act as coreceptors for fractalkine, and the integrin-binding defective mutant of fractalkine is an antagonist of CX3CR1. J. Immunol. 189, 5809–5819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamaji S., Saegusa J., Ieguchi K., Fujita M., Mori S., Takada Y. K., Takada Y. (2010) A novel fibroblast growth factor-1 (FGF1) mutant that acts as an FGF antagonist. PLoS One 5, e10273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mori S., Tran V., Nishikawa K., Kaneda T., Hamada Y., Kawaguchi N., Fujita M., Takada Y. K., Matsuura N., Zhao M., Takada Y. (2013) A dominant-negative FGF1 mutant (the R50E mutant) suppresses tumorigenesis and angiogenesis. PLoS One 8, e57927. [DOI] [PMC free article] [PubMed] [Google Scholar]