

Background: TOR complex 2 (TORC2) is a conserved protein complex that regulates multiple aspects of cell survival and proliferation.
Results: DNA metabolism and DNA damage response are impaired upon disruption of TORC2.
Conclusion: TORC2 affects replication-associated damages, independent of checkpoint activation.
Significance: TORC2 is required to maintain genome stability in fission yeast, a function that may be evolutionarily conserved.
Keywords: DNA Damage Response, Molecular Cell Biology, Signal Transduction, Stress, Yeast Genetics, S. pombe, TORC2, Fission Yeast, Rapamycin
Abstract
DNA damage can occur due to environmental insults or intrinsic metabolic processes and is a major threat to genome stability. The DNA damage response is composed of a series of well coordinated cellular processes that include activation of the DNA damage checkpoint, transient cell cycle arrest, DNA damage repair, and reentry into the cell cycle. Here we demonstrate that mutant cells defective for TOR complex 2 (TORC2) or the downstream AGC-like kinase, Gad8, are highly sensitive to chronic replication stress but are insensitive to ionizing radiation. We show that in response to replication stress, TORC2 is dispensable for Chk1-mediated cell cycle arrest but is required for the return to cell cycle progression. Rad52 is a DNA repair and recombination protein that forms foci at DNA damage sites and stalled replication forks. TORC2 mutant cells show increased spontaneous nuclear Rad52 foci, particularly during S phase, suggesting that TORC2 protects cells from DNA damage that occurs during normal DNA replication. Consistently, the viability of TORC2-Gad8 mutant cells is dependent on the presence of the homologous recombination pathway and other proteins that are required for replication restart following fork replication stalling. Our findings indicate that TORC2 is required for genome integrity. This may be relevant for the growing amount of evidence implicating TORC2 in cancer development.
Introduction
TOR is an atypical protein kinase that was isolated as the target of the immunosuppressive and anticancer drug rapamycin. TOR belongs to the PIKK (phosphatidylinositol kinase-related kinase) family of proteins and plays central roles in growth, proliferation, and survival (1, 2). Other members of this family include the ATM and ATR protein kinases, which have well established roles in maintaining genome integrity (3). Abnormal regulation of the human homologue, mTOR, is the cause of several human diseases, including cancer, multiple hamartoma syndrome, diabetes, and obesity (1, 2). TOR proteins can be found in two distinct evolutionarily conserved complexes, known as TORC13 and TORC2. TORC1 is the main target for inhibition by rapamycin, whereas TORC2 is insensitive or far less sensitive to the drug (4–6). The ability to inhibit TORC1 by rapamycin facilitated extensive studies of the TORC1 complex, whereas the cellular roles of TORC2 are less well understood. However, recent studies have revealed key roles of TORC2 in metabolism, cell survival, and proliferation (reviewed in Ref. 2). Accordingly, TORC2 deregulation has been implicated in cancer and metabolic disorders (1, 2). Several features of TORC2 appear highly conserved in evolution, including the composition of the subunits that make up the complex and several cellular functions, such as actin organization and protein synthesis (7). More recently, it was suggested that activation of TORC2 by association with the ribosomes is also conserved (8).
The fission yeast Schizosaccharomyces pombe contains two TOR homologues, Tor1 and Tor2 (9). These were numbered based on their order of discovery. Later, it was found that Tor1 interacts with Ste20 (Rictor) and Sin1 to form TORC2, whereas Tor2 interacts with Mip1 (Raptor) to form TORC1 (10, 11). As in human cells, S. pombe TORC1 is negatively regulated by the TSC complex (12, 13). TORC1 is essential for cell growth. Disruption of TORC1 (e.g. tor2-ts alleles) results in a phenotype that closely mimics nitrogen starvation in wild type cells, indicating a major positive role in growth and proliferation in response to nutrient availability (10, 11, 14–17). TORC2 is not essential for growth, and disruption of TORC2 (e.g. Δtor1) results in pleiotropic defects. These include an inability to respond to starvation and severe sensitivity to a variety of stresses, including osmotic, oxidative, and temperature stress; a delay in mitotic entry; and a decrease in amino acid uptake (18–24). Interestingly, in fission yeast, TORC1 and TORC2 oppositely regulate several cellular functions, including expression of nitrogen starvation-induced genes, sexual development, and mitotic entry (16, 25). However, an underlying mechanism(s) for TORC2 activity in cell biology is largely missing in fission yeast as well as in other model systems. TORC2 mediates most if not all of its functions via phosphorylation and activation of the AGC kinase, Gad8 (19, 26). Accordingly, deletion of gad8+ results in a very similar phenotype to that of Δtor1 mutants (19, 21, 26). More recently, it was also shown that in fission yeast, TORC2 is activated by the Rab family GTPase, Ryh1 (27).
Recently, our genome-wide expression profiling of Δtor1 cells revealed an extensive overlap between gene expression in Δtor1 and in cells mutated for chromatin remodelers (21). Consistently, we found that TORC2 is required for gene silencing at heterochromatic regions, such as the mating type locus and the subtelomeric regions. TORC2 mutant cells (Δtor1 or Δste20) or Δgad8 cells also show elongated telomeres (21). Like many chromatin-modulating mutants, the TORC2-Gad8 module is required for cell survival under DNA-damaging conditions. Loss of function of TORC2 or Gad8 results in severe sensitivity to chronic exposure to hydroxyurea (HU) or to methyl-methane sulfonate (MMS). Initial studies showed that TORC2 mutant cells transiently arrest cell cycle progression in response to HU but fail to dephosphorylate and activate Cdc2 upon removal of HU (21). Unexpectedly, about 40% of TORC2 mutant cells (Δtor1) arrested in HU with septa, indicating a delay in cytokinesis. Because this delay may represent an effect of HU that is not linked with DNA damage (28, 29), we set out to examine the effect of additional DNA-damaging agents on TORC2 mutant cells and to explore in more detail the DNA damage-sensitive phenotype of TORC2-Gad8 mutant cells.
We show here that, in striking contrast to the extreme sensitivity to replication stresses, such as HU, MMS, or camptothecin, TORC2-Gad8 mutant cells are not sensitive to ionizing radiation (IR), which produces double strand breaks that are repaired at the G2 phase of the cell cycle. We show that in the absence of TORC2-Gad8, cells spontaneously accumulate Rad52 foci, which may represent DNA damage sites (30, 31). Consistent with accumulation of DNA damage, we uncovered synthetic genetic interactions between mutations in TORC2-Gad8 and mutations in the homologous recombination (HR) pathway. TORC2 mutant cells are sensitive to chronic but not transient replication stress. This sensitivity is independent of checkpoint activation through Chk1 phosphorylation, but termination of the checkpoint signal is delayed. We suggest that the inability of TORC2 mutant cells to cope with chronic DNA-damaging conditions represents a failure to terminate the checkpoint signal, either through a direct mechanism or due to accumulation of unrepaired lesions.
EXPERIMENTAL PROCEDURES
Yeast Techniques
S. pombe strains are described in Table 1. All experiments were performed by using standard genetic and molecular yeast techniques as described (32). Yeast cells were cultured in YES medium at 30 °C, as described previously (9), unless otherwise indicated. For cell survival assays, HU (H8627, Sigma), MMS (129925, Sigma) or camptothecin (CPT) (C9911, Sigma) was added at the indicated concentrations. For UV and ionizing radiation, logarithmic growing cells were serially diluted, plated on YES medium, and exposed to UV using a UV Stratalinker 1800 cross-linker (Stratagene) or exposed to ionizing radiation using γ-radiation at 7.4 grays/min from a cesium-137 source.
TABLE 1.
Strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| TA000 | 972 h− | Laboratory stock |
| TA298 | tor1::his1+ leu1-32 ura4-D18 ade6-M216 his1-102 h+ pIRT2-tor1+ | Laboratory stock |
| TA390 | tor1::ura4+ ura4-D18 | Laboratory stock |
| TA788 | swi1::kanMX leu1-32 ura4-D18 h− | P. Russell |
| TA789 | swi3::kanMX leu1-32 ura4-D18 h+ | P. Russell |
| TA795 | tor1::ura4+ swi1::kanMX leu1-32 ura4-D18 | Laboratory stock |
| TA815 | rad3::ura+ leu1-32 ura4-D18 ade6-704 h− | P. Russell |
| TA817 | mus81::kanMX leu1-32 ura4-D18 h+ | P. Russell |
| TA818 | swi1::KanMX rad22-YFP::KanMX leu1-32 ura4-D18 h+ | P. Russell |
| TA819 | rad22-YFP::KanMX leu1-32 ura4-D18 h− | P. Russell |
| TA820 | chk1:9myc2HA6HA:ura4+ leu1-32 ura4-D18 h− | P. Russell |
| TA847 | tor1::ura4+ mus81::kanMX leu1-32 ura4-D18 | Laboratory stock |
| TA880 | tor1::his chk1:9myc2HA6HA:ura4+ leu1-32 ura4-D18 | Laboratory stock |
| TA881 | tor1::ura4+ rad22+YFP:kanMX leu1-32 ura4-D18 | Laboratory stock |
| TA891 | leu1-32 ura4-D18 ade6 M210 chr16 R23 h- | S. Whitehall |
| TA905 | rqh1::ura4+ leu1-32 ura4-D18 h− | YGRCa |
| TA991 | tor1::his1+ rqh1:: ura4+ ade6-M210 leu1-32 ura4-D18 his1-102 | Laboratory stock |
| TA1047 | top1::LEU2 leu1-32 ura4-D18 his1-102 h+ | Laboratory stock |
| TA1051 | top1::LEU2 rad3::ura4+ leu1-32 ura4-D18 | Laboratory stock |
| TA1058 | top1::LEU2 tor1::his1+ his1-102 leu1-32 ura4-D18 | Laboratory stock |
| TA1095 | tor1::ura4+ leu1-32 ura4D-18 ade6-M210 chr16 R23 | Laboratory stock |
| TA1106 | tor1::ura4+ swi1::KanMX rad22-YFP::KanMX leu1-32 ura4-D18 ade6-216 | Laboratory stock |
| TA1114 | ste20::kanMX6 leu1-32 ura4-D18 h- | K. Shiozaki |
| TA1132 | gad8::ura4+ ura4-D18 | Laboratory stock |
| TA1138 | sin1::kanMX6 | Laboratory stock |
| TA1148 | gad8::ura4+ leu1-32 ura4-D18 ade6-M216 h+ pREP1-gad8+ | Laboratory stock |
| TA1153 | ste20::kanMX6 leu1-32 ura4-D18 ade6-M210 h+ pREP1-ste20:myc | Laboratory stock |
| TA1172 | gad8::ura4+ rad22-YFP::KanMX leu1-32 ura4-D18 ade6-M216 | Laboratory stock |
| TA1174 | rad22::ura4+ ura4D-18 smt-0 h− | A. Carr |
| TA1183 | tor1::ura4+ swi3::kanMX leu1-32 ura4-D18 ade6-M210 h− | Laboratory stock |
| TA1186 | leu1-32 ura4-D18 his3-D1 ade6-M375int::pUC8/his3+/ade6-L469 h− | F. Osman |
| TA1196 | ste20::kanMX6 rad22-YFP::KanMX leu1-32 ura4-D18 ade6 | Laboratory stock |
| TA1215 | gad8::ura4+ leu1-32 ura4-D18 ade6-M210 chr16 R23 | Laboratory stock |
| TA1223 | rhp51::ura leu1-32 ura4-D18 smt-0 h− | P. Russell |
| TA1225 | mms22::KanMX leu1-32 ura4-D18 h+ | P. Russell |
| TA1226 | mms1::KanMX leu1-32 ura4-D18 h+ | P. Russell |
| TA1240 | ste20::kanMX6 leu1-32 ura4-D18 ade6-M210 chr16 R23 | Laboratory stock |
| TA1252 | ryh1::KanMX leu1-32 h− | K. Shiozaki |
| TA1253 | sat1::KanMX leu1-32 h− | K. Shiozaki |
| TA1254 | sat4::KanMX leu1-32 h− | K. Shiozaki |
| TA1280 | tor1::ura4+ leu1-32 ura4-D18 his3-D1ade6-M375int::pUC8/his3+/ade6-L469 | Laboratory stock |
| TA1283 | rhp51::ura4+ leu1-32 ura4-D18 his3-D1− ade6-M375int::pUC8/his3+/ade6-L469 smt-0 | Laboratory stock |
| TA1285 | ste20::kanMX6 leu1-32 ura4-D18 his3-D1 ade6-M375int::pUC8/his3+/ade6-L469 | Laboratory stock |
| TA1327 | gad8::ura4+ leu1-32 ura4-D18 his3-D1 ade6-M375int::pUC8/his3+/ade6-L469 | Laboratory stock |
| TA1360 | gad8::ura4 top1::LEU2 leu1-32 ura4-D18 | Laboratory stock |
| TA1501 | brc1::hphMX6 leu1-32 h+ | P. Russell |
| TA1550 | ste20::kanMX6 brc1::hphMX6 leu1-32 | Laboratory stock |
| TA1552 | gad8::ura4+ brc1::hphMX6 leu1-32 | Laboratory stock |
a Yeast Genetic Resource Center, Japan.
Microscopy
Fluorescence imaging of nuclei and septa was carried out as described previously (33). Cells were visualized using a Nikon eclipse E600 fluorescence microscope and photographed using a Nikon digital camera (DXM1200) and the ACT1 software. Rad52-YFP-expressing strains were cultured in yeast extract and imaged live using a Leica SP5 confocal microscope. For quantification of foci, at least 750 nuclei were scored in three independent experiments.
Western Blotting
For Western blot analysis, proteins were extracted with TCA as described previously (34) and resolved by SDS-PAGE using 8% acrylamide gels. Proteins were transferred to nitrocellulose membranes and blocked with 5% milk in TBST (0.05% Tween). Chk1 activation was detected by probing the membrane with anti-HA antibody (catalog no. 7392, Santa Cruz Biotechnology, Inc. (Santa Cruz, CA)). Cdc2 was detected by anti-PSTAIRE (catalog no. 53, Santa Cruz).
Spontaneous Recombination Frequency
Recombination frequencies were determined by fluctuation tests as described previously (35) with the following modifications. For each strain, 12 independent colonies were used for each assay, and each assay was repeated independently at least three times. For each assay, single colonies grown on YE medium were resuspended in sterile water, and cells were plated onto EMMG without adenine for the selection of Ade+ recombinants and onto complete EMMG (supplemented with adenine, uracil, leucine, and histidine at a final concentration of 225 mg/liter each) for determination of cell titers. The Ade+ recombinants on the selective plates were replica-plated onto EMMG lacking adenine and histidine to determine the proportion of conversion type (Ade+ His+) and deletion type (Ade+ His−) recombinants. For each strain, the average recombinant rate was determined using the median method as described (36).
Minichromosome 16 (Ch16) Loss Assay
Cells from individual Ade+ colonies were plated on adenine-limiting YE plates, incubated at 30 °C for 3–4 days and then at 4 °C for 1–2 overnight periods to allow deepening of the red color in Ade− colonies. The frequency of chromosome loss was determined as the number of colonies with a red sector equal to half of the colony divided by the sum of white and sectored colonies.
RESULTS
TORC2-Gad8 Is Required for Survival of DNA Damage in S Phase
We previously reported that Δtor1, Δste20, Δsin1, or Δgad8 strains, defective in the TORC2-Gad8 pathway, are highly sensitive to chronic exposure to HU or MMS (21) (see also Fig. 1). These two drugs are known to interfere with S phase progression, either by depletion of dNTPs in the case of HU or via blocking the progression of the replication fork by DNA alkylation in the case of MMS. We further assessed the sensitivity of TORC2-Gad8 mutant cells to additional DNA-damaging agents: CPT, UV, and IR. The cytotoxicity associated with CPT is highly S phase-specific. CPT stabilizes covalent DNA-topoisomerase I complexes by preventing the religation step of topoisomerase I, thus leading to collision of DNA replication forks with the drug-enzyme-DNA complex (37, 38). UV irradiation halts DNA replication by producing cyclobutane pyrimidine dimers that block the progression of replicative polymerases (39, 40). In contrast, the primary toxic effect of IR is the induction of double strand breaks, which are repaired in G2 phase (41). We found that deletion of each of the genes encoding specific TORC2 components resulted in cells that are highly sensitive to chronic exposure to CPT (Fig. 1A). The sensitivity of Δtor1 or Δgad8 deletion strains to CPT is rescued by deletion of top1+, which encodes topoisomerase I (Fig. 1B), indicating that TORC2 mutant cells are sensitive to CPT due to its toxic effect when associated with Top1. Deletion mutants in the TORC2-Gad8 pathway were also mildly sensitive to UV irradiation (Fig. 1A). In striking contrast, Δtor1, Δste20, Δsin1, or Δgad8 mutant strains are as resistant as wild type cells to high doses of IR, unlike the strong sensitivity of Δrad3 mutant cells to these conditions (Fig. 1A). Because HU, MMS, and CPT are known to impair fork replication progress, these findings suggest that the TORC2-Gad8 pathway is particularly required for tolerance of genotoxins that interfere with DNA replication. The mild sensitivity to UV, in contrast to strong sensitivity to HU, MMS, or CPT, is reminiscent of mutations in Rpa3, encoding replication protein 3 homologue in S. pombe, which was recently identified as critical for mediating RPA functions required for repair or tolerance of DNA lesions in S phase (42).
FIGURE 1.

The TORC2-Gad8 pathway is required for survival under chronic replicative stress. A–C, 10-fold serial dilution of logarithmic growing cells was plated onto YE-rich medium, containing the indicated DNA-damaging agents or irradiated by UV or IR. Plates were incubated at 30 °C for 3–4 days.
Recently, upstream regulators of the TORC2 complex were identified in fission yeast. Ryh1, a Rab GTPase, was found to regulate TORC2-dependent phosphorylation of Gad8, and the proteins Sat1 and Sat4 were found to act as a guanine nucleotide exchange factor complex for Ryh1 (43). Cells lacking Sat1, Sat4, or Ryh1 share several phenotypes with TORC2-Gad8 mutant cells, such as sterility, inability to arrest in G1 in response to nitrogen limitation, and sensitivity to various stress conditions (43, 44). Similar to TORC2-Gad8 mutant cells, the Δryh1, Δsat1, and Δsat4 strains were sensitive to CPT, HU, or MMS but not to IR (Fig. 1C). Thus, it is likely that Ryh1 and its guanine nucleotide exchange factor complex function upstream of TORC2 in inducing tolerance to replication stress. Because cells that lack Ryh1 or its guanine nucleotide exchange factor complex are not as sensitive as TORC2-Gad8 mutant cells to HU, additional upstream regulators may be involved.
TORC2-Gad8 Is Required for Inactivation of the DNA Damage Checkpoint
In contrast to the sensitivity of TORC2-Gad8 mutant cells to chronic exposure to low doses of MMS or CPT (Fig. 1A), Δtor1 or Δgad8 mutant cells did not lose viability in response to a short exposure to higher doses of these drugs (Fig. 2, A and B). The resistance of Δtor1 or Δgad8 mutant cells to short exposure to DNA-damaging drugs is in contrast to the rapid loss of viability of the Δrad3 mutant cells (Fig. 2, A and B). Δrad3 mutant cells rapidly lose viability in the presence of MMS or CPT because they fail to activate the DNA damage checkpoint and proceed into mitosis before their DNA is completely repaired (45). The insensitivity of TORC2 mutant cells to a relatively short exposure to DNA damage suggests that TORC2 is not required for the initial activation of the DNA damage checkpoint. Consistently, we found that Δtor1 or Δgad8 cells arrest growth in response to MMS or CPT, as elongated cells, indicative of a transient delay in entry into mitosis (46) (Fig. 2C).
FIGURE 2.

TORC2-Gad8 is required for deactivation of the DNA damage checkpoint but not for its activation. A and B, Tor1 or Gad8 are dispensable for maintaining cellular viability in response to short exposure to MMS (A) or CPT (B). Cells were grown to log phase and shifted to 0.03% MMS or to 30 μm CPT for 6 h. Samples were taken every hour to determine cell viability by assessing plating efficiency on rich medium. C, logarithmic growing cells were transferred to rich medium containing either 30 μm CPT or 0.03% MMS. Samples of the indicated strains, before and after a 6-h exposure to the drugs, were stained with Hoechst and Calcofluor to visualize nuclei and septa, respectively. D, Tor1 is dispensable for Chk1 phosphorylation in response to CPT. Wild type or Δtor1 cells containing an HA-tagged Chk1 were grown to log phase and treated with 30 μm CPT. Protein extract from each of the indicated time points was analyzed by Western blot. The resulting membranes were probed with anti-HA to visualize Chk1 and with anti-Cdc2 as a loading control. E, the dephosphorylation of Chk1 is delayed in a Δtor1 mutant. Cells were arrested for 3 h in 30 μm CPT, washed thoroughly, and resuspended in fresh medium. Protein samples from the indicated time points were analyzed by Western blot as described above.
Exposure of cells to DNA damage, at any phase of the cell cycle, activates the main DNA damage checkpoint kinase, Rad3 (ATR). This leads to the activation of downstream effector kinases (47). In response to CPT, Chk1 (also known as Chk1 in higher eukaryotes) becomes activated by phosphorylation and is essential for propagating the checkpoint activation signal (38). Δtor1 mutant cells show phosphorylation of Chk1 in response to CPT, although there is a slight delay in the kinetics of phosphorylation compared with wild type cells (Fig. 2D). Because TORC2 mutant cells do not lose viability upon short exposure to CPT (Fig. 2B) and show the expected cell cycle arrest phenotype (Fig. 2C), it appears that the checkpoint activation signal is properly propagated in these mutant cells. In contrast, upon removal of CPT, dephosphorylation of Chk1 is markedly delayed in Δtor1 mutant cells (Fig. 2E). This delay in dephosphorylating Chk1 may suggest that TORC2 mutant cells are defective in reentry into the cell cycle following DNA damage induction.
TORC2-Gad8 Mutant Cells Accumulate Spontaneous Nuclear Rad52 Foci
Rad52 (also referred to as Rad22 in S. pombe; homologue of Saccharomyces cerevisiae Rad52) is a DNA repair protein that binds to single-stranded DNA and plays a key role in DNA repair by homologous recombination. Rad52 stimulates DNA annealing and enhances Rad51 (also referred to as Rhp51 in S. pombe)-catalyzed strand invasion (48, 49). Accumulation of Rad52 nuclear foci under normal growth conditions occurs in cells defective for DNA damage repair or replication functions (30, 31) and is induced in wild type cells in response to various DNA damages.
Quantitative analysis of wild type cells showed that ∼10% of the nuclei contained at least one Rad52-YFP focus, in agreement with previous reports (50, 51). No wild type nuclei contained multiple Rad52-YFP foci (Fig. 3, A and B). In contrast, ∼29%, 24, and 29% of the nuclei in Δtor1, Δste20, and Δgad8 mutant cells contained at least one Rad52-YFP focus, respectively (Fig. 3, A and B). In addition, 9, 4, and 6% of the nuclei in Δtor1, Δste20, and Δgad8, respectively, displayed more than one focus per nucleus (Fig. 3B). The majority of cells in the S and early G2 phases of the cell cycle displayed Rad52-YFP foci (Fig. 3C), consistent with the idea that TORC2-Gad8 mutant cells accumulate Rad52-YFP foci because of spontaneous DNA damage that occurs during S phase. Alternatively, Rad52-YFP foci may form normally in the mutants during replication but disassemble with slow kinetics, leading to their apparent accumulation.
FIGURE 3.

Elevated levels of Rad52-YFP foci in TORC2-Gad8 mutants. A and B, spontaneous Rad52-YFP foci occur in Δtor1, Δste20, or Δgad8 cells. Cells were cultured in YE liquid medium at 30 °C until mid-log phase, stained with Hoechst and Calcofluor, and visualized live by confocal microscopy. The numbers represent the mean values of three independent experiments, with error bars representing the S.D. Bar, 20 μm. C, quantification of Rad52-YFP foci in nuclei representing different phases of the cell cycle as estimated from cellular and nuclear morphology. D, the assembly and disassembly of Rad52-YFP foci during a transient exposure to CPT. Cells were grown to mid-log phase and arrested in CPT. After 3 h, cells were collected and resuspended in fresh YE medium. Cells were photographed live, at the indicated time points, and the Rad52-YFP foci were counted as described above.
In order to distinguish between the above possibilities, we examined the dissolution rate of the Rad52-YFP foci by monitoring the time-dependent disappearance of CPT-induced Rad52-YFP foci. Following treatment with CPT, the wild type cells showed a sharp increase in the percentage of nuclei with Rad52-YFP foci, from an average of ∼13% under normal growth conditions up to ∼72% (Fig. 3D). By 6 h after removal of CPT, the percentage of nuclei displaying Rad52-YFP foci dropped to an average of ∼50%. Δtor1 or Δgad8 mutant cells also show an increase in the number of nuclei that contain Rad52-YFP foci following CPT treatment, from ∼33 to ∼86% for Δtor1 mutant cells or from ∼28 to ∼85% for Δgad8 mutant cells. Following CPT removal, Δtor1 or Δgad8 mutant cells show a similar rate of decrease in the number of Rad52-YFP foci as wild type cells, reaching their corresponding Rad52-YFP focus basal level by 16 h after the release from CPT arrest (Fig. 3D). Notably, Δtor1 or Δgad8 mutant cells show a delay of 2 h before disassembly of the foci. This delay may be associated with the delay in deactivation of Chk1 following the removal of CPT in Δtor1 cells (Fig. 2E).
Because neither Tor1 nor Gad8 play a central role in the disappearance of CPT-induced DNA damage foci, we suggest that TORC2 and Gad8 are not key factors in DNA damage repair. Rather, the accumulation of Rad52 foci in Δtor1 or Δgad8 mutant cells may represent defects in replication fork progression, which are lethal upon prolonged exposure to DNA replication stress.
Mutations in Components of TORC2-Gad8 Reduce the Rate of Spontaneous Recombination
To further probe into the mechanism that causes elevated levels of Rad52-YFP foci in TORC2-Gad8 mutants, we determined the frequency of spontaneous recombination in these mutants. For this purpose, we created Δtor1, Δste20, Δgad8, and Δrad51 mutant strains containing non-tandem direct repeats of ade6− heteroalleles flanking a functional his3+ gene (35). As shown in Fig. 4A, spontaneous mitotic intrachromosomal recombination can be scored by the recovery of ade6+ recombinants. These are classified into those obtained by gene conversion (Ade+ His+), and those created by an event that results in deletion (Ade+ His−) (35). A Δrad51 mutant strain, which served as a control, displayed a 3-fold increase in spontaneous recombination rate and almost completely lacked conversion type recombinants, as described previously (35). In contrast, TORC2-Gad8 mutant cells exhibited a 2.5–5-fold reduction in spontaneous recombination rate, compared with wild type cells (Fig. 4B). Δtor1, Δste20, and Δgad8 mutant cells are hyporecombinant for both conversion and deletion type recombinants. Therefore, the TORC2-Gad8 pathway is required for normal levels of mitotic recombination between direct repeats.
FIGURE 4.
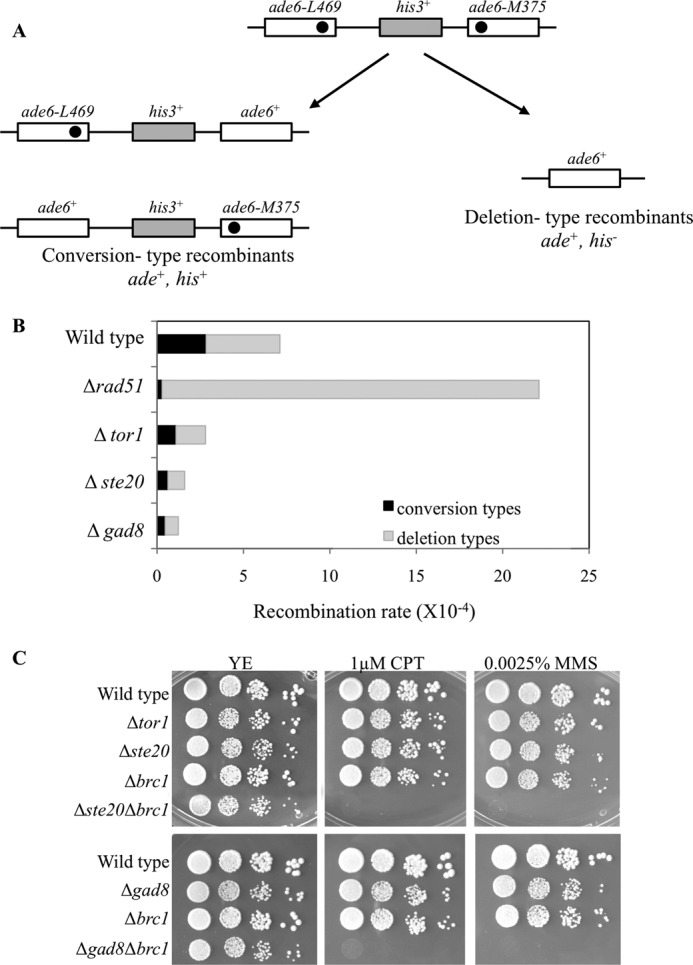
Spontaneous recombination rates are reduced in TORC2-Gad8 mutants compared with wild type. A, schematic representation of the recombination substrate and of the two types of the resulting Ade+ recombinants. Gene conversion results in Ade+ His+, whereas deletion leads to the formation of Ade+ His− colonies. B, recombination frequencies of the indicated strains (per 104 cells). Conversion types (black) or deletion types (gray) were determined by replica plating. C, genetic interactions between mutations in TORC2-Gad8 components and deletion of Brc1. 10-fold serial dilutions of logarithmic growing cells were platted on YE-rich medium, containing the indicated concentrations of MMS or CPT. Plates were incubated at 30 °C for 3–4 days.
Our data indicate that elevated levels of Rad52-YFP foci do not necessarily correlate with high recombination rates between direct repeats. Similar results were obtained from studies of Brc1, a six-BRCT domain protein (52). Δbrc1 cells show elevated levels of spontaneous Rad52-YFP foci (53) but reduced spontaneous recombination rates between direct repeats (52). Genetic interaction analysis between Δste20 or Δgad8 mutant cells and Δbrc1 revealed that the double mutant cells are viable but show additive effects with respect to sensitivity to MMS or CPT (Fig. 4C). Thus, TORC2-Gad8 works, at least partially, independently of Brc1.
The Homologous Recombination Pathway Is Essential in the Absence of TORC2-Gad8
Deletion of chk1+, cds1+, or mrc1+ in the background of Δtor1 resulted in double mutant cells that are viable but show stronger sensitivity to replication stress compared with each of the parental single mutant strains (21) (Table 2). Deletion of rad3 or rad17 in the background of Δtor1 resulted in the double mutant cells that are as sensitive to replication stress as the single mutants Δrad3 or Δrad17 (21) (Table 2), indicating an epistatic interaction between Tor1 and the main checkpoint genes.
TABLE 2.
Summary of genetic interactions involving Δtor1
Cells deleted for tor1+ were crossed with the indicated alleles. During vegetative growth, double mutant cells were phenotypically similar to the single mutant Δtor1 (None) or showed a synthetic lethal (SL) or sick (SS) interaction. If viable, double mutant strains were tested for their sensitivity to prolonged exposure to replication stress (HU, MMS, or CPT) and compared with the parental strains. The resulting double mutants showed negative genetic interactions (+) or strong negative genetic interactions (+++). Epistatic interactions were found with Δrad3 or Δrad17 in response to HU and MMS, as previously published (marked with an asterisk) (21).
| Allele | Function | Untreated | Replication stress |
|---|---|---|---|
| Δrad3* | ATR checkpoint kinase, activates Chk1 and Cds1 | None | Epistatis of rad3 |
| Δrad17* | RFC-related checkpoint protein | None | Epistatis of rad17 |
| Δchk1* | DNA damage checkpoint kinase | None | + |
| Δcds1* | DNA replication checkpoint kinase | None | + |
| Δmrc1* | Mediator of replication checkpoint 1 | None | + |
| Δswi1 | Replication fork protection complex subunit | None | + |
| Δswi3 | Replication fork protection complex subunit | None | + |
| Δrad52 | DNA recombination protein | SL | ND |
| Δrad51 | RecA family recombinase | SL | ND |
| Δrqh1 | RecQ type DNA helicase | None | + |
| Δmus81 | Holliday junction resolvase | SS | +++ |
| Δmms1 | DNA repair protein in complex with mms22 | SS | +++ |
| Δmms22 | DNA repair protein in complex with mms1 | SS | +++ |
| Δbrc1 | BRCT domain protein | None | +++ |
We extended our genetic interaction analysis by combining mutations in TORC2-Gad8 with mutations affecting different aspects of the DNA damage response: homologous recombination, rad52 and rad51 (also referred to as rhp51) (54); replication restart, mus81, mms1, mms22, and rqh1 (55–57); and fork stabilization, swi1 and swi3 (58). If viable, double mutant strains were tested for their sensitivity to prolonged exposure to replication stress and compared with the parental strains. Our findings are summarized in Table 2.
Most notably, we found significant negative interactions between deletion mutations in TORC2 and deletion mutations in rad52, rad51, mus81, mms1, or mms22. The negative genetic interactions between TORC2 mutants and Δrad51 or Δrad52 were the most severe, because tetrad analysis of the meiotic progeny revealed a highly synthetic sick to synthetic lethal phenotype of the resulting double mutant cells (Fig. 5A). Double mutant colonies did not form at the expected frequency, whereas double mutant spores that did germinate had a severe growth defect; the resulting colonies were extremely small and contained many elongated and misshapen cells (data not shown). Thus, we conclude that the major HR genes are essential for the viability of TORC2 or gad8 mutant cells.
FIGURE 5.

TORC2-Gad8 mutants show synthetic sick interactions with components of the homologous recombination pathway. A and B, tetrad analysis of heterozygous diploids with the indicated mutations. Representative spores from five asci are shown for each cross. C, strains were grown to log phase in rich medium and visualized by light microscopy. Bar, 20 μm. D and E, 10-fold serial dilution of logarithmic growing cells were plated on YE-rich medium in the presence or absence of the indicated DNA-damaging agents and incubated at 30 °C for 3–4 days.
Synthetic sick interactions were observed for crosses with Δmus81, Δmms1, and Δmms22 (Fig. 5, B–D). The resulting double mutant strain Δtor1Δmus81 produced small colonies (Fig. 5B, top) with a heterogeneous population that contained many elongated cells (Fig. 5C). In addition, Δtor1Δmus81 displayed a synergistic interaction in response to a prolonged exposure to MMS or CPT (Fig. 5D). Similar results were obtained for the Δtor1Δmms1 or Δtor1Δmms22 double mutants; double mutant colonies were significantly smaller than the corresponding single mutants (Fig. 5D) and displayed a significant increased sensitivity to prolonged replicative stress (Fig. 5D). The negative synthetic genetic interactions between TORC2-Gad8 mutant cells and Δrad52, Δrad51, Δmus81, Δmms1, or Δmms22, together with the finding of elevated levels of Rad52-YFP foci in TORC2-Gad8 mutant cells, suggest that TORC2-Gad8 mutant cells experience elevated levels of DNA damage during replication that depend on the HR pathway for repair.
In contrast, cells lacking Tor1 do not depend on the Rqh1 helicase for viability (Fig. 5B, bottom). The Rqh1 protein is a member of the RecQ DNA helicase family, homologous to the budding yeast Sgs1 protein and mammalian BLM and WRN (59). Δrqh1 mutants are hypersensitive to DNA-damaging agents and defective in the recovery from S phase arrest (59). The double mutant Δtor1Δrqh1 colonies are indistinguishable from the Δtor1 and the Δrqh1 single mutant colonies (Fig. 5B, bottom). In response to MMS, HU, or CPT the double mutant cells, Δtor1Δrqh1, showed only a mild additive sensitivity (Fig. 5E). Thus, it appears that in the absence of Tor1, cells accumulate DNA damage during replication that depends on the HR machinery for repair but not on Rqh1.
Swi1 and Swi3 represent another type of mechanism required for maintenance of genome stability. Swi1 and Swi3 form the replication fork protection complex. Deletion of swi1+ or swi3+ results in increased sensitivity to replication stress and elevated levels of spontaneous Rad52-YFP foci along with increased cell length (51, 58, 60). The Δtor1Δswi1 or Δtor1Δswi3 double mutant cells were indistinguishable from the parental single mutant parents with respect to cell length (data not shown). Moreover, Δtor1Δswi1 double mutants do not show an additive increase in the number of spontaneous Rad52-YFP foci when compared with the Δtor1 or Δswi1 single mutant cells (Fig. 6A). Thus, TORC2 and Swi1/Swi3 may work in a common pathway for the suppression of spontaneous DNA damage during cell proliferation. However, Δtor1Δswi1 or Δtor1Δswi3 double mutant cells were more sensitive to replication stress induced by HU, MMS, or CPT than the respective single mutants (Fig. 6B). This indicates that TORC2 and the fork protection complex play distinct roles in the presence of external DNA-damaging agents.
FIGURE 6.

Genetic interactions between TORC2 and the Swi1-Swi3 fork replication protection complex. A, mutations in tor1+ and swi1+ show epistasis with respect to spontaneous Rad52-YFP foci level. Strains growing logarithmically in rich medium were stained with Hoechst and imaged live. The percentage of cells containing Rad52-YFP foci was calculated as described in the legend to Fig. 3. B, Tor1 and Swi1 show additive sensitivity to replication stress.
Deletion of Tor1, Ste20, or Gad8 Causes an Increase in Minichromosome Loss Rate
Genomic instability may be manifested in an increase in chromosome loss (50, 61). Thus, we examined whether spontaneous Rad52-YFP foci observed in Δtor1, Δste20, or Δgad8 are translated into genomic instability, as was observed in mms22 or rad51 mutant cells (50, 62, 63). We used Δtor1, Δste20, and Δgad8 strains that carried a copy of the artificial Ch16. Ch16 is a highly stable 530-kb linear minichromosome that contains a centric region of chromosome III (64). Ch16 carries an ade6-M216 point mutation that is capable of complementing an ade6-M210 mutation, thus leading to an ade+ phenotype. The spontaneous loss of Ch16 was monitored in wild type, Δtor1, Δste20, or Δgad8 cells by plating colonies on medium with limiting amounts of adenine. Under these conditions, cells that have lost the minichromosome become auxotrophic for adenine and accumulate a red pigment. We scored half-sectored colonies in order to determine the frequency of minichromosome loss. We found that Δtor1, Δste20, and Δgad8 show spontaneous chromosome loss frequencies that are 10-fold higher than wild type (Table 3), indicating that TORC2 mutant cells are constantly experiencing genomic instability.
TABLE 3.
TORC2-Gad8 is required for the maintenance of an artificial minichromosome
Ch16-containing colonies were plated on adenine-limited plates, and the number of half-sectored colonies was determined. The number of colonies counted is indicated in parentheses.
| Strain | Ch16 loss ±S.D. | No. of colonies |
|---|---|---|
| % | ||
| Wild type | 0.02 ±0.02 | 14,071 |
| Δtor1 | 0.15 ±0.07 | 15,988 |
| Δste20 | 0.48 ±0.07 | 5841 |
| Δgad8 | 0.21 ±0.03 | 9941 |
DISCUSSION
Disruption of TORC2 in fission yeast results in cells that are unable to respond properly to variety of stress conditions, including nutritional, osmotic, oxidative, and temperature stresses. More recently, we discovered that TORC2 is also required for survival in the presence of the DNA-damaging agents HU or MMS (21). Here we explored the role of TORC2 under DNA-damaging conditions. We demonstrate that TORC2 is required to maintain genome stability during DNA replication and has a key role in cell survival under prolonged replication stress conditions. These new findings uncover new aspects of TORC2 biological functions and add a new player to the arena of signaling pathways that affect survival under DNA-damaging conditions.
In S. pombe, the DNA damage and replication stress checkpoints are both activated by Rad3 (ATR homologue), which in turn activates its downstream protein kinases, Chk1 (in response to MMS or CPT) or Cds1 (in response to HU) (47, 65, 66). Several observations support a model in which TORC2-Gad8 is dispensable for the initial stages of the DNA damage response. This includes cell cycle elongation of TORC2-Gad8 mutant cells in response to DNA damage (Fig. 2C), activation of Chk1 by phosphorylation in response to CPT (Fig. 2D), and an increase in Rad52-YFP foci in response to replication stress, indicative of proficient sensing of the damage at the level of the DNA (Fig. 3D). Consistently, unlike DNA damage checkpoint mutants, TORC2-Gad8 mutant cells are resistant to short exposure to DNA-damaging conditions and do not die due to illegitimate entry into mitosis.
An important observation is that transient exposure to replicative stress does not impair the viability of TORC2-Gad8 mutant cells. Additionally, TORC2-Gad8 mutant cells are insensitive to IR. The resistance of TORC2-Gad8 mutant cells to transient exposure to replicative stress is in contrast to the sensitivity of HR mutants, such as rad52 and rad51, to such conditions (67). Moreover, following a transient CPT treatment, which markedly elevates the number of Rad52 foci, we detected no major defects in returning to basal Rad52 foci levels in Δtor1 or Δgad8 mutant cells (Fig. 3D). Thus, it is unlikely that TORC2-Gad8 plays a major role in DNA damage repair. This leaves us with the question of why TORC2-Gad8 mutants are highly sensitive to continuous replication damage. We observed two major defects of TORC2-Gad8 mutant cells that are relevant for the DNA damage-sensitive phenotype. One is elevated spontaneous Rad52 foci, particularly in S phase (Fig. 3C), and the second is a delay in dephosphorylation and thus deactivation of Chk1 (Fig. 2E). The presence of Rad52 foci may represent defects in DNA replication (e.g. incompletion of replication, as has been suggested previously (52). The number of Rad52 foci is elevated to very high levels in TORC2-Gad8 mutant cells in response to replication stress (Fig. 3D). It is thus plausible that under repeated rounds of replication in the presence of replication stress, cells accumulate lesions that eventually prevent deactivation of the checkpoint and lead to cell death. The mechanism required for signal termination following DNA damage recovery is currently poorly understood. However, an interesting possibility is that dephosphorylation of Chk1 is dependent on Dis2, a specific protein phosphatase (PP1) (68). We plan to determine in future experiments whether Tor1 (TORC2) and Dis2 may functionally interact.
The sensitivity of tor1 mutant cells under prolonged exposure to replication stress is reminiscent of that of mutant genes affecting the replication fork protection complex, Swi1-Swi3. Like TORC2 mutant cells, Δswi1 exhibits an elevated level of Rad52-YFP foci (51, 58). In addition, similar to Δtor1, Δswi1 and Δswi3 are synthetic sick with a null mutation in Mus81 (Fig. 5B) (58). The level of Rad52-YFP foci is not further elevated in the double mutant Δtor1Δswi1 cells, suggesting that under normal growth conditions, TORC2 and the replication protection complex may work in a common pathway (Fig. 6A). However, loss of Swi1 or Swi3 induced Chk1 phosphorylation during vegetative growth (51), whereas no such activation was observed in TORC2 mutants (Fig. 2, D and E). Thus, the Rad52-YFP foci observed in TORC2-Gad8 mutant cells may represent a form of DNA damage that is not recognized by the checkpoint machinery. Under replicative stress, Δtor1Δswi1 cells show additive interactions, suggesting that these proteins may also work through independent pathways.
The phenotype of disruption of the TORC2-Gad8 pathway also resembles the phenotype of Δbrc1 mutant cells. Brc1 has also been implicated in response to replication-associated DNA damage (53). Δbrc1 mutant cells are sensitive to prolonged but not acute exposure to replication stressors and show an elevated level of spontaneous Rad52-YFP foci, which, as in TORC2 mutant cells, are not associated with activation of Chk1. In addition, as in TORC2 mutant cells, spontaneous recombination rates are decreased in Δbrc1 mutant cells (52). As suggested above, Rad52 foci may not represent sites of recombination but incompletion of replication under conditions of replication stress.
Our findings show that TORC2-Gad8 is critical for the suppression of genomic instability by preventing chromosome loss (Table 3). In the absence of Ste20, we observed a higher rate of minichromosome loss compared with Δtor1 or Δgad8 mutant cells. ste20+ and wat1+/pop3+, a common component for TORC1 and TORC2, have previously been isolated in a screen for genes that show negative genetic interactions with Separase/Cut1 (69). Cut1 is a conserved protease essential for anaphase progression (70). Recently, the temperature-sensitive allele tor1-D was shown to have negative interactions with cut1 (Separase) mutants, as well as being sensitive to Cut1 overexpression (25). This suggests that TORC2 and Separase/Cut1 share an essential function, consistent with our observed increase in chromosome loss rates (Table 3).
In conclusion, our studies suggest that the TORC2-Gad8 pathway is required for genomic instability, in particular during replication, and contributes to cell survival under constant replication stress. These novel roles of TORC2 in fission yeast may be conserved human cells and thus may be relevant for the growing amount of evidence implicating TORC2 in cancer development.
Acknowledgments
We thank P. Russell, A. M. Carr, K. Shiozaki, S. Whitehall, M. C. Whitby, F. Osman, and the Yeast Genetic Resource Center, Japan, for strains and members of the Kupiec laboratory for encouragement and support.
This work was supported by Association of International Cancer Research Grant 11-0281 and Open University of Israel Research Fund Grant 37076 (to R. W.).
- TORC1 and -2
- TOR complex 1 and 2, respectively
- HU
- hydroxyurea
- MMS
- methyl-methane sulfonate
- IR
- ionizing radiation
- HR
- homologous recombination
- CPT
- camptothecin
- Ch16
- minichromosome 16.
REFERENCES
- 1. Dazert E., Hall M. N. (2011) mTOR signaling in disease. Curr. Opin. Cell Biol. 23, 744–755 [DOI] [PubMed] [Google Scholar]
- 2. Laplante M., Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lovejoy C. A., Cortez D. (2009) Common mechanisms of PIKK regulation. DNA Repair 8, 1004–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sarbassov D. D., Ali S. M., Kim D. H., Guertin D. A., Latek R. R., Erdjument-Bromage H., Tempst P., Sabatini D. M. (2004) Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302 [DOI] [PubMed] [Google Scholar]
- 5. Sarbassov D. D., Ali S. M., Sengupta S., Sheen J. H., Hsu P. P., Bagley A. F., Markhard A. L., Sabatini D. M. (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168 [DOI] [PubMed] [Google Scholar]
- 6. Loewith R., Jacinto E., Wullschleger S., Lorberg A., Crespo J. L., Bonenfant D., Oppliger W., Jenoe P., Hall M. N. (2002) Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 10, 457–468 [DOI] [PubMed] [Google Scholar]
- 7. Wullschleger S., Loewith R., Hall M. N. (2006) TOR signaling in growth and metabolism. Cell 124, 471–484 [DOI] [PubMed] [Google Scholar]
- 8. Zinzalla V., Stracka D., Oppliger W., Hall M. N. (2011) Activation of mTORC2 by association with the ribosome. Cell 144, 757–768 [DOI] [PubMed] [Google Scholar]
- 9. Weisman R., Choder M. (2001) The fission yeast TOR homolog, tor1+, is required for the response to starvation and other stresses via a conserved serine. J. Biol. Chem. 276, 7027–7032 [DOI] [PubMed] [Google Scholar]
- 10. Matsuo T., Otsubo Y., Urano J., Tamanoi F., Yamamoto M. (2007) Loss of the TOR kinase Tor2 mimics nitrogen starvation and activates the sexual development pathway in fission yeast. Mol. Cell. Biol. 27, 3154–3164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hayashi T., Hatanaka M., Nagao K., Nakaseko Y., Kanoh J., Kokubu A., Ebe M., Yanagida M. (2007) Rapamycin sensitivity of the Schizosaccharomyces pombe tor2 mutant and organization of two highly phosphorylated TOR complexes by specific and common subunits. Genes Cells 12, 1357–1370 [DOI] [PubMed] [Google Scholar]
- 12. Aspuria P. J., Sato T., Tamanoi F. (2007) The TSC/Rheb/TOR signaling pathway in fission yeast and mammalian cells. Temperature sensitive and constitutive active mutants of TOR. Cell Cycle 6, 1692–1695 [DOI] [PubMed] [Google Scholar]
- 13. van Slegtenhorst M., Carr E., Stoyanova R., Kruger W. D., Henske E. P. (2004) Tsc1+ and tsc2+ regulate arginine uptake and metabolism in Schizosaccharomyces pombe. J. Biol. Chem. 279, 12706–12713 [DOI] [PubMed] [Google Scholar]
- 14. Alvarez B., Moreno S. (2006) Fission yeast Tor2 promotes cell growth and represses cell differentiation. J. Cell Sci. 119, 4475–4485 [DOI] [PubMed] [Google Scholar]
- 15. Nakashima A., Sato T., Tamanoi F. (2010) Fission yeast TORC1 regulates phosphorylation of ribosomal S6 proteins in response to nutrients and its activity is inhibited by rapamycin. J. Cell Sci. 123, 777–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weisman R., Roitburg I., Schonbrun M., Harari R., Kupiec M. (2007) Opposite effects of tor1 and tor2 on nitrogen starvation responses in fission yeast. Genetics 175, 1153–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takahara T., Maeda T. (2012) TORC1 of fission yeast is rapamycin-sensitive. Genes Cells 17, 698–708 [DOI] [PubMed] [Google Scholar]
- 18. Kawai M., Nakashima A., Ueno M., Ushimaru T., Aiba K., Doi H., Uritani M. (2001) Fission yeast tor1 functions in response to various stresses, including nitrogen starvation, high osmolarity, and high temperature. Curr. Genet. 39, 166–174 [DOI] [PubMed] [Google Scholar]
- 19. Matsuo T., Kubo Y., Watanabe Y., Yamamoto M. (2003) Schizosaccharomyces pombe AGC family kinase Gad8p forms a conserved signaling module with TOR and PDK1-like kinases. EMBO J. 22, 3073–3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Petersen J., Nurse P. (2007) TOR signalling regulates mitotic commitment through the stress MAP kinase pathway and the Polo and Cdc2 kinases. Nat. Cell Biol. 9, 1263–1272 [DOI] [PubMed] [Google Scholar]
- 21. Schonbrun M., Laor D., López-Maury L., Bähler J., Kupiec M., Weisman R. (2009) TOR complex 2 controls gene silencing, telomere length maintenance, and survival under DNA-damaging conditions. Mol. Cell. Biol. 29, 4584–4594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Urano J., Sato T., Matsuo T., Otsubo Y., Yamamoto M., Tamanoi F. (2007) Point mutations in TOR confer Rheb-independent growth in fission yeast and nutrient-independent mammalian TOR signaling in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 104, 3514–3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weisman R., Finkelstein S., Choder M. (2001) Rapamycin blocks sexual development in fission yeast through inhibition of the cellular function of an FKBP12 homolog. J. Biol. Chem. 276, 24736–24742 [DOI] [PubMed] [Google Scholar]
- 24. Weisman R., Roitburg I., Nahari T., Kupiec M. (2005) Regulation of leucine uptake by tor1+ in Schizosaccharomyces pombe is sensitive to rapamycin. Genetics 169, 539–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ikai N., Nakazawa N., Hayashi T., Yanagida M. (2011) The reverse, but coordinated, roles of Tor2 (TORC1) and Tor1 (TORC2) kinases for growth, cell cycle and separase-mediated mitosis in Schizosaccharomyces pombe. Open. Biol. 1, 110007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ikeda K., Morigasaki S., Tatebe H., Tamanoi F., Shiozaki K. (2008) Fission yeast TOR complex 2 activates the AGC-family Gad8 kinase essential for stress resistance and cell cycle control. Cell Cycle 7, 358–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tatebe H., Shiozaki K. (2010) Rab small GTPase emerges as a regulator of TOR complex 2. Small Gtpases 1, 180–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moynihan E. B., Enoch T. (1999) Liz1p, a novel fission yeast membrane protein, is required for normal cell division when ribonucleotide reductase is inhibited. Mol. Biol. Cell 10, 245–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stolz J., Caspari T., Carr A. M., Sauer N. (2004) Cell division defects of Schizosaccharomyces pombe liz1− mutants are caused by defects in pantothenate uptake. Eukaryot. Cell 3, 406–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lisby M., Rothstein R., Mortensen U. H. (2001) Rad52 forms DNA repair and recombination centers during S phase. Proc. Natl. Acad. Sci. U.S.A. 98, 8276–8282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alvaro D., Lisby M., Rothstein R. (2007) Genome-wide analysis of Rad52 foci reveals diverse mechanisms impacting recombination. PLoS Genet. 3, 2439–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moreno S., Klar A., Nurse P. (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194, 795–823 [DOI] [PubMed] [Google Scholar]
- 33. Forsburg S. L., Rhind N. (2006) Basic methods for fission yeast. Yeast 23, 173–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Foiani M., Marini F., Gamba D., Lucchini G., Plevani P. (1994) The B subunit of the DNA polymerase α-primase complex in Saccharomyces cerevisiae executes an essential function at the initial stage of DNA replication. Mol. Cell. Biol. 14, 923–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Osman F., Adriance M., McCready S. (2000) The genetic control of spontaneous and UV-induced mitotic intrachromosomal recombination in the fission yeast Schizosaccharomyces pombe. Curr. Genet. 38, 113–125 [DOI] [PubMed] [Google Scholar]
- 36. Lea D., Coulson C. (1949) The distribution of the numbers of mutants in bacterial populations. J. Genet. 49, 264–285 [DOI] [PubMed] [Google Scholar]
- 37. Hsiang Y. H., Lihou M. G., Liu L. F. (1989) Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 49, 5077–5082 [PubMed] [Google Scholar]
- 38. Wan S., Capasso H., Walworth N. C. (1999) The topoisomerase I poison camptothecin generates a Chk1-dependent DNA damage checkpoint signal in fission yeast. Yeast 15, 821–828 [DOI] [PubMed] [Google Scholar]
- 39. Sinha R. P., Häder D. P. (2002) UV-induced DNA damage and repair. A review. Photochem. Photobiol. Sci. 1, 225–236 [DOI] [PubMed] [Google Scholar]
- 40. Michel B., Grompone G., Florès M. J., Bidnenko V. (2004) Multiple pathways process stalled replication forks. Proc. Natl. Acad. Sci. U.S.A. 101, 12783–12788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rhind N., Russell P. (1998) The Schizosaccharomyces pombe S-phase checkpoint differentiates between different types of DNA damage. Genetics 149, 1729–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cavero S., Limbo O., Russell P. (2010) Critical functions of Rpa3/Ssb3 in S-phase DNA damage responses in fission yeast. PLoS Genet. 6, e1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tatebe H., Morigasaki S., Murayama S., Zeng C. T., Shiozaki K. (2010) Rab-family GTPase regulates TOR complex 2 signaling in fission yeast. Curr. Biol. 20, 1975–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kominami K., Seth-Smith H., Toda T. (1998) Apc10 and Ste9/Srw1, two regulators of the APC-cyclosome, as well as the CDK inhibitor Rum1 are required for G1 cell-cycle arrest in fission yeast. EMBO J. 17, 5388–5399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jimenez G., Yucel J., Rowley R., Subramani S. (1992) The rad3+ gene of Schizosaccharomyces pombe is involved in multiple checkpoint functions and in DNA repair. Proc. Natl. Acad. Sci. U.S.A. 89, 4952–4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boddy M. N., Furnari B., Mondesert O., Russell P. (1998) Replication checkpoint enforced by kinases Cds1 and Chk1. Science 280, 909–912 [DOI] [PubMed] [Google Scholar]
- 47. Carr A. M. (2002) DNA structure dependent checkpoints as regulators of DNA repair. DNA Repair 1, 983–994 [DOI] [PubMed] [Google Scholar]
- 48. Ostermann K., Lorentz A., Schmidt H. (1993) The fission yeast rad22 gene, having a function in mating-type switching and repair of DNA damages, encodes a protein homolog to Rad52 of Saccharomyces cerevisiae. Nucleic Acids Res. 21, 5940–5944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. New J. H., Sugiyama T., Zaitseva E., Kowalczykowski S. C. (1998) Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature 391, 407–410 [DOI] [PubMed] [Google Scholar]
- 50. Dovey C. L., Russell P. (2007) Mms22 preserves genomic integrity during DNA replication in Schizosaccharomyces pombe. Genetics 177, 47–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Noguchi E., Noguchi C., Du L. L., Russell P. (2003) Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol. Cell. Biol. 23, 7861–7874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bass K. L., Murray J. M., O'Connell M. J. (2012) Brc1-dependent recovery from replication stress. J. Cell Sci. 125, 2753–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Williams J. S., Williams R. S., Dovey C. L., Guenther G., Tainer J. A., Russell P. (2010) γH2A binds Brc1 to maintain genome integrity during S-phase. EMBO J. 29, 1136–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Raji H., Hartsuiker E. (2006) Double-strand break repair and homologous recombination in Schizosaccharomyces pombe. Yeast 23, 963–976 [DOI] [PubMed] [Google Scholar]
- 55. Boddy M. N., Gaillard P. H., McDonald W. H., Shanahan P., Yates J. R., 3rd, Russell P. (2001) Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107, 537–548 [DOI] [PubMed] [Google Scholar]
- 56. Dovey C. L., Aslanian A., Sofueva S., Yates J. R., 3rd, Russell P. (2009) Mms1-Mms22 complex protects genome integrity in Schizosaccharomyces pombe. DNA Repair 8, 1390–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stewart E., Chapman C. R., Al-Khodairy F., Carr A. M., Enoch T. (1997) rqh1+, a fission yeast gene related to the Bloom's and Werner's syndrome genes, is required for reversible S phase arrest. EMBO J. 16, 2682–2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Noguchi E., Noguchi C., McDonald W. H., Yates J. R., 3rd, Russell P. (2004) Swi1 and Swi3 are components of a replication fork protection complex in fission yeast. Mol. Cell. Biol. 24, 8342–8355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Win T. Z., Mankouri H. W., Hickson I. D., Wang S. W. (2005) A role for the fission yeast Rqh1 helicase in chromosome segregation. J. Cell Sci. 118, 5777–5784 [DOI] [PubMed] [Google Scholar]
- 60. Sommariva E., Pellny T. K., Karahan N., Kumar S., Huberman J. A., Dalgaard J. Z. (2005) Schizosaccharomyces pombe Swi1, Swi3, and Hsk1 are components of a novel S-phase response pathway to alkylation damage. Mol. Cell. Biol. 25, 2770–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hartwell L. H., Kastan M. B. (1994) Cell cycle control and cancer. Science 266, 1821–1828 [DOI] [PubMed] [Google Scholar]
- 62. Meister P., Poidevin M., Francesconi S., Tratner I., Zarzov P., Baldacci G. (2003) Nuclear factories for signalling and repairing DNA double strand breaks in living fission yeast. Nucleic Acids Res. 31, 5064–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Muris D. F., Vreeken K., Carr A. M., Murray J. M., Smit C., Lohman P. H., Pastink A. (1996) Isolation of the Schizosaccharomyces pombe RAD54 homologue, rhp54+, a gene involved in the repair of radiation damage and replication fidelity. J. Cell Sci. 109, 73–81 [DOI] [PubMed] [Google Scholar]
- 64. Niwa O., Matsumoto T., Chikashige Y., Yanagida M. (1989) Characterization of Schizosaccharomyces pombe minichromosome deletion derivatives and a functional allocation of their centromere. EMBO J. 8, 3045–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Osborn A. J., Elledge S. J., Zou L. (2002) Checking on the fork. The DNA-replication stress-response pathway. Trends Cell Biol. 12, 509–516 [DOI] [PubMed] [Google Scholar]
- 66. Humphrey T. (2000) DNA damage and cell cycle control in Schizosaccharomyces pombe. Mutat. Res. 451, 211–226 [DOI] [PubMed] [Google Scholar]
- 67. Sheedy D. M., Dimitrova D., Rankin J. K., Bass K. L., Lee K. M., Tapia-Alveal C., Harvey S. H., Murray J. M., O'Connell M. J. (2005) Brc1-mediated DNA repair and damage tolerance. Genetics 171, 457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. den Elzen N. R., O'Connell M. J. (2004) Recovery from DNA damage checkpoint arrest by PP1-mediated inhibition of Chk1. EMBO J. 23, 908–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yuasa T., Hayashi T., Ikai N., Katayama T., Aoki K., Obara T., Toyoda Y., Maruyama T., Kitagawa D., Takahashi K., Nagao K., Nakaseko Y., Yanagida M. (2004) An interactive gene network for securin-separase, condensin, cohesin, Dis1/Mtc1 and histones constructed by mass transformation. Genes Cells 9, 1069–1082 [DOI] [PubMed] [Google Scholar]
- 70. Oliveira R. A., Hamilton R. S., Pauli A., Davis I., Nasmyth K. (2010) Cohesin cleavage and Cdk inhibition trigger formation of daughter nuclei. Nat. Cell Biol. 12, 185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]