

Background: GIPr mediates insulin secretion upon GIP stimulation.
Results: Gipg013 is a highly specific and potent antagonist of GIPr with a fully characterized mode of action.
Conclusion: Gipg013 antagonizes GIPr in vivo, as exemplified by inhibition of GIP-induced insulin secretion.
Significance: This antagonizing antibody to the GIPr will be useful as a tool to further understand the biological roles of GIP.
Keywords: Antibody Engineering, Crystal Structure, Diabetes, G-Protein-coupled Receptors (GPCR), Phage Display, Antagonist, GIP Receptor (GIPr), Glucose-dependent Insulinotropic Polypeptide (GIP), Incretin
Abstract
Glucose-dependent insulinotropic polypeptide (GIP) is an endogenous hormonal factor (incretin) that, upon binding to its receptor (GIPr; a class B G-protein-coupled receptor), stimulates insulin secretion by beta cells in the pancreas. There has been a lack of potent inhibitors of the GIPr with prolonged in vivo exposure to support studies on GIP biology. Here we describe the generation of an antagonizing antibody to the GIPr, using phage and ribosome display libraries. Gipg013 is a specific competitive antagonist with equally high potencies to mouse, rat, dog, and human GIP receptors with a Ki of 7 nm for the human GIPr. Gipg013 antagonizes the GIP receptor and inhibits GIP-induced insulin secretion in vitro and in vivo. A crystal structure of Gipg013 Fab in complex with the human GIPr extracellular domain (ECD) shows that the antibody binds through a series of hydrogen bonds from the complementarity-determining regions of Gipg013 Fab to the N-terminal α-helix of GIPr ECD as well as to residues around its highly conserved glucagon receptor subfamily recognition fold. The antibody epitope overlaps with the GIP binding site on the GIPr ECD, ensuring competitive antagonism of the receptor. This well characterized antagonizing antibody to the GIPr will be useful as a tool to further understand the biological roles of GIP.
Introduction
Glucose-dependent insulinotropic peptide (GIP)2 is an incretin hormone released from intestinal K cells in response to food intake (1–3). GIP primarily circulates as a 42-amino acid peptide (GIP(1–42)) but is also present in a form lacking the C-terminal 12 amino acids (GIP(1–30)), which exerts very similar effects at β-cells (4). The receptor for GIP (GIPr) is expressed on pancreatic β-cells, where activation leads to insulin release (5). GIPr is also expressed in adipocytes, which respond to increased GIP levels with increased glucose uptake, fatty acid synthesis, and fatty acid incorporation into lipids within the adipocytes (6). Whereas the related incretin hormone GLP-1 has found widespread therapeutic use, the complex nature of GIP biology has meant that proposals for agonism (7) or antagonism of the GIPr (8) for the treatment of diabetes and obesity have yet to lead to therapeutic applications.
To understand the complex effects of GIP on plasma glucose and fat deposition, it would be desirable to have a specific potent antagonist with an extended half-life. To date, only low potency antagonists with a short half-life have been available. Truncated GIP peptide antagonists have been reported: GIP(3–42) (9), GIP(6–30) (10), and GIP(7–30), which has an IC50 of ∼100 nm in a cAMP generation assay (5).
The GIPr belongs to the glucagon receptor subfamily of class 2 (class B) GPCRs, a class that also includes the receptors for GLP1, glucagon, parathyroid hormone, calcitonin, corticotropin-releasing factor, and other therapeutically important peptide hormones (11). The isolation of neutralizing antibodies to GPCRs has proven difficult, due to the low proportion of the receptor exposed on the extracellular surface and the involvement of ligand binding to transmembrane regions in receptor activation. Only limited examples have been published to date (e.g. monoclonal antibodies to the class A receptors CXCR4 and S1P3, from mouse hybridomas (12, 13), and CCR5, from human scFv phage display libraries (14, 15)). For class B GPCRs, neutralizing monoclonal antibodies in complex with the glucagon receptor and neutralizing polyclonal antibodies to the GIPr have been reported (16–18). More recently, fully human monoclonal antibodies to glucagon and GLP1 receptors have been obtained by immunization of mice transgenic for human antibody genes (19, 20). Crystal structures of monoclonal antibodies in complex with the glucagon receptor have been reported (21). In this paper, we report an antagonist antibody derived from phage display libraries, Gipg013, that shows potent competitive neutralization of GIP activity at its receptor. Gipg013 should prove to be a useful tool for understanding the biological effects of GIP at the GIPr.
The crystal structure of GIP(1–42) in complex with the extracellular domain (ECD) of the GIPr demonstrated that the hormone binds in an α-helical conformation in a surface groove of the ECD largely through hydrophobic interactions (22). It has been proposed that the C-terminal part of GIP first interacts with the ECD, and this event then helps the binding of the N-terminal part of the peptide with the juxtamembrane region of the receptor and activation of the receptor. The binding of peptides to class B receptors shows some common structural features. Superimposition of the crystal structures of these class B GPCRs shows that the sandwich fold, consisting of an α-helix and two anti-parallel β-sheets linked by three disulfide bonds, is well conserved in the family, although sequence alignment shows less conservation (23). We have determined the crystal structure of the Gipg013 Fab in complex with the GIPr ECD and compared this with the structure for GIP in complex with the GIPr ECD.
EXPERIMENTAL PROCEDURES
GIPr ECD
The GIPr ECD with an N-terminal His6 and FLAG tag was expressed and purified as described previously (22) and biotinylated using EZ-link Sulfo-NHS-LC-Biotin (Perbio/Pierce, product no. 21335). Parthier (22) reported the Kd of GIP(1–42) for GIPr ECD as 1.1 μm as measured by calorimetry. The GIPr ECD preparation used in selections and screening was validated by competition with the cell surface GIPr on HEK293 cells for binding to GIP in the cAMP assay. An IC50 of 7.0 μm was obtained.
Cell Culture
Stable cell lines expressing human, mouse, rat, and dog GIP receptor (HEK293 human GIPr, mouse GIPr, rat GIPr, and dog GIPr) were generated in HEK293 cells. In brief, HEK293 cells were transfected with the expression vector pIRESneo3 containing the full-length GIP receptor gene of each species. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% FBS and 0.8 mg/ml Geneticin (G418) (Invitrogen) at 37 °C in a humidified environment containing 5% CO2. Cells were seeded every 2–4 days at a density to achieve 3–4 × 107 cells on the day of the assay. Cells were harvested using Accutase (PAA Laboratories GmbH), counted, and resuspended in a suitable volume of appropriate assay buffer to achieve correct cell density for either selection or assay procedures, as outlined below.
Phage Display Libraries
The combined spleen library (8.5 × 1010) has been described by Lloyd et al. (24) and was included in the phage selections reported here. A sublibrary of the combined spleen library, S5 (1.8 × 1010) was investigated separately and was applied as the source of V genes for the generation of a separate ribosome display library.
Construction of Naive PBL Phage Display Libraries
40-μg aliquots of the ribosome display PBL libraries were digested with NcoI and NotI to excise the scFv constructs and clone them into pCantab6 (25). The ligations were transformed into Escherichia coli TG1 by electroporation, yielding libraries of 6.0 × 108 and 8.0 × 108 cfu for the phage display PBLκ and phage display PBLλ libraries, respectively. 88 clones from each library were picked and sequenced to validate the quality of the libraries, revealing at least 75% functional clones.
Ribosome Display Libraries
Generation of PBL library has been described in detail previously (26). Here we have applied the two subsets (κ and λ) as individual libraries in the selections. Similar to the PBL library, the S5 library was generated by subcloning the variable genes from the phage display S5 and recombining into a new ribosome display library.
Display Library Selection
The phage display and ribosome display selections on soluble biotinylated GIPr ECD were carried out as described previously (26–31). In brief, the phage display selections were performed with biotinylated GIPr ECD at 200, 50, 25, and 10 nm in rounds 1–4, respectively, to increase selection pressure throughout the selection rounds. The ribosome display selections were performed with biotinylated GIPr ECD at 200, 200, 50, and 10 nm in rounds 1–4, respectively. To enable cell surface selections, the outputs from the fourth round of ribosome display selection were subcloned to phage display format, yielding libraries of 1.5 × 107, 7.9 × 106, and 1.1 × 107 for PBLλ, PBLκ, and S5, respectively. The cell surface selections were performed on HEK293 cells transformed with either an empty control construct or a construct for overexpressing the human GIP receptor and were performed with all of the subcloned fourth round ribosome display libraries and the second round phage display library selection outputs. The selections were monitored by screening outputs for scFv binding to GIPr ECD in phage ELISA as described previously (30).
Screening for GIPr Antagonism in Cell-based Activity Assay
The cAMP HTRF assay was based upon the ability of an antibody to inhibit the GIP/GIPr interaction (acting as an antagonist) and thus decrease cellular production of cAMP. The resultant assay signal is inversely related to the levels of cAMP generated. The cAMP dynamic 2 kit (Cisbio) was used according to the manufacturer's recommendations.
For the high throughput screen of scFv peripreps (∼10,500 clones, evenly picked across selections), HEK293 GIPr cells were resuspended in cell medium (DMEM, 10% FBS, 1.6% G418), containing 0.5 mm 3-isobutyl-1-methylanthine (Sigma), and 5 μl of cells were incubated with 2.5 μl of periplasmic preparations in a 384-well plate for 30 min at room temperature. Following this, 2.5 μl of GIP(1–42) peptide at 4 pm (an EC80 concentration) (Bachem) was added to samples, which were incubated for a further 30 min in order to produce an agonist response.
For profiling experiments, cells were incubated with a 2.5-μl dilution series of purified IgG or scFv (11-point, half-logarithmic dilutions) (32). The IC50 values were calculated by non-linear regression curve fitting using GraphPad Prism®, version 5.01.
Schild Analysis
The assay was carried out as the cAMP HTRF assay outlined above, with the following adjustments to measure dose-response curves at different antibody concentrations. Briefly, HEK293 human GIPr cells were incubated with a dilution series of a Gipg013 IgG antagonist starting from 3.75 μm (final concentration) or with assay buffer only (2.5 μl). Following incubation for 5 min, a dilution series of agonist (GIP peptide) (2.5 μl) was added to each concentration of Gipg013 IgG, and incubated for a further 30 min. EC50 values calculated for each antibody concentration and dose ratios determined using GraphPad Prism®. Subsequently, Schild plots were analyzed to yield pA2.
Binding Kinetic Estimation by Reflectometric Interference Spectroscopy
The Octet RED system (ForteBio) was used to determine the equilibrium dissociation constant (KD). The assays were performed in PBS supplemented with 10× kinetics buffer, using a volume of 200 μl for all incubations. The Super Streptavidin biosensors were loaded with biotinylated GIPr ECD at 1.2 μg/ml. All of the immobilized metal affinity chromatography-purified scFvs were assayed at ∼0.5 μm (12.5 μg/ml). The kinetic data sets were fitted using 1:1 Langmuir binding using Octet RED software to yield dissociation constants.
Purification of scFv and IgG
scFv was expressed in E. coli and purified from periplasmic extracts by immobilized metal affinity chromatography as described previously (33). For IgG conversion, the variable genes were cloned into the pEU vectors and expressed and purified as described previously (25).
Receptor Ligand Binding Assay
Fluorescent microvolume assay technology (FMAT) was used for the receptor ligand binding assay. Gipg013 or isotype control antibody NIP228 (0.25 μg/ml final concentration) was prepared in 384-well plates (10 μl). To these, 0.4 μg/ml (final concentration) AlexaFluor 647-labeled goat anti-human IgG H+L (Invitrogen) (10 μl) was added.
Stably transfected cell lines overexpressing either human, dog, mouse, or rat receptors for glucagon, GLP-1, or GIP were harvested and resuspended in Hanks' balanced salt solution (Invitrogen) containing 0.1% BSA (Sigma) and added to the 384-well assay plate (5000 cells in 20 μl). Plates were incubated in the dark at room temperature for 3 h. Subsequently, plates were read on the FMATTM 8100 HTS System (Applied Biosystems), and FL1 readings were plotted using GraphPad Prism®, version 5.01.
Receptor Ligand Competition Assay
FMAT was used for the receptor ligand competition assay. Dilution series of the antibodies were prepared in 384-well plates (10 μl). To this, 0.5 nm AlexaFluor 647-labeled GIP (Cambridge Research Biochemicals) (20 μl) was added. HEK293 human GIPr cells were harvested and resuspended in Hanks' balanced salt solution (Invitrogen) containing 0.1% BSA (Sigma), and added to the plate (10 μl). Plates were incubated in the dark at room temperature for 1–2 h. Subsequently, plates were read on the FMATTM 8100 HTS system (Applied Biosystems). For FMAT analysis, IC50 values were determined by non-linear regression curve fitting using GraphPad Prism®, version 5.01.
Binding Kinetics by Surface Plasmon Resonance
Real-time binding kinetics were analyzed by surface plasmon resonance using a Biacore 2000 Instrument. All reagents were purchased from BIAcore (Uppsala, Sweden). Experiments were carried out at 25 °C using a constant flow rate (30 μl/min) in running buffer (HBS-N; 10 mm HEPES, pH 7.4, 150 mm sodium chloride). Gipg013 IgG was immobilized on the sensor chip (CM5) at a low concentration using the amine coupling method as described by the manufacturer. Binding of GIPr ECD was observed at a range of concentrations (500, 250, 125, and 62.5 nm (concentrations used for the IgG experiment were 125, 62.5, 31.25, and 15.6 nm)). The sensor chip was regenerated with Biacore Regeneration Buffer (10 nm glycine-HCl, pH 1.5). Sensorgrams were analyzed using BIAevaluation software, version 3.0, using a 1:1 Langmuir binding model.
Crystallization, X-ray Data Collection, and Structure Solution
Gipg013 Fab (3.9 mg/ml) and GIPr ECD (0.77 mg/ml) protein samples were mixed at a 1:1 molar ratio in 0.1 m Tris buffer (0.1 m Tris, pH 7.6, 0.1 m NaCl). Using sparse matrix screens from Hampton Research and Molecular Dimensions (Suffolk, UK) in 96-well Corning plates (Corning Inc.) at 4 and 20 °C, preliminary crystallization conditions were identified. Sitting drop vapor diffusion experiments were performed using a Phoenix crystallization robot (Art Robbins Instruments). Gip013 Fab-GIPr ECD complex solution was mixed with reservoir solutions at a 1:1, 1:2, or 2:1 ratio (200-nl final volume), and the mixtures were equilibrated against 50 μl of reservoir solution. Crystallization conditions, data collection, and refinement statistics are summarized in Table 2. Crystals were flash-frozen in liquid nitrogen by adding 20% glycerol to the reservoir solution. Diffracting quality crystals were obtained from 0.02 m TAPS, pH 9.0, 30% (w/v) PEG 10,000.
TABLE 2.
Data processing and refinement statistics
| Unit cell parameters | |
| a, b, and c (Å) | a = 48.3, b = 109.9, c = 105.9 |
| α, β, and γ (degrees) | α = 90.0, β = 97.8, γ = 90.0 |
| Space group: | P21 |
| Resolution range | 47.71–3.0 Å |
| No. of molecules/asymmetric unit | 2 |
| No. of reflections | |
| Observed | 90,279 (13,110) |
| Unique | 21,867 (3167) |
| I/σ (I) | 7.8 (2.6) |
| Completeness (%) | 100.0 (100.0) |
| Rmerge (%)a | 16.0 (61.0) |
| Multiplicity | 4.1 (4.1) |
| Rcryst (%)b | 25.5 |
| Rfree (%)b | 31.1 |
| No. of protein atoms | 7639 |
| No. of chains | 6 |
| Root mean square deviation from ideal geometry | |
| Bond length (Å) | 0.005 |
| Bond angles (degrees) | 1.150 |
| B values (Å2) | |
| Wilson B | 48.89 |
| Average B | 48.22 |
| Ramachandran plot (%) | |
| Residues in preferred regions | 89.31 |
| Residues allowed regions | 10.58 |
| Residues in disallowed regions | 0.1 |
a Rmerge = ΣhklΣi‖Ii(hkl) − (I(hkl))‖/ΣhklΣiIi(hkl), where Ii(hkl) is the ith observation of reflection hkl and (I(hkl)) is the weighted average intensity for all observations i of reflection hkl. Values in parentheses refer to the highest resolution shell.
b Rcryst and Rfree = (Σ‖Fo| − |Fc‖)/(Σ|Fo|), where |Fo| is the observed structure factor amplitude and |Fc| is the calculated structure factor amplitude.
X-ray data were collected using a MAR-345dtb image plate detector (MAR Research, Hamburg, Germany) mounted on a rotating anode x-ray generator equipped with a Helios optical system (Microstar Generator, Bruker AXS). Data were processed to 3.0 Å, using the programs MOSFLM (34) and SCALA (35). The complex structure was solved by molecular replacement using the program PHASER (36). Polyalanine models of incretin-bound extracellular domain of a GPCR (PDB code 2QKH) and murine IGG1 λ antibody (PDB code 1GIG) were used for GIPr ECD and Gipg013 Fab fragment, respectively. Refinement was done using the programs PHENIX-Refine (36), REFMAC5 (37), and COOT (38) with 5% of data set aside to calculate Rfree. Density modification was performed using DM in the program suite CCP4 followed by refinement, applying TLS refinement. The final structure was validated using the program PROCHECK (39). Figures were prepared using the program PyMOL (40).
Static Insulin Secretion Assays in Dispersed Rat Islets
Following isolation, islets were cultured in 11 mm glucose RPMI overnight. Subsequently, islets were collected and allowed to sediment, medium was removed, 1 ml of TryplExpress (Invitrogen) was added, and the islets were dispersed for 5 min. The cells were washed and reconstituted in KRH (129 mm NaCl, 5.0 mm NaHCO3, 4.8 mm KCl, 1.2 mm KH2PO4, 1.2 mm MgSO4, 10 mm Hepes, 2.5 mm CaCl2, 0.1% BSA fraction V, 3 mm glucose). The cell density was adjusted to 1.43 × 104 cells/ml prior to incubation at 37 °C in 5% CO2. After 30 min, buffer was removed, and fresh KRH buffer was added to obtain the same density of cells, and 70 μl (1000 cells)/well was dispensed on a 96-well plate containing treatments, which was incubated at 37 °C in 5% CO2 for 60 min.
For the insulin quantification, the buffer was removed, and insulin was quantified using the insulin HTRF kit (CisBio). All samples were tested undiluted or diluted 1:2 on the low range in 384-well plates. All plates were read on an Envision plate reader. The raw data obtained from the insulin HTRF were expressed as ng/1000 cells/h. Results for each treatment group (n = 6) were averaged in Excel, S.E. was calculated.
Single Dose Pharmacokinetics Study with Gipg013 in Mice
Three groups of six female C57 mice were administrated Gipg013 at 3 mg/kg intravenously and 3 and 30 mg/kg Gipg0113 subcutaneously, respectively. Pharmacokinetics analysis was performed in a sandwich ELISA-based assay using mouse anti-human IgG, clone JDC-10 (Southern Biotech) for capture and HRP anti-human IgG, clone G18-145 (BD Pharmingen) for detection.
GIP-induced Insulin Secretion
Male Sprague-Dawley rats (350–480 g) were fasted for 6 h and anesthetized with Inactin® (120 mg/kg intraperitoneally). Body temperature was monitored with a rectal probe and maintained between 37.5 and 38.0 °C throughout the experiment. Animals were tracheotomized (polyethylene tubing PE 240), and catheters were placed in the right jugular vein (two PE10; one for substance administration and one for GIP delivery) and left carotid artery (PE50) for blood sampling.
The rats were given two identical 10-min periods of GIP infusion (50 pmol/kg/min) with 60 min between the periods. During each period, blood samples for insulin levels were collected at 0 (immediately before the start of GIP infusion) and 5, 10, 15, and 30 min after the start of GIP infusion. Vehicle (0.9% NaCl + 0.2% BSA, n = 6) or GIP013 (3 mg/kg or 30 mg/kg, n = 3) was administered as a bolus 30 min prior to start of the second GIP infusion. A blood sample was also collected 30 min prior to the first GIP infusion to obtain basal levels of glucose and insulin and 90 min after GIP013 infusion to determine substance plasma concentration. Insulin concentrations were measured using radioimmunoassay (rat insulin RIA kit; Linco Research, St. Charles, MO). The experiment was approved by the local ethics committee in Gothenburg, Sweden.
RESULTS
Selection of GIPr-antagonizing Antibodies
In order to generate antibodies specifically antagonizing the GIPr, scFv ribosome display and phage display libraries were selected for binding to purified GIPr ECD either as the sole enrichment method or followed by a selection step on GIPr-overexpressing cells. Selections were monitored for enrichment of clones binding to GIPr ECD and diversity. This identified 490 unique scFv clones binding to GIPr ECD. Phage and ribosome display selections generated populations of antibodies with different sequences, thereby supplementing each other in the generation of a large panel of binding clones (data not shown). Subsequently, selection outputs were screened for antibodies able to antagonize cAMP production, stimulated by the ligand GIP in a cell-based activity assay. The high throughput screen for inhibition of GIP-stimulated cAMP production identified 291 potential hits, of which 33 unique scFvs were confirmed in the profiling assay using purified scFv antibodies, with 23 clones showing full antagonism of the GIPr (IC50 values in the range of 4–178 nm). Binding kinetics of the scFvs to the GIPr ECD were investigated by reflectometric interference spectroscopy using an Octet RED instrument. Binding kinetics were estimated from a kinetic experiment at a single antibody concentration to provide a crude ranking of the clones, which displayed dissociation constants between 6 and 700 nm.
Conversion of Antibodies to IgG1 Format
The VH and VL genes from the scFv clones were incorporated into constructs for full-length antibodies in the IgG1 format and expressed in mammalian cells. Purified IgGs were reprofiled in the cell-based activity assay to yield IC50 values for the antagonism of the human GIPr. Two clones retained full antagonist activity, whereas several clones lost potency or were characterized as partial antagonists. The antibodies Gipg013 and Gipg133 yielded complete antagonistic profiles with IC50 values of 6 and 2 nm, respectively (Fig. 1, A and B). Upon IgG conversion, Gipg013 gained potency from 233 nm as scFv to 6 nm as IgG, whereas Gipg133 only showed a modest gain in potency from 9 nm as scFv to 2 nm as IgG. The amino sequence of the variable domains of the two antibodies is shown in Table 1.
FIGURE 1.

Antagonism of GIP-induced cAMP production in GIPr-overexpressing cell lines. Gipg013 and Gipg133 were characterized for GIPr antagonism in a cell-based cAMP HTRF assay, and data were plotted using nonlinear regression. A and B show antagonistic profiles from HEK293 cells overexpressing human, mouse, rat, and dog GIPr using Gipg013 and Gipg133 IgGs, respectively. C and D show antagonistic and agonistic profiles in the human GIPr assay for GIP(7–30), Pro3GIP, and GIP. Values have been normalized to the maximum activity of GIPr, which is defined by total cellular cAMP produced in the agonism assay or in the absence of peptide/IgG in the antagonism assay. Values shown are the mean ± S.E. (error bars) from duplicate wells, and data shown are representative of at least three separate experiments. Values shown are the mean ± S.E. from duplicate wells. ●, human; ■, mouse; ▾, rat; ▴, dog; ♦, isotype control IgG1; *, GIP; +, GIP(7–30); ×, Pro3GIP.
TABLE 1.
Sequence of Gipg013 and Gipg133
Protein sequence of VH and VL with the CDR sequences shown in boldface type.

Antagonism of Mouse, Rat, and Dog GIPr
To evaluate species cross reactivity, Gipg013 and Gipg133 IgGs were profiled in the cAMP HTRF assay using HEK293 cells overexpressing either mouse, rat, or dog GIPr. Gipg013 IgG was found to cross-react with all three receptor species, with IC50 values ranging from 8 to 19 nm. Gipg133 IgG antagonized the dog receptor with an IC50 of 5 nm, but it had no effect on either mouse or rat receptors (Fig. 1, A and B) and is likely to bind to a different epitope on GIPr than Gipg013.
Comparing Properties of Antibodies with Other GIP Antagonists
Gipg013 and Gipg133 were compared with GIP(7–30) as an exemplar of antagonists derived by truncation of GIP. GIP(7–30) showed a much weaker antagonism of the receptor with an IC50 of 230 nm in our cell-based activity assay (Fig. 1C). The data are consistent with the IC50 of 100 nm reported by Tseng et al. (5) using a rat GIPr cell line. Pro3GIP did not antagonize the GIP-induced cAMP response in our activity assay (Fig. 1C), whereas antagonism by Pro3GIP was reported by Gault et al. (41) in an assay under significantly different conditions (100 pm GIP rather than 1 pm as in our study). In contrast, Pro3GIP partially agonized the receptor in our assay, to 83% of the response with GIP, with an EC50 of 180 nm (Fig. 1D).
The specificity of the interaction of Gipg013 with the GIPr was assessed in FMAT cell binding assays using cell lines overexpressing human, mouse, rat, and dog GIPr or the related receptors GLP-1r and GCGr (Fig. 2A). As expected, binding was observed to the GIPr cell lines, with no detectable binding to any of the other cell lines.
FIGURE 2.
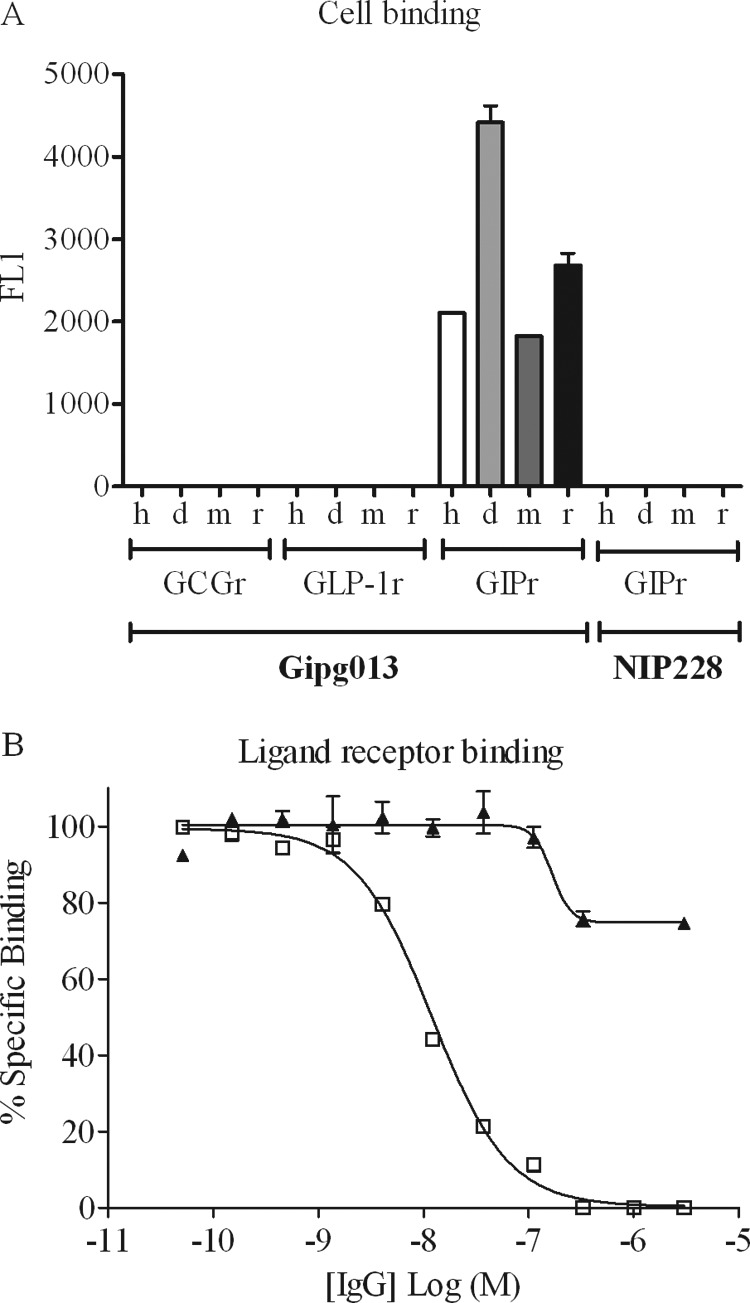
Antibody cell binding and inhibition of ligand binding. A, binding of Gipg013 to GIPr and related receptors on overexpressing cells. Shown is direct binding of 0.25 μg/ml Gipg013 IgG to human (h), dog (d), mouse (m), or rat (r) orthologs of GCGr, GLP-1r, or GIPr. Control for nonspecific binding on GIPr orthologs was NIP228_TM at 0.25 μg/ml. Values shown are the mean ± S.E. from duplicate wells, and data shown are representative of two separate experiments for the human receptors and a single experiment for the rodent and canine receptors. B, receptor ligand competition assay showing IC50 determination of Gipg013 IgG binding to GIPr-overexpressing cells. □, Gipg013; ▴, isotype control IgG1. Values shown are the mean ± S.E. (error bars) from duplicate wells, and data shown are representative of four separate experiments.
Receptor Ligand Competition Assay with Gipg013
The competitive binding of Gipg013 to the GIPr was explored further in a receptor ligand competition assay. The IC50 for Gipg013 IgG was determined by competing a fixed concentration (below IC50 for GIP) of an AlexaFluor 647-labeled GIP peptide with increasing levels of the IgG antibody for binding to GIPr-overexpressing cells in an FMAT assay. Gipg013 IgG was shown to have an IC50 of 17.2 ± 6 nm for displacement of GIP from human GIPr-overexpressing cells (Fig. 2B). In comparison, the ligand GIP and the peptide antagonist GIP(7–30) gave IC50 values of 4.0 and 83 nm, respectively (data not shown), in good agreement with the 7 and 200 nm reported by Tseng et al. (5) using displacement of 125I-GIP bound to L293 cells.
Characterization of GIPr Antagonism by Gipg013
Production of cAMP was measured as a function of GIP concentration in the absence or presence of a range of fixed Gipg013 concentrations. The dose-response curves as a function of GIP concentrations (Fig. 3A) were used for Schild regression analysis based on the EC50 values (Fig. 3B). The slope of the Schild plot (0.99 ± 0.08) confirmed that the antibody is competitive with the ligand GIP on the receptor. pA2 analysis of the Schild plot yielded a Kd of 6.8 ± 0.6 nm.
FIGURE 3.
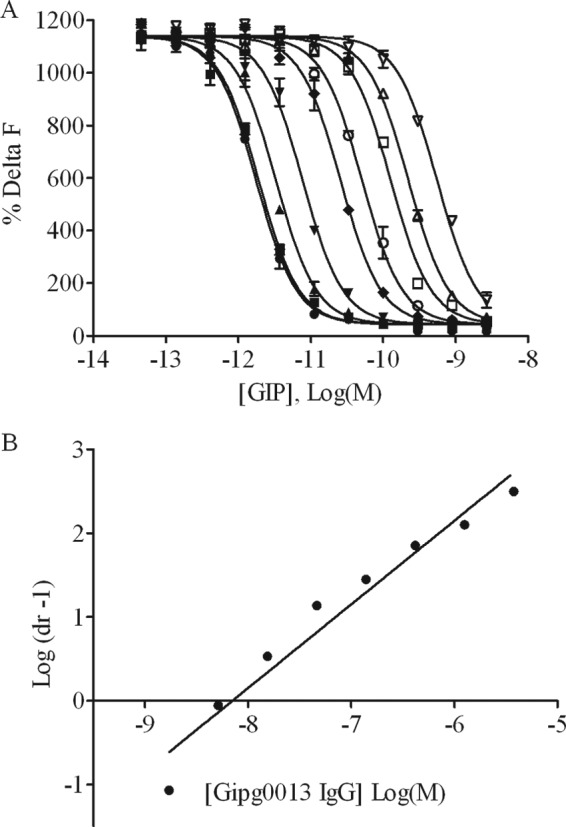
Analysis of Gipg013 antagonism of GIPr. A, GIP dose-response curves in the presence of Gipr013. The nonlinear regression plot of the dose-effect curve for GIP was determined in the presence of various concentrations of Gipg013: 3750 nm (▿), 1250 nm (▵), 417 nm (□), 139 nm (○), 46 nm (♦), 15 nm (▾), 5 nm (▴), 1.7 nm (■), and 0 nm (●). B, Schild plot analysis of dose-response curves. The Schild plot intersects the abscissa at pA2 (= KD). Values shown are the mean ± S.E. (error bars) from duplicate wells, and data shown are representative of three separate experiments.
Binding Kinetics of GipG013 Binding to Gipr ECD
The binding kinetics of the Gipg013 IgG were characterized in detail using surface plasmon resonance on a Biacore instrument. In contrast to the Kd determinations with the Octet instrument, analysis was performed with the antibody immobilized on the Biacore chip and the GIPr ECD in solution. The IgG was immobilized at a low concentration (∼139 resonance units), and the binding interaction was analyzed at six different concentrations of GIPr ECD. The data gave a good fit when processed assuming a 1:1 Langmuir binding interaction. The kinetic analysis yielded association and dissociation rates of 1.59 × 105 m−1 s−1 and 3.69 × 10−3 s−1, respectively, and a Kd of 23 nm, which is comparable with the values of 7.45 × 104 m−1 s−1, 1.88 × 10−3 s−1, and 25 nm measured for the Gipg013 scFv by reflectometric interference spectroscopy on the Octet RED. There was no dependence on whether the antibody was immobilized, as in surface plasmon resonance, or the GIPr ECD was immobilized, as for reflective interference spectroscopy. Therefore, the binding of the Gipg013 IgG to the GIPr does not seem to have a pronounced avidity effect. The difference in antagonistic potency between the scFv (IC50 = 233 nm) and the IgG (IC50 = 6 nm) could be caused mainly by steric effects.
Generation and Characterization of Gipg013 Fab
For the crystallization of Gipg013 with the GIPr ECD, we generated the Fab fragment of the antibody by papain digestion. As expected, the Gipg013 Fab retained binding to GIPr ECD, with a Kd of 40 nm measured using BIAcore, and its ability to antagonize GIPr in our cell-based activity assay, with an IC50 of 102 nm, comparable with the Kd of 25 nm and IC50 of 233 nm for the scFv, respectively.
Overall Crystal Structure
The Gipg013 Fab-GIPr ECD complex crystallized in space group P21, and the structure was refined to a final Rcryst and Rfree of 25.5 and 31.1%, respectively. The asymmetric unit contains two complexes, of which complex 1 (designated A-PQ, where A represents the GIPr ECD, and P and Q are the heavy and light chains of Gipg013 Fab, respectively) has an average temperature factor of 51.17 (〈BA〉) for the GIPr ECD as compared with complex 2 (designated B-CD, where B represents the GIPr ECD, and C and D are the heavy and light chains of Gipg013 Fab, respectively), where GIPr ECD has an average temperature factor of 94.57 (〈BB〉). Initial phasing resulted in only complex 1. After several refinement cycles, GIPr ECD in complex 2 could be modeled manually. Considering this factor, the remaining results and discussion involve complex 1 unless stated otherwise. Final refinement parameters are summarized in Table 2.
Crystal Structure of Gipg013 Fab in Complex with GIPr ECD
The crystal structure reveals the interface between the CDR loops of the Gipg013 Fab and the GIPr ECD (Fig. 4A). The overall structure of GIPr ECD is similar to the structure of the GIPr ECD in the GIPr ECD-GIP(1–42) complex (PDB code 2QKH) (21). The structures of the GIPr ECDs overlap with a root mean square deviation of 0.799 Å2 (Fig. 4B). The final model of Gipg013 Fab is composed of a heavy chain with 206 residues (Gln3–Val217) and a light chain with 211 residues (Ser1–Glu213) and shows the antibody binding site comprising residues from the complementarity-determining region loops outlined in Table 1.
FIGURE 4.

Crystal structure of GIPr ECD and Gipg013 Fab complex. A, overall structure of the complex where GIPr ECD and Gipg013 Fab heavy chain and light chains are represented in gray, cyan, and magenta schematics, respectively (PDB code 4HJ0). B, GIPr ECD-GIP(1–42) (PDB code 2QKH) superposed on GIPr ECD-Gipg013 Fab crystal structure. GIPr ECDs overlap with a root mean square deviation of 0.799 Å2. GIP(1–42) and GIPr ECD (2QKH) are shown in orange and salmon schematics. C, glucagon family recognition fold is highly conserved. The three clusters (viz cluster 1 (Trp71, Val99, Arg101, and Trp109 in orange atomic color mode), cluster 2 (Trp39, Tyr42, and Phe65 in yellow atomic color mode), and cluster 3 (Tyr68, Pro85, Tyr87, Leu88, and Trp90 in cyan atomic color mode)) are a characteristic feature of class B GPCR N-terminal extracellular domain. Asp66 in green atomic color mode is a highly conserved residue involved in stabilizing the structure. The three disulfide links are shown in magenta atomic color mode. D, CDRs of heavy chain play a vital role in complex formation. H-CDR1 (Tyr32) creates a network of hydrogen bond interactions with Arg113 and His115 of GIPr ECD. It is further aided by Ser31 (H-CDR1) interacting with Glu119 and Asp107 (H-CDR3) with Tyr68 of GIPr ECD. E, L-CDR1 and L-CDR2 of Gipg013 Fab light chain make a series of hydrogen bond interactions with N-terminal α-helix of GIPr ECD. In D and E, GIPr ECD, Gipg013 Fab heavy chain (H-CDRs) and light chains (L-CDRs) are represented in white, cyan, and magenta atomic color modes, respectively.
The Glucagon Receptor Subfamily Fold of GIPr ECD
The GIPr ECD exhibits a three-layer α-β-βα fold, typical of the glucagon receptor subfamily of class B GPCRs (42), with three clusters of intramolecular interactions (Fig. 4C). GIPr ECD is a compact molecule with an N-terminal α1-helix (Ala32–Ala52) situated by the side of a central core created by two anti-parallel β-sheets. Further, each β-sheet comprises two β-strands (β1, Ser64–Phe65 and Cys70–Trp71; β2, Ala78–Ser83 and Phe98–Cys103). At the C-terminal end, two short helices, α2 (His91–Val94) and α3 (Thr116–Cys118), are present. The whole structure is stabilized by three disulfide links provided by Cys46–Cys70, Cys61–Cys103, and Cys84–Cys118 residues, which are a characteristic feature of the N-terminal domain of class B GPCRs (43) (Fig. 4C).
The overall fold of the GIPr ECD in complex with Gipg013 Fab correlates to the GIPr prototype described by Parthier et al. (22), but there are differences in detailed interactions. The glucagon family recognition fold in the Fab complex has the highly conserved aspartate (Asp66) at its center, creating backbone amide interactions with Tyr68 and Val69 unlike in the GIPr ECD-GIP(1–42) complex, where Asp66 has amide interactions with Met67, Tyr68, and Val69 (22). Furthermore, Asp66 provides stability to the GIPr ECD by side chain hydrogen bond interaction with -NH1 of Arg113 (bond length, 3.39 Å) and electrostatic interactions with Trp71. Asp66 does not form a salt bridge to Arg101 as observed in the case of CRFR-2β ECD (Asp65) (44).
Epitope of Gipg013 Fab on GIPr ECD
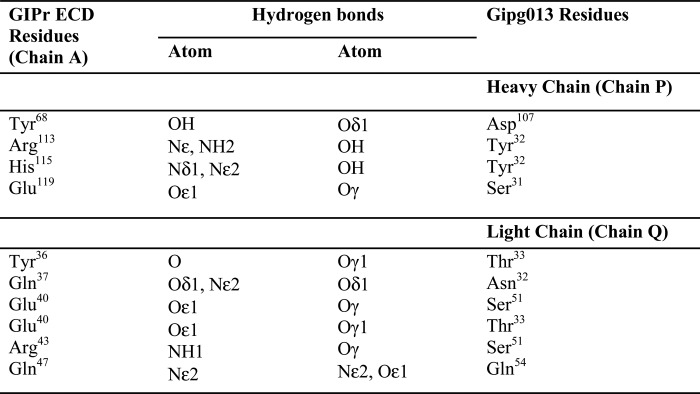
Gipg013 Fab binds GIPr ECD through a series of hydrogen bond interactions between the antibody CDRs and GIPr ECD, as shown in Table 3. The Gipg013 Fab heavy chain plays a crucial role in complex formation. The residue Tyr32 of H-CDR1 interacts with Arg113 and His115 of GIPr ECD through the side chain -OH. Furthermore, the interactions of Ser31 (H-CDR1) with Glu119 as well as Asp107 (H-CDR3; Asp101 in Kabat numbering) with Tyr68 stabilize the complex (Fig. 4D). The light chain CDRs further create stable interactions for the complex. L-CDR1 (Asn32 and Thr33) and L-CDR2 (Ser51 and Gln54) of Gipg013 Fab provide more hydrogen bond interactions to the N-terminal helix of GIPr ECD, as shown in Fig. 3E. The side chain of Asn32 (-Oδ1) interacts with the side chain of Gln37 (-Nϵ2) (GIPr ECD). In addition to this, Thr33 (-OH) interacts with the residues Tyr36 (-OH) and Glu40 (-Oϵ1) of GIPr ECD through side chain contacts.
TABLE 3.
Hydrogen bond interactions between GIPr ECD and Gipg013 Fab, complex 1 (A-PQ)
γ, δ, ϵ is the standard nomenclature for atom position on the amino acid.
From analysis of the accessible surface area and the buried surface area using the program PISA, a buried surface area of 5210 Å2 was calculated for the GIPr ECD-Gipg013 Fab complex. This includes ∼3100 Å2 of buried surface area at the interface of the Gipg013 Fab heavy and light chains. At the antibody binding site, there is a buried surface area of 1050 Å2 on the GIPr surface and 690 and 366 Å2 on the heavy and light chains of Gipg013 Fab, respectively, giving a total buried surface area of 2106 Å2. This contrasts with 1250 Å2 buried in the GIPr ECD-GIP(1–42) complex with 625 Å2 buried on each of the GIPr and GIP(1–42) surfaces. Thus, Gipg013 Fab has a large epitope on the GIPr ECD, binding to residues overlapping the binding site for the ligand GIP as well as to further residues burying a larger surface on the GIPr ECD. This confers higher stability on the GIPr ECD-Gipg013 Fab complex compared with the GIPr ECD-GIP(1–42) complex, as indicated by calculated free energy values (Table 4). Thus, Gipg013 Fab has a large epitope on the GIPr ECD, binding to residues overlapping the binding site for the ligand GIP as well as to further residues burying a larger surface on the GIPr ECD.
TABLE 4.
Comparison of the estimates of accessible surface area and the buried surface area in the GIPr ECD-Gipg013 Fab and GIPr ECD-GIP(1–42)complexes using PISA
| Complex | Accessible surface area | Buried surface area | ΔGinta | ΔGdissb |
|---|---|---|---|---|
| Å2 | Å2 | kcal/m | kcal/m | |
| GIPr ECD- Gipg013 Fab, A-PQc | 23,740 | 5210 | −34.0 | 4.0 |
| GIPr ECD- Gipg013 Fab, B-CDc | 24,300 | 5110 | −39.5 | 7.1 |
| GIPr ECD-GIP(1–42), PDB code 2QKH | 7920 | 1250 | −7.9 | −0.3 |
a Solvation free energy gain upon formation of the assembly, in kcal/m.
b Free energy of assembly dissociation, in kcal/m, where ΔGdiss > 0 indicates a thermodynamically stable complex.
c A-PQ and B-CD denote the complex chains in the asymmetric unit of the crystal structure of GIPr ECD- Gipg013 Fab, where A and B are GIPr ECDs, and PQ and CD are Fab fragments of Gipg013. When the interface of heavy and light chains of Gipg013 Fab is excluded in the complex for free energy calculations, complex 1 (A-PQ) has a ΔGint and ΔGdiss of −11.9 and 2.4, respectively. Complex 2 (B-CD) has a ΔGint and ΔGdiss of −12.1 and 2.2, respectively, indicating a stable complex compared with GIPr ECD-GIP(1–42).
Inhibition of GIP Enhancement of Glucose-stimulated Insulin Secretion
Inhibition of GIP enhancement of glucose-stimulated insulin secretion was studied in isolated rat pancreatic islet cells. GIP stimulated further the increase in insulin secretion in response to raising the glucose concentration from 3 to 11 nm, with greater enhancement as the GIP concentration increased to 100 nm. At all concentrations of GIP, this further stimulation by GIP was essentially abolished by the addition of the antibody Gipg013 (600 nm) (Fig. 5A). This suppressed GIP stimulation was also highly significant at 300 nm Gipg013 (Fig. 5B). In a separate experiment, there was an 81% reduction of GIP-induced insulin secretion at 200 nm Gipg013 (p < 0.004) compared with the no antibody sample, but at concentrations of Gipg013 of 60 nm or lower, the difference was not statistically significant (data not shown). Thus, Gipg013 inhibits GIP-induced insulin secretion in isolated pancreatic islet cells with increased effects at higher concentrations.
FIGURE 5.
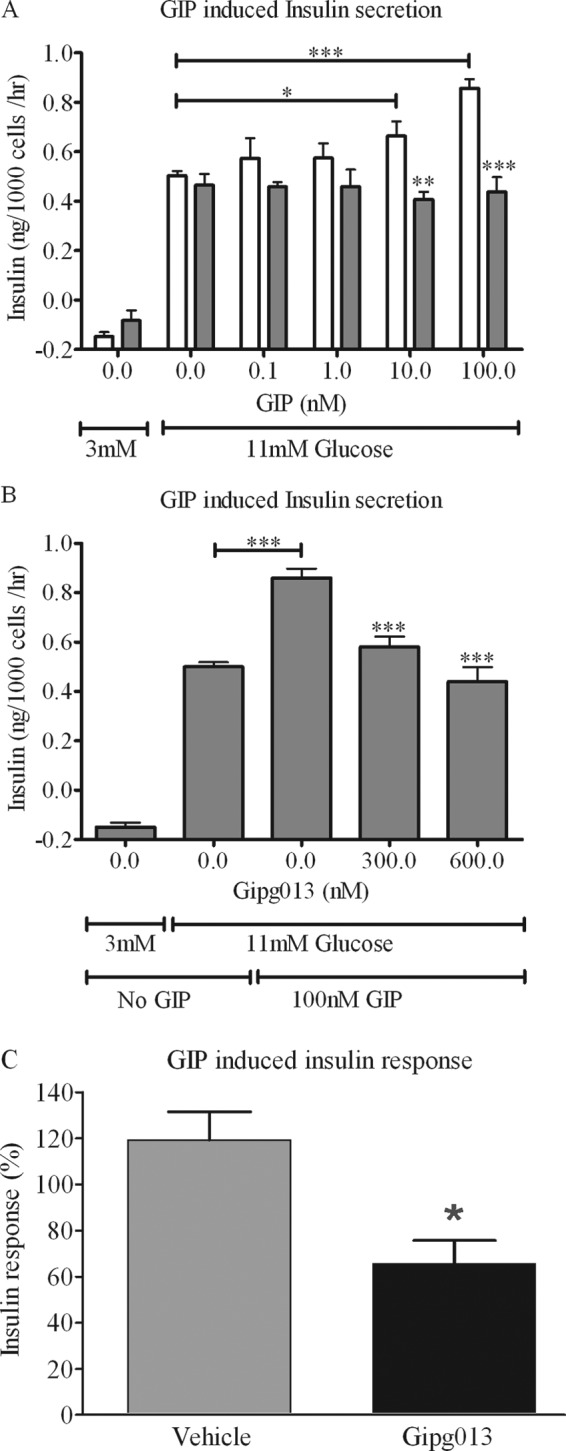
Inhibition of GIP-induced insulin secretion. A and B show inhibition of GIP stimulation in a glucose-stimulated insulin secretion assay with dispersed rat islets. A, effect of GIP on top of the glucose-stimulated insulin secretion (open bars) and the inhibition of this effect with 600 nm Gipg013 (filled bars). B, the GIP enhanced insulin secretion was inhibited with Gipg013 at concentrations of both 300 and 600 nm (*, p < 0.05; **, p < 0.01; ***, p < 0.001). C, inhibition of GIP-induced insulin secretion in vivo. Insulin response was measured as the relative insulin AUC0–30 min between period 1 and 2 for GIP-induced insulin secretion in anesthetized rats, AUC1 before and AUC2 30 min after Gipg013 infusion. Insulin response = AUC2/AUC1 × 100. n = 6 for vehicle and n = 3 for Gipg013. *, p < 0.05 between vehicle and Gipg013. Error bars, S.E.
In Vivo Half-life of Gipg013
To demonstrate the extended half-life of Gipg013, the antibody was dosed to C57 mice for evaluation of pharmacokinetics. The half-life of antibody was ∼10 days, as expected for the human IgG1 format (45) and considerably longer than the half-life in the range of minutes predicted for GIP(7–30). The antibody was well tolerated, with no adverse events observed in a repeated dosing scheme of subcutaneous administration of 30 mg/kg every 5 days for 5 weeks. Serum samples were collected 99 h after the final dose, and an average antibody concentration of 5 μm was determined, in good agreement with the expected concentration from pharmacokinetic modeling. The antibody in the serum samples was active, with no loss of activity when compared with control serum added to 5 μm Gipg013 in both the ligand receptor competition assay and the cell-based activity assay (data not shown).
Inhibition of GIP-induced Insulin Secretion
Inhibition of GIP-induced insulin secretion by GIPr antagonism with Gipg013 was assessed in a rat infusion model. Prior to the experiment, it was established that the magnitude of the insulin response to a 10-min GIP infusion differed between animals but was similar when repeated in the same animal. Therefore, the first GIP infusion period served as the control to the second, Gipg013-treated period.
The area under the insulin response curve during 30 min from the start of the 10-min GIP infusion (AUC0–30 min) was significantly suppressed by Gipg013 (Fig. 5C). 3 and 30 mg/kg Gipg013 induced a similar suppression of insulin secretion, indicating that the maximum response was reached already at the lower dose, and therefore these doses were grouped together in the analysis. Average plasma concentration 90 min after dose was 97 and 1150 mg/liter (0.6 and 7 μm) for 3 and 30 mg/kg, respectively. The plasma levels of antibody at both doses were considerably higher than the total dose of GIP delivered to the mice during the infusion (500 pmol/kg, ∼15 nm). Thus, Gipg013 suppressed GIP-induced insulin secretion in vivo.
DISCUSSION
In this study, we have generated a highly specific and potent antagonistic antibody against GIPr. The antibody Gipg013 is a competitive antagonist with respect to GIP-stimulated cAMP production at the receptor, giving a Kd of 6.8 nm by Schild analysis. Gipg013, specifically antagonizes the GIPr, with no detectable inhibition of the related GLP1r and GCGr. It is equally potent on human, mouse, rat, and dog GIP receptors and will therefore be a useful pharmacological tool for elucidating biological effects at the GIPr in model systems. Insight into the mechanism of antagonism has been provided by the crystal structure of Gipg013 Fab bound to the extracellular domain of the GIPr. The epitope for the antibody overlaps with the docking site of the GIP peptide observed in the crystal structure of the complex of GIP with the GIPr ECD (22).
GIP binds the soluble GIPr ECD very weakly (IC50 ∼1 μm (22)) compared with the full-length receptor expressed in cells. Wheeler et al. (46) reported a Kd of 200 pm for human GIP and an IC50 of 2.6 nm for displacement by human GIP of 125I-GIP from full-length GIP receptor expressed in CHO cells, similar to the IC50 of 4 nm for GIP displacement used in this study. The increased affinity for the full-length receptor arises from extra binding interactions as follows. GIP binds first to the ECD through its α-helical region between residues 12 and 30; the N-terminal region of GIP is then thought to interact with the membrane-associated portion of the GIPr, leading to stimulation of cAMP production (22). In contrast, the antibody binds exclusively to the GIPr ECD, showing equally high affinity to the free GIPr ECD and the cell-bound form.
There is a wide ranging footprint of Gipg013 Fab over GIPr ECD in the complex, leading to a larger buried surface area (Fig. 6). Several hydrogen bonds along with electrostatic interactions play a major role in forming the stable complex between Gipg013 Fab and GIPr ECD. In the case of the GIPr ECD-GIP(1–42) complex, Gln30, Ala32, Pro89, and Arg113 of GIPr ECD are involved in complex formation. On the other hand, the GIPr ECD-Gipg013 Fab complex is formed through interactions of Tyr36, Gln37, Glu40, Arg43, and Gln47 as well as Tyr68, Arg113, His115, and Glu119. This explains the higher affinity of the Gipg013 Fab toward GIPr ECD as compared with GIP(1–42).
FIGURE 6.

Gipg013 Fab exhibits high affinity toward GIPr ECD. Shown are footprints of GIP(1–42) and Gipg013 Fab on GIPr ECD domains. The buried surface area is greater in the GIPr ECD-Gipg013 Fab complex (see Table 4), and the interacting residues are labeled and shown on a green shaded surface. Only four residues are involved in hydrogen bond interactions with GIP(1–42).
Gipg013 does not show any detectable inhibition of or binding to GLP-1r and GCGr, which exhibit a sequence identity with GIPr ECD of 36% for GLP1r and 42% for GCGr. Comparison of the sequences of human GIPr, GLP1r, and GCGr with the epitope for Gipg013 Fab (Table 5A) not only reveals the basis for this selectivity but defines the critical interactions for the affinity of Gipg013 Fab for GIPr. Tyr68 and Arg113, which are important in complex formation with Gipg013 Fab, are highly conserved in all these human hormone receptor ECDs. Indeed, the homologous residues (Tyr65 and Arg111) contribute to the epitope of an antagonist antibody to the GCGr (21). However, although the majority of interacting residues are similar to GIPr, in GLP1r, valine replaces Tyr36 and leucine replaces His115. GIPr His115, which provides stronger hydrogen bond interactions through -Nδ1 and -Nϵ2 to the side chain of VH CDR1 Tyr32, may play a critical role in the affinity of Gipg013 Fab. Tyr36 of GIPr is required for the side chain interaction with Thr33 of VL CDR1, and replacement of Tyr36 with valine in GLP1r effectively removes the contact. Similarly, in GCGr, alanine and phenylalanine replace His115 and Tyr36, respectively, supporting the definition of these residues as crucial contacts for Gipg013 Fab. Multiple sequence alignment of human, dog, mouse, and rat GIPr (Table 5B) reveals that all of the residues involved in binding to the antibody are conserved between human and dog sequences, and seven of eight are conserved in mouse and rat sequences. The Gly for Arg43 substitution found in the mouse and rat GIPr and the human GCGr removes a hydrogen bond interaction with -Oγ of Ser51 through side chain atoms but has little or no effect on the antibody potency or binding. The antagonism by Gipg013 at GIPr of different species is therefore explained by the observed crystal structure contacts. The properties of Gipg013 IgG make it suitable for use in animal pharmacology and toxicology models.
TABLE 5.
Multiple sequence alignments
A, alignment of ECDs of human hormone receptors GIPr, GLP1r, and GCGr. The numbering is as per the crystal structure of GIPr ECD in complex with Gipg013 Fab. Highlighted residues are involved in the binding to Gipg013 Fab. Residues in boldface type are conserved residues. Protein sequences were obtained from the following Uniprot entries: human GIPr, P48546; human GCGr, P47871; human GLP-1r, P43220. B, alignment for comparison of contact residues for Gipg013 Fab in GIPr ECD of human, dog, mouse, and rat GIPr. The numbering is as per the crystal structure of GIPr ECD in complex with Gipg013 Fab. Highlighted residues are involved in the binding to Gipg013 Fab. Residues in boldface type are conserved residues. Protein sequences were obtained from the following Uniprot entries: human GIPr, P48546; canine GIPr, E2RIK5; mouse GIPr, Q0P543; rat GIPr, P43219.

The strategy used to generate Gipg013 favors the selection of antibodies that compete for ligand binding and should be generally applicable to the isolation of antagonistic antibodies against class B GPCRs, such as the GLP1 and glucagon receptors. It is proposed that the C-terminal helical portion of GIP first interacts with the GIPr ECD, and this event then helps the binding of the N-terminal part of the peptide with the juxtamembrane region of the receptor and activation of the receptor. Selection of antibody phage display libraries on purified receptor extracellular domain enriches for antibodies that bind to the same region of the receptor as the ligand. Further selection of the outputs on cells overexpressing the receptor enriches for antibodies that recognize the receptor in its native conformation and therefore antagonize GIP action in a native context. In previous studies in our laboratory where naive antibody phage display libraries have been directly selected only on GPCR-overexpressing cell lines, only modest enrichment of antibodies specific for receptors has been obtained, and consequently neutralizing antibodies remained rare in selection outputs. The selection on purified ECD will enrich for binders to the target and largely eliminate background usually associated with cell selections, thereby enriching primarily specific antibodies recognizing the native receptor. The complex steric effects involved in antagonism by the antibodies are indicated by the reduction in antagonistic activity for many antibodies when converting antibody clones from the monomeric scFv to the dimeric IgG format. Upon conversion, only two of the 30 clones retained full antagonist activity, including Gipg013. Several IgG antibodies were partial antagonists, whereas the rest lost all activity. This emphasizes the value of the strategy deployed here, where a large panel of clones was screened as IgG.
The biological role of GIP and the interaction with GIPr have been extensively studied, but uncertainty remains about the predicted therapeutic effects of modulating GIP action to regulate plasma glucose and fat deposition in adipocytes. The role in glucose regulation through insulin secretion suggests a therapeutic potential for GIPr agonism in type 2 diabetes (7). However, these patients are resistant to the insulinotropic effects of GIP (47). Transgenic mice overexpressing GIP exhibited reduced diet-induced obesity, although excessively elevated levels led to GIP resistance (48). Conversely, reduced body weight gain has been observed in diet-induced obesity models in a GIPr−/− knock-out mouse (49), upon immunization with GIP to generate antagonistic antibodies, and upon administration of the GIPr peptide antagonist Pro3GIP or ablation of GIP-producing K cells (50–53).
However, the phenotypes observed cannot be definitively linked to the actions of GIP at its receptor. Alternative mechanisms and pathways may compensate for the ablation of the receptor in genetic knock-out models (54–56). Some antibodies generated by immunization could have the effect of extending the half-life of GIP in plasma by acting as a carrier, and this effect may overcome neutralization by other antibodies, as has been observed with MCP-1 (13). Furthermore, Pro3GIP, which has been proposed to antagonize GIPr (32), is an agonist of GIPr-mediated cAMP production in the assays reported in this study, consistent with the response noted by others (57). Finally, a small molecule antagonist of the GIPr, which has recently been reported to have an IC50 of 2.5 μm in a GIP-dependent cAMP assay, also retains some antagonistic activity at the glucagon receptor (58).
We suggest that a competitive antagonist at the receptor will more efficiently block signaling through the GIPr in vivo, especially because incretins show pulsative high intensity signaling. Gipg013 does indeed antagonize the GIPr and efficiently inhibit GIP-stimulated insulin secretion, as demonstrated in our rat in vitro and in vivo models. We have confirmed the extended half-life of Gipg013 and demonstrated antagonistic activity in serum after more than 4 days. The competitive mechanism, high potency, and selectivity combined with the extended half-life make Gipg013 an attractive antagonist of GIPr for future chronic pharmacological studies.
Acknowledgments
We gratefully acknowledge Rainer Rudolph for advice on the production of GIPr ECD. Colleagues at AstraZeneca supported this work with the generation of GIPr ECD and the in vivo studies. Research teams at MedImmune Cambridge supported this work with sequencing and scFv and IgG antibody production. Furthermore, Julie Douthwaite and Susan Kunze established the Reconstituted Ribosome Display system used in this work. We thank the crystallization facility platform established by the National Center of Competence in Research Structural Biology Program.
The atomic coordinates and structure factors (code 4HJ0) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- GIP
- glucose-dependent insulinotropic polypeptide
- CDR
- complementarity-determining region
- FMAT
- fluorescencent microvolume assay technology
- GCGr
- glucagon receptor
- GIPr
- GIP receptor
- GLP-1r
- GLP-1 receptor
- ECD
- extracellular domain
- GPCR
- G-protein-coupled receptor
- HTRF
- homogeneous time-resolved fluorescence
- PBL
- peripheral blood lymphocyte
- scFv
- single chain fragment variable
- VH
- variable fragment heavy chain
- VL
- variable fragment light chain
- TAPS
- 3-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}-1-propanesulfonic acid
- PDB
- Protein Data Bank.
REFERENCES
- 1. Brubaker P. L. (2007) Incretin-based therapies. Mimetics versus protease inhibitors. Trends Endocrinol. Metab. 18, 240–245 [DOI] [PubMed] [Google Scholar]
- 2. Holst J. J., Vilsbøll T., Deacon C. F. (2009) The incretin system and its role in type 2 diabetes mellitus. Mol. Cell. Endocrinol. 297, 127–136 [DOI] [PubMed] [Google Scholar]
- 3. Nauck M. A. (2009) Unraveling the science of incretin biology. Eur. J. Intern. Med. 20, S303–S308 [DOI] [PubMed] [Google Scholar]
- 4. Hinke S. A., Manhart S., Pamir N., Demuth H., Gelling R. W., Pederson R. A., McIntosh C. H. (2001) Identification of a bioactive domain in the amino-terminus of glucose-dependent insulinotropic polypeptide (GIP). Biochim. Biophys. Acta 1547, 143–155 [DOI] [PubMed] [Google Scholar]
- 5. Tseng C. C., Kieffer T. J., Jarboe L. A., Usdin T. B., Wolfe M. M. (1996) Postprandial stimulation of insulin release by glucose-dependent insulinotropic polypeptide (GIP). Effect of a specific glucose-dependent insulinotropic polypeptide receptor antagonist in the rat. J. Clin. Invest. 98, 2440–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Getty-Kaushik L., Song D. H., Boylan M. O., Corkey B. E., Wolfe M. M. (2006) Glucose-dependent insulinotropic polypeptide modulates adipocyte lipolysis and reesterification. Obesity 14, 1124–1131 [DOI] [PubMed] [Google Scholar]
- 7. Meier J. J., Nauck M. A., Schmidt W. E., Gallwitz B. (2002) Gastric inhibitory polypeptide. The neglected incretin revisited. Regul. Pept. 107, 1–13 [DOI] [PubMed] [Google Scholar]
- 8. Flatt P. R. (2008) Dorothy Hodgkin Lecture 2008. Gastric inhibitory polypeptide (GIP) revisited. A new therapeutic target for obesity-diabetes? Diabet. Med. 25, 759–764 [DOI] [PubMed] [Google Scholar]
- 9. Gault V. A., Parker J. C., Harriott P., Flatt P. R., O'Harte F. P. (2002) Evidence that the major degradation product of glucose-dependent insulinotropic polypeptide, GIP(3–42), is a GIP receptor antagonist in vivo. J. Endocrinol. 175, 525–533 [DOI] [PubMed] [Google Scholar]
- 10. Gelling R. W., Coy D. H., Pederson R. A., Wheeler M. B., Hinke S., Kwan T., McIntosh C. H. (1997) GIP(6–30amide) contains the high affinity binding region of GIP and is a potent inhibitor of GIP1–42 action in vitro. Regul. Pept. 69, 151–154 [DOI] [PubMed] [Google Scholar]
- 11. Foord S. M., Bonner T. I., Neubig R. R., Rosser E. M., Pin J. P., Davenport A. P., Spedding M., Harmar A. J. (2005) International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol. Rev. 57, 279–288 [DOI] [PubMed] [Google Scholar]
- 12. McKnight A., Wilkinson D., Simmons G., Talbot S., Picard L., Ahuja M., Marsh M., Hoxie J. A., Clapham P. R. (1997) Inhibition of human immunodeficiency virus fusion by a monoclonal antibody to a coreceptor (CXCR4) is both cell type- and virus strain-dependent. J. Virol. 71, 1692–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harris G. L., Creason M. B., Brulte G. B., Herr D. R. (2012) In vitro and in vivo antagonism of a G protein-coupled receptor (S1P3) with a novel blocking monoclonal antibody. PloS One 7, e35129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mirzabekov T., Kontos H., Farzan M., Marasco W., Sodroski J. (2000) Paramagnetic proteoliposomes containing a pure, native, and oriented seven-transmembrane segment protein, CCR5. Nat. Biotechnol. 18, 649–654 [DOI] [PubMed] [Google Scholar]
- 15. Osbourn J. K., Earnshaw J. C., Johnson K. S., Parmentier M., Timmermans V., McCafferty J. (1998) Directed selection of MIP-1 α neutralizing CCR5 antibodies from a phage display human antibody library. Nat. Biotechnol. 16, 778–781 [DOI] [PubMed] [Google Scholar]
- 16. Buggy J., Rossomando A., MacDougall M., Mierz D., Wunderlich D., Yoo-Warren H. (1996) Human glucagon receptor monoclonal antibodies. Antagonism of glucagon action and use in receptor characterization. Horm. Metab. Res. 28, 215–219 [DOI] [PubMed] [Google Scholar]
- 17. Lewis J. T., Dayanandan B., Habener J. F., Kieffer T. J. (2000) Glucose-dependent insulinotropic polypeptide confers early phase insulin release to oral glucose in rats. Demonstration by a receptor antagonist. Endocrinology 141, 3710–3716 [DOI] [PubMed] [Google Scholar]
- 18. Wright L. M., Brzozowski A. M., Hubbard R. E., Pike A. C., Roberts S. M., Skovgaard R. N., Svendsen I., Vissing H., Bywater R. P. (2000) Structure of Fab hGR-2 F6, a competitive antagonist of the glucagon receptor. Acta Crystallogr. D Biol. Crystallogr. 56, 573–580 [DOI] [PubMed] [Google Scholar]
- 19. Gu W., Winters K. A., Motani A. S., Komorowski R., Zhang Y., Liu Q., Wu X., Rulifson I. C., Sivits G., Jr., Graham M., Yan H., Wang P., Moore S., Meng T., Lindberg R. A., Véniant M. M. (2010) Glucagon receptor antagonist-mediated improvements in glycemic control are dependent on functional pancreatic GLP-1 receptor. Am. J. Physiol. Endocrinol. Metab. 299, E624–E632 [DOI] [PubMed] [Google Scholar]
- 20. Yan H., Gu W., Yang J., Bi V., Shen Y., Lee E., Winters K. A., Komorowski R., Zhang C., Patel J. J., Caughey D., Elliott G. S., Lau Y. Y., Wang J., Li Y. S., Boone T., Lindberg R. A., Hu S., Véniant M. M. (2009) Fully human monoclonal antibodies antagonizing the glucagon receptor improve glucose homeostasis in mice and monkeys. J. Pharmacol. Exp. Ther. 329, 102–111 [DOI] [PubMed] [Google Scholar]
- 21. Koth C. M., Murray J. M., Mukund S., Madjidi A., Minn A., Clarke H. J., Wong T., Chiang V., Luis E., Estevez A., Rondon J., Zhang Y., Hötzel I., Allan B. B. (2012) Molecular basis for negative regulation of the glucagon receptor. Proc. Natl. Acad. Sci. U.S.A. 109, 14393–14398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parthier C., Kleinschmidt M., Neumann P., Rudolph R., Manhart S., Schlenzig D., Fanghänel J., Rahfeld J. U., Demuth H. U., Stubbs M. T. (2007) Crystal structure of the incretin-bound extracellular domain of a G protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 104, 13942–13947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kumar S., Pioszak A., Zhang C., Swaminathan K., Xu H. E. (2011) Crystal structure of the PAC1R extracellular domain unifies a consensus fold for hormone recognition by class B G-protein coupled receptors. PloS One 6, e19682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lloyd C., Lowe D., Edwards B., Welsh F., Dilks T., Hardman C., Vaughan T. (2009) Modelling the human immune response. Performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng. Des. Sel. 22, 159–168 [DOI] [PubMed] [Google Scholar]
- 25. McCafferty J., Fitzgerald K. J., Earnshaw J., Chiswell D. J., Link J., Smith R., Kenten J. (1994) Selection and rapid purification of murine antibody fragments that bind a transition-state analog by phage display. Appl. Biochem. Biotechnol. 47, 157–171; discussion 171–153 [DOI] [PubMed] [Google Scholar]
- 26. Ravn P. (2012) Selection of lead antibodies from naive ribosome display antibody libraries. Methods Mol. Biol. 805, 213–235 [DOI] [PubMed] [Google Scholar]
- 27. Finlay W. J., Cunningham O., Lambert M. A., Darmanin-Sheehan A., Liu X., Fennell B. J., Mahon C. M., Cummins E., Wade J. M., O'Sullivan C. M., Tan X. Y., Piche N., Pittman D. D., Paulsen J., Tchistiakova L., Kodangattil S., Gill D., Hufton S. E. (2009) Affinity maturation of a humanized rat antibody for anti-RAGE therapy. Comprehensive mutagenesis reveals a high level of mutational plasticity both inside and outside the complementarity-determining regions. J. Mol. Biol. 388, 541–558 [DOI] [PubMed] [Google Scholar]
- 28. Groves M., Lane S., Douthwaite J., Lowne D., Rees D. G., Edwards B., Jackson R. H. (2006) Affinity maturation of phage display antibody populations using ribosome display. J. Immunol. Methods 313, 129–139 [DOI] [PubMed] [Google Scholar]
- 29. Hawkins R. E., Russell S. J., Winter G. (1992) Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J. Mol. Biol. 226, 889–896 [DOI] [PubMed] [Google Scholar]
- 30. Vaughan T. J., Williams A. J., Pritchard K., Osbourn J. K., Pope A. R., Earnshaw J. C., McCafferty J., Hodits R. A., Wilton J., Johnson K. S. (1996) Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat. Biotechnol. 14, 309–314 [DOI] [PubMed] [Google Scholar]
- 31. Villemagne D., Jackson R., Douthwaite J. A. (2006) Highly efficient ribosome display selection by use of purified components for in vitro translation. J. Immunol. Methods 313, 140–148 [DOI] [PubMed] [Google Scholar]
- 32. Edwards B. M., Barash S. C., Main S. H., Choi G. H., Minter R., Ullrich S., Williams E., Du Fou L., Wilton J., Albert V. R., Ruben S. M., Vaughan T. J. (2003) The remarkable flexibility of the human antibody repertoire. Isolation of over one thousand different antibodies to a single protein, BLyS. J. Mol. Biol. 334, 103–118 [DOI] [PubMed] [Google Scholar]
- 33. Ravn P., Danielczyk A., Jensen K. B., Kristensen P., Christensen P. A., Larsen M., Karsten U., Goletz S. (2004) Multivalent scFv display of phagemid repertoires for the selection of carbohydrate-specific antibodies and its application to the Thomsen-Friedenreich antigen. J. Mol. Biol. 343, 985–996 [DOI] [PubMed] [Google Scholar]
- 34. Leslie A. G. W. (1992) Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography [Google Scholar]
- 35. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 36. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Murshudov G. N., Vagin A. A., Lebedev A., Wilson K. S., Dodson E. J. (1999) Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr. D Biol. Crystallogr. 55, 247–255 [DOI] [PubMed] [Google Scholar]
- 38. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 39. Laskowski R. A., Moss D. S., Thornton J. M. (1993) Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 231, 1049–1067 [DOI] [PubMed] [Google Scholar]
- 40. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrodinger, LLC, New York [Google Scholar]
- 41. Gault V. A., O'Harte F. P., Harriott P., Flatt P. R. (2002) Characterization of the cellular and metabolic effects of a novel enzyme-resistant antagonist of glucose-dependent insulinotropic polypeptide. Biochem. Biophys. Res. Commun. 290, 1420–1426 [DOI] [PubMed] [Google Scholar]
- 42. Pioszak A. A., Xu H. E. (2008) Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 105, 5034–5039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bazarsuren A., Grauschopf U., Wozny M., Reusch D., Hoffmann E., Schaefer W., Panzner S., Rudolph R. (2002) In vitro folding, functional characterization, and disulfide pattern of the extracellular domain of human GLP-1 receptor. Biophys. Chem. 96, 305–318 [DOI] [PubMed] [Google Scholar]
- 44. West A. P., Jr., Llamas L. L., Snow P. M., Benzer S., Bjorkman P. J. (2001) Crystal structure of the ectodomain of Methuselah, a Drosophila G protein-coupled receptor associated with extended lifespan. Proc. Natl. Acad. Sci. U.S.A. 98, 3744–3749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu L., Stadheim A., Hamuro L., Pittman T., Wang W., Zha D., Hochman J., Prueksaritanont T. (2011) Pharmacokinetics of IgG1 monoclonal antibodies produced in humanized Pichia pastoris with specific glycoforms. A comparative study with CHO produced materials. Biologicals 39, 205–210 [DOI] [PubMed] [Google Scholar]
- 46. Wheeler M. B., Gelling R. W., McIntosh C. H., Georgiou J., Brown J. C., Pederson R. A. (1995) Functional expression of the rat pancreatic islet glucose-dependent insulinotropic polypeptide receptor. Ligand binding and intracellular signaling properties. Endocrinology 136, 4629–4639 [DOI] [PubMed] [Google Scholar]
- 47. Vilsbøll T., Krarup T., Madsbad S., Holst J. J. (2002) Defective amplification of the late phase insulin response to glucose by GIP in obese Type II diabetic patients. Diabetologia 45, 1111–1119 [DOI] [PubMed] [Google Scholar]
- 48. Kim S. J., Nian C., Karunakaran S., Clee S. M., Isales C. M., McIntosh C. H. (2012) GIP-overexpressing mice demonstrate reduced diet-induced obesity and steatosis, and improved glucose homeostasis. PloS One 7, e40156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miyawaki K., Yamada Y., Ban N., Ihara Y., Tsukiyama K., Zhou H., Fujimoto S., Oku A., Tsuda K., Toyokuni S., Hiai H., Mizunoya W., Fushiki T., Holst J. J., Makino M., Tashita A., Kobara Y., Tsubamoto Y., Jinnouchi T., Jomori T., Seino Y. (2002) Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat. Med. 8, 738–742 [DOI] [PubMed] [Google Scholar]
- 50. Fulurija A., Lutz T. A., Sladko K., Osto M., Wielinga P. Y., Bachmann M. F., Saudan P. (2008) Vaccination against GIP for the treatment of obesity. PloS One 3, e3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McClean P. L., Irwin N., Cassidy R. S., Holst J. J., Gault V. A., Flatt P. R. (2007) GIP receptor antagonism reverses obesity, insulin resistance, and associated metabolic disturbances induced in mice by prolonged consumption of high-fat diet. Am. J. Physiol. Endocrinol. Metab. 293, E1746–E1755 [DOI] [PubMed] [Google Scholar]
- 52. Althage M. C., Ford E. L., Wang S., Tso P., Polonsky K. S., Wice B. M. (2008) Targeted ablation of glucose-dependent insulinotropic polypeptide-producing cells in transgenic mice reduces obesity and insulin resistance induced by a high fat diet. J. Biol. Chem. 283, 18365–18376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ugleholdt R., Pedersen J., Bassi M. R., Füchtbauer E. M., Jørgensen S. M., Kissow H. L., Nytofte N., Poulsen S. S., Rosenkilde M. M., Seino Y., Thams P., Holst P. J., Holst J. J. (2011) Transgenic rescue of adipocyte glucose-dependent insulinotropic polypeptide receptor expression restores high fat diet-induced body weight gain. J. Biol. Chem. 286, 44632–44645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hansotia T., Baggio L. L., Delmeire D., Hinke S. A., Yamada Y., Tsukiyama K., Seino Y., Holst J. J., Schuit F., Drucker D. J. (2004) Double incretin receptor knockout (DIRKO) mice reveal an essential role for the enteroinsular axis in transducing the glucoregulatory actions of DPP-IV inhibitors. Diabetes 53, 1326–1335 [DOI] [PubMed] [Google Scholar]
- 55. Irwin N., McClean P. L., Patterson S., Hunter K., Flatt P. R. (2009) Active immunisation against gastric inhibitory polypeptide (GIP) improves blood glucose control in an animal model of obesity-diabetes. Biol. Chem. 390, 75–80 [DOI] [PubMed] [Google Scholar]
- 56. Irwin N., Montgomery I. A., Flatt P. R. (2012) Evaluation of the long-term effects of gastric inhibitory polypeptide-ovalbumin conjugates on insulin resistance, metabolic dysfunction, energy balance and cognition in high-fat-fed mice. Br. J. Nutr. 108, 46–56 [DOI] [PubMed] [Google Scholar]
- 57. Tschöp M. H., DiMarchi R. D. (2012) Outstanding Scientific Achievement Award Lecture 2011. Defeating diabesity. The case for personalized combinatorial therapies. Diabetes 61, 1309–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nakamura T., Tanimoto H., Mizuno Y., Tsubamoto Y., Noda H. (2012) Biological and functional characteristics of a novel low-molecular weight antagonist of glucose-dependent insulinotropic polypeptide receptor, SKL-14959, in vitro and in vivo. Diabetes Obes. Metab. 14, 511–517 [DOI] [PubMed] [Google Scholar]