

Background: Liver receptor homolog 1 (LRH-1, NR5A2) regulates functions of liver, intestines, and pancreas; its aberrant activity is associated with tumorigenesis.
Results: Our work identifies the first antagonists of LRH-1.
Conclusion: The identified ligands inhibit LRH-1 transcriptional activity, diminishing expression of the receptor's target genes.
Significance: LRH-1 inhibitors could be used for analyses of the receptor's biological mechanisms and for development of cancer therapeutics.
Keywords: Hormone Receptors, Ligand-binding Protein, Molecular Docking, Nuclear Receptors, Transcription Regulation, LRH-1, NR5A2, Antagonist, Inhibitor
Abstract
Liver receptor homolog 1 (nuclear receptor LRH-1, NR5A2) is an essential regulator of gene transcription, critical for maintenance of cell pluripotency in early development and imperative for the proper functions of the liver, pancreas, and intestines during the adult life. Although physiological hormones of LRH-1 have not yet been identified, crystallographic and biochemical studies demonstrated that LRH-1 could bind regulatory ligands and suggested phosphatidylinositols as potential hormone candidates for this receptor. No synthetic antagonists of LRH-1 are known to date. Here, we identify the first small molecule antagonists of LRH-1 activity. Our search for LRH-1 modulators was empowered by screening of 5.2 million commercially available compounds via molecular docking followed by verification of the top-ranked molecules using in vitro direct binding and transcriptional assays. Experimental evaluation of the predicted ligands identified two compounds that inhibit the transcriptional activity of LRH-1 and diminish the expression of the receptor's target genes. Among the affected transcriptional targets are co-repressor SHP (small heterodimer partner) as well as cyclin E1 (CCNE1) and G0S2 genes that are known to regulate cell growth and proliferation. Treatments of human pancreatic (AsPC-1), colon (HT29), and breast adenocarcinoma cells T47D and MDA-MB-468 with the LRH-1 antagonists resulted in the receptor-mediated inhibition of cancer cell proliferation. Our data suggest that specific antagonists of LRH-1 could be used as specific molecular probes for elucidating the roles of the receptor in different types of malignancies.
Introduction
Liver receptor homolog 1, commonly known as LRH-15 or NR5A2, is a member of the nuclear receptor family of regulatory transcription factors (1). In adults, this protein is expressed primarily in liver, intestine, and pancreas, where it controls expression of proteins maintaining cholesterol and bile acid homeostasis as well as production of pancreatic enzymes (1, 2). LRH-1 is also expressed in the ovary and breast adipose tissue where it controls biosynthesis of steroids (3, 4). LRH-1 is vital in early development as it maintains a pool of undifferentiated embryonic stem (ES) cells by controlling expression of two master transcription factors, POU5F1 (known as OCT3/4) and NANOG (5–7). Recent studies demonstrated that LRH-1 can substitute for POU5F1 in derivation of induced pluripotent stem cells (7).
Because of its decisive role in cell differentiation, LRH-1 is linked to multiple developmental pathways, including Hedgehog (8) and Wnt/β-catenin (6, 9, 10) signaling. In particular, LRH-1 enhances transcription of multiple genes controlled by the regulatory Wnt/β-catenin cascade. The established transcriptional targets of LRH-1 paired with β-catenin include CCND1 and CCNE1 genes as well as MYC genes known for controlling cell differentiation, growth, and proliferation (6, 7, 9). Because these developmental pathways and associated genes are re-activated during tumorigenesis (11–16), an aberrant activity of LRH-1 is linked to different types of malignancies, including breast and endometrial cancers as well as intestinal tumors and cancer of the pancreas (17–24). The LRH-1 receptor is also implicated in development of various metabolic disorders related to insufficient liver and pancreas functions (25–27). Because of the critical roles of this receptor in human physiology and pathophysiology, identification of specific regulatory ligands, modulators of LRH-1 transcriptional activity, is extremely important.
LRH-1 is classed as an orphan nuclear receptor because its activating hormones (physiological agonists) have not yet been identified. Crystallographic and biochemical studies presented compelling evidence that LRH-1 could bind regulatory ligands (27–32) and suggested phosphatidylinositols as potential hormone candidates for this receptor (29). Studies in mice showed that dilauroyl phosphatidylcholine stimulates LRH-1 activity, increasing bile acid levels, lowering hepatic lipids, and improving glucose homeostasis (27, 28). LRH-1 is also regulated via post-translational modifications, including phosphorylation and sumoylation (33, 34). Specifically, phosphorylation of the regulatory hinge region (connecting the ligand- and DNA-binding domains of LRH-1) by MAPK/ERK stimulates the receptor's transcriptional activity (33), whereas sumoylation of this region results in receptor inhibition (34). Known transcriptional regulators of LRH-1 include co-activators steroid receptor co-activators (SRCs), CREB-binding protein (CBP), and peroxisome proliferator-activated receptor γ co-activator-1α (PGC-1a) as well as co-repressors silencing mediator of retinoid and thyroid hormone receptors (SMRT), SHP, prospero-related homeobox 1 (PROX1), and dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1 (DAX1) (1, 35, 36). No synthetic antagonists of LRH-1 are available to date.
Here, we describe the first synthetic antagonists of LRH-1. Candidate modulators have been identified using screening by molecular docking against a model of the LRH-1 ligand-binding domain (LBD) in an antagonized conformation. This computational screening was followed by direct binding, transcription, and cell proliferation studies in vitro. The results described and discussed in this work suggest that specific antagonists of LRH-1 could be developed for studies of the receptor's biological mechanisms as well as therapeutic treatments.
EXPERIMENTAL PROCEDURES
Molecular Docking Calculations
DOCK3.6 (37–40) was used to screen a library of commercially available compounds against a model of LRH-1 LBD in a transcriptionally inactive conformation. The flexible ligand sampling algorithm in DOCK3.6 superimposes atoms of the docked molecule onto spheres matching a defined binding site; these spheres represent favorable positions for individual ligand atoms (39, 40). Fifty matching spheres mimicking the inside of the receptor ligand-binding pocket (LBP) were used for the molecular docking calculations. The positions of the spheres were dictated by the conformation of the phospholipid bound in the LRH-1 LBP (PDB code 1YUC (30)) and were re-adjusted manually to increase sampling at the opening in the receptor's molecular surface created by deletion of its helix H12 (see “Results”). The accuracy of ligand sampling is determined by the bin size, bin size overlap, and distance tolerance; these three parameters were set to 0.2, 0.1, and 1.4 Å, respectively, for both the binding site matching spheres and the docked molecules. For ligand conformations passing an initial steric filter, a physics-based scoring function was used to evaluate the fit to the binding site. For the best scoring ligands and conformations, 100 steps of rigid-body minimization were carried out prior to assignment of the final score. The score for each conformation was calculated as the sum of the receptor-ligand electrostatic and van der Waals interaction energies, corrected for ligand desolvation; the latter three terms were deduced from pre-calculated grids, as described previously (37). Partial charges from the united atom AMBER force field (41) were used for all receptor atoms (except for Val-406, for which the polarity of the backbone atoms was increased by adding +0.4 and −0.4 electrons to the partial charges of hydrogen and oxygen atoms, respectively, of the peptide bond).
A library of 5.2 million commercially available molecules from the ZINC database (42) was screened against the LRH-1 LBD model. The screen included two sets of compounds; the first set was composed of compounds with molecular weight (Mr) less than 400 (with predicted logP value less than 4, and less than 10 rotatable bonds); the second set included molecules with Mr between 350 and 400 and a predicted logP value between 4 and 5. Prior to docking, subsets of up to 1000 conformations for each molecule were prepared using the program OMEGA (OpenEye Scientific Software). Partial atomic charges and transfer free energies for each ligand atom have been calculated using AMSOL (43) and van der Waals parameters determined using an all-atom potential from AMBER (44).
Protein Expression and Purification
Recombinant nuclear receptors LRH-1 and SF-1 were expressed and purified using similar methods. In brief, cDNA encoding human LRH-1 LBD (amino acids 294–541) was cloned into pRSF-2 Ek/LIC (Novagen) vector containing the N-terminal His6 tag followed by tobacco etch virus protease cleavage site. The recombinant protein was expressed in BL21(DE3) cells using standard methods (induction with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside followed by overnight cell culturing at 16 °C) and purified using Ni2+-nitrilotriacetic acid affinity column (Qiagen) followed by size exclusion chromatography (HiLoad 16/60 Superdex 200, GE Healthcare) in buffer containing 20 mm Tris, pH 8.0, 150 mm NaCl, 5 mm DTT, 10% glycerol and 2 mm CHAPS.
cDNA encoding human SF1 LBD (hSF-1, amino acids 218–461) was cloned into pET-46 Ek/LIC (Novagen) vector containing His6 tag followed by a tobacco etch virus protease cleavage site. The recombinant protein was expressed and purified as described above.
Mutagenesis
cDNA encoding wild type hLRH-1 LBD (amino acids 294–541) in pRSF-2 vector (Novagen) and the QuikChange site-directed mutagenesis kit (Stratagene) were used for generating mutants A349F (forward and reverse primers 5′-GGG CTT ATG TGC AAA ATG TTC GAT CAA-3′ and 5′-GGA GAA GAG AGT TTG ATC GAA CAT TTT-3′) and A349W (forward and reverse primers 5′-GGG CTT ATG TGC AAA ATG TGG GAT CAA-3′ and 5′-GGA GAA GAG AGT TTG ATC CCA CAT TTT-3′). The introduced mutations were verified by sequencing, and the mutant proteins were expressed and purified as described above for wild type LRH-1.
Differential Scanning Fluorimetry (DSF)
Protein stability in the presence and the absence of tested compounds was assessed using the DSF method, MxPro3005P qRT-PCR detection system (Stratagene) in a 96-well format. Sypro-Orange dye (Invitrogen) was used to monitor the fluorescence, with carboxyfluorescein (FAM) filter for fluorescence excitation (492 nm) and carboxy-X-rhodamine (ROX) filter for fluorescence emission (610 nm). The DSF spectra for purified wild type and mutant variants of hLRH-1 LBD (10 μm) were recorded using screening buffer (TBS) with added Sypro-Orange dye (1:2000 dilution), in the presence of individual compounds (100 μm) or 1% DMSO (control). Tested sample mixtures (final volume 50 μl) were heated gradually, from 25 to 96 °C, at the rate of 2 °C/min, and the corresponding fluorescence was recorded following every 1 °C increase. The melting temperature (Tm) for each sample was deduced by the KaleidaGraph program (Synergy) from the first derivative of the corresponding denaturation curve generated by the MxPro QPCR software (Stratagene).
Surface Plasmon Resonance
SPR was used for quantification of direct binding of compounds 3 and 3d2 to hLRH-1 LBD. Measurements were performed on a Biacore T100 instrument, with a running buffer of 20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 5 mm DTT, 5% DMSO, and 0.05% Tween 20, at 10 °C. The purified LRH-1 protein (either wild type or mutant LBD) was covalently immobilized to the surface of a CM5 biosensor chip to a surface density of about 3000 resonance units, using standard amine coupling chemistry. The individual compounds at 0.8–15 μm concentrations were injected over immobilized LRH-1 and reference surfaces, and binding response sensorgrams were solvent-corrected against running buffer with DMSO concentrations ranging from 4.9 to 5.1%. The corresponding equilibrium dissociation constants (Kd) were determined using steady-state analysis of the compounds' binding affinities, assuming 1:1 ligand-protein stoichiometry. Prior to evaluations of binding affinities of compounds, the functionality of immobilized LRH-1 protein was confirmed by demonstrating its high affinity interactions with a peptide DAX1–3 corresponding to amino acids 140PRQGSILYSLLTSSK154 of the receptor's transcriptional co-regulator DAX-1.
Evaluation of the effects of compounds 3 and 3d2 on binding of DAX1–3 peptide to LRH-1 was performed using a Biacore T200 instrument at 25 °C, with the running buffer described above. The purified LRH-1 protein was covalently immobilized to the surface of a CM5 biosensor chip to a surface density of about 1000 resonance units, using standard amine coupling chemistry. Solutions of DAX1–3 peptide at 100 nm concentration in the presence of either 5% DMSO (solvent control) or individual compounds at different concentrations (0.063–40 μm) were injected over immobilized LRH-1 and reference surfaces; binding response sensorgrams were recorded and quantified using the Biacore T200 software.
Fluorescence Anisotropy Assay
Fluorescence polarization ligand binding assay was used to determine whether compounds 3 and 3d2 bind human estrogen hormone receptor α. The assay was performed using the PolarscreenTM ERα competitor assay, green kit (Invitrogen), according to the manufacturer's protocol. Serial dilutions of estradiol (E2, positive control) and individual compounds in DMSO were prepared and transferred to the wells of a black OptiPlateTM 384F plate (PerkinElmer Life Sciences) containing E2 Screening Buffer. Following addition of FluormoneTM E2-ERα complex to each well (4.5 nm E2, 15 nm ERα), the assay plate was incubated for 2 h in the dark, and polarization values were measured using a EnVision® multilabel reader (PerkinElmer Life Sciences). All measurements were done in triplicate and the data fit using Prism software (GraphPad Software).
Transactivation Assays
Two (Tet)-inducible HEK293 cell lines expressing full-length hLRH-1 or hSF-1 receptors, respectively (32), were plated into 12-well tissue culture plates in batches of 105 cells. After 24 h, tetracycline (Sigma) at a final concentration of 10 nm was added to each well to induce the expression of hLRH-1 or hSF-1. Three hours after the induction, cells were treated with either individual compounds (at concentrations 1–10 μm) or DMSO (0.1%, control). Following 24 h of incubation with compounds, cells were lysed, total RNA was isolated and the corresponding cDNA synthesized, and mRNA levels for G0S2 (in cells expressing hLRH-1) or NR0B2 (encoding SHP, in cells expressing hSF-1) genes in each sample were assessed by qPCR (see under “RNA Purification, cDNA Synthesis and qPCR Analysis”).
For a transactivation assay with estrogen hormone receptor α (45), transient co-transfections of HeLa cells with vectors encoding either Gal4 DNA-binding domain (DBD) or Gal4 DBD-hERα LBD fusion (gift from Dr. S. Ayers, The Methodist Hospital Research Institute, Houston, TX), both at 10 ng/well, constructs for Gal4-E1B promoter linked to a luciferase reporter gene (200 ng/well) and actin β-galactosidase (10 ng/well, internal control) were performed in batches of 105 cells seeded into 12-well tissue culture plates. The transfections were done using FuGENE HD transfection reagent (Promega), and the transfection efficiencies were assessed by measuring the corresponding activity of β-galactosidase. At 3 h after the transfections, cells were treated with either DMSO (0.1%, control) or individual compounds at different concentrations, in the presence of E2 (10 nm), in the medium containing no fetal bovine serum. Following 24 h of incubation, luciferase activities in each well were assessed using the luciferase assay system (Promega) relative to the control. Cells transfected with Gal4 DBD vector served as a control for ERα-independent effects.
For a transcription assay with androgen hormone receptor (46), transient co-transfections of HeLa cells with vectors encoding either Gal4 DBD or Gal4 DBD-hAR LBD fusion (both at 10 ng/well), constructs for GK1 reporter (200 ng/well) and actin β-galactosidase (10 ng/well, internal control) were performed in batches of 105 cells seeded into 12-well tissue culture plates. The transfections were done using TransFectin lipid reagent (Bio-Rad), and the transfection efficiencies were assessed by measuring the corresponding activity of β-galactosidase. Three hours after the transfections, cells were treated with either DMSO (0.1%, control) or compounds 3 or 3d2 at different concentrations, in the absence or the presence of dihydrotestosterone (1 μm). Following 24 h of incubation, luciferase activities in each well were assessed using the luciferase assay system (Promega) relative to the control. Cells transfected with Gal4 DBD vector served as a control for AR-independent effects.
For a transcription assay with thyroid hormone receptor β (46), transient co-transfections of HeLa cells with vectors encoding either Gal4 DBD or Gal4 DBD-hTRβ LBD fusion (both at 10 ng/well), constructs for GK1 reporter (200 ng/well), and actin β-galactosidase (10 ng/well, internal control) were performed in batches of 105 cells seeded into 12-well tissue culture plates. The transfections were done using TransFectin lipid reagent (Bio-Rad), and the transfection efficiencies were assessed by measuring the corresponding activity of β-galactosidase. After 3 h, cells were treated with either DMSO (0.1%, control) or compounds 3 or 3d2 at different concentrations, in the absence or the presence of T3 (1 μm). Following 24 h of incubation, luciferase activities in each well were measured using the luciferase assay system (Promega) relative to the control. Cells transfected with Gal4 DBD vector served as a control for TRβ-independent effects.
RNA Purification, cDNA Synthesis, and qPCR Analysis
Total RNA from different cell samples was isolated using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. cDNA was synthesized from 500 ng of total RNA at 42 °C for 60 min in the presence of random primers (Invitrogen) using the SuperScript-II reverse transcriptase (Invitrogen). Quantitative PCR amplification of mRNA for G0S2, NR0B2, CCNE1, and RPS9 (ribosomal protein S9 gene, internal control) was performed in triplicates using the Mx3005P real time PCR system (Stratagene) and the SYBR Green I dye for detection (Stratagene). Specific oligonucleotides used for these experiments were as follows: for CCNE1 (PPH00131A-200, SABiosciences), G0S2 (5′-CAGAGAAACCGCTGACATCTAGAA-3′ and 5′-CAGCAAAACTCAATCCCAAACTC-3′, IDT), NR0B2 (PPH05889A-200, SABiosciences), and RPS9 (5′-AAGGCCGCCCGGGAACTGCTGAC-3′ and 5′-ACCACCTGCTTGCGGACCCTGATA-3′, IDT). To control for external contamination, no template control and no reverse transcription control were included in each run. The amplification curves were analyzed with the Mx3005P software using the comparative cycle threshold (Ct) method. Relative quantification of the target mRNAs was evaluated after normalization of Ct values with respect to the RPS9 levels.
Promiscuous Inhibition Test
To exclude fortuitous inhibition by tested compounds due to their possible colloidal aggregation, dynamic light scattering method (DLS) was employed to assess particle formation in solutions of tested compounds. In addition, a standard enzymatic assay was performed to detect any unspecific inhibition of AmpC β-lactamase by these ligands. For DLS experiments, concentrated DMSO stocks of compounds were diluted with filtered buffer used for quantitative Biacore-based direct binding assay (20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 5 mm DTT, 5% DMSO, 0.05% Tween 20). Measurements were made using a DynaPro MS/X instrument (Wyatt Technology), with a 55-milliwatt laser (100% power) at 826.6 nm and a 90° detector angle. Inhibition of AmpC β-lactamase was assessed in 50 mm potassium phosphate, pH 7.0. Individual compounds (100 μm solutions in 1% DMSO) were incubated with 1 nm β-lactamase for 5 min, and reactions were initiated by adding the substrate CENTA (Chromothin, Tydock Pharma). To assess enzyme inhibition, the absorbance at 405 nm was recorded for 5 min using a spectrophotometer (Agilent). The assay was performed in 1-ml cuvette duplicates, with controls measuring enzyme activity in the presence of solvent (1% DMSO).
Cell Line Maintenance
Human pancreatic cancer cell lines AsPC-1 and L3.3 were kindly provided by Dr. M. McMahon (Helen Diller Family Comprehensive Cancer Center, University of California at San Francisco); cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2, in DMEM (University of California at San Francisco Cell Culture Facility (CCF)) supplemented with 10% fetal bovine serum (FBS, Hyclone), 1× l-glutamine, and 1× Pen/Strep antibiotics (from 100× stocks, CCF). Human breast cancer cells T47D were purchased from the CCF and maintained in RPMI 1640 media (CCF) with 10% FBS (Hyclone), 1× Pen/Strep antibiotics, and 0.2 IU/ml insulin (CCF). Human breast cancer cells MDA-MB-458 were purchased from the CCF and maintained in Leibovitz's L-15 medium without NaHCO3 (CCF) supplemented with 10% FBS (Hyclone), 1× Pen/Strep antibiotics, 1× l-glutamine (CCF), and 3.7 g/liter NaHCO3 (CCF). Human colon adenocarcinoma cells HT-29 were purchased from the CCF and maintained in McCoy's 5A medium (CCF) supplemented with 10% FBS (Hyclone) and 1× Pen/Strep antibiotics (CCF). Tet-inducible HEK293 cell lines expressing either hLRH-1 or hSF-1 receptors were kindly provided by Dr. H. Ingraham (Dept. of Cellular and Molecular Pharmacology, University of California at San Francisco) and maintained in DMEM (Invitrogen) supplemented with 10% Tet-negative FBS (Hyclone), 1× Pen/Strep antibiotics (CCF), 5 μg/ml blasticidin (Invitrogen), and 50 μg/ml hygromycin (Invitrogen); for cells expressing hSF-1, extra 1× sodium pyruvate (CCF) and 1× nonessential amino acids solution (CCF) were added to the medium. HeLa cells were kindly provided by Phuong Nguyen (Fletterick Laboratory, Department of Biochemistry and Biophysics, University of California at San Francisco) and maintained in DMEM H-21 (CCF) supplemented with 10% charcoal, dextran-stripped FBS (Hyclone), and 1× Pen/Strep antibiotics (CCF). All cells were passaged when they reached 80% confluence and harvested using a solution containing 0.05% trypsin and EDTA.
Cell Proliferation Assays and Cytotoxicity Measurements
For cell proliferation assays, cells were plated in 96-well microtiter plate triplicates, at a density of 104 cells/ml. Three hours after the plating, cells were treated with individual compounds at different concentrations or with DMSO (control); 24, 48, 72 and 96 h following the treatments, cell proliferation in each well was quantified using the CellTiter-Glo reagent (Promega). Cell proliferation rates for treated cells were compared with those of the control cells. For cytotoxicity measurements, cells were plated in 96-well microtiter plate triplicates, at a density of 104 cells/ml; cells were then treated with different concentrations of tested compounds or DMSO (control), and 24 h following the treatments, cytotoxicity was assessed using the CytoTox-Glo cytotoxicity assay reagent (Promega).
RESULTS
Modeling of the hLRH-1 LBD in a Transcriptionally Inactive Conformation
All available crystal structures of the LRH-1 LBD represent the receptor in its active state (with the C-terminal helix H12 tightly packed against helices H3, H4–H5, and H11, poised for interactions with transcriptional co-activators) (28–32). Furthermore, all known LRH-1 ligands, identified either in structural or functional studies, are receptor agonists stabilizing its active conformation (27–32). Thus, to enable identification of LRH-1 antagonists via structure-based virtual screening, we generated a model of a transcriptionally inactive state of the receptor LBD. Based on the structural similarity between LRH-1 and estrogen receptor α (ERα) LBDs (Fig. 1, A and B), we hypothesized that these receptors could be antagonized in a similar manner. Numerous antagonists have been developed against ERα, and independent atomic resolution structures of ERα have been determined with individual antagonists bound in the receptor's ligand-binding pocket (47–49) (illustrated by ERα bound by receptor antagonist 4-hydroxytamoxifen (PDB code 3ERT (47)) in Fig. 1C). The ERα antagonists typically have a hydrophobic core, which is connected to a bulky and often polar “side chain.” Whereas the hydrophobic core binds in the ligand-binding cavity of the receptor, the side chain, which cannot be contained within the pocket, protrudes out of the cavity. As a consequence, helix H12 is sterically hindered from aligning in the proper agonist conformation, precluding binding of co-activators to ERα. By analogy, the inactive state for LRH-1 LBD was modeled by undocking helix H12 (amino acids 524–538) from a structure of the receptor LBD in transcriptionally active conformation (PDB code 1YUC (30)) (Fig. 1D). Because different orientations for H12 have been observed for estrogen receptor bound by different antagonists (47–50) (including those that completely abolished the association between H12 and the rest of the LBD (50)), helix H12 of LRH-1 was omitted from the model. In addition, side chains of Leu-517 and Asp-350 were re-positioned in the LRH-1 LBD to match their counterparts in the structure of antagonist-bound ERα (Fig. 1, C and D). These structural re-arrangements are thought to facilitate binding of a potential antagonist to the LRH-1 LBD, in particular, by enabling favorable electrostatic interactions between the antagonist's polar moiety and the re-oriented carboxylate group of Asp-350. In the active conformation of LRH-1, this conserved residue (Asp-351 in ERα, Fig. 1, A and B) registers and stabilizes the proper orientation of helix H12 by making hydrogen bonds with the main chain of H12 at its base.
FIGURE 1.

Design of a model for LRH-1 ligand-binding pocket in an antagonized state. A, architecture of the pocket of ERα with bound agonist. A schematic model for ERα polypeptide chain (ERα LBD bound by R,R-5,11-cis-diethyl-5,6,11,12-tetrahydrochrysene-2,8-diol, PDB code 1L2I) is shown in gray; structural elements forming the pocket (H3, H4–H5, H11, and H12) are indicated; helix H12 docked in the “active” conformation is highlighted in magenta. Bound agonist is shown as a stick model. B, architecture of the hormone-binding pocket of LRH-1 in transcriptionally active state. A schematic model for LRH-1 polypeptide chain (PDB code 1YUC) is shown in gray, with structural elements forming the pocket indicated. Helix H12 docked in the active conformation is highlighted in magenta. C, architecture of the hormone-binding pocket of ERα with antagonist 4-hydroxytamoxifen bound in the pocket. Structural elements forming the ligand-binding pocket of ERα (PDB code 3ERT) are indicated. Bound antagonist is shown as a stick model. An alternative conformation for side chain of Asp-351 facilitating binding interactions of ERα with 4-hydroxytamoxifen is indicated. D, model for the hormone-binding pocket of LRH-1 in transcriptionally inactive state. Undocked helix H12 is omitted from the model. Alternative conformations for side chains of Leu-517 and Asp-350 predicted to facilitate binding interactions of LRH-1 with potential ligands-antagonists are indicated.
Virtual Screening and Compound Selection
DOCK3.6 (37–40) was used to screen 5.2 million commercial compounds from the ZINC database (42) against our model of the inactive state of hLRH-1 LBD. For each screened molecule, ∼2800 orientations have been sampled, and for each of these, ∼18,000 conformations were scored. The molecules were ranked based on the sum of their van der Waals and electrostatic interaction energies corrected for ligand desolvation (see under “Experimental Procedures”). The top ranked 10,000 compounds (0.2% of the compound library) were then post-processed to identify molecules that could not be accommodated by the active conformation of the receptor (PDB code 1YUC (30)) and that had the potential to displace helix H12 from its active orientation. This was accomplished by selecting compounds capable of forming a favorable electrostatic interaction with the LRH-1 residue Asp-350 (shown for two docked candidate compounds in Fig. 2); the analogous electrostatic bridge is a characteristic feature of many ERα-antagonist complexes (indicated in Fig. 1C for ERα bound by 4-hydroxytamoxifen). Following visual inspection of the resulting top-ranked 1000 compounds (0.02% of the initial library content), eight were selected for experimental evaluations (compounds 1–8, Table 1). The criteria for this manual selection process were as follows: 1) clear structural complementarity between a considered compound and the receptor ligand-binding pocket; 2) a strong predicted interaction of a candidate compound with the LRH-1 Asp-350 (to favor antagonism), and 3) immediate availability of compounds for experimental evaluations.
FIGURE 2.

Predicted mode of binding for the identified LRH-1 antagonists. A and B, schematic model for LRH-1 polypeptide chain in the vicinity of the hormone-binding pocket is shown in gray, with structural elements forming the pocket indicated. Docked receptor antagonists (compound 3 in A and compound 3d2 in B) are shown as color-coded stick models. Side chains of Leu-517 and Asp-350 predicted to facilitate binding interactions of LRH-1 with ligands-antagonists are indicated. C and D, mutations in the LRH-1 ligand-binding pocket (A349F and A349W, shown in magenta and pink) predicted to interfere with binding of compounds 3 and 3d2 to the receptor (shown in C and D, respectively); these mutants were used as negative controls in in vitro direct binding assays.
TABLE 1.
Candidate compounds selected from the molecular docking screen
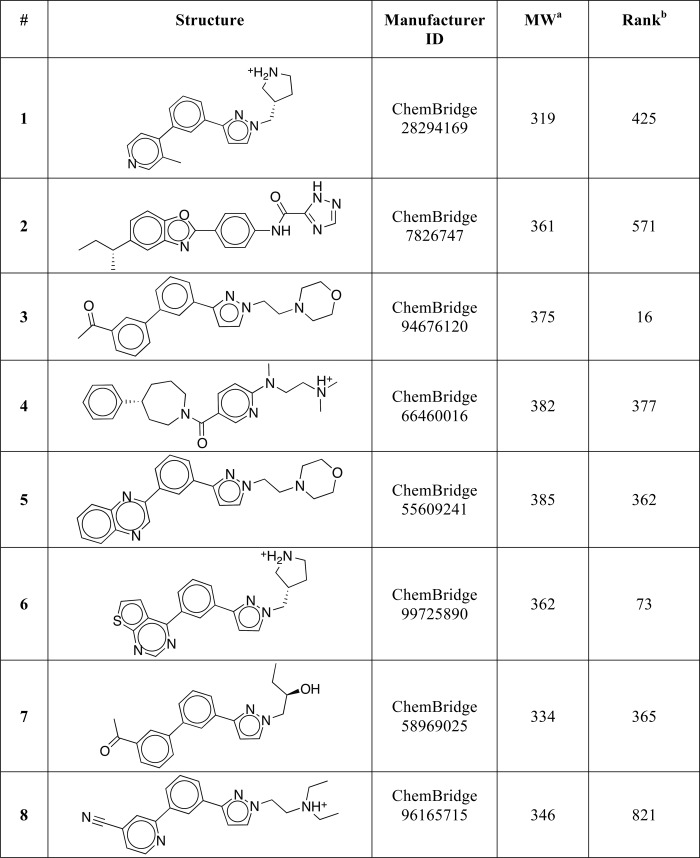
a MW is molecular weight.
b Ranking of the compound in the virtual screen.
Three Out of Eight Predicted Antagonists Bind Directly to hLRH-1 LBD
Using the DSF method, we analyzed binding of each of the eight selected compounds to the purified recombinant hLRH-1 LBD. Preceding these experiments, the proper folding and functionality of the purified LRH-1 protein were assessed by the DSF and fluorescence anisotropy methods. Using the DSF-based quality control, we analyzed the protein's melting curve and determined its transition temperature (Tm). This analysis showed that in the presence of 1% DMSO (solvent control), hLRH-1 LBD has a stable base line, a transition at 53.4 ± 0.1 °C (reference Tm), and a sloped denatured base line. Following the DSF-based protein folding control, the functionality of the hLRH-1 LBD was confirmed using quantitative SPR method, to measure binding and release of the receptor's co-regulator peptide. This analysis showed that a co-regulator peptide DAX1–3 140PRQGSILYSLLTSSK154, which corresponds to the nuclear receptor box 3 motif of nuclear receptor DAX1, binds to hLRH-1 LBD with high affinity (Kd = 90 ± 1 nm), attesting to the DSF-based evidence of the proper receptor LBD folding; the latter is necessary to maintain a proper architecture of the receptor co-regulator binding site (AF-2), which is targeted by the DAX1–3 peptide.
When tested in the DSF experiments, three out of the eight candidate receptor antagonists (compounds 3, 5, and 7, purchased from the ChemBridge library and used at 100 μm in DSF assay) were able to shift the hLRH-1 LBD transition temperature (Tm) significantly. In the presence of compounds 3, 5, and 7, the hLRH-1 LBD Tm was 51.1 ± 0.1 °C, 51.8 ± 0.3 °C, and 50.6 ± 0.2 °C, respectively (Fig. 3A); no nonspecific interactions between the compounds and the fluorescent dye were detected. Notably, all three compounds shifted the receptor Tm downward, indicating that the LRH-1 LBD is destabilized upon binding of these ligands (Fig. 3A; all DSF-based analyses were performed using freshly purified protein; measurements were taken in triplicates and repeated for three different batches of the protein). The observed downward shift of Tm could be explained by the presumed mode of binding of the tested compounds. The preceding molecular docking experiments predicted that binding of these molecules into the hLRH-1 ligand-binding pocket would cause detachment of helix H12 from the LBD core (Figs. 1 and 2), resulting in receptor destabilization and deactivation.
FIGURE 3.
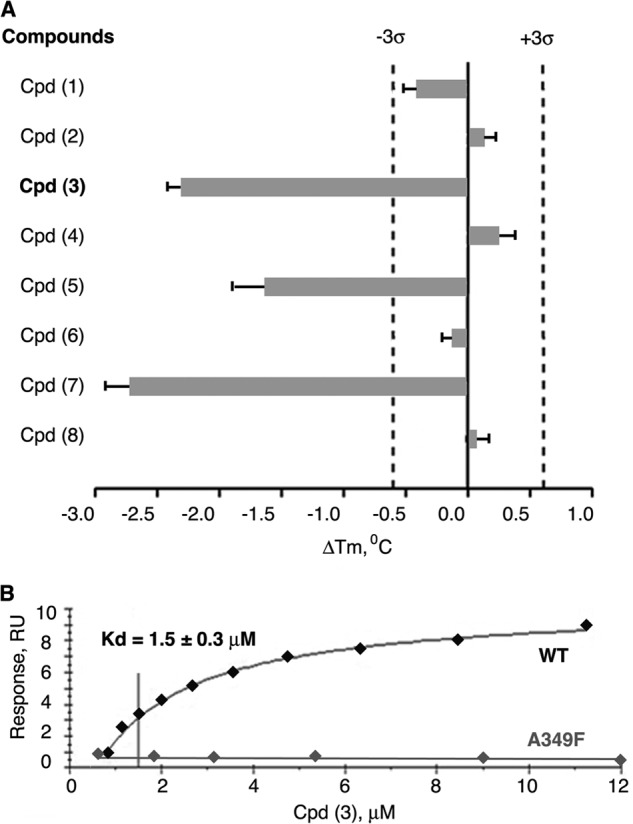
Results of direct binding assays for eight selected LRH-1 ligands with predicted antagonistic properties. A, melting temperature shifts for LRH-1 LBD treated with candidate compounds. Three out of eight receptor antagonist candidates (compounds 3, 5, and 7) shift the melting temperature (Tm) of hLRH-1 LBD, demonstrating direct binding to the receptor. For each tested compound, averaged data are shown as horizontal bars; experimental errors and 3σ thresholds are indicated with solid and dashed lines. Compound 3 demonstrating antagonistic effect in transcription assay is indicated in boldface. B, evaluation of binding affinity of compound 3 in Biacore-based assay. The purified LRH-1 protein (either wild type or mutant variants A349F/W) was covalently immobilized to the surface of a CM5 chip, solutions of compound 3 at 0.8–15 μm concentrations were injected over immobilized LRH-1 and reference surfaces, and dose-dependent steady-state responses were recorded and measured relative to the reference. The equilibrium dissociation constant (Kd, indicated for wild type LRH-1 LBD) was determined using steady-state analysis of binding affinities, assuming 1:1 ligand-protein stoichiometry. No dose-dependent binding was observed for LRH-1 ligand pocket mutant A349F under similar conditions (negative control, indicated).
Compound 3 Inhibits Transcriptional Activity of hLRH-1
To verify that these ligands not only bind to LRH-1 but also deactivate the receptor upon binding, the transcriptional activity of LRH-1 was assessed in the absence and the presence of compounds 3, 5, and 7. HEK 293 cells expressing Tet-inducible hLRH-1 were employed to detect effects of the ligands on the receptor activity; this cellular system has been designed and used previously for assessment of transcriptional activity of nuclear receptors LRH-1 and SF-1 in the presence of synthetic small molecule agonists (32). Following the induction of LRH-1 expression with tetracycline (Tet-On LRH-1(+) in Fig. 4A) and treatments of HEK293 cells with either individual compounds or DMSO (0.1%, solvent control), the mRNA levels for the endogenous G0S2 gene (G0/G1 switch gene, a transcriptional target of LRH-1 (32)) were evaluated in the compound-treated cells relative to the control. In these experiments, only compound 3 (1-(3′-{1-[2-(4-morpholinyl)ethyl]-1H-pyrazol-3-yl}-3-biphenylyl)ethanone; ranked number 16 in the preceding molecular docking screen, Table 1) inhibited the transcriptional activity of hLRH-1. Treatments with this compound lowered the G0S2 mRNA levels with an IC50 value of 5 ± 1 μm (Fig. 4A). No changes in the levels of LRH-1 mRNA were observed as a result of these treatments (Tet-On LRH-1(+), Fig. 4B). Neither compound 5 nor 7 displayed any antagonist activity in these transcriptional studies (data not shown). To prove that the observed transcriptional effect by compound 3 is LRH-1-mediated, the analogous experiment was performed in noninduced HEK293 cells; under these conditions, no changes in the levels of G0S2 transcripts were detected in cells treated with compound 3 compared with the control (Fig. 4A, Tet-Off LRH-1(−)). All measurements were taken in triplicates and repeated in three independent experiments.
FIGURE 4.

Inhibition of transcriptional activity of LRH-1 by compound 3. A and B, compound 3 inhibits transcriptional activity of LRH-1. HEK293 cells harboring Tet-inducible expression vector encoding full-length LRH-1 were treated with either DMSO (0.1%, solvent control) or compound 3 at different concentrations (indicated). The experiments were performed both in the presence and the absence of Tet (indicated as Tet-On LRH-1(+) and Tet-Off LRH-1(−) in A and B). Following 24-h treatments, levels of mRNA for G0S2 (LRH-1 target gene, shown in A) and mRNA for LRH-1 (shown in B) in all cell samples were evaluated by qPCR. For each concentration point, data are shown relative to control (0.1% DMSO), as average of three independent measurements, with experimental errors shown as black lines. The corresponding IC50 value was calculated using Prism 5 software and is indicated in A. C and D, compound 3 diminishes binding of DAX1–3 peptide to LRH-1 in Biacore-based assay. The purified LRH-1 LBD protein was covalently immobilized to the surface of a CM5 chip, and solutions of DAX1–3 peptide at 100 nm concentration were injected over immobilized LRH-1 and reference surfaces in the presence of either 5% DMSO (solvent control) or different concentrations (0.063–40 μm) of Cpd 3. Dose-dependent steady-state responses were recorded and measured relative to the reference (shown in C). Quantification of the Biacore data is shown in D; gray bars indicate binding of DAX1–3 peptide in the presence of different concentrations of Cpd 3 relative to control; experimental errors are shown as black lines. RU, response units.
Assessment of Binding Affinity of Compound 3
The affinity of binding interactions between compound 3 and LRH-1 LBD was determined by employing the quantitative SPR method. For this analysis, the purified LRH-1 protein (either wild type LBD or the ligand pocket mutants A349F/A349W) was covalently immobilized to the surface of a CM5 biosensor chip (for details, see “Experimental Procedures”), and solutions of compound 3 at concentrations ranging from 0.8 to 15 μm were injected over immobilized LRH-1 and reference surfaces. Compound 3 was shown to bind to wild type LRH-1 LBD, producing a steady-state SPR response in a dose-dependent manner. Curve-fitting analysis of the SPR response isotherm estimated a Kd value of 1.5 ± 0.3 μm (Fig. 3B). No dose-dependent binding was observed for LRH-1 ligand pocket mutants A349F and A349W under similar conditions (shown for A349F in Fig. 3B). These mutants were prepared based on the structure of LRH-1 with the docked candidate antagonists (Fig. 2) and used as a negative control in these experiments, as the bulky amino acid substitutions in the receptor's pocket were expected to interfere with the predicted mode of ligand binding (Fig. 2, C and D). Prior to testing the mutant proteins in direct binding assays, their proper fold and stability were assessed by the DSF; no change in the transition temperature was detected for either protein variant compared with wild type LRH-1 LBD.
Further SPR analyses revealed that binding of compound 3 to LRH-1 LBD diminishes the receptor's interactions with co-regulator DAX1–3 peptide in a dose-dependent manner (Fig. 4, C and D). Being consistent with the results of transcriptional studies (Fig. 4, A and B), this observation substantiates the expected mechanism of receptor deactivation as a result of its binding to the proposed antagonist.
Selection of Compound 3 Analogs
Encouraged by the results of direct binding and transcriptional studies, we evaluated a series of commercially available analogs of compound 3 by docking them into the previously described model of LRH-1 (Fig. 1). Following these experiments, four additional top-ranked compounds with predicted receptor-specific antagonistic properties (Table 2) were purchased and analyzed in the DSF and transcriptional assays described above. The DSF studies demonstrated that one of the four candidate ligands (compound (3d2), 4-(3-{1-[2-(dimethylamino)ethyl]-1H-pyrazol-3-yl}phenyl)-N,N-5,6-tetramethyl-2-pyrimidinamine) shifted the transition temperature of the LRH-1 LBD significantly (from 53.4 ± 0.1 °C to 51.4 ± 0.1 °C, see Fig. 5A). Similar to the original compound 3, binding of this analog to hLRH-1 resulted in receptor destabilization, likely due to the detachment of helix H12 from the LBD core. This destabilization model was predicted by the molecular docking experiments (Fig. 2, A and B). All DSF measurements were taken in triplicates, using three different batches of freshly purified hLRH-1 LBD protein; no nonspecific interactions between any of the compound analogs and the fluorescent dye were detected in these experiments. Direct binding of analog (3d2) to LRH-1 has been confirmed using quantitative Biacore-based analysis. For wild type receptor, the equilibrium response data fit well to a simple 1:1 protein-ligand isotherm, with an estimated Kd value of 1.8 ± 0.4 μm (Fig. 5B). Under similar conditions, no binding was detected for the receptor ligand pocket mutants A349F and A349W (negative control, shown for A349F in Fig. 5B). Similar to compound 3 and consistent with the proposed mechanism of allosteric regulation, binding of this analog to wild type LRH-1 LBD inhibited the receptor's interactions with co-regulator DAX1–3 peptide (Fig. 6, A and B).
TABLE 2.
Structures of analogs of compound 3
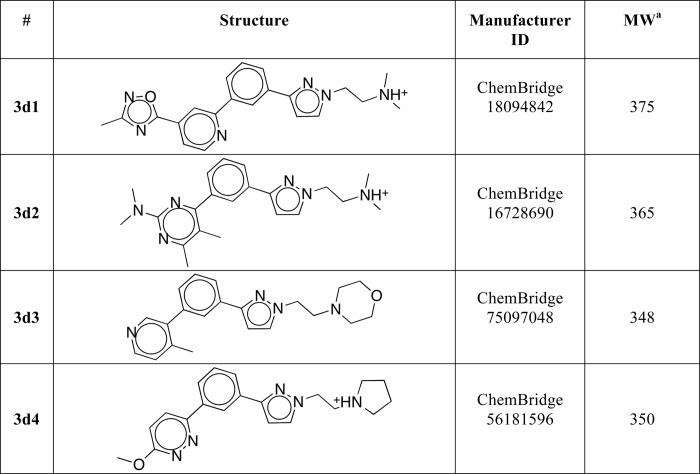
a MW is molecular weight.
FIGURE 5.
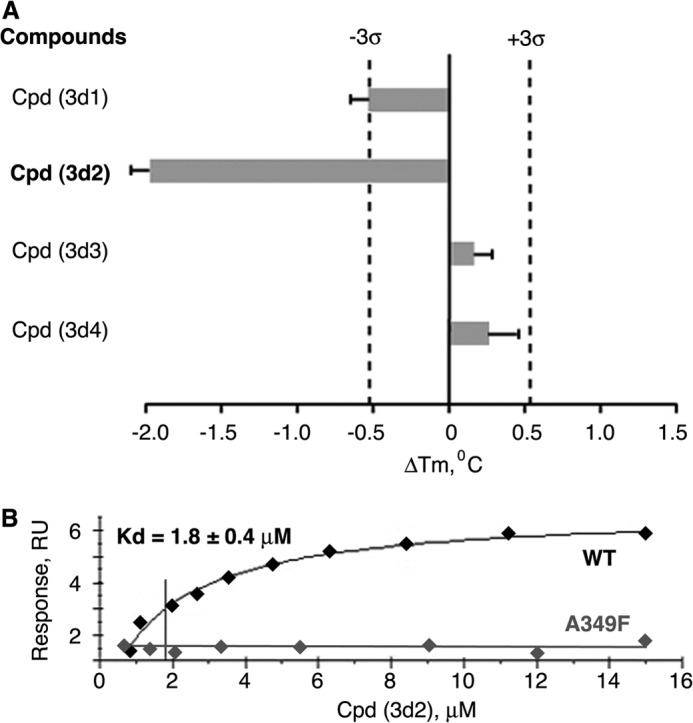
Results of direct binding assays for selected analogs of compound 3. A, melting temperature shifts for LRH-1 LBD treated with analog compounds. One out of four selected compound analogs (compound 3d2) shifts the melting temperature (Tm) of hLRH-1 LBD, demonstrating direct binding to the receptor. For each tested compound, averaged data are shown as horizontal bars; experimental errors and 3σ thresholds are indicated with solid and dashed lines. Analog 3d2 demonstrating antagonistic effect in transcription assay is indicated in boldface. B, assessment of binding affinity of analog 3d2 in Biacore-based analysis. The purified LRH-1 protein (either wild type or mutant variants A349F/W) was covalently immobilized to the surface of a CM5 chip; solutions of compound 3d2 at 0.8–15 μm concentrations were injected over immobilized LRH-1 and reference surfaces, and dose-dependent steady-state responses were recorded and measured relative to the reference. The equilibrium dissociation constant (Kd, indicated for wild type LRH-1 LBD) was determined using steady-state analysis of binding affinities, assuming 1:1 ligand-protein stoichiometry. Under similar conditions, no binding was observed for LRH-1 ligand pocket mutant A349F (negative control, indicated).
FIGURE 6.

Inhibition of transcriptional activity of LRH-1 by compound 3d2. A and B, compound 3d2 diminishes binding of DAX1–3 peptide to LRH-1 in Biacore-based assay. The purified LRH-1 LBD protein was covalently immobilized to the surface of a CM5 chip, and solutions of DAX1–3 peptide at 100 nm concentration were injected over immobilized LRH-1 and reference surfaces in the presence of either 5% DMSO (solvent control) or different concentrations (0.063–40 μm) of Cpd 3d2. Dose-dependent steady-state responses were recorded and measured relative to the reference (shown in A). Quantification of the Biacore data is shown in B; gray bars indicate binding of DAX1–3 peptide in the presence of different concentrations of Cpd 3d2 relative to control; experimental errors are shown as black lines. C and D, compound 3d2 inhibits transcriptional activity of LRH-1. HEK293 cells harboring Tet-inducible expression vector encoding full-length LRH-1 were treated with either DMSO (0.1%, solvent control) or compound 3d2 at different concentrations (indicated). The experiments were performed both in the presence and the absence of Tet (indicated as Tet-On LRH-1(+) and Tet-Off LRH-1(−) in C and D). Following 24-h treatments, levels of mRNA for G0S2 (shown in C) and LRH-1 (shown in D) in all samples were evaluated by qPCR. For each concentration point, data are shown relative to control (0.1% DMSO), as average of three independent measurements, with experimental errors shown as black lines. The corresponding IC50 value was calculated using Prism 5 software and is indicated in C. RU, response unit.
Following direct binding assessments by the DSF and SPR methods, the potential of compound 3d2 to inhibit transcriptional activity of hLRH-1 was evaluated using the Tet-inducible HEK293 cellular model described above. These experiments demonstrated that compound 3d2 suppresses the transcriptional activity of hLRH-1, lowering the G0S2 mRNA levels in the treated cells compared with the control, with an IC50 of 6 ± 1 μm (Tet-On LRH-1(+), Fig. 6C). No changes in the levels of the receptor mRNA were observed as a result of these treatments (Tet-On LRH-1(+), Fig. 6D). Similar to the original compound 3, this analog did not exhibit any LRH-1 independent activity at or under 10 μm (Tet-Off LRH-1(−), Fig. 6C).
Assessing Specificity of Compounds 3 and 3d2
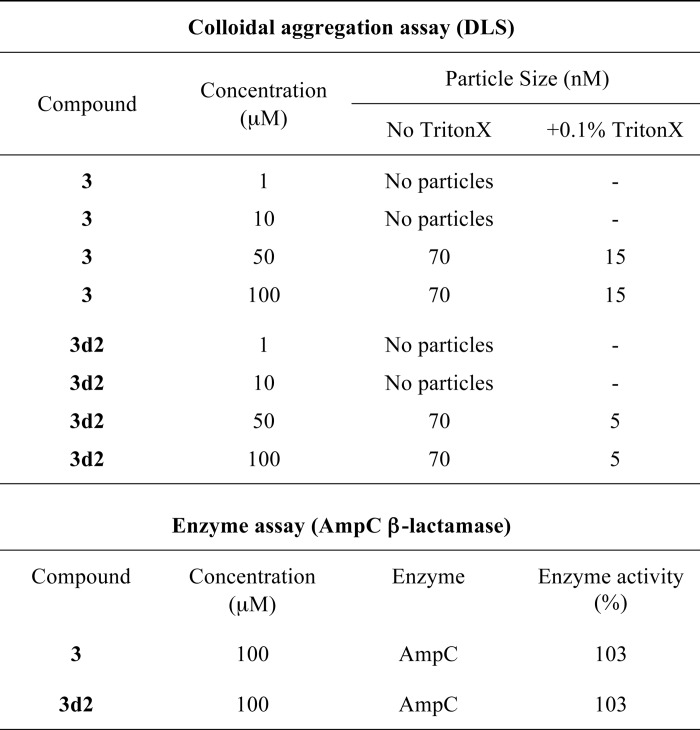
To investigate a possibility that the inhibition by compounds 3 and 3d2 might be caused by their colloidal particles and thus be an artifact (51), the two ligands were evaluated by DLS and counter-screened against two model enzymes, AmpC β-lactamase and cruzain, used for the assessment of this effect (52). No colloidal particles were detected for either compound at the range of concentrations (1–10 μm) used in the transcriptional and quantitative direct binding assays (Table 3). Although particles were observed for compounds 3 and 3d2 at 50-100 μm concentrations by DLS (Table 3), these resembled precipitate, which is thought not to confer inhibition (51). Furthermore, no inhibition by either molecule at concentrations up to 100 μm was detected in the counter-screen against AmpC β-lactamase (Table 3). Although modest, up to 30%, inhibition of cruzain was observed at a higher concentration of compounds (500 μm, data not shown), this was not reversed by the addition of the nonionic detergent Triton X-100. Taken together, these observations suggest that these two ligands are not promiscuous inhibitors.
TABLE 3.
Evaluation of compounds 3 and 3d2 for artifactual inhibition due to colloidal aggregation
Results of DLS experiments are shown for different concentrations of compounds 3 and 3d2 in the absence or presence of 0.1% Triton X-100 (indicated). Results of analysis of enzymatic activity of AmpC β-lactamase (BlaAmpC) in the presence of either compound are shown relative to solvent control (DMSO).
Following the promiscuous inhibition tests, we examined whether compounds 3 and 3d2 exert any effects on transcriptional activities of other nuclear receptors. Using published methods described under “Experimental Procedures,” transactivation by four different nuclear receptors, steroidogenic factor 1 (SF-1, a close structural and functional homolog of LRH-1) as well as more distant receptors ERα, AR, and TRβ, was assessed in the absence and the presence of these compounds (Figs. 7 and 8). For an accurate comparison between LRH-1 and SF-1, the effects of compounds on transcriptional activity of SF-1 were evaluated using a cellular model, Tet-inducible HEK293 cells expressing SF-1 receptor, similar to that employed for the analogous experiments with LRH-1. Following the induction of SF-1 expression with tetracycline and treatments of HEK293 cells with either individual compounds or DMSO (0.1%, solvent control), the mRNA levels for the endogenous G0S2 gene (a shared transcriptional target of LRH-1 and SF-1 (32)) were evaluated in the compound-treated cells relative to the control (Fig. 7, A and B); no compound-mediated changes in the SF-1 activity were detected in these tests under similar conditions (compare Figs. 4, A and B, 6, C and D, and 7, A and B). Complementing these results, no significant shifts in melting temperature of hSF-1 LBD were recorded following treatments of the purified protein with either compound in the DSF assay (Fig. 7C). Combined structural differences between hLRH-1 and hSF-1 ligand-binding pockets in the vicinity of the docked ligands (shown for Cpd 3d2 model in Fig. 7D), might explain the discriminating binding of compounds to these receptors. In particular, more rigid Leu-265 and Val-348 residues of SF-1 (substituting for more accommodating Met-345 and Met-428 in LRH-1) could disfavor binding of the tested antagonists to SF-1.
FIGURE 7.

Assessing specificity of compounds 3 and 3d2. Neither compound 3 (A) nor its analog 3d2 (B) affect transcriptional activity of hSF-1. HEK293 cells harboring Tet-inducible expression vector encoding full-length SF-1 were treated with either DMSO (0.1%, solvent control) or individual compounds at indicated concentrations (shown in A for Cpd 3 and in B for Cpd 3d2). The experiments were performed both in the presence and the absence of Tet (indicated as Tet-On SF-1(+) and Tet-Off SF-1(−) in A and B). Following 24-h treatments, levels of mRNA for G0S2 gene in all cell samples were evaluated by qPCR. For each concentration point, data are shown relative to control (0.1% DMSO), as average of three independent measurements, with experimental errors shown as black lines. C, melting temperature shifts for hSF-1 LBD treated with compounds 3 and 3d2. Neither compound demonstrates any significant effect on the melting temperature of the receptor. For each tested compound, averaged data are shown as horizontal bars; experimental errors and 3σ thresholds are indicated with solid and dashed lines. D, differences between hLRH-1 and hSF-1 LBPs. Selected structural elements forming the two receptors LBPs are superposed and shown in blue for SF-1 and in gray for LRH-1. Different amino acid residues in the vicinity of a docked ligand (shown for Cpd 3d2 as predicted by modeling) are indicated.
FIGURE 8.
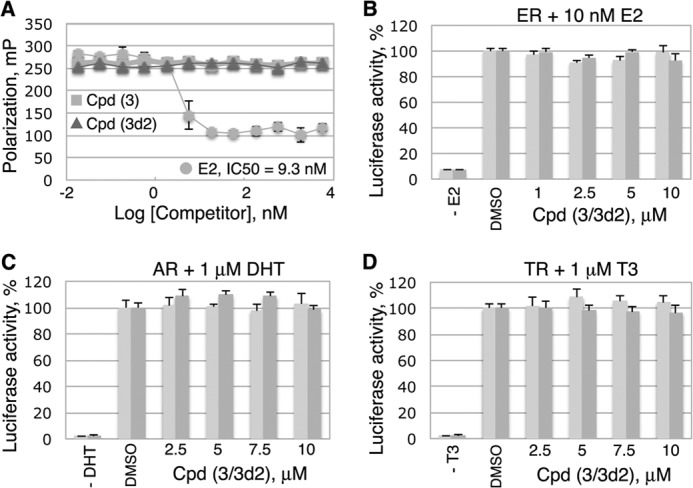
Assessing specificity of compounds 3 and 3d2. Neither compound 3 nor its analog 3d2 inhibits transcriptional activities of ERα, AR, or TRβ receptors. A, compounds 3 and 3d2 do not bind to ERα LBD in a competitive fluorescence polarization ligand binding assay. The assay was performed in a black multiwell plate using the PolarscreenTM ERα competitor assay (Invitrogen) with FluormoneTM E2-ERα complex, in the presence of either E2 (positive control) or individual compounds at different concentrations (indicated). Error bars indicate standard deviations from the mean values of triplicate measurements. B, transactivation assays with ERα in the absence or the presence of 10 nm E2 demonstrated no detectable changes in the transcriptional activity of the receptor following treatments with either Cpd 3 or Cpd 3d2 (see “Experimental Procedures” for details). C and D, analogous transactivation assays with AR and TRβ receptors (shown in C and D, see “Experimental Procedures” for details) were performed in the absence or the presence of either 1 μm dihydrotestosterone (indicated for AR in C) or 1 μm T3 (shown for TRβ in D). No detectable compound-mediated effects were observed in these experiments. B–D, light and dark gray bars represent data for Cpd 3 and Cpd 3d2, respectively. All measurements were done in triplicates; the corresponding data are shown as average, with experimental errors indicated.
Because selected features of the ligand-driven structural dynamics observed for ERα receptor were implemented in the model of antagonized LRH-1 LBD (Fig. 1), we tested whether the identified compounds bind to ERα. The fluorescence polarization data showed that although E2 (positive control, Fig. 8A) replaced a fluorescently labeled E2 bound to ERα with an estimated Kd of 9.3 nm, neither compound 3 nor 3d2 was capable of displacing the fluormone E2 ligand from ERα at concentrations up to 10 μm. Consistent with these direct binding results, no effects of compounds on transactivation by ERα were observed in a reporter-based transcription assay (Fig. 8B).
Effects of the probes on transcriptional activities of two additional nuclear receptors, AR and TRβ, were analyzed in a similar luciferase reporter-based assay (Fig. 8, C and D), as described previously (46). These transcriptional studies presented no evidence of any specific probe-mediated changes in the transcriptional activities of either of the tested receptors. Based on these combined data, we conclude that the identified inhibitors bind to the LRH-1 receptor and inhibit its transcriptional activity preferentially.
Treatments with LRH-1 Antagonists Inhibit Cancer Cell Proliferation in Vitro
Because multiple LRH-1 gene targets (including CCNE1 (encoding cyclin E1), CCND1 (encoding cyclin D1), and c-MYC genes) are known to control cell growth and proliferation (7, 9), we investigated whether treatments of cells with identified receptor antagonists affect cell proliferation in vitro. Our previous work demonstrated that selective inhibition of LRH-1 transcription by siRNA arrests growth and proliferation of human pancreatic cancer cells (24). This receptor-mediated anti-proliferative effect was observed in four different pancreatic ductal adenocarcinoma cell lines, including AsPC-1, which express high levels of LRH-1 (24). This study shows that treatments of AsPC-1 cells with compounds 3 and 3d2 result in a similar dose-dependent inhibition of cell proliferation (Fig. 9, A and B; concentrations of compounds associated with ∼50% inhibition of cell proliferation are indicated). Notably, no significant anti-proliferative effects were observed in pancreatic cancer cells L3.3 (Fig. 8, C and D) that do not express the LRH-1 receptor at a detectable level (24). In concert with these data, inhibition of transcription of LRH-1 target genes NR0B2 and CCNE1 (encoding SHP and cyclin E1, in light and dark gray, Fig. 9, E and F) was detected in AspC-1 but not in L3.3 cells following these treatments. No general cytotoxicity was encountered for either compound at the concentrations used for these experiments (Fig. 10, A–D).
FIGURE 9.
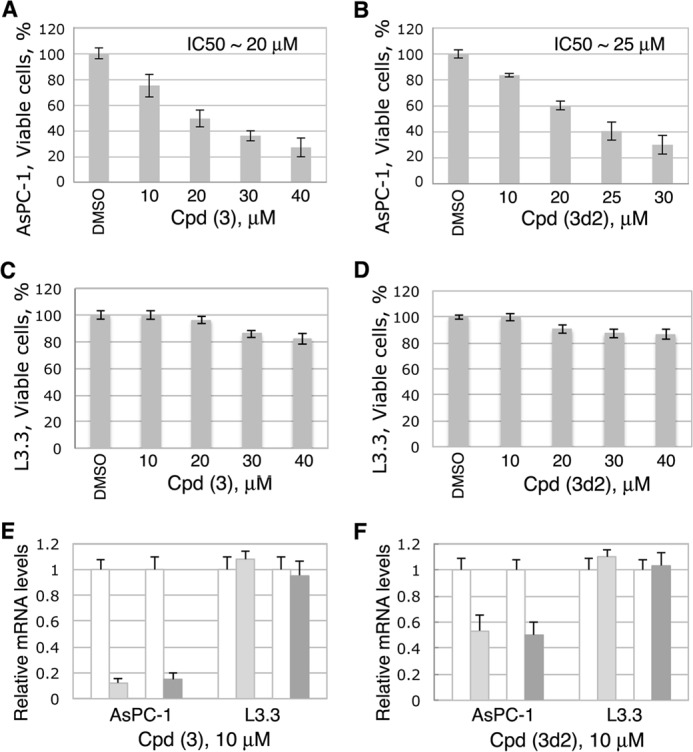
LRH-1 antagonists inhibit proliferation of pancreatic cancer cells AsPC-1 (LRH-1-positive) but not L3.3 cells (LRH-1-negative). A–D, cell proliferation rates for both pancreatic cancer cells were measured and compared in the absence and the presence of different concentrations of compounds 3 (A and C) and 3d2 (B and D) relative to control (0.1% DMSO). The corresponding IC50 values are indicated. Evaluations of general cytotoxic effects for both compounds in these cells were performed in parallel and are shown in Fig. 10. E and F, effects of compounds 3 (E) and 3d2 (F) on transcription of the receptor target genes NR0B2 (encoding SHP) and CCNE1 (encoding cyclin E1, CycE1) in AsPC-1 and L3.3 cells. Cell samples were analyzed by qPCR for the relative levels of mRNA corresponding to SHP and Cyc E1 following treatments with individual compounds at 10 μm concentration. Controls in white correspond to cells treated with solvent (0.1% DMSO); light and dark gray bars show the levels of mRNA for SHP and Cyc E1 in cells treated with indicated compounds. Data are shown as average of three independent measurements, with experimental errors indicated.
FIGURE 10.
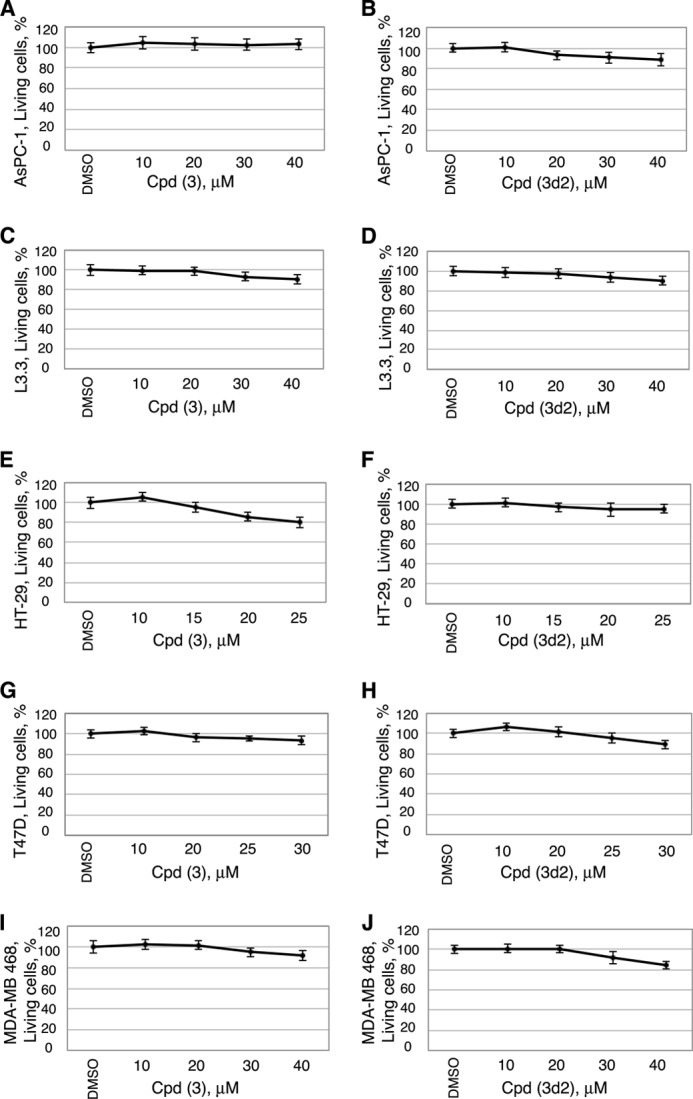
Cytotoxicity data for human cancer cells treated with compounds 3 and 3d2. Shown are data for pancreatic cancer cells AsPC-1 (A and B) and L3.3 (C and D), colon cancer cells HT-29 (E and F), and breast cancer cells T47D (G and H) and MDA-MB 468 (I and J). Viability measurements for cells treated with different concentrations of compounds are shown relative to control (0.1% DMSO). For each cell line, cytotoxicity was assessed 24 h following the addition of compounds, using the CytoTox-Glo cytotoxicity assay reagent (Promega). All data are shown as average of three independent measurements, and experimental errors are indicated as black lines (mean ± S.D.).
We confirmed the observed anti-proliferative effects of both inhibitors in three additional human cancer cell lines known to express LRH-1: HT-29 (colon adenocarcinoma, Fig. 11, A and B) and ER-positive and -negative breast adenocarcinoma cells T47D (Fig. 11, C and D) and MDA-MB-468 (Fig. 11, E and F). Previous independent studies demonstrated that disabling of LRH-1 by either receptor-specific siRNA or genetic manipulations results in inhibition of growth and proliferation of breast (20, 21) and colon (17) cancer cells. Complementing these findings, our work reveals that proliferation rates of both breast and colon carcinoma cells are significantly compromised following treatments with LRH-1 antagonists (corresponding concentrations of compounds associated with ∼50% inhibition of cell proliferation are indicated in Fig. 11, A–F). No significant general cytotoxicity has been detected for either compound at the tested concentration range in either cell line (Fig. 10). These results support the idea that the observed anti-proliferative effects of the probes are receptor-mediated and specific. Our data demonstrate that different types of malignant cells expressing LRH-1 are sensitive to treatments with the receptor-specific inhibitors and that growth and proliferation of LRH-1-positive cancer cells could be markedly decreased following such treatments.
FIGURE 11.

LRH-1 antagonists inhibit proliferation of LRH-1-positive colon and breast cancer cells in a dose-dependent manner. Cell proliferation rates for colon (A and B) as well as ER-positive (C and D) and ER-negative (E and F) breast cancer cells were measured and compared in the absence and the presence of different concentrations of compounds 3 and 3d2 relative to control (0.1% DMSO). For each cell line, concentrations of compounds associated with ∼50% inhibition of cell proliferation are indicated. Evaluations of general cytotoxic effects for both compounds in these cells were performed in parallel and are shown in Fig. 10.
DISCUSSION
This work describes the identification and characterization of the first selective synthetic antagonists of human nuclear receptor LRH-1. To date, physiological hormones for this receptor have not been identified, and its regulatory mechanisms triggered by ligand binding remain largely unknown. Both synthetic and naturally occurring agonists (27–32), including phosphatidylinositol di- and triphosphates, the proposed hormone candidates (29), have been reported for this receptor. However, identification of small molecules, antagonists of LRH-1, evaded previous search efforts. We attribute the success in the discovery of LRH-1-specific inhibitors to an efficient screening strategy, which combined high throughput computer-assisted search for compounds with preferred structural characteristics with the following in vitro direct binding and functional assays for compound selection and validation. Such combinations of the protein-centric computational approaches with experimental verifications of the top-ranked hits have proven to be successful for identification of specific ligands for different protein targets in the past (53–57).
A challenge that the LRH-1 target presented was the absence of a structure for the receptor in its “inactive state,” which would allow docking of potential antagonists into the hormone-binding pocket. We addressed this problem by creating a model for the ligand-binding pocket of LRH-1 that would be compatible with binding of receptor-specific antagonists (Figs. 1 and 2). This model implemented structural features that facilitated binding of ligands-antagonists by human estrogen hormone receptor, for which numerous independent atomic resolution structures have been determined with different antagonists bound in the hormone-binding pocket (47–50). As a result, three out of eight candidate molecules selected for experimental verifications (compounds 3, 5, and 7, 40% of pre-selected hits, Table 1) were shown to bind to LRH-1 LBD directly, changing the melting temperature (Tm) of the protein in the DSF-based binding assay (Fig. 3). Notably, all three compounds destabilized the receptor upon binding, shifting its Tm downwards (Fig. 3, as indicated). The observed destabilizing effect was expected based on the predicted mode of binding for the presumed receptor antagonists that required undocking of helix H12 from the core LBD to facilitate their binding. Necessary exchange of phospholipids, fortuitous ligands-agonists commonly co-purified with recombinant LRH-1 LBD (29–31), for ligands-antagonists was expected to result in the receptor destabilization as well. Importantly, one of the three ligands shown to interact with the receptor directly, compound 3 (Fig. 3), compromised interactions of LRH-1 LBD with co-regulator peptide in direct binding studies (Fig. 4, C and D), most likely via distortion of the co-activator binding region of the receptor (AF-2), as was predicted by the molecular docking. Consistent with the predicted mode of binding of this ligand into the hormone-binding pocket of LRH-1, bulky amino acid substitutions in the pocket (A349F and A349W, Fig. 2C) abolished its binding to the receptor (shown for A349F in Fig. 3B). As expected, compound 3 also inhibited transactivation by LRH-1 in cell-based transcription assays (Fig. 4, A and B). No inhibitory effects of compound 3 on transcriptional activities of closely related and more distant receptor homologs SF-1, ERα, AR, and TRβ have been observed in these studies (Figs. 7 and 8).
Computer-assisted search for chemical analogs of compound 3 identified four additional candidate molecules (Table 2) with similar antagonistic mode of binding to the receptor (illustrated for compounds 3 and 3d2 in Fig. 2). One on these analogs, compound 3d2, was proven to bind and inhibit the transcriptional activity of LRH-1 selectively (Figs. 5–8). Similar to the original compound 3, analog 3d2 was shown to bind to the receptor's hormone pocket, destabilizing the LBD upon binding and shifting its transition temperature (Tm) downwards (Fig. 5). Furthermore, as was predicted by the molecular docking, binding of this ligand antagonized the receptor, diminishing its interactions with co-regulator peptide (shown for DAX1–3 in Fig. 6, A and B). Consequently, compound 3d2 exerted receptor-specific antagonistic effect on transcriptional activity of LRH-1 in cell-based experiments (Fig. 6, C and D). No effects on transcription by other nuclear receptors, including the closest structural and functional homolog of LRH-1, nuclear receptor SF-1 (Fig. 7), as well as ERα, AR, and TRβ (Fig. 8) were detected in these studies. These data, combined with the analogous results for the original compound 3 (Figs. 3, 4, 7, and 8, demonstrate that the identified receptor antagonists target LRH-1 preferentially. Raloxifene-based analogs were recently shown to antagonize LRH-1 (58); however, these also inhibit the ERα receptor; selectivity of these compounds for LRH-1 over SF-1 was not tested.
To confirm that the observed binding and related inhibitory effects of compounds 3 and 3d2 are not artificial (caused by their unspecific colloidal aggregation (51)), formation of colloidal particles for the identified receptor antagonists was assessed by DLS. No colloidal particles were detected for either compound at the range of concentrations (1–10 μm) used in the transcriptional and quantitative direct binding assays (Table 3). Although particles were observed for both molecules at 50-100 μm concentrations (most likely due to limited solubility and precipitation of these compounds in aqueous solutions), no unspecific inhibition by the probes at this concentration range was detected in a standard test for promiscuous inhibition (based on analysis of enzymatic activity of AmpC β-lactamase (52), Table 3). These data, combined with specificity controls used for transcription and direct binding assays (Figs. 7 and 8), show that the two LRH-1 antagonists are not promiscuous inhibitors and that their biological effects are mediated by specific target-ligand interactions.
The identification of compound 3d2 in the second round of the search elevated the success rate of our computer-assisted discovery of specific inhibitors of LRH-1 to ∼17% (two novel inhibitors as a result of testing of 12 top-ranked candidates). For comparison, independent search for LRH-1 antagonists using the Prestwick Chemical Library (Illkirch, France) of 1120 drug-like chemicals, for which no computer-assisted selection of preferred candidates have been made, resulted in identification of only one compound (<0.1% of tested chemicals) capable of binding to the receptor (data not shown; all compounds were tested using the same DSF-based method for direct binding followed by the analogous transcription assay). The identified ligand was not pursued, however, as it cross-reacted with other nuclear receptors and was shown to bind to unrelated protein targets. This comparative analysis demonstrates that the success rate for an unbiased search for receptor-specific ligands approaches zero, unless a massive number of chemicals is evaluated for desired effects in high throughput experimental screenings, which require special machinery, considerable material resources, and substantial human effort (53).
In this work, the efficiency of the search for receptor antagonists was increased by incorporating a virtual computer-assisted high throughput filtering step, which 1) substantially reduced the number of compounds needed for functional analyses and 2) enriched the pool of pre-selected molecules with potential LRH-1-specific ligands. An unusual aspect of this filtering step was our use of a non-native model for the LRH-1 receptor that was based on experimental observations for the estrogen receptor. As a result, two novel LRH-1-specific antagonists have been identified in the following in vitro direct binding and functional assays with the input of only 12 pre-selected, top-ranked small molecules. We note that only eight candidate compounds were selected for initial experimental verifications out of 1000 top-ranked candidate molecules (see “Results”). Thus, it is plausible that more specific receptor antagonists, including those with entirely different chemotypes, could be identified if a substantially larger fraction of the candidate molecules had been tested.
The LRH-1 inhibitors identified in this work bind and antagonize receptor-mediated transcriptional activity selectively, with IC50 values of 5 ± 1 and 6 ± 1 μm (Figs. 4 and 6). Because no significant general cytotoxicity for these compounds is observed at these and higher concentrations (Fig. 10), these probes could be used as molecular tools for deciphering the roles of LRH-1 in different cellular contexts. For example, previous research demonstrated that selective blocking of LRH-1 function by either receptor-specific siRNA or genetic manipulations results in inhibition of growth and proliferation of cancerous cells expressing the receptor; the latter include breast (20, 21) and colon (17) cancer as well as pancreatic adenocarcinoma cells (24). The analogous, anti-proliferative effects have been observed for the epithelial cells of intestinal crypts in mice with loss-of-function mutation in the LRH-1 gene (9). The inhibitory effects are tracked to the attenuation of receptor target genes controlling cell growth, proliferation, and differentiation (9, 17, 24). Our data demonstrate that similar specific, receptor-mediated anti-proliferative effects are observed after treatments of LRH-1-positive cancer cells with the identified receptor antagonists (shown for pancreatic cancer cells in Fig. 9 and for colon and breast cancer cells in Fig. 11). We emphasize that ER-positive as well as ER-negative breast cancer cells, which both express LRH-1, are shown to be sensitive to treatments with the receptor antagonists (Fig. 11, C–F). These observations suggest that inhibition of LRH-1 might present a plausible route for controlling growth and proliferation of breast cancer cells that do not respond to selective estrogen receptor modulators.
Based on the results of this work, we propose that the identified LRH-1 inhibitors could be used as molecular probes for elucidating the roles of the receptor in different physiological and pathophysiological processes; in particular, they may be useful for studying developmental mechanisms as well as formation and progression of cancers of breast, colon, and pancreas. We expect that once fully characterized and optimized, LRH-1-specific antagonists could be developed into future drugs for molecular targeted therapies of these and possibly other diseases driven by this receptor.
Acknowledgments
We thank Drs. M. McMahon, S. Gysin, and H. Ingraham for providing necessary experimental reagents; Dr. C. Filgueira for assistance with transactivation and fluorescence polarization assays for ERα receptor, and R. Villagomez for assistance with mutagenesis of LRH-1 receptor.
This work was supported, in whole or in part, by National Institutes of Health Grants GM59957 (to B. K. S.) and R01 DK078075 and R21 CA140751 (to R. J. F.). This work was also supported by Department of Defense Grant W81XWH-12-1-0396 (to R. J. F.), by a fellowship from the California Breast Cancer Research Program (to C. B.), and a fellowship from the Knut and Alice Wallenberg Foundation (to J. C.).
- LRH-1
- liver receptor homolog 1
- LBD
- ligand-binding domain
- ERα
- estrogen receptor α
- PDB
- Protein Data Bank
- Tet
- tetracycline
- TRβ
- thyroid hormone receptor β
- DSF
- differential scanning fluorimetry
- E2
- estradiol
- SPR
- surface plasmon resonance
- AR
- androgen receptor
- Cpd
- compound
- CCF
- Cell Culture Facility
- Pen/Strep
- penicillin/streptomycin
- h
- human
- DLS
- dynamic light scattering
- LBP
- ligand-binding pocket
- qPCR
- quantitative PCR
- AmpC
- AmpC β-lactamase
- SHP
- small heterodimer partner.
REFERENCES
- 1. Fayard E., Auwerx J., Schoonjans K. (2004) LRH-1: an orphan nuclear receptor involved in development, metabolism, and steroidogenesis. Trends Cell Biol. 14, 250–260 [DOI] [PubMed] [Google Scholar]
- 2. Fayard E., Schoonjans K., Annicotte J.-S., Auwerx J. (2003) Liver receptor homolog 1 controls the expression of carboxyl ester lipase. J. Biol. Chem. 278, 35725–35731 [DOI] [PubMed] [Google Scholar]
- 3. Kim J. W., Peng N., Rainey W. E., Carr B. R., Attia G. R. (2004) Liver receptor homolog-1 regulates the expression of steroidogenic acute regulatory protein in human granulosa cells. J. Clin. Endocrinol. Metab. 89, 3042–3047 [DOI] [PubMed] [Google Scholar]
- 4. Clyne C. D., Kovacic A., Speed C. J., Zhou J., Pezzi V., Simpson E. R. (2004) Regulation of aromatase expression by the nuclear receptor LRH-1 in adipose tissue. Mol. Cell. Endocrinol. 215, 39–44 [DOI] [PubMed] [Google Scholar]
- 5. Gu P., Goodwin B., Chung A. C., Xu X., Wheeler D. A., Price R. R., Galardi C., Peng L., Latour A. M., Koller B. H., Gossen J., Kliewer S. A., Cooney A. J. (2005) Orphan nuclear receptor LRH-1 is required to maintain Oct4 expression at the epiblast stage of embryonic development. Mol. Cell. Biol. 25, 3492–3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wagner R. T., Xu X., Yi F., Merrill B. J., Cooney A. J. (2010) Canonical Wnt/β-catenin regulation of liver receptor homolog-1 mediates pluripotency gene expression. Stem Cells 28, 1794–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heng J. C., Feng B., Han J., Jiang J., Kraus P., Ng J.-H., Orlov Y. L., Huss M., Yang L., Lufkin T., Lim B., Ng H.-H. (2010) The nuclear receptor Nr5a2 can replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell Stem Cell 6, 167–174 [DOI] [PubMed] [Google Scholar]
- 8. Sheela S. G., Lee W. C., Lin W. W., Chung B. C. (2005) Zebrafish ftz-f1a (nuclear receptor 5a2) functions in skeletal muscle organization. Dev. Biol. 286, 377–390 [DOI] [PubMed] [Google Scholar]
- 9. Botrugno O. A., Fayard E., Annicotte J.-S., Haby C., Brennan T., Wendling O., Tanaka T., Kodama T., Thomas W., Auwerx J., Schoonjans K. (2004) Synergy between LRH-1 and β-catenin induces G1 cyclin-mediated cell proliferation. Mol. Cell 15, 499–509 [DOI] [PubMed] [Google Scholar]
- 10. Yumoto F., Nguyen P., Sablin E. P., Baxter J. D., Webb P., Fletterick R. J. (2012) Structural basis of coactivation of liver receptor homolog-1 by β-catenin. Proc. Natl. Acad. Sci. U.S.A. 109, 143–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clevers H. (2006) Wnt/β-Catenin signaling in development and disease. Cell 127, 469–480 [DOI] [PubMed] [Google Scholar]
- 12. Villanueva A., Newell P., Chiang D. Y., Friedman S. L., Llovet J. M. (2007) Genomics and signaling pathways in hepatocellular carcinoma. Semin. Liver Dis. 27, 55–76 [DOI] [PubMed] [Google Scholar]
- 13. Saif M. W., Chu E. (2010) Biology of colorectal cancer. Cancer J. 16, 196–201 [DOI] [PubMed] [Google Scholar]
- 14. Thayer S. P., di Magliano M. P., Heiser P. W., Nielsen C. M., Roberts D. J., Lauwers G. Y., Qi Y. P., Gysin S., Fernández-del Castillo C., Yajnik V., Antoniu B., McMahon M., Warshaw A. L., Hebrok M. (2003) Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 425, 851–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pasca di Magliano M., Biankin A. V., Heiser P. W., Cano D. A., Gutierrez P. J., Deramaudt T., Segara D., Dawson A. C., Kench J. G., Henshall S. M., Sutherland R. L., Dlugosz A., Rustgi A. K., Hebrok M. (2007) Common activation of canonical Wnt signaling in pancreatic adenocarcinoma. PLoS One 2, e1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takebe N., Warren R. Q., Ivy S. P. (2011) Breast cancer growth and metastasis: interplay between cancer stem cells, embryonic signaling pathways, and epithelial-to-mesenchymal transition. Breast Cancer Res. 13, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schoonjans K., Dubuquoy L., Mebis J., Fayard E., Wendling O., Haby C., Geboes K., Auwerx J. (2005) Liver receptor homolog 1 contributes to intestinal tumor formation through effects on cell cycle and inflammation. Proc. Natl. Acad. Sci. U.S.A. 102, 2058–2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sidler D., Renzulli P., Schnoz C., Berger B., Schneider-Jakob S., Flück C., Inderbitzin D., Corazza N., Candinas D., Brunner T. (2011) Colon cancer cells produce immunoregulatory glucocorticoids. Oncogene 30, 2411–2419 [DOI] [PubMed] [Google Scholar]
- 19. Wang S. L., Zheng D. Z., Lan F. H., Deng X. J., Zeng J., Li C. J., Wang R., Zhu Z. Y. (2008) Increased expression of hLRH-1 in human gastric cancer and its implication in tumorigenesis. Mol. Cell. Biochem. 308, 93–100 [DOI] [PubMed] [Google Scholar]
- 20. Annicotte J. S., Chavey C., Servant N., Teyssier J., Bardin A., Licznar A., Badia E., Pujol P., Vignon F., Maudelonde T., Lazennec G., Cavailles V., Fajas L. (2005) The nuclear receptor liver receptor homolog-1 is an estrogen receptor target gene. Oncogene 24, 8167–8175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thiruchelvam P. T., Lai C. F., Hua H., Thomas R. S., Hurtado A., Hudson W., Bayly A. R., Kyle F. J., Periyasamy M., Photiou A., Spivey A. C., Ortlund E. A., Whitby R. J., Carroll J. S., Coombes R. C., Buluwela L., Ali S. (2011) The liver receptor homolog-1 regulates estrogen receptor expression in breast cancer cells. Breast Cancer Res. Treat. 127, 385–396 [DOI] [PubMed] [Google Scholar]
- 22. Chand A. L., Herridge K. A., Thompson E. W., Clyne C. D. (2010) The orphan nuclear receptor LRH-1 promotes breast cancer motility and invasion. Endocr. Relat. Cancer 17, 965–975 [DOI] [PubMed] [Google Scholar]
- 23. Dubé C., Bergeron F., Vaillant M. J., Robert N. M., Brousseau C., Tremblay J. J. (2009) The nuclear receptors SF1 and LRH1 are expressed in endometrial cancer cells and regulate steroidogenic gene transcription by cooperating with AP-1 factors. Cancer Lett. 275, 127–138 [DOI] [PubMed] [Google Scholar]
- 24. Benod C., Vinogradova M. V., Jouravel N., Kim G. E., Fletterick R. J., Sablin E. P. (2011) Nuclear receptor liver receptor homolog 1 (LRH-1) regulates pancreatic cancer cell growth and proliferation. Proc. Natl. Acad. Sci. U.S.A. 108, 16927–16931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sonoda J., Pei L., Evans R. M. (2008) Nuclear receptors: Decoding metabolic disease. FEBS Lett. 582, 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mataki C., Magnier B. C., Houten S. M., Annicotte J. S., Argmann C., Thomas C., Overmars H., Kulik W., Metzger D., Auwerx J., Schoonjans K. (2007) Compromised intestinal lipid absorption in mice with a liver-specific deficiency of liver receptor homolog 1. Mol. Cell. Biol. 27, 8330–8339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee J. M., Lee Y. K., Mamrosh J. L., Busby S. A., Griffin P. R., Pathak M. C., Ortlund E. A., Moore D. D. (2011) A nuclear receptor dependent phosphatidylcholine pathway with antidiabetic effects. Nature 474, 506–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Musille P. M., Pathak M. C., Lauer J. L., Hudson W. H., Griffin P. R., Ortlund E. A. (2012) Antidiabetic phospholipid–nuclear receptor complex reveals the mechanism for phospholipid-driven gene regulation. Nat. Struct. Mol. Biol. 19, 532–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Krylova I. N., Sablin E. P., Moore J., Xu R. X., Waitt G. M., MacKay J. A., Juzumiene D., Bynum J. M., Madauss K., Montana V., Lebedeva L., Suzawa M., Williams J. D., Williams S. P., Guy R. K., Thornton J. W., Fletterick R. J., Willson T. M., Ingraham H. A. (2005) Structural analyses reveal phosphatidylinositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell 120, 343–355 [DOI] [PubMed] [Google Scholar]
- 30. Ortlund E. A., Lee Y., Solomon I. H., Hager J. M., Safi R., Choi Y., Guan Z., Tripathy A., Raetz C. R., McDonnell D. P., Moore D. D., Redinbo M. R. (2005) Modulation of human nuclear receptor LRH-1 activity by phospholipids and SHP. Nat. Struct. Mol. Biol. 12, 357–363 [DOI] [PubMed] [Google Scholar]
- 31. Wang W., Zhang C., Marimuthu A., Krupka H. I., Tabrizizad M., Shelloe R., Mehra U., Eng K., Nguyen H., Settachatgul C., Powell B., Milburn M. V., West B. L. (2005) The crystal structures of human steroidogenic factor-1 and liver receptor homologue-1. Proc. Natl. Acad. Sci. U.S.A. 102, 7505–7510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Whitby R. J., Stec J., Blind R. D., Dixon S., Leesnitzer L. M., Orband-Miller L. A., Williams S. P., Willson T. M., Xu R., Zuercher W. J., Cai F., Ingraham H. A. (2011) Small molecule agonists of the orphan nuclear receptors steroidogenic factor-1 (SF-1, NR5A1) and liver receptor homolog-1 (LRH-1, NR5A2). J. Med. Chem. 54, 2266–2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee Y. K., Choi Y. H., Chua S., Park Y. J., Moore D. D. (2006) Phosphorylation of the hinge domain of the nuclear hormone receptor LRH-1 stimulates transactivation. J. Biol. Chem. 281, 7850–7855 [DOI] [PubMed] [Google Scholar]
- 34. Lee M. B., Lebedeva L. A., Suzawa M., Wadekar S. A., Desclozeaux M., Ingraham H. A. (2005) The DEAD-Box protein DP103 (Ddx20 or Gemin-3) represses orphan nuclear receptor activity via SUMO modification. Mol. Cell. Biol. 25, 1879–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qin J., Gao D. M., Jiang Q. F., Zhou Q., Kong Y. Y., Wang Y., Xie Y. H. (2004) Prospero-related homeobox (Prox1) is a co-repressor of human liver receptor homolog-1 and suppresses the transcription of the cholesterol 7-α-hydroxylase gene. Mol. Endocrinol. 18, 2424–2439 [DOI] [PubMed] [Google Scholar]
- 36. Sablin E. P., Woods A., Krylova I. N., Hwang P., Ingraham H. A., Fletterick R. J. (2008) The structure of co-repressor Dax-1 bound to its target nuclear receptor LRH-1. Proc. Natl. Acad. Sci. U.S.A. 105, 18390–18395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Irwin J. J., Shoichet B. K., Mysinger M. M., Huang N., Colizzi F., Wassam P., Cao Y. (2009) Automated docking screens: a feasibility study. J. Med. Chem. 52, 5712–5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mysinger M. M., Shoichet B. K. (2010) Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Model. 50, 1561–1573 [DOI] [PubMed] [Google Scholar]
- 39. Lorber D. M., Shoichet B. K. (1998) Flexible ligand docking using conformational ensembles. Protein Sci. 7, 938–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lorber D. M., Shoichet B. K. (2005) Hierarchical docking of databases of multiple ligand conformations. Curr. Top. Med. Chem. 5, 739–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weiner S. J., Kollman P. A., Case D. A., Singh U. C., Ghio C., Alagona G., Profeta S., Weiner P. (1984) A new force-field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 106, 765–784 [Google Scholar]
- 42. Irwin J. J., Shoichet B. K. (2005) ZINC–A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 45, 177–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li J. B., Zhu T. H., Cramer C. J., Truhlar D. G. (1998) New class IV charge model for extracting accurate partial charges from wave functions. J. Phys. Chem. A 102, 1820–1831 [Google Scholar]
- 44. Weiner S. J., Kollman P. A., Nguyen D. T., Case D. A. (1986) An all atom force-field for simulations of proteins and nucleic acids. J. Comput. Chem. 7, 230–252 [DOI] [PubMed] [Google Scholar]
- 45. Wilkinson J. M., Hayes S., Thompson D., Whitney P., Bi K. (2008) Compound profiling using a panel of steroid hormone receptor cell-based assays. J. Biomol. Screen. 13, 755–765 [DOI] [PubMed] [Google Scholar]
- 46. Estébanez-Perpiñá E., Moore J. M., Mar E., Delgado-Rodrigues E., Nguyen P., Baxter J. D., Buehrer B. M., Webb P., Fletterick R. J., Guy R. K. (2005) The molecular mechanisms of co-activator utilization in ligand-dependent transactivation by the androgen receptor. J. Biol. Chem. 280, 8060–8068 [DOI] [PubMed] [Google Scholar]
- 47. Shiau A. K., Barstad D., Loria P. M., Cheng L., Kushner P. J., Agard D. A., Greene G. L. (1998) The structural basis of estrogen receptor/co-activator recognition and the antagonism of this interaction by tamoxifen. Cell 95, 927–937 [DOI] [PubMed] [Google Scholar]
- 48. Brzozowski A. M., Pike A. C., Dauter Z., Hubbard R. E., Bonn T., Engström O., Ohman L., Greene G. L., Gustafsson J. A., Carlquist M. (1997) Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 389, 753–758 [DOI] [PubMed] [Google Scholar]
- 49. Wu Y. L., Yang X., Ren Z., McDonnell D. P., Norris J. D., Willson T. M., Greene G. L. (2005) Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol. Cell 18, 413–424 [DOI] [PubMed] [Google Scholar]
- 50. Pike A. C., Brzozowski A. M., Walton J., Hubbard R. E., Thorsell A. G., Li Y. L., Gustafsson J. A., Carlquist M. (2001) Structural insights into the mode of action of a pure antiestrogen. Structure 9, 145–153 [DOI] [PubMed] [Google Scholar]
- 51. Coan K. E., Shoichet B. K. (2008) Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc. 130, 9606–9612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Doak A. K., Wille H., Prusiner S. B., Shoichet B. K. (2010) Colloid formation by drugs in simulated intestinal fluid. J. Med. Chem. 53, 4259–4265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Doman T. N., McGovern S. L., Witherbee B. J., Kasten T. P., Kurumbail R., Stallings W. C., Connolly D. T., Shoichet B. K. (2002) Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J. Med. Chem. 45, 2213–2221 [DOI] [PubMed] [Google Scholar]
- 54. Powers R. A., Morandi F., Shoichet B. K. (2002) Structure-based discovery of a novel, non-covalent inhibitor of AmpC β-lactamase. Structure 10, 1013–1023 [DOI] [PubMed] [Google Scholar]
- 55. Huang D., Lüthi U., Kolb P., Cecchini M., Barberis A., Caflisch A. (2006) In silico discovery of β-secretase inhibitors. J. Am. Chem. Soc. 128, 5436–5443 [DOI] [PubMed] [Google Scholar]
- 56. Carlsson J., Yoo L., Gao Z. G., Irwin J. J., Shoichet B. K., Jacobson K. A. (2010) Structure-based discovery of A2A adenosine receptor ligands. J. Med. Chem. 53, 3748–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schapira M., Raaka B. M., Das S., Fan L., Totrov M., Zhou Z., Wilson S. R., Abagyan R., Samuels H. H. (2003) Discovery of diverse thyroid hormone receptor antagonists by high-throughput docking. Proc. Natl. Acad. Sci. U.S.A. 100, 7354–7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rey J., Hu H., Kyle F., Lai C. F., Buluwela L., Coombes R. C., Ortlund E. A., Ali S., Snyder J. P., Barrett A. G. (2012) Discovery of a new class of liver receptor homolog-1 (LRH-1) antagonists: Virtual screening, synthesis, and biological evaluation. ChemMedChem 7, 1909–1914 [DOI] [PubMed] [Google Scholar]