

Background: Oxidative thiol modifications are thought to be one of the major effects of peroxynitrite on proteins.
Results: Quantitative redox proteomics identified proteins thiol-modified by peroxynitrite, and cells lacking these proteins show an impaired recovery.
Conclusion: Thiol modifications caused by peroxynitrite in Escherichia coli are highly specific for a small number of selected proteins.
Significance: Thiol modifications regulate the activity of proteins under peroxynitrite stress.
Keywords: Microbiology, Nitrosative Stress, Proteomics, Redox, Thiol, Peroxynitrite
Abstract
Peroxynitrite is a highly reactive chemical species with antibacterial properties that are synthesized in immune cells. In a proteomic approach, we identified specific target proteins of peroxynitrite-induced modifications in Escherichia coli. Although peroxynitrite caused a fairly indiscriminate nitration of tyrosine residues, reversible modifications of protein thiols were highly specific. We used a quantitative redox proteomic method based on isotope-coded affinity tag chemistry and identified four proteins consistently thiol-modified in cells treated with peroxynitrite as follows: AsnB, FrmA, MaeB, and RidA. All four were required for peroxynitrite stress tolerance in vivo. Three of the identified proteins were modified at highly conserved cysteines, and MaeB and FrmA are known to be directly involved in the oxidative and nitrosative stress response in E. coli. In in vitro studies, we could show that the activity of RidA, a recently discovered enamine/imine deaminase, is regulated in a specific manner by the modification of its single conserved cysteine. Mutation of this cysteine 107 to serine generated a constitutively active protein that was not susceptible to peroxynitrite.
Introduction
Oxidative and nitrosative stress, caused by increased levels of reactive oxygen and nitrogen species (ROS4 and RNS), leads to cellular damage and is considered to be a major factor in many pathologies. The response of cells to oxidative and nitrosative stressors such as hydrogen peroxide, superoxide, and nitric oxide has been extensively studied in the past, and much of our knowledge about the mechanisms of these responses has been discovered in model organisms such as Escherichia coli (1–3). As more of these studies were conducted, it became apparent that ROS and RNS do not only have damaging effects on the cell but also play roles in cellular signaling and regulation. Thus, the concept of “redox regulation” emerged. Many of the regulatory mechanisms discovered are mediated through proteins that have redox-sensitive cysteines (4). Cysteine is predestined as a sensor of redox changes in the cellular environment, because its thiol group cannot only be modified easily through oxidation but several of these modifications, including sulfenic acids, disulfides, thiolations, and nitrosylations, are reversible in vivo as well. This led to a paradigmatic model; a redox-regulated protein typically contains one or several redox-sensing cysteines, which are in the free thiol state in the reducing environment of the cytosol. When the cell encounters exogenous or endogenous oxidative and/or nitrosative stress, the redox-sensitive cysteine(s) become oxidized, which leads to a change in protein structure, frequently inactivating the protein in question but sometimes activating a function that is required under the redox stress conditions encountered by the cell. Redox-regulated proteins include metabolic enzymes, regulators, kinases, chaperones, and other key players in cellular signaling (1, 5–7). Once the cell has overcome the redox stress, cellular systems such as the glutaredoxin or thioredoxin system reduce the redox-sensing cysteines and return the protein's activity back to “normal.” This regulation is typically highly specific, and proteins have been shown to be oxidized exclusively by selected nitrosative or oxidative agents (8, 9). Even very strong oxidants such as hydrogen peroxide, which, given its high redox potential of +1349 mV (10), should be capable of oxidizing virtually every thiol in a cell, only have relatively few specific targets in the cell. This selectivity can be explained by the fact that thiol-redox reactions are governed much more by kinetic constraints than by thermodynamics.
To expand our knowledge of cysteines affected by oxidative and nitrosative stressors, we looked in this study at the effects of peroxynitrite (ONOO−) on E. coli. ONOO− is a highly reactive species with antibacterial properties that can be synthesized in high concentrations (up to 1 mm min−1) by immune cells (11). Immune cells release this RNS upon contact with invading microorganisms. Peroxynitrite is the product of the recombination of the reactive oxygen species superoxide anion (O2˙̄) and the reactive nitrogen species nitric oxide (NO•) and seems to be more potent than the single species by themselves (12). Some of its molecular effects are known as follows: peroxynitrite causes damage to lipids, DNA, and proteins. In other organisms, it has been shown that proteins are modified through tyrosine nitration and that thiol modifications are another source of protein damage and/or regulation by peroxynitrite (13). In a recent study in E. coli, the effect of ONOO− on the transcriptome was examined (14). Interestingly, typical oxidative stress response genes were up-regulated, although genes involved in a nitrosative stress response were largely unaffected. In our study, we particularly examined oxidative and nitrosative modifications of E. coli proteins under peroxynitrite stress conditions. Although tyrosine nitration can be detected in a major part of E. coli proteins visible on a protein gel, our detailed mass spectrometry-based analysis of modified cysteines revealed that only a small subset of E. coli proteins is modified at cysteine residues in vivo. These proteins were AsnB, FrmA, MaeB, and RidA. Interestingly, all four proteins had positive effects on the growth of E. coli under peroxynitrite stress conditions. We studied the implications of the thiol modifications in the protein RidA in more detail. RidA was recently discovered to act as enamine/imine deaminase (15) and has not yet been implicated in nitrosative stress. It contains a single cysteine (Cys-107) that is conserved in many species. We show that this protein is specifically affected by RNS. RidA is an enzyme that accelerates the formation of 2-ketobutyrate from threonine catalyzed by the threonine deaminase IlvA. In in vitro experiments, we could show that Cys-107 of RidA has a regulatory function, is specifically modified by nitrosative stressors, and its oxidation inhibits its catalytic activity. However, Cys-107 is not directly involved in the catalytic function of RidA, and a mutation to serine results in a fully active but “redox-dead” enzyme.
EXPERIMENTAL PROCEDURES
Cell Growth and Maintenance
E. coli strains (Table 1) were grown on LB plates for routine maintenance and in liquid MOPS minimal media (16) supplemented with 10 μm thiamine at 37 °C and 125 rpm in a water bath for physiological stress experiments. MOPS minimal media were buffered at pH 7.4. Cell density was monitored spectrophotometrically at 600 nm. When an OD600 of ∼0.4 was reached, the culture was split into subcultures of 20 ml, and peroxynitrite was added to the subcultures to a final concentration between 500 μm and 3 mm. Because of the instability of peroxynitrite, the concentration of the commercially available peroxynitrite stock solution in 0.3 m NaOH (Cayman, Ann Arbor, MI) was determined spectrophotometrically immediately before the experiment using the extinction coefficient of ϵ302 = 1670 m−1 cm−1. Volumes of ONOO− stock added to the culture did not exceed 2.5% (v/v), and the identical volume of 0.3 m NaOH was used as vehicle control in experiments.
TABLE 1.
Overview of E. coli strains used in this study
Donor strains were obtained from the Keio collection at the National BioResource Project (National Institute of Genomics, Japan) (17).
| E. coli strain | Genotype | Source/donor strain for P1 transduction |
|---|---|---|
| MG1655 | F− λ− ilvG− rfb-50 rph-1 | ATCC strain 700926, American Type Culture Collection/− |
| BL21 (DE3) | F− ompT− gal− dcm− lon− hsdSB (rB− mB−)λ (DE3 (lacI lacUV5-T7 gene 1 ind1 sam7 nin5)) | Stratagene, Santa Clara, CA/− |
| CL043 | MG1655 asnB::Kan | This work/E. coli JW0660 |
| CL044 | MG1655 frmA::Kan | This work/E. coli JW0347 |
| CL045 | MG1655 maeB::Kan | This work/E. coli JW2447 |
| CL046 | MG1655 ridA::Kan | This work/E. coli JW5755 |
| CL048 | BL21 (DE3) ridA::Kan | This work/E. coli JW5755 |
| CL053 | BL21 (DE3) pCL001 (ridA in pET11-a) | This work/− |
| CL054 | CL048 pCL002 (ridA Cys-107Ser in pET11-a) | This work/− |
Preparation of Deletion Strains
The deletion strains CL043 to CL046 and CL048 (see Table 1) were obtained by P1 transduction of gene knock-outs from the Keio Collection from the National BioResource Project (National Institute of Genomics, Japan) (17) into E. coli K12 MG1655 and BL21(DE3). The P1 transduction procedure was performed as described elsewhere (18). The successful transduction of the gene knock-out of interest was verified by PCR with both strain-specific primers (Table 2) and kanamycin resistance cassette-specific primers as described (17). The Lac phenotype, which distinguishes donor and recipient strain, was tested on MacConkey agar.
TABLE 2.
Primers used to verify the successful knock-out of genes in strains used in this study
| Gene | Primer | Sequence (5′ → 3′) |
|---|---|---|
| asnB | U_asnB | GGATTCGAACCTGTGACCC |
| D_asnB | CCAGCTGGATTTACCCGTC | |
| Internal asnB to U | GCAGCAGGTTTCTGATCAGC | |
| Internal asnB to D | GCTGATCAGAAACCTGCTGC | |
| frmA | U_frmA | CCTGATCAACCATGATTTCCG |
| D_frmA | CCAGTACTCCGGAAGAGAAG | |
| Internal frmA to U | CACGCATACCATGAGCCTG | |
| Internal frmA to D | CAAATGCAACGGCAGCACG | |
| maeB | U_maeB | GTTAGCGTCATAATGCCAATTG |
| D_maeB | CGAATTCCTTCCCAGGTCG | |
| Internal maeB to U | GATCAGGATCTGAGCTTTGG | |
| Internal maeB to D | CCAAAGCTCAGATCCTGATC | |
| ridA | U_ridA | CGTTAAGCTATTTCCCGTGGC |
| D_ridA | GCACGGTAATTGACCATATCC | |
| Internal ridA to U | GGTCAGATCCCGGTAAATCC | |
| Internal ridA to D | GGATTTACCGGGATCTGACC |
Detection of Nitrotyrosine by Western Blot
E. coli MG1655 was stressed with 1, 2, or 3 mm peroxynitrite, and 0.3 m NaOH was used as a vehicle control. Immediately before and 5, 16, and 60 min after stress treatment, 1-ml aliquots were harvested by centrifugation (16,100 × g, 4 °C, 2 min) and washed twice with 1 ml of ice-cold 2× phosphate-buffered saline (PBS), pH 7.4. The 2× PBS buffer was prepared by a 1:5 dilution of 10× PBS stock (137 mm sodium chloride, 2.7 mm potassium chloride, 10 mm disodium hydrogen phosphate, and 1.8 mm potassium dihydrogen phosphate). The cell pellets were resuspended in ice-cold 2× PBS in a volume that was adjusted to the OD600 (80 μl at an OD600 of 0.4). The samples were lysed in 1× NuPAGE SDS sample buffer and 1× NuPAGE reducing agent at 95 °C for 10 min. Additionally, the samples were sonicated in a VialTweeter sonicator (Hielscher Ultrasonics, Teltow, Germany) for 15 s at a cycle of 0.5 s at an amplitude of 100% to shear DNA. Of each sample, 15 μl were applied to an SDS-PAGE 4–12% BisTris polyacrylamide gel (Invitrogen). As control, 10 μl of a 1 μg/μl nitrotyrosine/BSA solution (Sigma) was applied. The electrophoresis was performed under reducing conditions according to the protocol provided by the manufacturer. Proteins were transferred to a nitrocellulose membrane using the iBlot Gel Transfer System (Invitrogen). Subsequently, the nitrotyrosine antigen was detected with monoclonal rabbit anti-nitrotyrosine antibodies (Sigma) diluted 1:1000 in 2% milk powder in 1× TBS and a secondary goat anti-rabbit IgG horse radish peroxidase conjugate (Bio-Rad) diluted 1:2000 in 2% milk powder in 1× TBS.
NOxICAT Labeling of Protein Extracts
The quantification of the thiol redox proteome was performed according to the recently described NOxICAT method (19). Briefly, the NOxICAT method selectively labels reduced cysteines in a complex sample with light ICAT reagent (20) and reversibly oxidized cysteines with heavy ICAT under low oxygen conditions.
Bacterial cultures grown in MOPS minimal medium were exposed to 1 and 3 mm peroxynitrite for 16 and 60 min in the medium. MOPS minimal medium is buffered at pH 7.4. Initial steps of the sample preparation were carried out under low light conditions, due to the known UV sensitivity of S-nitrosylated proteins. To prevent artifactual thiol oxidation, samples were harvested in argon-flushed containers, and buffers used were equilibrated in small volumes (5 ml) for at least 4 h in an anaerobic chamber (Coy Labs, Ann Arbor, MI) prior to use. For each sample, 2 ml of cell culture were harvested by centrifugation (16, 100 × g, 2 min, 4 °C) and washed twice with ice-cold anaerobic 2× PBS. The 2× PBS buffer was prepared by 1:5 dilution of 10× PBS stock (137 mm sodium chloride, 2.7 mm potassium chloride, 10 mm disodium hydrogen phosphate, and 1.8 mm potassium dihydrogen phosphate). The cell pellet was then resuspended in an anaerobic mixture of 80 μl of denaturing alkylation buffer (6 m urea, 0.5% (w/v) SDS, 10 mm EDTA, 200 mm Tris-HCl, pH 8.5) and the content of one vial of cleavable light ICAT reagent (AB Sciex, Framingham, MA) dissolved in 20 μl of acetonitrile (ACN). Cells were disrupted in this alkylation mix by sonication in a pre-chilled (4 °C) VialTweeter instrument (Hielscher Ultrasonics, Teltow, Germany) with a cycle of 0.5 s at an amplitude of 90% three times for 1 min interrupted by a 1-min incubation on ice. The sample was then briefly spun down (30 s, 4 °C, 16,100 × g) and incubated at 1300 rpm for 2 h at 37 °C in the dark in a thermomixer (Eppendorf, Hamburg, Germany).
Proteins were precipitated by adding 400 μl of pre-chilled (−20 °C) acetone and incubated overnight at −20 °C. After centrifugation (16,100 × g, 4 °C, 60 min), the protein pellet was washed twice by rinsing with 400 μl of pre-chilled acetone (−20 °C). Subsequently, the protein pellet was dried and then dissolved in a mixture of 80 μl of denaturing alkylation buffer, 2 μl of 50 mm tris(2-carboxyethyl)phosphine hydrochloride (TCEP) as a thiol-reducing agent, and the contents of one vial of cleavable heavy ICAT dissolved in 20 μl of ACN. The sample was incubated at 1300 rpm in a thermomixer for 2 h at 37 °C. After incubation, proteins were precipitated with 400 μl of prechilled acetone and stored for 4 h at −20 °C. After centrifugation (16,100 × g, 4 °C, 60 min), the protein pellet was washed and dried as described above. The resulting pellet was dissolved in 80 μl of denaturing buffer from the ICAT methods development kit and 20 μl of ACN. The sample was mixed with 100 μl of 0.125 μg/μl trypsin solution (Invitrogen) and incubated 12–16 h at 37 °C without shaking. The ICAT-labeled peptides were purified by cation exchange and avidin affinity with materials provided with the ICAT methods development kit according to the manufacturer's instructions. The eluate was concentrated to dryness in a vacuum centrifuge and re-dissolved in 5% triisopropylsilane in trifluoroacetic acid (TFA) and incubated for 2 h at 37 °C and 300 rpm. Subsequently, the samples were concentrated to dryness in a vacuum centrifuge.
LC-MS/MS Analysis of Complex Samples
LC-MS/MS experiments of at least four biologically independent replicates were performed. The vehicle control and 16-min time point were both repeated four times. The LC-MS/MS data files associated with this manuscript may be downloaded from ProteomeCommons.org Tranche using the following hash: ZTgDWKANYpkMpwyAh7eoFbGOxKkEnVUGYBHR3O8AEqCfbcCQSjwkGiBcNdTJM0eq6Cf7r/QayY/DBHRXiVWe9O9N2vcAAAAAAAACBQ==.
Briefly, NOxICAT-labeled peptides were separated by reverse phase nano-LC and detected by MS/MS with Fourier transform mass spectrometry in an LTQ Orbitrap instrument (Thermo Fisher Scientific, Waltham, MA). Identification and quantification were performed with Mascot (version 2.2.0, MatrixScience, London, UK) against the EcoProt database (21) and MSQuant version 1.4.3 (22).
For LC-MS/MS analysis, the samples were re-dissolved in 40 μl of 0.1% TFA. LC of 15 μl of the peptide sample was performed using an HP Ultimate 3000 system (Dionex, Sunnyvale, CA) equipped with a column oven (60 °C), a variable wavelength UV detector (214 nm), and a loading pump and micro pump system. For enrichment and desalting, the samples were first loaded onto a 300-μm × 5-mm Acclaim PepMap100 C18 micro-precolumn with 3-μm particle size (Dionex). The loading and washing steps were performed by the loading pump system, with a mixture of 95% solvent A (0.1% TFA) and 5% solvent B (0.1% TFA, 50% ACN (v/v)) at a flow rate of 30 μl/min for 10 min. The pre-column was then connected to a 75-μm × 25-cm Acclaim PepMap 100 C18, 3 μm, 100-Å column (Dionex), where the peptide sample was loaded with 95% solvent A (0.1% formic acid (v/v)) and 5% solvent B (0.1% formic acid, 84% ACN) at a flow rate of 0.400 μl/min by the micro pump system. Peptides were eluted with a linear gradient of 5% B to 40% B over 120 min at a flow rate of 0.400 μl/min. Mass spectra were obtained on line by an LTQ Orbitrap XL and an LTQ Orbitrap Velos instrument (Thermo Fisher Scientific, Waltham, MA). Fourier transform mass spectrometry was performed in a mass/charge range of 300–2000 with a resolution of 30,000 (LTQ Orbitrap XL) or 60,000 (LTQ Orbitrap Velos). The 6 or 20 most intense peaks (LTQ Orbitrap XL or LTQ Orbitrap Velos, respectively, minimal signal intensity 1500, charge range +2 to +4) in each MS spectrum were selected for MS/MS fragmentation (collision energy 35 V) in collision-induced dissociation mode. The exclusion list size was set to 500, with an exclusion duration time of 35.00 s.
Data Analysis-Protein Quantification Using MSQuant
For quantifying the redox state of thiol groups, the LC-MS/MS data were evaluated by MSQuant version 1.4.3 (8, 22). MSQuant allows visualization and validation of peptide identification results of the mass spectrometry data and supports relative protein quantification based on ICAT. To be able to use the MSQuant software version 1.4.3 in our context, we additionally installed the following programs: Internet Explorer version 6.0 (Microsoft Corp., Redmond, WA), Xcalibur version 2.0.7, BioWorks Browser 3.3 (both Thermo Fisher Scientific), and DTASuperCharge version 1.17 (22). The data analysis was divided into three steps.
Data Analysis Step 1. File Conversion by DTASuperCharge Version 1.17
In the first processing step, DTASuperCharge software extracted the peak list from the raw data file and converted it into an .mgf file (mascot generic file). The chosen parameters in DTA processing were “N-2 if phosphorylation loss” for precursor mass determination with a precursor tolerance of 0.01 and a bad precursor cutoff at 15 Da. Furthermore, the box “use charge state from DTA file” was checked.
Data Analysis Step 2. Protein/Peptide Identification with Mascot and Construction of .html Result File
Protein/peptide identification was accomplished using the Mascot search engine (version 2.2.0, Matrix Science, London, UK). Each MS/MS spectrum was searched against an E. coli-specific decoy database based on the EcoProt database (21). The following parameters were used for the Mascot search. Trypsin specificity was selected with one potentially missed cleavage; Light ICAT, Heavy ICAT, and Oxidation (M) were chosen as variable modifications. Furthermore, only MS/MS spectra from peptides with a charge from +1 to +4 were used for identification. We used a peptide tolerance of ±15 ppm and a MS/MS tolerance of ±0.4 Da. To generate an .html result file for use in MSQuant, the mascot search result was converted to “peptide summary” with the following parameters: significance threshold p < 0.05, the “max number of hits” was cut below 5% false discovery rate, and the ions score cutoff was set to 15. The boxes “show pop-ups,” “MudPit scoring,” and “require bold red” were checked. The generated peptide summary was saved as “webpage, complete” .html file with Internet Explorer 6.0.
Data Analysis Step 3. Quantification with MSQuant Software
After adding the .html result file and the corresponding .RAW file to MSQuant, both files were “associated.” Before starting the MSQuant workflow, the parameters for correlation settings and options were set according to supplemental Table 1. The protein list created by MSQuant was quantified using the MSQuant quantification mode. All quantitations were manually verified, and for further quantitation, values in ratio heavy ICAT/light ICAT below 0.01 were set to 0.01. Peptides were selected as candidate peptide if a difference in the fold change of >4 in the peroxynitrite-treated sample compared with both control samples (untreated control and vehicle control) was observed in all biological replicates. The statistical significance was additionally determined using Student's t test with the TTEST function of Excel for Mac 2011 version 14.2.5 with the following parameters: tails 2 and type 3 (Microsoft Corp., Redmond, WA).
Cloning and Mutagenesis of RidA
The RidA gene was amplified from genomic DNA by PCR with the primers ridA_for (5′-CATCATATGAGCAAAACTATCGC-3′) and ridA_rev (5′-GGAGGATCCTTCTTAGCGACGA-3′). The PCR product was cloned into pET11a (Stratagene, Santa Clara, CA) using the NdeI and BamHI restriction sites, generating pCL001. The C107S mutant was created from pCL001 by QuikChange mutagenesis, using the mutagenic primers C107S_for (5′-TTCCCGGCACGTTCTTCCGTTGAAGTTGCC-3′) and C107S_rev (5′-GGCAACTTCAACGGAAGAACGTGCCGGGAA-3′), yielding the mutagenized plasmid pCL002.
Overexpression of RidA
For overexpression, E. coli BL21 CL053 cells, carrying pCL001, and E. coli CL054 cells, carrying pCL002, were grown overnight without shaking in five flasks, each containing 1 liter of LB media with 200 mg/liter ampicillin. The next morning, the cultures were grown shaking at 125 rpm until an OD600 of 0.5 was reached. Protein expression was induced by addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside to the cell culture. E. coli cells were harvested 5 h later by centrifugation at 2000 × g and at 4 °C for 30 min.
Purification of RidA
The cell pellet was washed once with 40 ml of cold low salt buffer (40 mm HEPES-KOH, pH 7.5, 40 mm KCl, 2 mm DTT) and resuspended in a mixture of 100 ml of ice-cold low salt buffer and 2 ml of EDTA-free protease inhibitor mixture (Roche Applied Science). Cells were lysed by passing the cell suspension three times through a TS 0.75 constant cell disruption system at 1.8 kbar and 4 °C (Constant Systems Ltd., Daventry, UK) followed by the addition of 2 ml of Protease Inhibitor Mixture and PMSF to a final concentration of 1 mm. The cell lysate was centrifuged at 6700 × g and 4 °C for 1 h. The supernatant was separated from the pellet and vacuum filtered through a 0.2-μm filter. The filtrate was loaded onto a four times 5-ml HiTrap Q FF column (GE Healthcare) pre-equilibrated with low salt buffer. Subsequently, 150 ml of low salt buffer were applied to the column. The flow-through, containing RidA, was concentrated to 5 ml by using a Vivaspin 20 PES, MWCO 5000 concentrator system (Sartorius Stedim Biotech, Göttingen, Germany). The protein solution was loaded onto a size exclusion Superdex 75 26/60 column (GE Healthcare), pre-equilibrated with 40 mm potassium phosphate buffer, pH 7.5, 200 mm potassium chloride, 2 mm DTT, running on an ÄKTA purifier FPLC system (GE Healthcare). Fractions containing purified RidA were combined and concentrated using the Vivaspin 20 concentrator system. RidA aliquots were stored at −80 °C.
In Vitro Treatment of RidA with Oxidative and Nitrosative Stressors
For treatment of RidA with oxidative and nitrosative stressors, the storage buffer of purified RidA was replaced by 100 mm potassium phosphate buffer, pH 7.3, containing no reducing agent using a V-membrane Vivaspin 20 concentrator system. RidA (70 μm) was incubated with nitrosative and oxidative stressors at 37 °C and 300 rpm in a thermomixer at molar ratios of 1:1 and 1:10. Incubation times varied depending on the stressor (peroxynitrite, 5 min; diamide and hydrogen peroxide, 20 min; and S-nitrosylated glutathione, 30 min). The oxidative and nitrosative stressors were removed by NAP5-Sephadex G-25 columns (GE Healthcare) equilibrated with 100 mm potassium phosphate buffer, pH 7.3. Protein concentration was determined spectrophotometrically using an extinction coefficient of ϵ280 = 2980 m−1 cm−1 for the monomer of RidA, calculated by the ProtParam web tool (23). Reduction with ascorbate was performed at 37 °C and 300 rpm in the presence of 1 mm ascorbate for 2 h.
Analytical Gel Filtration
Analytical gel filtration was carried out in 50 mm phosphate buffer, 0.15 m sodium chloride, pH 7.0, on a Superdex 75 10/300 GL column connected to an ÄKTA purifier FPLC system. The column was calibrated with a commercially available mix containing 3 mg·ml−1 conalbumin (75 kDa), 3 mg·ml−1 carbonic anhydrase (29 kDa), 3 mg·ml−1 ribonuclease (13.7 kDa), 3 mg·ml−1 aprotinin (6.5 kDa), and 4 mg·ml−1 ovalbumin (44 kDa) (GE Healthcare). Of each sample, 100 μl were loaded onto the column, and the separation was performed at a flow rate of 0.5 ml·min−1.
Ellman's Assay to Determine Free Thiols in RidA
Protein samples in 100 mm potassium phosphate buffer, pH 7.3, and 1 mm EDTA were analyzed in a 3.5-ml QS-macro cuvette (10 mm) (Hellma Analytics, Müllheim, Germany) equipped with a magnetic stir bar in a temperature-controlled Jasco V-550 spectrophotometer at 20 °C (Jasco, Tokyo, Japan). 5,5′-Dithiobis-2-nitrobenzoic acid (Ellman's reagent) was added to a final concentration of 150 μm, and the absorption at 412 nm was monitored until no further increase in absorption was detectable. Free thiols were determined using the extinction coefficient of 2-nitro-5-thiobenzoate of ϵ412 = 14,150 m−1·cm−1 (24).
Ellman's Assay in Crude Extracts
5 ml of cells in MOPS minimal medium, treated 16 min with 1 mm diamide, 1 mm GSNO, and 1 mm peroxynitrite were washed twice and resuspended in 1 ml of 2× PBS. Cells were lysed by sonication as described. Crude extract (500 μl) was diluted with 500 μl of 2× PBS, and 100 μl of 15 mm 5,5′-dithiobis-2-nitrobenzoic acid (Ellman's reagent) solution was added. The absorption at 412 nm was monitored in a temperature-controlled Jasco V-550 spectrophotometer at 25 °C (Jasco, Tokyo, Japan) for all samples, including the control (untreated cells) in parallel kinetics mode, until the absorption reached a constant level. A412 of the control was set to 100%, and the A412 of the other samples was expressed as percentage of the control.
SDS-Gel Electrophoresis
Samples of RidA were incubated with 10 mm iodoacetamide in 0.1 m potassium phosphate buffer, pH 7.3, to avoid changes in the redox state of Cys-107 through air oxidation. The modified protein was then mixed with the appropriate volume of 5× SDS loading buffer (10% SDS, 50% glycerol, 300 mm Tris, pH 7.0, 0.05% bromphenol blue). Reducing buffer additionally contained 25% 2-mercaptoethanol. Samples were incubated for 10 min at 95 °C and then separated on a NuPAGE 4–12% BisTris SDS gel (Invitrogen). Gels were stained using Coomassie Brilliant Blue as described previously (25).
Isoelectric Focusing (IEF) Gel Electrophoresis
IEF separation was performed on a Novex IEF gel, pH 3–7 (Invitrogen). RidA samples were mixed with the appropriate amount of 2× IEF sample buffer, pH 3–7 (Invitrogen). Of each sample, 15 μl was loaded onto the gel. Electrophoresis was performed according to the manufacturer's instructions.
Mass Spectrometry of Full-length RidA
RidA was treated with peroxynitrite and GSNO as described. Stressors were removed by ZipTip C18 micro columns (Millipore, Billerica, MA) according to the instruction manual. RidA was eluted from the ZipTips with 0.1% trifluoroacetic acid and 70% acetonitrile. The sample was concentrated to dryness in a vacuum centrifuge and re-dissolved in 50 μl 0.1% formic acid. The protein sample was analyzed with an LTQ Orbitrap Velos system (Thermo Fisher Scientific, Waltham, MA) in positive ion mode directly injecting the sample into the mass spectrometer using a metal needle at a source voltage of 1.60 kV and a source current of 0.48 μA.
Threonine Deaminase Assay
RidA and RidA C107S were treated with different stressors as described. The threonine deaminase assay was performed as described elsewhere (15). Product formation was monitored in at least three independent experiments spectrophotometrically at 230 nm in a 3.5-ml QS-macrocuvette (10 mm) equipped with a magnetic stir bar in a temperature-controlled Jasco V-550 spectrophotometer at 20 °C. RidA or RidA C107S at a concentration of 1.8 μm, 0.9 μm IlvA, and 20 μm pyridoxal phosphate in 1× reaction buffer (50 mm MES, 50 mm HEPES, 50 mm TAPS, pH 7.5) were stirred in the cuvette, and the background absorption of this solution was measured for a short time (<1 min) at 230 nm. To start the reaction, threonine was added to a final concentration of 10 mm, and the formation of 2-ketobutyrate was followed for 300 s. The statistical significance in differences in activity was determined using Student's t test with the TTEST function of Excel for Mac 2011, version 14.2.5, with the following parameters: tails, 2, and type 2 (Microsoft Corp.).
RidA Abundance and Steady State
For RidA abundance and steady state analysis, 1 ml of bacterial culture was harvested at an OD of 0.4 (5 min, 4 °C, 16,100 × g) and the supernatant discarded. The cell pellet was resuspended in 40 μl of TE buffer, pH 8.0 (10 mm Tris, 1 mm EDTA), and cell disruption was carried out by sonication as described. After a short centrifugation step, 10 μl of reducing SDS loading buffer was added to the sample, and the sample was heated for 10 min at 95 °C. Of the sample, 10 μl (corresponding to 108 cells) were loaded on a 4–12% BisTris SDS gel. Purified RidA (10 μl) with known amounts of 0.01 to 66 ng were loaded onto the same SDS gel for calibration purposes. After SDS-PAGE separation, proteins were transferred to a nitrocellulose membrane using the iBlot Gel Transfer System (Invitrogen). Subsequently the membrane was incubated in a specific polyclonal rabbit anti-RidA antibody diluted 1:2000 in 2% milk powder in 1× TBS. The anti-RidA antibody was detected by the secondary IRDye 680LT goat anti-rabbit IgG (H+L) (Li-COR Biosciences, Bad Homburg, Germany) diluted 1:5000 in 2% milk powder in 1× TBS. RidA bands were visualized in the Odyssey Infrared Imaging System (Li-COR Biosciences).
RidA Stability
Cell cultures were treated with chloramphenicol (final concentration 200 μg/ml) to inhibit de novo protein synthesis. Aliquots of 1 ml were harvested at the indicated time points from peroxynitrite-stressed and untreated cultures and immediately frozen in liquid nitrogen until they could be further processed. The aliquots were thawed on ice, and cells were pelleted at 4 °C for 5 min at 16,100 × g. The cells were resuspended in 40 μl of TE buffer, pH 8.0, and lysed by sonication as described. RidA was detected via Western blot as described.
Quantification of RidA Abundance
The abundance of RidA was determined in quantitatively scanned Western blots by measuring the integrated density in selections of the bands 24 × 21 pixels in size using the Analysis tool in Photoshop CS5 extended (Adobe Systems Inc., San Jose, CA). A selection of identical size in an empty lane served as background and was subtracted from the values of the individual bands. The densities of the signals derived from purified RidA were plotted against the known quantity of RidA in those bands. These values were used as input in the GROWTH function of Excel for Mac 2011 version 14.2.5 to calculate the amount of RidA from the density detected in the neighboring cell lysates bands. The average from three experiments was used to calculate the molecules per cell and the effective molarity of RidA in the medium.
Western Blot-based Detection of Protein Glutathionylation and Sulfenylation
Sample harvest was carried out as described, and glutathionylation was detected on nonreducing and reducing SDS-polyacrylamide gels using commercially available anti-glutathione monoclonal antibodies (Virogen, Watertown, MA) at a dilution of 1:1000 in 2% milk powder in 1× TBS as described. Nonreduced and reduced glutathione/ovalbumin (Biotrend, Cologne, Germany) with a concentration of 0.2 μg/μl served as control. Similarly, sulfenylation was detected with commercially available anti-cysteine sulfenic acid polyclonal antibodies (Millipore, Temecula, CA) diluted 1:5000 in 2% milk powder in 1× TBS. Hydrogen peroxide-treated GAPDH served as a control.
RESULTS
Peroxynitrite Affects E. coli Growth in a Dose-dependent Manner
To get an idea about the influence of peroxynitrite on E. coli growth, we added increasing amounts of ONOO− (500 μm to 3 mm) to an exponentially growing cell culture in MOPS minimal medium (16). Upon addition of 1 mm or more peroxynitrite, the cells immediately ceased growth. The length of the growth arrest increased with the quantity of peroxynitrite added. To test if peroxynitrite is bactericidal under these conditions, we checked cell survival. The number of viable cells remained constant in cultures treated with concentrations of up to 2 mm; a concentration of 3 mm eventually led to cell death (Fig. 1). After a lag phase, the cultures resumed growth at a rate comparable with growth before treatment, indicating that E. coli can overcome peroxynitrite stress even at fairly high concentrations (Fig. 1).
FIGURE 1.
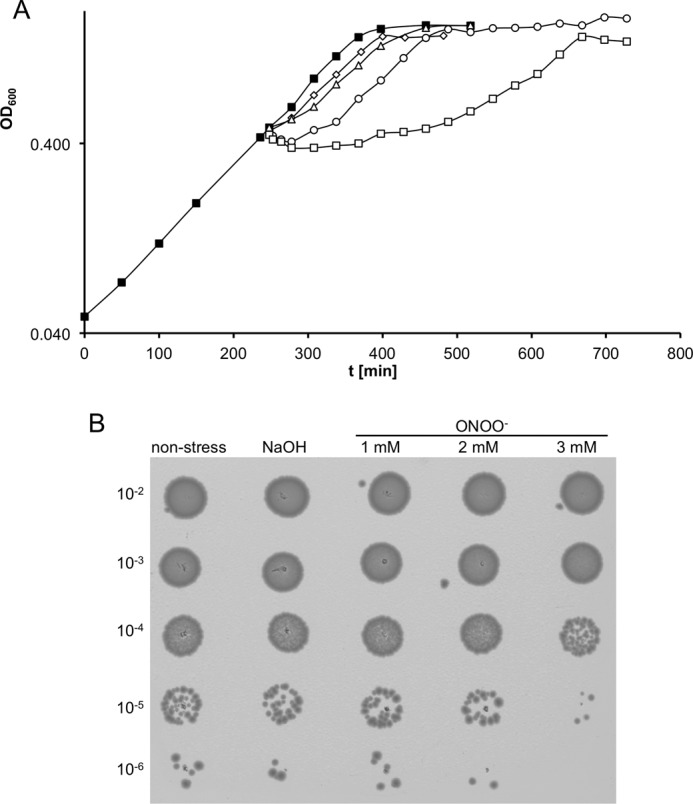
Growth and survival of E. coli wild type cells under different peroxynitrite concentrations (500 μm to 3 mm). A, E. coli MG1655 wild type was grown at 37 °C in MOPS medium to an OD600 of about 0.4. Then the culture was split into five subcultures. The subcultures were stressed with different peroxynitrite concentrations as follows: unstressed control (■); 500 μm (♢); 1 mm (▵), 2 mm (○); 3 mm (□). B, E. coli MG1655 wild type was treated for 16 min with different concentrations of peroxynitrite. Untreated cells and cells treated with an identical volume of 0.3 m NaOH served as (vehicle) control. Cells were diluted and plated onto LB agar plates.
Peroxynitrite Nitrosates Tyrosine Residues in E. coli Proteins
To analyze the molecular cause of the growth stop, we focused on specific protein modifications. Peroxynitrite is known to target side chains of tyrosines in proteins (26). The exposure of protein tyrosines to ONOO− leads to the formation of 3-nitrotyrosine. We used antibodies specific to 3-nitrotyrosine to detect these modifications in Western blots. Proteins in E. coli showed varying amounts of tyrosine nitration in a dose-dependent manner. Based on the blots, we did not detect a specificity for selected proteins (Fig. 2). It seems that nitrotyrosine formation, an irreversible modification, indiscriminately damages proteins. This damaging effect seems to be instantaneous (even after 5 min the modification was complete and no further increase was observed) and could be one of the molecular reasons of the observed growth arrest.
FIGURE 2.

Detection of tyrosine nitration in E. coli wild type under different peroxynitrite concentrations in vivo. E. coli MG1655 wild type was grown in MOPS medium to an absorbance of 0.4 and exposed to peroxynitrite (1–3 mm) or 0.3 m NaOH as vehicle control. Samples were taken 5, 16, and 60 min after the addition of peroxynitrite. Tyrosine nitration was detected with 3-nitrotyrosine-specific antibodies, and nitrated BSA at a concentration of 1 μg/μl served as positive control.
A Thiol-based Redox Proteomics Approach to Quantify the Oxidation of E. coli Proteins
An emerging mechanism of the regulation of protein activity under oxidative and nitrosative stress conditions is regulation by reversible thiol modification. Recently, we have developed a proteomic approach to identify and quantify these modifications (8). This approach is based on the differential modification of reduced and oxidized thiols in a complex protein sample with the isotopically different but chemically identical 12C (light) and 13C (heavy) forms of the ICAT reagent (8). It allows for the quantification of the ratio of the reduced and oxidized fraction of hundreds of thiols in the cell in a single experiment. We have now adapted this protocol to study modifications caused by reactive nitrogen species (Fig. 3) (19). The use of TCA in our original protocol could potentially lead to artifactual thiol modification during sample preparation due to nitric oxide released from nitrite under acidic conditions. Nitrite is a major degradation product of peroxynitrite (27), and we have observed the phenomenon of artifactual oxidation in samples derived from cells treated with NO• in a prior study (9). To avoid the use of acidic conditions, our modified approach prevents the oxidation of samples by the use of oxygen-free buffer and by working in an anaerobic chamber or under a semi-anaerobic argon atmosphere.
FIGURE 3.

NOxICAT redox proteomics technique. A hypothetical cellular protein is depicted in its fully reduced (nonstress conditions) or partially modified form (stress conditions). Light ICAT modification of reduced thiols is performed under anaerobic conditions to prevent thiol oxidation by air. The presence of denaturing agents facilitates the reaction of the light ICAT with free thiol groups in the protein (step I). Subsequently, all reversibly oxidized cysteines are reduced with TCEP and labeled with heavy ICAT (step II). Then protein samples are digested by trypsin and purified using the biotin-affinity tag of the ICAT reagent. The biotin-affinity tag is cleaved after purification (step III). NOxICAT-labeled peptides are then analyzed by LC-MS.
To assess the reliability of the modified method, we first investigated samples obtained before addition of peroxynitrite. In accordance with previous experiments, cysteines from periplasmic proteins, such as OmpA, which form structural disulfides appeared exclusively oxidized. The majority of cysteines from cytoplasmic proteins, however, were identified in their reduced form (Fig. 4). When compared with our previous studies with a TCA-based oxidation quench, cysteines from cytoplasmic proteins identified in this study often showed lower oxidation rates, and in many cases a peak corresponding to an oxidized form was not detectable at all. Proteins, which use a catalytic thiol-disulfide exchange reaction in their catalytic cycle, such as glutaredoxin 3 (GrxC), however, could be detected in an equilibrium of the reduced and oxidized form, which reflected their in vivo redox state (Fig. 4). In total, we could reliably identify and quantify up to 700 thiol-containing peptides in a sample (supplemental Table 2).
FIGURE 4.
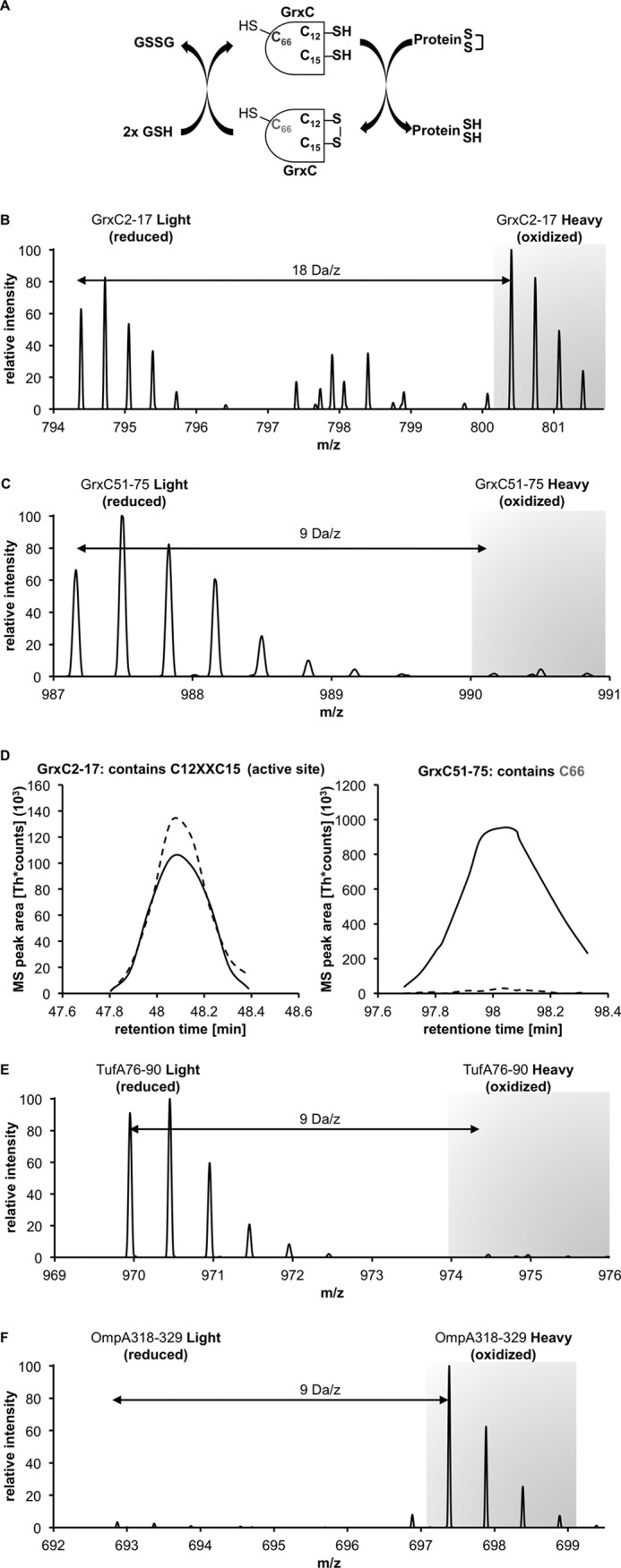
Oxidation state of cysteines of cytosolic and periplasmic proteins under nonstress growth conditions. A, GrxC (glutaredoxin 3) is an enzyme that forms a disulfide bond between Cys-12 and Cys-15 during its active cycle. The nonconserved C66 is not involved in the catalytic cycle. B, NOxICAT method allows us to observe the steady state of this enzyme. The parental ion MS of the peptide identified as GrxC 2–17 shows the reduced and oxidized form 2 × 9 Da = 18 Da apart. The intensities correspond to the redox state of this antioxidant enzyme system. C, peptide containing C66, however, is almost exclusively present in its reduced form. D, redox state of both peptides can be quantified with MSQuant using the elution profile of these peptides. The solid line represents the reduced (light) form, and the dashed line represents the oxidized (heavy) form. E, NOxICAT detects cytosolic cysteines that are not involved in catalytic cycles, such as Cys-82 from TufA (EF-Tu, one of the highest abundant proteins in the E. coli cytoplasm) almost exclusively in their reduced form, confirming the dogma of a reducing cytosol. F, OmpA, a major periplasmic protein is detected as fully oxidized in the NOxICAT assay, demonstrating the ability of NOxICAT to detect structural disulfides present in periplasmic proteins.
Peroxynitrite Treatment Leads to Small but Significant Changes in the Thiol Redox Proteome of E. coli
Next, we challenged exponentially growing E. coli MG1655 in MOPS minimal medium (buffered at pH 7.4) with a sublethal dose of 1 mm peroxynitrite and harvested samples immediately before and 16 and 60 min after addition of the stressor or the respective volume of a vehicle control containing the identical volume of 0.3 m NaOH.
To identify proteins that are affected by peroxynitrite treatment, we compared the thiol oxidation state of the identified proteins under ONOO− treatment to that of untreated samples. In four proteins we could detect a significant increase in thiol oxidation under peroxynitrite stress conditions after 16 min (Fig. 5 and Table 3). These were glutamine-hydrolyzing asparagine synthase B, AsnB (modified cysteine, Cys-2), NADP-dependent malic enzyme, MaeB (Cys-642), glutathione-dependent formaldehyde dehydrogenase, FrmA (Cys-106), and a newly described enamine/imine deaminase RidA (Cys-107). The redox state of the identified cysteines in these proteins was at least 4-fold more oxidized under peroxynitrite stress when compared with nonstress conditions (Table 3). The oxidized fraction of the thiol groups in these proteins was highest after 16 min of stress treatment and decreased 60 min after stress treatment, indicating a potential in vivo reversibility of the observed thiol modifications and an adaptation of the cell to peroxynitrite stress (Table 3). This decrease in oxidation also coincides with the resumption of growth of the bacterial culture during the first 30 min after stress treatment (Fig. 1). In three of the proteins identified, the cysteine affected was highly conserved (Table 3). The identified redox-sensitive Cys-2 of AsnB, for example, is located in the active site of the N-terminal region of this protein (28). Asparagine synthase B belongs to the Ntn- (N-terminal nucleophile) enzyme category of amidotransferases and catalyzes the ATP-dependent synthesis of asparagine from aspartic acid and glutamine. The oxidative modification of Cys-2 in AsnB would block the ability of Cys-2 to act as the active-site nucleophile and thus prevent hydrolysis of glutamine to glutamate and ammonia (29). Interestingly, in a previous study we found that GltD (GOGAT small subunit), another enzyme that utilizes glutamine as nitrogen source for amino acid biosynthesis, was inhibited by NO• (9).
FIGURE 5.

Fourier transform mass spectrometry of MaeB, peptide 629–647, which harbors the redox-sensitive Cys-642. NADP-dependent malic enzyme (MaeB) is a protein sensitive toward peroxynitrite stress in E. coli. In untreated E. coli cells (nonstress), the cysteine is almost exclusively found in its reduced form (>99%). In cells treated 16 min with 1 mm peroxynitrite (stress), the redox-sensitive cysteine of MaeB is 18% oxidized.
TABLE 3.
Identified peroxynitrite-sensitive proteins and fold change of the oxidation of the identified redox-active cysteine 16 and 60 min after addition of 1 mm peroxynitrite and 16 min after addition of 3 mm peroxynitrite to the media
Peptide and ICAT-modified cysteine as identified by MS/MS. Protein function was according to EcoGene (21). Stress sensitivity of a mutant was according to observed growth behavior. Average fold change and p values were from data generated in at least four independent experiments.
| Peptide | Protein function | ICAT-modified cysteine | % oxidized |
Fold change after 16 min 1 mm ONOO− (p value) | Fold change after 60 min 1 mm ONOO− | Fold change after 16 min 3 mm ONOO− | Stress sensitivity of mutant | |
|---|---|---|---|---|---|---|---|---|
| Nonstress (vehicle control) | 1 mm ONOO− | |||||||
| AsnB (2–18) | Asparagine synthaseB (glutamine-hydrolyzing) | Cys-2a | 1.75 | 19.59 | 11.2 | 3.3 | 7.4 | + |
| p < 0.01 | ||||||||
| FrmA (103–110) | Glutathione-dependent formaldehyde dehydrogenase | Cys-106a | 2.10 | 8.76 | 4.2 | 2.3 | 2.9 | ++ |
| p < 0.05 | ||||||||
| MaeB (629–647) | Malic enzyme, NADP-dependent; NADP-ME | Cys-642 | 1.42 | 15.80 | 11.1 | 5.0 | 12.7 | + |
| p < 0.05 | ||||||||
| RidA (106–115) | Enamine/imine deaminase, reaction intermediate detoxification | Cys-107a | 1.38 | 6.44 | 4.7 | 2.5 | 10.8 | + |
| p < 0.05 | ||||||||
a This denotes a conserved cysteine based on conserved domain database families cd00352 (AsnB), cd08300 (FrmA), and cd00448 (RidA) (46).
Cells Lacking Proteins Modified upon Peroxynitrite Treatment Struggle to Overcome Peroxynitrite Stress
Thiol modifications have been shown to play a role in the regulation of the activity of proteins involved in the oxidative and nitrosative stress defense. We hypothesized that at least two of the proteins we found could play such a protective role under peroxynitrite stress. MaeB, NADP-dependent malic enzyme, could provide NADPH for reducing systems such as glutathione oxidoreductase and thioredoxin reductase. FrmA, glutathione-dependent formaldehyde dehydrogenase, has been shown to have a thiol-denitrosylating activity for GSNO, and it has been proposed that its NADH-dependent GSNO reductase activity is the main function of this enzyme (30). To test if the proteins we identified indeed play a role in the nitrosative stress defense of E. coli, we tested growth of knock-out strains in genes encoding the four candidate proteins in the presence of peroxynitrite. In all four knock-out strains, we observed an impaired recovery of cell growth, especially under severe nitrosative stress (3 mm) when compared with wild type (Fig. 6). A follow-up global experiment at these higher concentrations revealed that the four proteins are modified to a similar extent when compared with 1 mm peroxynitrite (Table 3).
FIGURE 6.

Cell growth of E. coli wild type versus E. coli ΔasnB, ΔfrmA, ΔmaeB, and ΔridA deletion strains in the presence of 3 mm peroxynitrite. E. coli wild type (■) and E. coli deletion strains, ΔasnB (♢), ΔfrmA (▵), ΔmaeB (○), and ΔridA (□), were grown at 37 °C in MOPS medium until an OD600 of about 0.4 was reached. Then each strain was exposed to 3 mm peroxynitrite (dashed line). Cultures treated with 0.3 m NaOH served as controls (continuous line). Deletion strains showed reproducibly a significantly longer growth inhibition when compared with E. coli wild type.
This indicated to us a direct or indirect role of the identified genes in cell defense mechanisms under peroxynitrite stress. Similar phenotypes in other organisms have been shown for FrmA; Saccharomyces cerevisiae cells lacking glutathione-dependent formaldehyde dehydrogenase show an impaired survival under GSNO and peroxynitrite stress (30). This indicates that formation of intracellular GSNO could be one of the molecular effects of peroxynitrite treatment.
Proteins Modified upon Peroxynitrite Treatment Are Not Abundant Enough to Act as an Effective Sacrificial Peroxynitrite Sink
We furthermore considered if the proteins we found could act as a peroxynitrite sink. Under this premise, the proteins would render peroxynitrite harmless by a thiol-based sacrificial reaction. The lack of the correspondent sink capacity in a mutant could then explain the impaired recovery.
However, in a study on protein abundance in E. coli grown in MOPS medium, the same medium we were using, the number of AsnB copies was determined with ∼233 per cell (31). E. coli has a typical cell density of 5·108 cells per ml of medium at an OD600 of 0.4. Because peroxynitrite rapidly permeates membranes (32), this would correspond to an effective concentration of ∼194 fm in the medium, almost 7 orders of magnitude below the concentration of peroxynitrite. In this same study, the effective media concentration of MaeB was determined to be ∼592 fm. Unfortunately, Lu et al. (31) did not determine the abundance of RidA and FrmA. But even the most abundant protein found in their study, the 50 S ribosomal subunit protein L7/L12 (RplL) with ∼109,420 copies per cell, only has an effective concentration of 91 nm in the medium.
This discrepancy between the molarity of the stressor and proteins in E. coli makes the function as a sacrificial sink unlikely. Additionally, neither of the genes encoding the proteins we found to be thiol-modified were significantly regulated in a transcriptomic study of the response of E. coli toward 300 μm peroxynitrite (14).
Nevertheless, the potential function as a sink could be based on a very high turnover of the proteins we found. We therefore determined the abundance of RidA and its stability under peroxynitrite stress conditions. The half-life of RidA is predicted by the ProtParam web tool to be >10 h in E. coli based on the “N-end rule” (23). We tested the stability of RidA under peroxynitrite stress in Western blots in cells treated with chloramphenicol to prevent de novo synthesis of proteins. Although we determined the steady state concentration of RidA to be at 5.5 nm in the medium (∼6663 molecules per cell) under control conditions, its half-life was substantially greater than 90 min. We could not detect any decline in protein concentration 90 min after the addition of chloramphenicol, the longest time point we tested. Under peroxynitrite stress conditions, there was no significant change in steady state abundance, in concordance with the transcriptomic study by McLean et al. (14). Additionally, we did not observe a diminished half-life of RidA under ONOO− stress conditions, a prerequisite of a function as an effective passive sink (Fig. 7).
FIGURE 7.
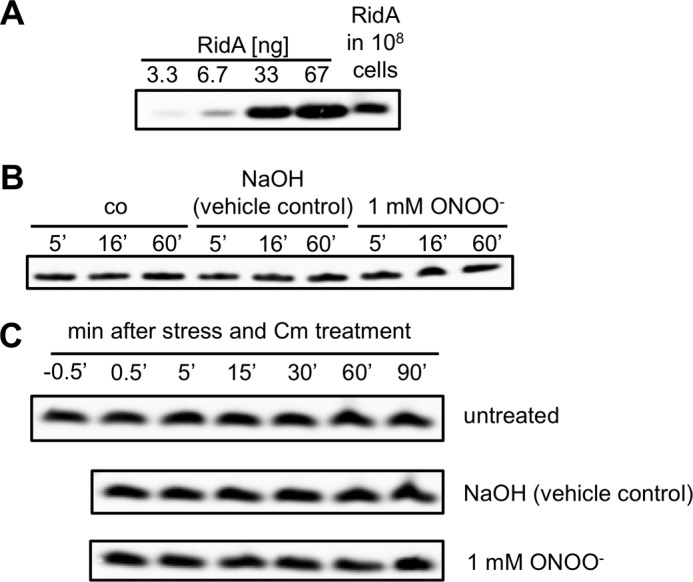
Steady state levels and stability of RidA. A, determination of the cellular steady state level of RidA. Purified RidA was applied for calibration purposes in the 1st 4 lanes. The lysate of 108 cells was applied in the 5th lane. RidA was detected with a RidA-specific antibody. B, comparison of steady state levels of unstressed and peroxynitrite stressed cells after various time points of stress. RidA was detected in cell lysates with a RidA-specific antibody. C, stability of RidA in untreated cells and cells subjected to peroxynitrite stress. Chloramphenicol (Cm) was added to the cells to inhibit de novo protein synthesis. Samples were harvested at the time points indicated, and RidA was detected in the lysates with a RidA-specific antibody.
Cys-107 of RidA Is Highly Susceptible to Modification by Peroxynitrite in Vitro
Until now, RidA has not been linked to the nitrosative stress response. As the function of this homotrimeric protein has been discovered only recently, we investigated its role under peroxynitrite stress. RidA belongs to the YjgF/YER057c/UK114 superfamily, which is highly conserved across a wide range of organisms. This protein family has a variety of assigned functions, ranging from translational inhibition and activation of a proteinase, to nuclease and chaperone activity (33–36). Recent studies identified an enamine/imine deaminase activity of RidA in Salmonella enterica (15). The presence of RidA leads to a faster release of ammonia from a reactive nitrogen enamine/imine intermediate generated by dehydration of threonine through threonine deaminase IlvA, the first step of isoleucine biosynthesis. Cys-107 is the sole cysteine in E. coli RidA and is conserved in several bacterial YjgF/YER057c/UK114 family members.
We overexpressed and purified RidA from E. coli to homogeneity and tested its susceptibility to oxidation by peroxynitrite in vitro. With a spectrophotometric method based on Ellman's reagent, we could show a substantial decrease in free thiols in RidA treated at a pH of 7.3 with equimolar amounts of peroxynitrite. Treatment of RidA with a 10-fold excess of peroxynitrite led to the complete disappearance of free thiols (Fig. 8A). Based on structural considerations, Cys-107 is thought to be located in a ligand pocket located at the subunit interfaces of the RidA trimer (37). A modification of this cysteine residue could potentially disrupt the trimeric structure of RidA. Because RidA contains a single cysteine, we considered the formation of an intersubunit disulfide bond, which then would lead to the formation of covalently linked dimers. However, on a nonreducing SDS-PAGE, we did not observe a significant portion (<9%) of disulfide-linked dimers of ONOO−-treated RidA (Fig. 9A). In agreement with this finding, analytical gel filtration demonstrated that RidA, even when modified by peroxynitrite, remains in its trimeric state. We could, however, force disulfide-linked dimer formation by the specific disulfide bond-forming reagent diamide, which presumably leads to non-native higher molecular weight aggregates that could be observed as an unspecific shoulder in the gel filtration chromatograms (Fig. 9).
FIGURE 8.
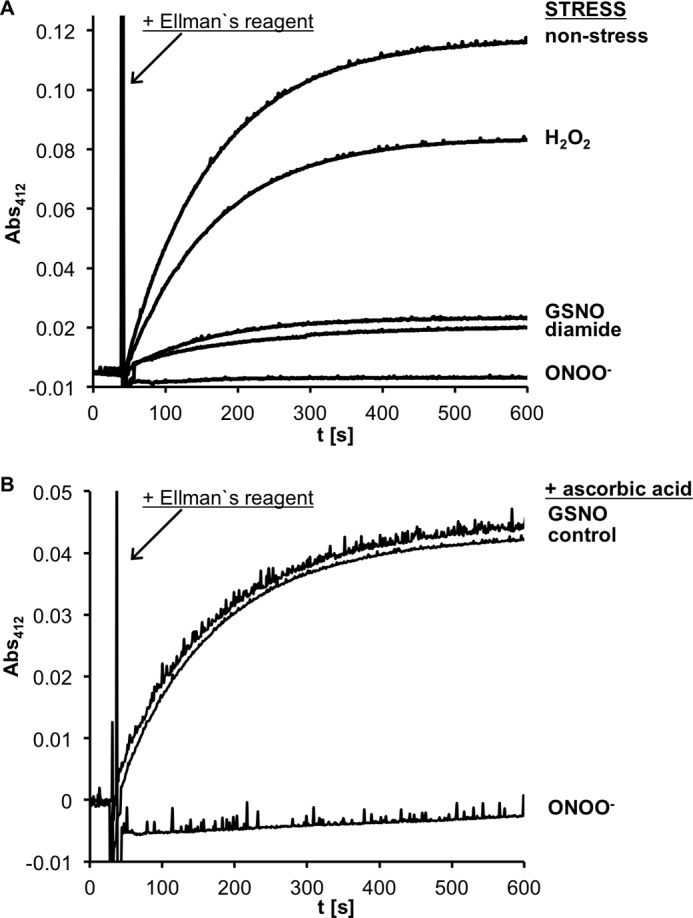
Oxidation of Cys-107 of RidA by different oxidative and nitrosative stressors and reversibility by ascorbate of these oxidations. RidA (70 μm) was incubated with nitrosative (peroxynitrite, GSNO) and oxidative stressors (hydrogen peroxide, diamide) at a molar ratio of 1:10 in potassium phosphate buffer at pH 7.3 for different lengths of times (peroxynitrite, 5 min; GSNO, 30 min; hydrogen peroxide and diamide, 20 min). Untreated RidA served as a control. A, concentration of free thiol groups in the samples was quantified spectroscopically by monitoring the release of thio-nitrobenzoic acid from 5,5′-dithiobis-2-nitrobenzoic acid (Ellman reagent) at 412 nm and 20 °C. B, reversibility of the thiol modifications of Cys-107 upon treatment with GSNO and peroxynitrite stress was analyzed by subsequent ascorbate reduction. Nonstressed and stressed RidA samples were incubated with 1 mm ascorbate for 2 h.
FIGURE 9.

Multimeric states of RidA upon nitrosative and oxidative stress. A, potential dimer formation of RidA upon intermolecular disulfide bond formation analyzed via SDS-PAGE. B, analysis of the native trimeric state of RidA by gel filtration. A, RidA (70 μm) was treated with ROS and RNS stressors at molar ratios of 1:1 and 1:10 for different periods of time (5 min for peroxynitrite, 20 min for diamide and hydrogen peroxide, and 30 min for GSNO) in phosphate buffer at pH 7.3. Untreated RidA served as control. The reaction was stopped by addition of 1 mm iodoacetamide and nonreducing SDS loading buffer. The samples were analyzed on an SDS gel. B, 100 μl of each sample was analyzed by size exclusion chromatography. The trimeric state of RidA was deduced from the apparent mass (∼35,600 Da) by comparison with the calibration of the column with a standard mix of proteins of known size as follows: conalbumin (75 kDa, 1), ovalbumin (45 kDa, 2), carbonic anhydrase (29 kDa, 3), ribonuclease (13.7 kDa, 4), and protinin (6.5 kDa, 5).
RidA Is Specifically Modified by Reactive Nitrogen Species but Not H2O2
Because RidA has not been described as susceptible to oxidative stress and because we have not observed RidA as a target of reactive oxygen species in previous global studies, we hypothesized that reactive nitrogen species could have a higher affinity for Cys-107 of RidA than reactive oxygen species. To test our hypothesis, we treated RidA with another reactive nitrogen species, GSNO, and the classical reactive oxygen species hydrogen peroxide. Indeed, although H2O2 only led to a very modest decrease in free thiols in RidA, GSNO was much more effective in modifying Cys-107 (Fig. 8A). Both stressors did not cause a disruption of the homotrimer, and less than 6% of disulfide-linked species were observed on a nonreducing SDS-PAGE (Fig. 9).
Treatment of RidA with Reactive Nitrogen Species Leads to S-Nitrosylation and Overoxidation of Cys-107
To test if modifications of Cys-107 induced by GSNO and peroxynitrite are reversible, we treated RidA that had been subjected to these nitrosative stressors with ascorbate, which is thought to be a specific reductant of S-nitrosylations. Ascorbate fully restored the level of unmodified thiols in the GSNO-treated sample to 1 cysteine per subunit, but it was not able to reduce peroxynitrite-treated RidA (Fig. 8B). Based on the lack of inter-subunit disulfides and the reversibility of GSNO modifications, we assumed that GSNO treatment of RidA leads to S-nitrosylation. In contrast, the irreversibility of peroxynitrite-induced modifications suggests an overoxidation of RidA Cys-107 to a sulfinic or sulfonic acid. We tested our assumption by isoelectric focusing electrophoresis of RidA treated with different stressors. Although the proteins treated with other stressors migrated similar to the untreated protein, we saw a clear shift in the isoelectric point of peroxynitrite-treated RidA toward a more acidic position. This indicated that we indeed overoxidized RidA by the addition of peroxynitrite in vitro (Fig. 10). The lack of a shift in the GSNO-treated sample made a thiol nitrosylation more likely than a glutathionylation, because the addition of glutathione should lower the isoelectric point of RidA slightly through the addition of the ammonia and carboxy groups from glutathione (Fig. 10).
FIGURE 10.

Overoxidation of RidA Cys-107 by peroxynitrite treatment in vitro. RidA (70 μm) was incubated with different RNS and ROS stressors at molar ratios of 1:1 and 1:10 in phosphate buffer at pH 7.3 for different lengths of time depending on the stressor used (5 min for peroxynitrite, 20 min for diamide and hydrogen peroxide, and 30 min for GSNO). RidA without stressor served as a control. Of each sample, 15 μl were loaded onto an IEF gel, pI 3–7.
To confirm these observations, we determined the full-length mass of untreated and peroxynitrite- or GSNO-treated RidA. GSNO-treated RidA showed an increase in mass of 29 Da, indicative of a thiol nitrosylation, and RidA showed an addition of 32 and 48 Da, indicating the presence of a mixture of sulfinic and sulfonic acid (Fig. 11), which was consistent with our data from Ellman's assay, the nonreducing SDS-PAGE, and the IEF gel. Our proteomic NOxICAT assay is limited to detecting reversible thiol modifications, ruling out that the observed oxidized form of RidA in vivo is a sulfinic or sulfonic acid. We assume that in vivo the effective concentration of peroxynitrite is lower than in our in vitro assay and we potentially observe an intermediate (e.g. a sulfenylation), which we do not detect in vitro. Alternatively, the potential formation of GSNO in the cell could lead to S-nitrosylation of RidA.
FIGURE 11.

Details of deconvoluted MS spectra of full-length RidA modified by nitrosative stressors. Purified RidA (70 μm) was incubated with peroxynitrite and GSNO at a molar ratio of 1:10 for 5 min (peroxynitrite) and 30 min (GSNO) in phosphate buffer at pH 7.3. Untreated RidA served as a control.
Cys-107 Is Not Required for RidA Activity but Its Oxidation Eliminates Its Enamine/Imine Deaminase Activity
In a coupled enzyme assay, we tested the role of Cys-107 in the catalysis of enamine/imine deamination by RidA and the effect of the thiol modification on RidA activity. For this, we purified E. coli threonine deaminase IlvA and measured the formation of 2-ketobutyrate from threonine. Addition of RidA accelerated the formation of 2-ketobutyrate as described previously for the Salmonella enzyme (15). When treated with peroxynitrite, the accelerating effect of RidA was eliminated (Fig. 12). Hydrogen peroxide treatment had no significant effect on activity, although the inhibition by GSNO and diamide was substantially lower than by ONOO− (Fig. 13). To test if cysteine 107 is directly required for the catalytic action of the enzyme, we mutated it to serine. Interestingly, RidA C107S was as active as the wild type enzyme, demonstrating that the cysteine is not directly involved in enzymatic activity. This RidA C107S mutant could no longer be deactivated by peroxynitrite treatment (Fig. 12), demonstrating the regulatory function of Cys-107.
FIGURE 12.

RidA wild type and RidA C107S mutant enamine/imine deaminase activity. In this assay, IlvA converts its substrate threonine to an enamine/imine intermediate, which spontaneously decays to 2-ketobutyrate. The enamine/imine deaminase RidA accelerates this process. The 2-ketobutyrate formation can be measured spectrophotometrically at 230 nm. We analyzed the rate of 2-ketobutyrate formation in the presence and absence of RidA and RidA treated with peroxynitrite at a 1:10 molar ratio in phosphate buffer at pH 7.3 (A) and RidA C107S mutant and RidA C107S mutant treated with peroxynitrite (B). The threonine deaminase assay includes 0.9 μm IlvA, 7.5 mm threonine, and 1.8 μm RidA. The assay in the absence of threonine served as a control. The acceleration in 2-ketobutyrate formation was quantified by determining the difference in the ΔOD230/min in the presence and absence of RidA (ΔΔOD230/min) (C). The difference in the acceleration of 2-ketobutyrate formation between the untreated and peroxynitrite-treated RidA was highly significant (**, p < 0.01), whereas the activity of the RidA C107S mutant was not affected by peroxynitrite treatment.
FIGURE 13.
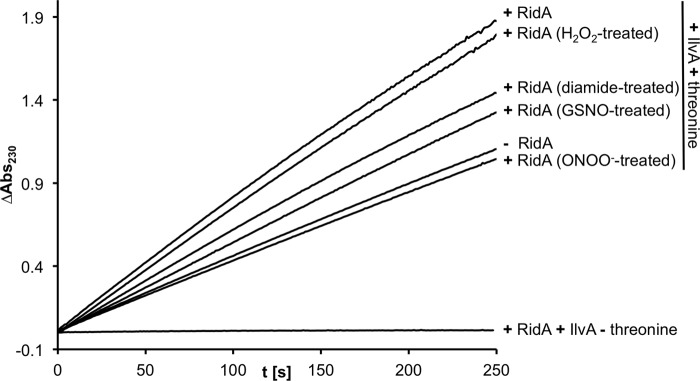
Threonine deaminase assay in the presence of RidA. RidA was modified with different nitrosative stressors (ONOO−, GSNO) and oxidative stressors (diamide, hydrogen peroxide). Only ONOO− inactivates RidA completely.
DISCUSSION
In biological systems, peroxynitrite arises most likely through the recombination of the NO• and O2˙̄ radical. It is a highly reactive species, and once formed, it decomposes in aqueous solution mostly to •OH, NO2•, and nitrite. Many other, often radical, decay products have been described, and ONOO− can directly form adducts, which are significantly more instable than peroxynitrite itself (for reviews of the chemistry and biological chemistry of peroxynitrite, see Refs. 27, 38). To study this somewhat complex oxidative/nitrosative stressor, we tested the effect of peroxynitrite on E. coli cells. We show here that concentrations of up to 2 mm are sublethal to this Gram-negative bacterium in MOPS minimal medium. Toxicity of peroxynitrite seems to be largely influenced by the presence of scavenging compounds in the medium. In particular carbonate, which is most likely present in media containing growing E. coli, has been shown to lower the antibacterial effectiveness of peroxynitrite (39).
On a molecular level, the ONOO− treatment leads to protein modification. We found that tyrosine nitration, a well known effect of peroxynitrite exposure (13), is fairly unspecific and affects a large number of E. coli proteins. This modification is thought to be irreversible and probably damages proteins irreparably. The dose-dependent protein tyrosine modifications are potentially contributing to the inhibition of E. coli by this nitrosative and oxidative stressor.
In contrast to the massive tyrosine nitration, oxidative thiol modifications caused by peroxynitrite are highly specific. We used an ICAT-based quantitative proteomic approach to determine changes in the thiol modifications of E. coli proteins. The proteins that we found to be cysteine-modified increased their oxidation up to 11.2-fold upon peroxynitrite treatment; however, the fraction of the reversibly oxidized portion made up only 19.6% of the individual protein. This low fraction can be explained by the reducing environment of the bacterial cytosol, where oxidized proteins are thought to be immediately reduced by dedicated and redundant systems, such as the thioredoxins and glutaredoxins. The fact that we can quantify the amount of both oxidized and reduced peptide highlights the advantage of our ratiometric ICAT-based approach to identifying thiol modifications. Methods that rely solely on the oxidized portion of a given protein for identification and quantification could lead to a gross overestimation of the significance of the rather small change in the absolute amount of oxidized protein.
It should be noted that the ICAT-based method we employ in this study has some shortcomings. First, we are unable to distinguish between different reversible modifications. Disulfide bonds, sulfenic acids, S-nitrosylations, glutathionylations, etc. are all reduced by TCEP and therefore will be labeled with heavy ICAT. Second, oxidized cysteines are expected to be more than an order of magnitude less present when compared with reduced cysteines in highly reducing cell compartments such as the bacterial cytoplasm. Because of the limited dynamic range of an MS-based quantitation, some oxidized cysteines can remain undetected (40). Third, overoxidized, irreversibly modified cysteines will escape our analysis. These shortcomings can be circumvented in part by other redox proteomics methods that are geared toward specific modifications or can identify overoxidized cysteines, for example, but these will then have different limitations. Extensive reviews of the advantages, limitations, and principles of redox proteomic methods can be found in Refs. 41, 42.
In this study, we identified four proteins that showed a significantly higher oxidation in peroxynitrite-treated cells. This relatively low number is somewhat surprising, because thiol modifications are considered to be one of the main effects of peroxynitrite on proteins (13). We have additionally used Ellman's assay to determine free thiols in crude cell extracts treated with different oxidative and nitrosative stressors (Fig. 14). In these experiments we did find a drop in free accessible thiols in cells treated with peroxynitrite when compared with untreated cells and cells treated with GSNO. This could indeed hint at overoxidation of cysteines caused by peroxynitrite, which would escape our ICAT-based approach.
FIGURE 14.
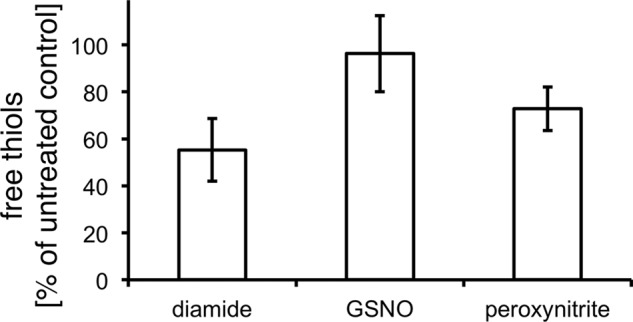
Determination of free thiol groups in crude cell extracts of cells treated 16 min with 1 mm peroxynitrite, 1 mm GSNO, and 1 mm diamide as compared with untreated cells. Levels of free thiols in control samples were set to 100%.
Another explanation would be the formation of inherently unstable sulfenic acids that are lost or modified otherwise (e.g. by glutathionylation) during the lysis and protein denaturation involved in the ICAT labeling process. Sulfenic acid formation is most likely the initial product of the reaction of peroxynitrite with thiols. We therefore used specific antibodies to detect peroxynitrite-induced sulfenylation or glutathionylation of proteins in Western blots (Fig. 15). In these experiments, we could not detect a change in the sulfenylation or glutathionylation state of E. coli proteins in response to peroxynitrite stress, in line with our ICAT-based experiments. Nevertheless, these experiments cannot exclude a loss of sulfenylation detection in our experiments based on the methodological limitations typical for iodoacetamide-based probes, especially in light of the fact that these probes can react to some extent with sulfenic acids (43). Therefore we must stress that our approach, as any other focused approach, will not be able to reveal the full extent of every possible modification caused by peroxynitrite or ONOO−-related reaction products.
FIGURE 15.

Detection of glutathionylated and sulfenylated proteins in cells treated with peroxynitrite. A, Western blot of protein extracts of bacterial cell cultures treated with peroxynitrite and NaOH (vehicle control) with a monoclonal anti-glutathione antibody. Glutathionylated ovalbumin served as positive control, and DTT-treated glutathionylated ovalbumin served as a negative control. Reducing gels were compared with nonreducing gels to account for unspecific binding of the antibody. B, Western blot of protein extracts of bacterial cell cultures treated with peroxynitrite and NaOH (vehicle control) with an anti-cysteine sulfenic acid antibody. Hydrogen peroxide-treated GAPDH served as positive control, and DTT-reduced hydrogen peroxide treated GAPDH served as a negative control. Reducing gels were compared with nonreducing gels to account for unspecific binding of the antibody. Hydrogen peroxide treatment of E. coli cells served as positive control.
In a proteomic approach to identify E. coli proteins that are thiol-modified upon NO• treatment of the cells, we identified 10 proteins that were significantly higher oxidized than under control conditions (9). There is no overlap with proteins identified in this study, which shows that peroxynitrite and NO• are not easily interconverted. This suggests that “nitrosative stress” is an umbrella term for a set of stresses that is actually highly diverse. Furthermore, exogenous reactive nitrogen species seem not to cause extensive reversible thiol modifications. This is in contrast to a recent study that demonstrates the occurrence of high endogenous protein S-nitrosylation in E. coli under anaerobic growth conditions, especially in the absence of the regulator OxyR (3).
Nevertheless, the proteins we found are critical during stress conditions, and knock-out mutants in genes encoding those proteins all showed growth defects under peroxynitrite stress. FrmA has been described as GSNO reductase and is required for the maintenance of SNO homeostasis (30), although MaeB (malic enzyme) is a key enzyme in the metabolic shift from NADH to NADPH (44). NADPH is a cofactor of major antioxidant enzymes, such as thioredoxin reductase or glutathione oxidoreductase.
Stoichiometric considerations brought us to exclude a role for these proteins as purely sacrificial sinks for peroxynitrite. Several studies on the protein abundance in E. coli and our own studies suggest that the abundance of these proteins is too low to act as a passive sacrificial sink (31, 45). It is possible that some of the proteins we found work as part of a system that actively detoxifies peroxynitrite in a mechanism that re-reduces the cysteines, e.g. with help of the glutathione system or other co-factors or that these proteins work as a sink for a highly toxic intermediate that is generated in small quantities under peroxynitrite stress.
We studied the relevance of the modifications found in vivo in more detail in experiments with purified RidA. The sole cysteine of RidA is highly susceptible toward nitrosative stressors in vitro. Overoxidation of Cys-107 in RidA by peroxynitrite eliminates the enamine/imine deaminase activity of this protein almost completely. The overall structure of RidA is not disrupted by the oxidative cysteine modification, and the peroxynitrite-inactivated protein retains its native trimeric state as shown by gel filtration. Interestingly, a cysteine to serine mutant of this protein retains activity but is no longer susceptible to peroxynitrite stress. This raises the question if the modifications observed in vivo and in vitro constitute protein damage or a regulative event. Growth tests demonstrated that under peroxynitrite stress, the presence of RidA is clearly advantageous for the cell. Unfortunately, a ridA mutant strain complemented with ridA from a plasmid did not overcome ONOO− stress better than a strain complemented with an empty plasmid, although we tried several different expression systems (data not shown). This could mean that RidA needs to be present at a specific dose to be effective in protecting cells under peroxynitrite stress conditions. RidA is a member of the YjgF/YER057c/UK114 superfamily (cd00448 in the NCBI conserved domain database (46)). Cys-107 is conserved within some but not all YjgF/YER057c/UK114 subfamilies. Because Cys-107 is not needed for catalytic activity, the high level of conservation points toward a regulatory function, although we do not yet fully understand the role of this protein in the oxidative/nitrosative stress response.
The specificity of peroxynitrite toward only a small number of cysteines demonstrates that oxidative cysteine modification by peroxynitrite is a process driven mostly by kinetic factors. Given the highly oxidizing redox potential of the radicals that can form during peroxynitrite decomposition, the reaction of virtually any reduced cysteine within the E. coli cell with peroxynitrite or its decay products should be thermodynamically highly favorable. However, we observe that most cysteines are not affected by peroxynitrite, which clearly shows that these reactions are kinetically controlled. We and others could show in redox proteomic studies that other reactive species such as hydrogen peroxide, sodium hypochlorite, and nitric oxide also have a particular set of cysteines with which they can react (8, 9, 47–49). This kinetic aspect of reactions of thiols with oxidants is probably one of the mechanistic underpinnings that allows for variation in redox regulation and warrants further study by redox proteomics experiments.
Supplementary Material
Acknowledgments
We thank Lu H. Thanh and Wiebke Karad for their help in establishing the in vitro studies of RidA activity and Anna Kusnezova and Jakob Eller for help in establishing the Ellman assays. Our special thanks go to Dr. Julia Bandow for carefully reading the manuscript and the many helpful suggestions.
This work was supported in part by a grant from the “NRW-Rückkehrerprogramm” of the German State of North Rhine-Westphalia (to L. I. L.).

This article contains supplemental Tables 1 and 2.
- ROS
- reactive oxygen species
- ACN
- acetonitrile
- FTMS
- Fourier transform mass spectrometry
- GSNO
- S-nitrosylated glutathione
- ICAT
- isotope coded affinity tag
- IEF
- isoelectric focusing
- NO•
- nitric oxide
- ONOO−
- peroxynitrite
- O2˙̄
- superoxide anion
- RNS
- reactive nitrogen species
- TCEP
- tris(2-carboxyethyl)phosphine
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- TAPS
- 3-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}-1-propanesulfonic acid.
REFERENCES
- 1. Zheng M., Aslund F., Storz G. (1998) Activation of the OxyR transcription factor by reversible disulfide bond formation. Science 279, 1718–1721 [DOI] [PubMed] [Google Scholar]
- 2. Zheng M., Storz G. (2000) Redox sensing by prokaryotic transcription factors. Biochem. Pharmacol. 59, 1–6 [DOI] [PubMed] [Google Scholar]
- 3. Seth D., Hausladen A., Wang Y.-J., Stamler J. S. (2012) Endogenous protein S-nitrosylation in E. coli: regulation by OxyR. Science 336, 470–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Antelmann H., Helmann J. D. (2011) Thiol-based redox switches and gene regulation. Antioxid. Redox Signal. 14, 1049–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brandes N., Schmitt S., Jakob U. (2009) Thiol-based redox switches in eukaryotic proteins. Antioxid. Redox Signal. 11, 997–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burgoyne J. R., Madhani M., Cuello F., Charles R. L., Brennan J. P., Schröder E., Browning D. D., Eaton P. (2007) Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science 317, 1393–1397 [DOI] [PubMed] [Google Scholar]
- 7. Jakob U., Muse W., Eser M., Bardwell J. C. (1999) Chaperone activity with a redox switch. Cell 96, 341–352 [DOI] [PubMed] [Google Scholar]
- 8. Leichert L. I., Gehrke F., Gudiseva H. V., Blackwell T., Ilbert M., Walker A. K., Strahler J. R., Andrews P. C., Jakob U. (2008) Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. U.S.A. 105, 8197–8202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brandes N., Rinck A., Leichert L. I., Jakob U. (2007) Nitrosative stress treatment of E. coli targets distinct set of thiol-containing proteins. Mol. Microbiol. 66, 901–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wood P. M. (1988) The potential diagram for oxygen at pH 7. Biochem. J. 253, 287–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ischiropoulos H., Zhu L., Beckman J. S. (1992) Peroxynitrite formation from macrophage-derived nitric oxide. Arch. Biochem. Biophys. 298, 446–451 [DOI] [PubMed] [Google Scholar]
- 12. Beckman J. S., Beckman T. W., Chen J., Marshall P. A., Freeman B. A. (1990) Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 87, 1620–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alvarez B., Radi R. (2003) Peroxynitrite reactivity with amino acids and proteins. Amino Acids 25, 295–311 [DOI] [PubMed] [Google Scholar]
- 14. McLean S., Bowman L. A., Sanguinetti G., Read R. C., Poole R. K. (2010) Peroxynitrite toxicity in Escherichia coli K12 elicits expression of oxidative stress responses and protein nitration and nitrosylation. J. Biol. Chem. 285, 20724–20731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lambrecht J. A., Flynn J. M., Downs D. M. (2012) Conserved YjgF protein family deaminates reactive enamine/imine intermediates of pyridoxal 5′-phosphate (PLP)-dependent enzyme reactions. J. Biol. Chem. 287, 3454–3461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Neidhardt F. C., Bloch P. L., Smith D. F. (1974) Culture medium for enterobacteria. J. Bacteriol. 119, 736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K. A., Tomita M., Wanner B. L., Mori H. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sternberg N. L., Maurer R. (1991) Bacteriophage-mediated generalized transduction in Escherichia coli and Salmonella typhimurium. Methods Enzymol. 204, 18–43 [DOI] [PubMed] [Google Scholar]
- 19. Lindemann C., Leichert L. I. (2012) Quantitative redox proteomics: the NOxICAT method. Methods Mol. Biol. 893, 387–403 [DOI] [PubMed] [Google Scholar]
- 20. Gygi S. P., Rist B., Gerber S. A., Turecek F., Gelb M. H., Aebersold R. (1999) Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 17, 994–999 [DOI] [PubMed] [Google Scholar]
- 21. Zhou J., Rudd K. E. (2013) EcoGene 3.0. Nucleic Acids Res. 41, D613–D624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mortensen P., Gouw J. W., Olsen J. V., Ong S.-E., Rigbolt K. T., Bunkenborg J., Cox J., Foster L. J., Heck A. J., Blagoev B., Andersen J. S., Mann M. (2010) MSQuant, an Open Source Platform for Mass Spectrometry-Based Quantitative Proteomics. J. Proteome Res. 9, 393–403 [DOI] [PubMed] [Google Scholar]
- 23. Wilkins M. R., Gasteiger E., Bairoch A., Sanchez J. C., Williams K. L., Appel R. D., Hochstrasser D. F. (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552 [DOI] [PubMed] [Google Scholar]
- 24. Creighton T. E. (1990) in Protein Structure, A Practical Approach (Creighton T. E., ed) 1st Ed. pp. 155–166, IRL Press at Oxford University Press, Oxford [Google Scholar]
- 25. Wong C., Sridhara S., Bardwell J. C., Jakob U. (2000) Heating greatly speeds Coomassie Blue staining and destaining. BioTechniques 28, 426–428 [DOI] [PubMed] [Google Scholar]
- 26. Ischiropoulos H., Zhu L., Chen J., Tsai M., Martin J. C., Smith C. D., Beckman J. S. (1992) Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch. Biochem. Biophys. 298, 431–437 [DOI] [PubMed] [Google Scholar]
- 27. Goldstein S., Lind J., Merényi G. (2005) Chemistry of peroxynitrites as compared to peroxynitrates. Chem. Rev. 105, 2457–2470 [DOI] [PubMed] [Google Scholar]
- 28. Larsen T. M., Boehlein S. K., Schuster S. M., Richards N. G., Thoden J. B., Holden H. M., Rayment I. (1999) Three-dimensional structure of Escherichia coli asparagine synthetase B: A short journey from substrate to product. Biochemistry 38, 16146–16157 [DOI] [PubMed] [Google Scholar]
- 29. Boehlein S. K., Richards N. G., Schuster S. M. (1994) Glutamine-dependent nitrogen transfer in Escherichia coli asparagine synthetase B. Searching for the catalytic triad. J. Biol. Chem. 269, 7450–7457 [PubMed] [Google Scholar]
- 30. Liu L., Hausladen A., Zeng M., Que L., Heitman J., Stamler J. S. (2001) A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 410, 490–494 [DOI] [PubMed] [Google Scholar]
- 31. Lu P., Vogel C., Wang R., Yao X., Marcotte E. M. (2007) Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 25, 117–124 [DOI] [PubMed] [Google Scholar]
- 32. Marla S. S., Lee J., Groves J. T. (1997) Peroxynitrite rapidly permeates phospholipid membranes. Proc. Natl. Acad. Sci. U.S.A. 94, 14243–14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmiedeknecht G., Kerkhoff C., Orsó E., Stöhr J., Aslanidis C., Nagy G. M., Knuechel R., Schmitz G. (1996) Isolation and characterization of a 14.5-kDa trichloroacetic-acid-soluble translational inhibitor protein from human monocytes that is up-regulated upon cellular differentiation. Eur. J. Biochem. 242, 339–351 [DOI] [PubMed] [Google Scholar]
- 34. Melloni E., Michetti M., Salamino F., Pontremoli S. (1998) Molecular and functional properties of a calpain activator protein specific for μ-isoforms. J. Biol. Chem. 273, 12827–12831 [DOI] [PubMed] [Google Scholar]
- 35. Morishita R., Kawagoshi A., Sawasaki T., Madin K., Ogasawara T., Oka T., Endo Y. (1999) Ribonuclease activity of rat liver perchloric acid-soluble protein, a potent inhibitor of protein synthesis. J. Biol. Chem. 274, 20688–20692 [DOI] [PubMed] [Google Scholar]
- 36. Farkas A., Nardai G., Csermely P., Tompa P., Friedrich P. (2004) DUK114, the Drosophila orthologue of bovine brain calpain activator protein, is a molecular chaperone. Biochem. J. 383, 165–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Volz K. (1999) A test case for structure-based functional assignment: the 1.2 Å crystal structure of the yjgF gene product from Escherichia coli. Protein Sci. 8, 2428–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ferrer-Sueta G., Radi R. (2009) Chemical biology of peroxynitrite: kinetics, diffusion, and radicals. ACS Chem. Biol. 4, 161–177 [DOI] [PubMed] [Google Scholar]
- 39. Hurst J. K., Lymar S. V. (1997) Toxicity of peroxynitrite and related reactive nitrogen species toward Escherichia coli. Chem. Res. Toxicol. 10, 802–810 [DOI] [PubMed] [Google Scholar]
- 40. Held J. M., Gibson B. W. (2012) Regulatory control or oxidative damage? Proteomic approaches to interrogate the role of cysteine oxidation status in biological processes. Mol. Cell. Proteomics 11, R111.013037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Butterfield D. A., Dalle-Donne I. (2012) Redox proteomics. Antioxid. Redox Signal. 17, 1487–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leichert L. I., Müller A. (2013) in Oxidative Stress and Redox Regulation (Jakob U., Reichmann D., eds) pp. 157–186, Springer, Dordrecht [Google Scholar]
- 43. Conte, Lo M., Carroll K. S. (2012) in Oxidative Stress and Redox Regulation (Jakob U., Reichmann D., eds) pp. 1–42, Springer, Dordrecht, Heidelberg, New York, London [Google Scholar]
- 44. Singh R., Lemire J., Mailloux R. J., Appanna V. D. (2008) A novel strategy involved anti-oxidative defense: The conversion of NADH into NADPH by a metabolic network. PLoS ONE 3, e2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ishihama Y., Schmidt T., Rappsilber J., Mann M., Hartl F. U., Kerner M. J., Frishman D. (2008) Protein abundance profiling of the Escherichia coli cytosol. BMC Genomics 9, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marchler-Bauer A., Lu S., Anderson J. B., Chitsaz F., Derbyshire M. K., DeWeese-Scott C., Fong J. H., Geer L. Y., Geer R. C., Gonzales N. R., Gwadz M., Hurwitz D. I., Jackson J. D., Ke Z., Lanczycki C. J., Lu F., Marchler G. H., Mullokandov M., Omelchenko M. V., Robertson C. L., Song J. S., Thanki N., Yamashita R. A., Zhang D., Zhang N., Zheng C., Bryant S. H. (2011) CDD: A Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brandes N., Reichmann D., Tienson H., Leichert L. I., Jakob U. (2011) Using quantitative redox proteomics to dissect the yeast redoxome. J. Biol. Chem. 286, 41893–41903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang H., Wang S., Lu Y., Alvarez S., Hicks L. M., Ge X., Xia Y. (2012) Proteomic analysis of early-responsive redox-sensitive proteins in Arabidopsis. J. Proteome Res. 11, 412–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wolf C., Hochgräfe F., Kusch H., Albrecht D., Hecker M., Engelmann S. (2008) Proteomic analysis of antioxidant strategies of Staphylococcus aureus: Diverse responses to different oxidants. Proteomics 8, 3139–3153 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.