

Background: Replication-independent (RI) histone H3.3 chromatin assembly is a highly conserved but incompletely understood genome regulatory mechanism.
Results: When RI H3.3 chromatin assembly is blocked, H3.3 accumulates at its incorporation site with sense and antisense RNA.
Conclusion: H3.3 recruitment is RNA-mediated and chaperone-independent.
Significance: Understanding the temporal and spatial organization of RI H3.3 chromatin assembly is crucial for understanding genome regulation.
Keywords: Antisense RNA, Chromatin Regulation, Chromatin Remodeling, Gene Expression, Gene Regulation, Gene Silencing, Microscopy, Nuclear Organization, RNA, Transcription
Abstract
Unlike the core histones, which are incorporated into nucleosomes concomitant with DNA replication, histone H3.3 is synthesized throughout the cell cycle and utilized for replication-independent (RI) chromatin assembly. The RI incorporation of H3.3 into nucleosomes is highly conserved and occurs at both euchromatin and heterochromatin. However, neither the mechanism of H3.3 recruitment nor its essential function is well understood. Several different chaperones regulate H3.3 assembly at distinct sites. The H3.3 chaperone, Daxx, and the chromatin-remodeling factor, ATRX, are required for H3.3 incorporation and heterochromatic silencing at telomeres, pericentromeres, and the cytomegalovirus (CMV) promoter. By evaluating H3.3 dynamics at a CMV promoter-regulated transcription site in a genetic background in which RI chromatin assembly is blocked, we have been able to decipher the regulatory events upstream of RI nucleosomal deposition. We find that at the activated transcription site, H3.3 accumulates with sense and antisense RNA, suggesting that it is recruited through an RNA-mediated mechanism. Sense and antisense transcription also increases after H3.3 knockdown, suggesting that the RNA signal is amplified when chromatin assembly is blocked and attenuated by nucleosomal deposition. Additionally, we find that H3.3 is still recruited after Daxx knockdown, supporting a chaperone-independent recruitment mechanism. Sequences in the H3.3 N-terminal tail and αN helix mediate both its recruitment to RNA at the activated transcription site and its interaction with double-stranded RNA in vitro. Interestingly, the H3.3 gain-of-function pediatric glioblastoma mutations, G34R and K27M, differentially affect H3.3 affinity in these assays, suggesting that disruption of an RNA-mediated regulatory event could drive malignant transformation.
Introduction
Nucleosomes, which are composed of ∼150 base pairs of DNA wrapped around an octamer of the four core histone proteins (H2A, H2B, H3, and H4), are both the basic packaging unit of eukaryotic genomes and the interface through which cells regulate DNA-based processes, including transcription, replication, and repair (1). The bulk of the histone proteins are synthesized during S phase, when they are utilized for replication-dependent chromatin assembly. However, in addition to these canonical histones, all eukaryotes have variants of H2A and H3, which are synthesized throughout the cell cycle. CENP-A is the H3 variant deposited into centromeres and is required for proper kinetochore assembly (2). Histone H3.3, which differs from the replication-dependent variants, H3.1 and H3.2, by 5 and 4 amino acids, respectively, is incorporated into chromatin through replication-independent (RI)3 mechanisms. Mammalian H3.3 is identical to the deduced ancestral form of H3 from the metazoan clade, and both Saccharomyces cerevisiae and Schizosaccharomyces pombe have only one form of H3, which resembles the RI variant (3). However, the essential function of RI H3 chromatin assembly is still not well understood.
Initially, H3.3 was thought to regulate transcriptionally active chromatin because the first reports were of its incorporation into ribosomal and induced heat shock genes (4, 5). However, it is now known that H3.3 is also incorporated into silent chromatin and that its assembly at these distinct locations is regulated by different chaperones (6–8). The metazoan-specific H3.3 chaperone, Daxx (death-associated protein), together with the SNF2-like ATP-dependent chromatin-remodeling factor, ATRX (α-thalassemia mental retardation X-linked), are required for H3.3 incorporation into pericentric heterochromatin, embryonic stem cell telomeres, and a transcriptionally repressed CMV promoter-regulated transgene array (9–12). The H3.3 chaperone, HIRA, directs its incorporation into specific single copy genes (11), and, together with UBN1, ASF1, and CABIN1, HIRA incorporates H3.3 into the genomes of senescing cells (13, 14). HIRA and the SNF2-like chromatin-remodeling factor, chromodomain helicase DNA-binding protein 1 (CHD1), are both required for H3.3 incorporation into the Drosophila male pronucleus (15–17). Additionally, CHD2 regulates H3.3 assembly at MyoD and myogenic regulatory sequences and is also required for differentiation (18).
The diversity of RI H3.3 chromatin assembly factors and the incorporation of H3.3 into both active and silent loci indicate that H3.3 regulates a wide array of genomic events, but they do not explain how H3.3 is recruited to these sites or what functionally unites them. A function for H3.3 in epigenetic transcriptional events could, however, provide the missing links because transcription regulates both euchromatin and heterochromatin. At single copy genes, H3.3 incorporation correlates with mRNA expression and RNA polymerase II density (19–22). Additionally, the enrichment of H3.3 at both transcription start and termination sites suggests that deposition could be regulated by events that occur at the boundaries of transcription units (11, 14, 18, 21–26).
Transcription is also linked to RI H3.3 chromatin assembly at heterochromatin. Loss of ATRX and Daxx is a hallmark of cells positive for the alternative lengthening of telomeres (ALT) pathway, a telomerase-independent telomere maintenance mechanism, and in both ALT-positive and ATRX-knock-out cells, expression of telomeric repeat-containing RNA (TERRA) is increased (11, 27–29). Disruption of Daxx, ATRX, and H3.3 also deleteriously affects transcription and heterochromatin regulation at pericentric heterochromatin (10, 30). The identification of H3.3 gain-of-function driver mutations in pediatric glioblastoma (31, 32) further suggests that the ATRX/DAXX/H3.3 axis has an essential function in chromatin organization.
We recently reported that the Daxx and ATRX pathway is required for both transcriptional repression and H3.3 incorporation into a chromatinized array of a CMV promoter-regulated reporter transgene (i.e. the site of transcription) (9). When this pathway is disrupted, transcription is rapidly induced from the array, and the incorporation of H3.3 into nucleosomes is blocked. Interestingly, H3.3 is still recruited to the activated transcription site, but it does not co-localize with DNA-binding factors. Instead, we report here that it co-localizes with sense and antisense RNA, which suggests that it is recruited to its incorporation sites through an RNA-mediated mechanism. Sequences in the N-terminal tail and the αN helix mediate both its recruitment to the activated site and an in vitro interaction with double-stranded RNA (dsRNA), indicating that this is the minimal region required for recruitment. In these assays, the H3.3 pediatric glioblastoma driver mutation, G34R, increases its affinity, and the K27M mutation reduces it, suggesting that disease etiology may be due, in part, to the disruption of an RNA regulatory event.
EXPERIMENTAL PROCEDURES
Plasmids
Cherry/CFP-tTA-ER, ER-tTA, YFP-MS2, H3.3-YFP, and pLU-H3.3–3XFLAG (9, 33, 34), H3.3 ΔN-tail-YFP(36–135) (35) (gift of S. Henikoff and K. Ahmad), and the GST-ADAR1 double-stranded RNA binding domain (ADAR1 dsRBD) construct (36) (gift of K. Nishikura) were described previously. H3.1-YFP and H3.2-YFP were made using overlapping PCR to produce the 7-amino acid linker in the H3.3-YFP construct (9). All of the other YFP-H3.3 histone deletion constructs were subjected to PCR and cloned into YFP-N1 (BglII/HindIII). H3.3 N-tail-αN was cloned into pGEX 4T-3 (SalI/NotI). Point mutations were introduced using the QuikChange site-directed mutagenesis kit (Agilent Technologies). YFP-Daxx-C3 was made by cloning the cDNA (gift of Roger Everett) into YFP-C3 (XhoI/SacII). H3.3-CFP was made by cloning H3.3 into CFP-N1 (BglII/HindIII). The plasmid template for in vitro RNA transcription, pBlueScriptII-Rbβglobin, was constructed by inserting the rabbit β-globin intron 2 slicing cassette into pBluescript II (BamHI/XhoI) (primers, 5′-CGCGGATCCGTGAGTTTGGGGGACCCTTGATTG-3′ and 5′-CCGCTCGAGTTTCCTTTATTAGCCAGAAGTCAGATGC-3′).
Cell Culture, Transfections, and Imaging
The U2OS cell lines, 2-6-3 and 2-6-3/YFP-MS2/rtTA were described previously (9, 33, 34). Activation was induced in 2-6-3/rtTA/YFP-MS2 cells by 1 μg/ml doxycycline. Activation using Cherry/CFP-tTA-ER and ER-tTA was induced with 1 μm 4-hydroxytamoxifen. Transfections were performed as described previously (37). Imaging and analysis were done as described previously (34, 37).
Computational Image Analysis of Time Course Imaging
Live cell imaging and computational analyses were described previously (34). In each H3.3-YFP time lapse series, the transcription site was manually selected as the region where Cherry-tTA-ER could first be clearly detected or where H3.3-YFP accumulated before activation. Error bars represent S.D. across 13 sets of normalized data. These data sets, collected over the course of ∼4.5 h, included longer asymptote regions than in previous analyses (34). Therefore, the data fit a logistical model at earlier time points but needed an additional linear term to account for continuous accumulation at later time points. The following equation and terms were used to calculate intensity,
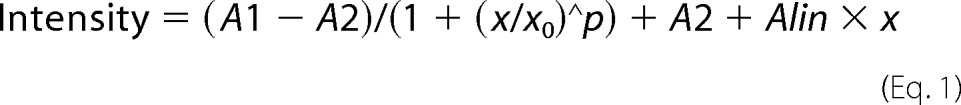 |
where A1 represents initial intensity, A2 is final intensity, x0 is the center (time at which the logistical term has reached halfway between A1 and A2), p is power, and Alin is a linear term to adjust for continued accumulation.
The timing of both the initial accumulation and the end of rapid accumulation for both H3.3-YFP and Cherry-tTA-ER were determined from the logistic fit. The accumulation start point was defined as the time at which the logistical term of the intensity passed 5% of the difference between the initial and final values, whereas the end of rapid accumulation was defined as the time at which the logistical term reached 95% of the final value. 45 cells were imaged in a total of 36 movies from six coverslips over 6 days of imaging. Cells that went out of focus during imaging, developed a saturated signal at the transcription site, bleached, or showed any kind of aberrant physiological features were not analyzed. The analysis was done on 13 cells (∼29% of total collected).
Strand-specific High Throughput Sequencing
Cells grown to ∼80% confluence were scraped from dishes in hypotonic TMS buffer (20 mm Hepes, pH 7.5, 2.5 mm MgCl2, 250 mm sucrose, 0.1 mm PMSF, and RNasin), incubated on ice for 25 min, and then spun at 3,600 rpm (10 min) to collect nuclei. RNA was extracted from purified nuclei using TRIzol (Invitrogen) according to the manufacturer's protocol. RNA samples were run on the Agilent Bioanalyzer RNA Pico-chip to assess ribosomal RNA contamination, which was estimated to be 4.7%. Sequencing libraries were prepared from 35 ng of RNA using Epicenter's ScriptSeqTM version 2 RNA-Seq library preparation kit. Libraries were reassessed by the Bioanalyzer high sensitivity DNA assay and sequenced using the Solexa GAIIx platform in a 36-base pair sequencing run. Unique sequencing reads were aligned against the transgene reference sequence using the Bowtie version 2.0 algorithm, allowing multiple matches without mismatches. The number of exact 36-bp subsequent copies throughout the transgene sequence was used for normalization to represent the expected transcription level of repeated regions.
RNA Fluorescence in Situ Hybridization (RNA FISH)
The RNA FISH protocol was adapted from Edith Heard (Curie Institute). Coverslips were mounted on slides as described previously (34). RNAFISH probes were synthesized and labeled with CY3 as described previously (38).
The following strand-specific probes were used: rabbit β-globin intron 2 sense RNA, GGCAGGATGATGACCAGGGTGTAGTTGTTTCTACCAATAAGAATATTTCC; rabbit β-globin intron 2 antisense RNA, GGAAATATTCTTATTGGTAGAAACAACTACACCCTGGTCATCATCCTGCC; rabbit β-globin exon 3 sense RNA, TTTGGCAGAGGGAAAAAGATCTCAGTGGTATTTGTGAGCCAGGGCATTGG; rabbit β-globin exon 3 antisense RNA, CCAATGCCCTGGCTCACAAATACCACTGAGATCTTTTTCCCTCTGCCAAA; rabbit β-globin 3′ genomic sequence sense RNA, CAGGGGGCTGTTTCATATACTGATGACCTCTTTATAGCCAACCTTTGTTC; rabbit β-globin 3′ genomic sequence antisense RNA, GAACAAAGGTTGGCTATAAAGAGGTCATCAGTATATGAAACAGCCCCCTG; bacterial plasmid sequence sense RNA, GCCTCAAAATGTTCTTTACGATGCCATTGGGATATATCAACGGTGGTATA; bacterial plasmid sequence antisense RNA, TATACCACCGTTGATATATCCCAATGGCATCGTAAAGAACATTTTGAGGC.
RT-PCR and Quantitative PCR Analysis
For each strand-specific reaction, 1 μg of TRIzol (Invitrogen)-isolated RNA was treated with DNase I (Promega, Madison, WI), heated to 65 °C (15 min), cooled on ice (1 h), heated again at 65 °C (15 min), cooled on ice (5 min), and purified with the RNeasy kit (Qiagen, Valencia, CA). Strand-specific RT reactions were done using OmniScript (Qiagen) followed by quantitative PCR with SYBR Green (Sigma) using the 7500 Fast Real-time PCR system (Applied Biosystems, Invitrogen). For analysis of H3.3 mRNA levels, RT was done with random hexamers. Quantitative PCR data were analyzed using the 2(-ΔCt) method according to AB guidance using GAPDH for normalization.
The following primer pairs were used: 5′-GGTGCAGGCTGCCTATCAG-3′ and 5′-TTTGTGAGCCAGGGCATTG-3′ (rabbit β-globin exon 3); 5′-ATGGAAATCCCATCACCATCTT-3′ and 5′-CGCCCCACTTGATTTTGG-3′ (hGAPDH); 5′-AATCGACCGGTGGTAAAGCA-3′ and 5′-CTTGCGAGCGGCTTTTGTA-3′ (hH3F3A); 5′-TGTGTGCCATCCACGCTAA-3′ and 5′-CGAGCCAACTGGATGTCTTTG-3′ (hH3F3B).
Knockdown Analyses
Daxx knockdown was performed using siDAXX SiGENOME Smart Pool and siLuc as the control (Dharmacon). HIRA was knocked down using siHIRA 2 and 4 (Dharmacon) as described previously (39). Transfections were done using Lipofectamine (Invitrogen) for 72 h. Daxx and HIRA protein levels were analyzed by immunofluorescence staining, as described previously (34), using the following antibodies: Daxx D7810 (Sigma) (1:4000) and HIRA (39) (gift of Peter Adams). Images were taken using the exact same software settings to compare Daxx and HIRA levels in cells. H3.3 knockdown was done using the following shRNA constructs (Sigma): shH3F3A (TRCN0000139066), shH3F3B (TRCN0000062389), and the control pLKO.1. Lentiviruses were prepared in 293-T cells as described previously (40). For knockdown, 1 × 106 2-6-3/rtTA/YFP-MS2 cells were plated in a 10-cm dish and infected the next day with the viruses. 24 h after infection, cells were split into 10-cm dishes (1:3). At 48 h postinfection, puromycin was added (0.5 μg/ml), and at 72 h, transcription was induced for 3 h by the addition of doxycycline (1 μg/ml). Cells were lysed in TRIzol (Invitrogen) for preparation of RNA or SDS lysis buffer for Western blotting.
Immunoblotting
Immunoblot analysis was done with the following antibodies: GFP (1:1000; Roche Applied Science), FLAG (1:10,000; M2; Sigma), histone H3.3 (1:1000; 09-838; EMD Millipore), and γ-tubulin (1:1000; Sigma).
In Vitro dsRNA Binding Assay
GST Fusion Protein Expression
pGEX constructs were transformed into Rosetta cells (Novagen) and grown at 37 °C to A600 0.5–0.6. Cultures were induced with 0.1 mm of isopropyl 1-thio-β-d-galactopyranoside for 4 h at 18 °C. Bacteria were pelleted and stored at −80 °C. For lysis, pellets were thawed on ice and resuspended in lysis buffer (25 mm Tris, 0.5 m KCl, 10% glycerol, 5 mm β-mercaptoethanol, 1 mm PMSF, pH 7.2) plus 1× CelLyticTM B cell lysis reagent (Sigma-Aldrich). After shearing bacterial DNA with needles, lysates were cleared by centrifugation (30,000 for 30 min), flash-frozen, and stored at −80 °C.
In Vitro Transcription
For transcription of sense (S) and antisense (AS) RNA, pBlueScriptII-Rbβ-globin digested with BamHI (S RNA) and XhoI (AS RNA) was gel-purified with a gel extraction kit (Invitrogen), treated with Proteinase K, and phenol/chloroform (Ambion)-extracted. RNA was transcribed using the MAXIscript in vitro transcription T7/T3 kit (Ambion), treated with TURBO DNase (Ambion), stopped with RQ1 DNase stop solution (Promega), and purified using TRIzol reagent (Ambion) according to the company's protocol with the following modification. The ethanol wash step was carried out at 14,000 rpm for 20 min at 4 °C, and the RNA pellets were resuspended in 30 μl of nuclease-free H2O.
Double-stranded RNA Preparation
dsRNA (41) was prepared fresh for each experiment. In vitro transcribed sense and antisense RNA were hybridized at a 1:1 ratio in 80% formamide, 40 mm PIPES (pH 6.7), 0.4 m NaCl, and 1 mm EDTA heated to 95 °C for 5 min and then incubated at 47.5 °C overnight. The reaction was RNase A (10 μg/ml)-treated at 37 °C for 30 min, and dsRNA was purified using the RNeasy minikit (Qiagen) and quantified by a spectrophotometer.
In Vitro RNA-binding Assay
Protein supernatants were incubated with glutathione Superflow beads (Clontech) for 90 min at 4 °C with rocking. Following two washes with 250 μl of resuspension buffer, half of the protein-bound beads were removed for SDS-PAGE analysis. The remainder was resuspended in 300 μl of binding buffer (10 mm Tris-HCl, pH 7.5, 150 mm KCl, 1 mm MgCl2, 25 μl/ml protease inhibitor mixture, 100 units/ml RNasin) and incubated with 100 ng of freshly prepared dsRNA at 4 °C with rocking for 2 h. Protein-RNA-bound beads were washed three times with washing buffer 1 (10 mm Tris-HCl, pH 7.5, 500 mm KCl, 1 mm MgCl2), one time with washing buffer 2 (10 mm Tris-HCl, pH 7.5, 350 mm KCl, 1 mm MgCl2), one time with washing buffer 3 (10 mm Tris-HCl, pH 7.5, 150 mm KCl, 1 mm MgCl2), and twice with 1× PBS (pH 7.4). The protein-RNA-bound beads were transferred to fresh tubes after the first wash. RNA was extracted with 1 ml of TRIzol (Ambion) according to the company's protocol with the following modification. The ethanol wash step was carried out twice (14,000 rpm, 20 min; 4 °C). RNA pellets were resuspended in 40 μl of nuclease-free H2O.
RNase III Treatment
Five microliters of RNA extracted from the RNA-binding experiment were digested by ShortCut® RNase III (New England Biolabs) according to the company's protocol but at half of the suggested reaction volume. Products were purified with the RNeasy minikit (Qiagen) and analyzed by qRT-PCR.
Quantitative RT-PCR Analysis
RNA extracted from the RNA-binding experiment was used as template for RT reaction using Superscript II (Invitrogen) according to the company's protocol but at half of the suggested reaction volume. Quantitative PCR was done with SYBR Green 10 (Sigma) using the AB 7500 system. Relative RNA levels were determined by calculating 2(-ΔCt) with respect to the Ct value of the GST-H3.3 N tail-αN sample. RNA binding levels were calculated by dividing RNA levels by protein levels and normalization to the GST-H3.3 N tail-αN sample. Proteins from GST-bound beads were run on an SDS-polyacrylamide gel, which was stained and imaged with ImageReader LAS3000 (FujiFilm) so that protein band intensity could be measured (Multi Gauge software) and used to normalize the in vitro dsRNA binding data. Protein levels were defined as the intensity difference between the protein band and the gel background.
RESULTS
Fluorescent Fusion Proteins Allow the Visualization of Specific Gene Elements at a Transcription Site
To investigate how gene regulatory events are coordinated at a molecularly defined transcription site, we utilize an inducible CMV promoter-regulated transgene, which allows DNA, RNA, and protein to be visualized in single living cells (Fig. 1A) (33). When stably integrated into a cell's genome, the transgene forms a chromatinized multicopy array that can be identified by expressing fluorescently tagged proteins, which bind to sequence elements in the transgene (9, 33). The transgene-binding proteins can be fused to different colored proteins (CFP/YFP/Cherry) and expressed in different combinations with tagged regulatory factors to evaluate the temporal and spatial organization of transcription, chromatin structure, and regulatory factor recruitment.
FIGURE 1.

Sense and antisense RNA is transcribed from the CMV promoter-regulated transgene in the ATRX-null U2OS cell line. A, diagram of the inducible transgene drawn to scale. Expression of the Cherry-lac repressor allows the transgene array to be visualized. Transcription is induced from the minimal CMV promoter by the activators Cherry-tTA-ER and ER-tTA in the presence of 4-hydroxytamoxifen (4-OHT) and rtTA in the presence of doxycycline (Dox). The transcribed RNA encodes CFP fused to a peroxisomal targeting signal (SKL). The RNA is visualized by YFP-MS2, which binds to the stem loops in the transcript. The 3′-end of the transcription unit is composed of the intron 2 splicing unit from the rabbit β-globin gene. The recruitment of YFP-tagged factors to the array can be monitored by co-expression with an array-binding protein. B, quantification of Cherry-tTA-ER (red) and H3.3-YFP (green) recruitment to the transgene array during activation in single cells. 4-Hydroxytamoxifen was added immediately after the first time point (∼0 min). Images were collected every 5 min for 4.5 h. Measured intensities were normalized to the high and low plateau values and fitted to a model (solid black line) containing logistic (dashed lines) and linear (dotted lines) parameters. The initial accumulation (downward pointing arrow) is the point when the logistic component of the curve deviates 5% from the initial base line. The end of rapid accumulation (upward pointing arrow) is the point when the logistic component reaches 95% of the final base line. The graph is the average of 13 independent cells. Error bars, S.D. Supplemental Movie 1 shows a representative time series. Table 1 summarizes the logistical and linear parameter values used in the equation to calculate intensity. C, strand-specific qRT-PCR analysis of total RNA collected from U2OS 2-6-3/YFP-MS2/rtTA cells 0 and 3 h after activation with doxycycline using a primer pair located in rabbit β-globin exon 3. Results are the average of at least three independent experiments. S.D. values, in the form of error bars, and p values, calculated using unpaired Student's t test, are presented in the graphs. D, strand-specific high throughput sequencing analysis of nuclear RNA isolated from 2-6-3/YFP-MS2/rtTA cells 3 h after activation with doxycycline. The upper bar depicts the structure of the transgene. Relative levels for sense (blue) and antisense (orange) transgene expression tags are shown below it. The black and green bar plot indicates the number of unique aligned sequencing reads across the transgene normalized by the number of repeated copies in the transgene segments. The heat map is color-coded for black to indicate the unique sequence regions; the green intensity is proportional to the repeat copy number in the transgene. The line plot at the bottom represents unnormalized data. Red rectangles below the transgene show the location of the strand-specific RNA FISH probes used in Fig. 2A.
The array site can be visualized by expressing the lac repressor protein, which binds to the lac operator repeats (Fig. 1A). Transcription is activated using a modified form of the tetracycline transcriptional activator (tTA). Fusion to the estrogen receptor (ER) hormone-binding domain (tTA-ER) retains it in the cytoplasm until the addition of 4-hydroxytamoxifen induces its entry into the nucleus, where it binds to the TRE repeats, activates transcription, and permits the visualization of the transcription site (34). Activation can also be induced by the reverse tetracycline transcriptional activator (rtTA) in the presence of doxycycline. The RNA transcript encodes CFP fused to a peroxisomal targeting signal (SKL), 24 repeats of the MS2 bacteriophage translational operator, and the rabbit β-globin intron 2 splicing unit. It can be visualized through the interaction of the MS2 coat protein with the MS2 translational operator repeats. The appearance of the CFP-SKL protein in the peroxisomes in the cytoplasm confirms that the mRNA has been properly processed and exported.
Initially, we introduced a stable multicopy array of this transgene (Fig. 1A) into U2OS cells (cell line 2-6-3) (33), which are ATRX null (27, 42). The Daxx and ATRX pathway is required for the transcriptional repression of the CMV promoter (9). Therefore, in U2OS cells, the transgene array can be rapidly activated (33, 34). Additionally, in the absence of ATRX, H3.3 is not specifically incorporated into the nucleosomes of the array chromatin (9). H3.3 is still, however, strongly recruited to the activated array, but it does not co-localize with any of the proteins that bind to the transgene DNA, which suggests that H3.3 accumulates at a step upstream of nucleosome deposition and that this system can be used to identify its mechanism of recruitment.
Single-cell Live Cell Imaging Suggests that H3.3 Is Recruited to Its Incorporation Sites through an RNA-mediated Mechanism
To begin to decipher the mechanism through which H3.3 is recruited to the array during activation, we imaged the activator, Cherry-tTA-ER, and H3.3-YFP in single U20S 2-6-3 cells (Fig. 1B and supplemental Movie 1). The data were then modeled (solid line) using a function composed of logistical and linear terms to account for the influence of binding site cooperativity and saturation as well as the possibility that the number of H3.3 binding sites increases over time (43, 44) (Fig. 1B and Table 1). Consistent with our previous analysis, the activator, Cherry-tTA-ER, accumulated at the array ∼7 min after induction (34). The dominance of the logistical term (dashed line) in the fit suggests that binding is regulated by cooperative mechanisms. After the first activator molecules bind, a pioneer round of transcription and/or chromatin remodeling may be required to make additional sites accessible (45–47). Cherry-tTA-ER levels plateau ∼2 h postinduction, which is when its binding sites become saturated.
TABLE 1.
Summary of logistical and linear parameter values and the error in the kinetic analysis of Cherry-tTA-ER and H3.3-YFP recruitment to transcription site during activation
Shown is a summary of the parameter values used in the equation to calculate intensity (see “Experimental Procedures” in the analysis of Cherry-tTA-ER and H3.3-YFP during activation. Graphs are shown in Fig. 1B
| Parameter | Cherry-tTA-ER | H3.3-YFP |
|---|---|---|
| A1 | −1.51 ± 1.47 | −0.80 ± 0.61 |
| A2 | 80.4 ± 2.9 | 23.7 ± 6.4 |
| x0 | 2.15 ± 0.16 | 99.85 ± 5.18 |
| p | 32.4 ± 1.1 | 4.1 ± 1.0 |
| Alin | 0.08 ± 0.01 | 0.28 ± 0.02 |
In contrast to the activator, Cherry-tTA-ER, H3.3-YFP does not begin to accumulate until ∼50 min postinduction, which suggests that its recruitment requires the initiation of another regulatory event (Fig. 1B). The large contribution of the linear function (dotted line) to the H3.3 fit also indicates that its binding sites increase over time. Because H3.3 accumulation correlates with transcriptional activation, which results in the continuous synthesis of RNA, this suggests that H3.3 could be recruited to its incorporation sites through an RNA-mediated mechanism.
Sense and Antisense RNA Are Transcribed from the Transgene
To test the hypothesis that histone H3.3 is recruited to its incorporation sites through an RNA-mediated mechanism, we first characterized the RNA transcribed from the transgene array using three independent strand-specific techniques: qRT-PCR, high throughput sequencing (HTS), and RNA FISH. Using total RNA and a primer pair located in rabbit β-globin exon 3 (Fig. 1A), we detected an ∼20-fold increase in S RNA and an ∼4-fold increase in AS RNA after 3 h of activation (Fig. 1C). This shows that, in addition to the mRNA encoding CFP-SKL (33), AS RNA is also transcribed from the array.
To profile the distribution of S and AS RNA along the transgene, we used strand-specific HTS (Fig. 1D). The qRT-PCR analysis indicated that, in total RNA, the S transcript is ∼40 times more enriched compared with the AS RNA (Fig. 1C). To obtain a portrait of the RNA distribution most representative of the transcripts physically associated with the array, we sequenced RNA isolated from nuclei. Both S and AS RNA transcripts were detected from all regions of the transgene, including the rabbit genomic DNA 3′ to the transcription unit and the bacterial plasmid sequence, indicating widespread transcriptional activity. Although the mechanism of AS transcription is unknown, it is interesting to note that S and AS RNA as well as H3.3 are all enriched in the regions 3′ to single copy genes (11), making it possible that they are functionally connected.
Histone H3.3 Is Recruited to Accumulations of Sense and Antisense RNA at the Activated Transgene Array
The qRT-PCR and HTS analyses indicate that both S and AS RNA are transcribed from the transgene during activation (Fig. 1, C and D). Therefore, we also wanted to evaluate their localization in cells using strand-specific RNA FISH (Fig. 2). Using a probe in rabbit β-globin exon 3, which hybridizes to the S strand (Fig. 2A), we detected co-localization with the MS2 protein both at the transcription site and throughout the nucleoplasm (Fig. 2B, a–d). The intensity profiles (Fig. 2B, d, h, l, and p) graph the pixel intensity measurements along the line drawn through the array (note the yellow line in enlarged insets in the merged images in c, g, k, and o). The complete overlap between the red (exon sense probe) and green (YFP-MS2) signals in the intensity profile graph (Fig. 2B, d) is consistent with their binding to adjacent elements in the S RNA (Figs. 1A and 2A). Labeling with the complementary exon antisense probe revealed that the AS RNA is retained at the transcription site (Fig. 2B, e–h). The lack of complete co-localization between the exon AS probe and YFP-MS2 confirms that they recognize different strands (note the lack of complete co-localization between the red and green peaks in the intensity profile graph (Fig. 2B, h)).
FIGURE 2.

H3.3 is recruited to sense and antisense RNA at the activated transgene array in U2OS cells. A, diagram of the 3′-end of the transcription unit (Fig. 1A) showing the locations of the strand-specific RNA FISH probes. B, localization of the exon sense (a–d) and exon antisense (e–h) RNA FISH probes at the activated array in relation to YFP-MS2. Shown is localization of the exon sense (i–l) and exon antisense (m–p) RNA FISH probes at activated arrays in relation to H3.3-YFP. Yellow lines in enlarged merge insets show the path through which the red and green intensities were measured in the intensity profiles (d, h, l, and p). Asterisks mark the start of the measured lines. Scale bar, 5 μm. Scale bar in enlarged inset, 1 μm. C, quantitative image analysis of S and AS RNA levels using the probes shown in A at arrays activated for 3 h. Error bars, S.E. p values, calculated using unpaired t test, are presented in the graphs. n values are as follows: intron, S, n = 19; AS, n = 26; exon, S, n = 21; AS, n = 27; genomic, S, n = 18; AS = 19; plasmid, S, n = 17; AS, n = 15.
Quantitative image analysis of S and AS RNA accumulations at activated arrays using probes that cover the region from the intron to the genomic DNA/bacterial plasmid junction (Fig. 2A) indicates that these transcripts also remain associated with the array (Fig. 2C; Fig. 1D shows locations of the RNA FISH probe in relation to HTS data). RNA polymerase II transcription typically terminates ∼600 to a few thousand nucleotides downstream of poly(A) sites (48). In rabbit β-globin, transcription proceeds for ∼600 nucleotides past the poly(A) site and decreases over the next ∼500 nucleotides (49). Interestingly, S RNA levels diminish as the distance from the poly(A) site increases, and AS RNA levels remain fairly constant (Fig. 2C), suggesting the possibility of different transcriptional mechanisms.
Taken together, these results indicate that S and AS RNA accumulate at the activated transgene array in the ATRX-null U2OS cells and, therefore, that it could serve as a recruitment platform for H3.3. To test this idea, we labeled activated cells expressing H3.3-YFP with the Exon S and AS probes. H3.3 co-localizes significantly with both S and AS RNA at the site (Fig. 2B, i–p), which supports the hypothesis that H3.3 is recruited to its incorporation sites through an RNA-mediated mechanism.
H3.1, H3.2, and H3.3 All Contain the Signal Required for RNA-mediated Recruitment to the RI Chromatin Assembly Site
To determine whether H3.3 is specifically recruited to the S and AS RNA at the activated transgene array, we evaluated the recruitment of the replication-dependent variants, H3.1 and H3.2, which differ from H3.3 by 5 and 4 amino acids, respectively (Fig. 3A). Most of these differences are located in helix α2 and regulate chaperone-specific interactions, including the association between H3.3 and Daxx (12). Interestingly, H3.1 and H3.2 are also strongly recruited to the activated array in the ATRX-null U2OS cells in a pattern and percentage indistinguishable from H3.3 (Fig. 3, B–D). Like H3.3, neither H3.1 nor H3.2 co-localizes with the activator, Cherry-tTA-ER, consistent with recruitment in the absence of chromatin assembly (9) (Fig. 3C, d and h; note the lack of complete overlap between the red and green peaks in the graph of the intensity signals). This result indicates that the region that mediates H3.3 recruitment to the activated array is conserved in all three variants.
FIGURE 3.

H3.1, H3.2, and H3.3 are all strongly recruited to the activated transgene array in U2OS cells. A, diagram of the domain structure of the H3 variants showing the locations of the amino acid differences between them. B, Western blot analysis of the YFP-tagged H3 variants using a GFP antibody. γ-Tubulin is used as a loading control. C, localization of H3.1-YFP (a–d) and H3.2-YFP (e–h) at the activated transgene array in relation to the activator, Cherry-tTA-ER. Yellow lines in the enlarged merge insets show the path through which the red and green intensities were measured in the intensity profiles (d and h). Asterisks mark the start of the measured line. Scale bar, 5 μm. Scale bars in enlarged inset, 1 μm. D, percentage of cells in which the histone H3 variants were recruited to the activated transgene array. 100 cells were counted in three independent experiments. S.D. values in the form of error bars are shown in the graph.
The finding that histones and chaperones interact with high affinity in biochemical assays suggested that chaperones transport histones to their incorporation sites (6–8). Because we are able to visualize a Daxx and ATRX-regulated transcription site in single cells during transcriptional activation when chromatin assembly is blocked, we wanted to determine whether chaperones were required for H3.3 recruitment to it. The recruitment of H3.1, H3.2, and H3.3 to the array in the ATRX-null U2OS cell line indicates that ATRX is not required (Fig. 3, C and D) (9, 33). The accumulation of H3.1 and H3.2 (Fig. 3, C and D), which do not interact with Daxx (12), suggests that Daxx is also not required. The recruitment of H3.3 after Daxx knockdown further supports this conclusion (Fig. 4A). Previously, we showed that HIRA is not enriched at this CMV promoter-regulated array (9), consistent with reports that Daxx and HIRA regulate mutually exclusive sites (11, 50). Here, we show that H3.3 is still recruited after HIRA knockdown, further indicating that HIRA is not required for H3.3 accumulation (Fig. 4B). The lack of co-localization between H3.3-YFP and CFP-Daxx indicates that they occupy functionally distinct regions at the activated array (Fig. 4C). Taken together, these results indicate that H3.3 is recruited to this transcription site through a chaperone-independent mechanism.
FIGURE 4.

H3.3 recruitment to the activated array in U2OS cells is chaperone-independent. A, H3.3-YFP recruitment to the CFP-tTA-ER-activated array in Daxx (a–d) and control (e–g) siRNA-treated cells. Daxx levels were assessed by imaging immunofluorescently labeled cells using the exact same settings. B, H3.3-YFP recruitment to the CFP-tTA-ER-activated array in control (a–d) and (e–g) HIRA siRNA-treated cells. HIRA levels were assessed by imaging immunofluorescently labeled cells using the exact same settings. C, localization of YFP-Daxx (a), Cherry-tTA-ER (b), and H3.3-CFP/CFP-SKL (c) in activated cells. Yellow lines in enlarged merge insets show the path through which the red, green, and blue intensities were measured in the intensity profiles (A and B, d and h; C, e). Asterisks mark the start of the measured lines. Scale bar, 5 μm. Scale bar in enlarged inset, 1 μm.
N-terminal Tail Sequences and the αN Helix Mediate H3.3 Recruitment to the Activated Transgene Array
To identify the region of H3.3 that mediates its recruitment to the activated transgene array, we evaluated the localization of a series of deletion constructs (Fig. 5). The recruitment of H3.3 ΔN-tail indicates that the first 35 amino acids are not required (Fig. 5, A and C, a–d). The recruitment of H3.3 N-term, H3.3 N-tail-αN, and H3.3 αN (Fig. 5, A and C, e–p) identified amino acids 36–63 as the minimal region required for H3.3 recruitment. The fact that this region is 100% conserved between H3.1, H3.2, and H3.3 explains why they all similarly accumulate (Fig. 3). The finding that the H3.3 α1-α2-α3 and H3.3 C-term constructs, which include the Daxx-interacting region in helix α2 (12), did not accumulate (Fig. 5, A and C, q–x) further supports a chaperone-independent recruitment mechanism.
FIGURE 5.

H3 N-tail amino acids and helix αN mediate H3.3 recruitment to the activated transgene array in U2OS cells. A, diagram of YFP-tagged H3.3 deletion constructs. Percentage of cells in which the H3.3 deletion constructs are recruited to the activated transgene array in U2OS cells. 100 cells were counted in three independent experiments. S.D. values are shown. B, Western blot analysis of the YFP-tagged H3.3 deletion constructs using a GFP antibody. γ-Tubulin is used as a loading control. C, localization of YFP-tagged H3.3 deletion constructs at the activated transgene array in relation to the activator, Cherry-tTA-ER. Yellow lines in enlarged merge insets show the path through which the red and green intensities were measured in the intensity profiles (d, h, l, p, t, and x). Asterisks mark the start of the measured line. Scale bar, 5 μm. Scale bars in enlarged inset, 1 μm.
Gain-of-function H3.3 Pediatric Glioblastoma Driver Mutations, G34R and K27M, Differentially Affect H3.3 Recruitment to the Activated Transgene Array
Within the nucleosome, the H3 histones interact with dsDNA through the solvent-exposed positively charged amino acids in the N-terminal tail and αN helix (51, 52). Fig. 6B shows the relationship of the Lys and Arg residues in this region (amino acids 37–56) with dsDNA in the nucleosomal structure. Because we speculate that H3.3 is recruited to its incorporation sites through an RNA-mediated mechanism and the region required for recruitment has nucleic acid binding properties, this suggested that it might also interact with RNA. To further analyze the H3.3 recruitment mechanism, we converted the Lys and Arg residues in the H3.3 N-tail-αN-YFP construct to alanine (Fig. 6A; red letters with asterisks indicate the residues mutated in the four-point mutant (4PTM) construct; Fig. 6D) and used quantitative image analysis to evaluate their effects on accumulation. All of the Lys/Arg to Ala conversions, except R53A, reduced H3.3 N-tail-αN-YFP accumulation, indicating that these residues mediate interactions with nucleic acids and/or proteins that are important for recruitment (Fig. 6C).
FIGURE 6.

Quantitative single-cell image analysis of H3.3 recruitment to the activated transgene array in U2OS cells. A, diagram of the amino acid sequence of the H3.3 N-tail-αN construct showing the locations of the point mutations analyzed in the single-cell recruitment assay. The locations of the pediatric glioblastoma driver mutations, K27M and G34R, are shown in purple. The amino acids converted to alanine are shown in red. Asterisks mark the amino acids mutated in the four-point mutant (4PTM) construct. Orange shading demarcates the αN helix. B, crystal structure of histone H3 from amino acids 37–60 (salmon) in relation to dsDNA (green) in the nucleosome (Protein Data Bank code 3LJA). The lysine and arginine residues changed to alanine are numbered. C, quantitative single-cell image analysis of the total intensity of the YFP-tagged H3.3 N-tail-αN constructs at the activated transgene array. Error bars, S.E. n and p values, calculated using unpaired t test, are presented in the chart below. D, Western blot analysis of the YFP-tagged H3.3 N-tail-αN constructs using a GFP antibody. Tubulin is used as a loading control. E, quantitative single-cell image analysis of the total intensity of the YFP-tagged H3.3 constructs at the activated transgene array. Error bars, S.E. n values are presented below the graph, and p values, calculated using unpaired t test, are presented in the graph. F, Western blot analysis of full-length YFP-tagged H3.3 constructs using a GFP antibody. Tubulin is used as a loading control.
By using the H3.3 N-tail-αN construct for this analysis, we were also able to evaluate the effects of the H3.3 pediatric glioblastoma driver mutations, K27M and G34R, on recruitment (Fig. 6, A and D) (31, 32). Strikingly, they significantly affected accumulation despite not being within the minimal 36–63 amino acid region; G34R increased accumulation, and K27M decreased it (Fig. 6C). Because their effects on H3.3 N-tail-αN recruitment were significant, we also evaluated their effects on the accumulation of full-length H3.3, which revealed a similar trend (Fig. 6, E and F). Due to the need to reduce the laser power by 80% to acquire unsaturated images of the full-length constructs, it is not possible to compare them to the deletion constructs on the same graph (Fig. 6, C and E). However, the higher intensity signals of the full-length constructs compared with H3.3 N-tail-αN suggest that the C-terminal amino acids increase the affinity of H3.3 for the site. Taken together, the finding that the H3.3 pediatric glioblastoma mutations differentially affect H3.3 recruitment to the activated array (Fig. 6, C and E) suggests that they may interfere with mechanistic events upstream of nucleosomal deposition.
Gain-of-function H3.3 Pediatric Glioblastoma Mutations, G34R and K27M, Differentially Affect H3.3 Binding to Double-stranded RNA in Vitro
The co-localization of H3.3 with S and AS RNA at the activated array (Fig. 2B) and the reduction in H3.3 N-tail-αN accumulation when the Lys and Arg residues were converted to Ala (Fig. 6C) suggested that this region might interact directly with dsRNA. To test this, we established an in vitro binding assay using GST-tagged bacterially expressed proteins (Fig. 7, A and C) and in vitro transcribed dsRNA from the ∼700-nucleotide rabbit β intron 2 splicing unit at the 3′-end of the inducible transgene (Fig. 1A). Indeed, in this assay, H3.3 N-tail-αN interacts with dsRNA, supporting the hypothesis that such an interaction could mediate H3.3 recruitment to RI incorporation sites. To confirm the specificity of this interaction, we treated the RNA bound to the wild-type H3.3 N-tail-αN and ADAR1 double-stranded RNA binding domain (ADAR1 dsRBD) construct with RNase III, which degrades dsRNA. RNase III treatment significantly reduced the signals, indicating that, in this assay, H3.3 N-tail-αN and ADAR1 dsRBD interact specifically with dsRNA (Fig. 7D).
FIGURE 7.

H3.3 N-tail-αN binds to double-stranded RNA in vitro. A, diagram of the amino acid sequence of the H3.3 N-tail-αN region showing the locations of the point mutations analyzed in the in vitro dsRNA binding assay. The location of the pediatric glioblastoma driver mutations, K27M and G34R, are shown in purple. The amino acids converted to alanine are shown in red. Asterisks mark the amino acids mutated in the four-point mutant (4PTM) construct. Orange shading demarcates the αN helix. B, measurement of dsRNA binding levels in the in vitro assay normalized to wild-type (WT) H3.3 N-tail-αN RNA and protein levels. GST is the negative control, and the ADAR1 dsRBD is the positive control. S.D. values, in the form of error bars, are presented in the graphs. p values, calculated using unpaired t test, are presented in the chart. C, Coomassie-stained gel of the purified bacterially expressed GST fusion proteins used in the binding assay. D, analysis of RNase III-treated WT H3.3 N-tail-αN and ADAR1 dsRBD-bound RNA normalized to WT RNA and protein levels. S.D. values, in the form of error bars, and p values, calculated using unpaired t test, are presented in the graphs.
Although the affinity of H3.3 N-tail-αN for dsRNA was ∼3.5 times lower compared with ADAR1 dsRBD, the interaction is specific because binding to GST was negligible (Fig. 7B). Compared with the H3.3 N-tail-αN four-point mutant construct (4PTM) (Fig. 7A; red asterisks mark the mutated sites) the single Lys/Arg to Ala conversions did not significantly reduce RNA binding, perhaps because of the compensatory effects of the other positively charged residues in this region (Fig. 7B). Consistent with the single-cell recruitment assay (Fig. 6, C and E), G34R increased dsRNA binding and K27M reduced it, which further supports the hypothesis that they affect an RNA regulatory event(s) in the RI chromatin assembly pathway.
H3.3 Regulates Sense and Antisense Transcription
Our analyses suggest that H3.3 is recruited to its incorporation sites through a chaperone-independent RNA-mediated mechanism. To determine whether H3.3 regulates transcriptional events at the transgene array, we evaluated the effects of overexpression and knockdown on S and AS RNA levels (Fig. 8). Expression of the FLAG-tagged H3.3 wild-type and K27M and G34R mutant constructs induced an ∼2-fold increase in S RNA levels but did not appreciably affect AS transcription compared with the active control (Fig. 8A). The mechanism through which H3.3 increases S expression is currently not known, but this result suggests a mechanistic interaction with the transcriptional machinery (Fig. 8A). Although in the single-cell imaging assay, the G34R and K27M mutations significantly affected H3.3 accumulation (Fig. 6E), it is likely that their impact on transcription, as detected by qRT-PCR, is not significant because all of the constructs are highly expressed, and measurements are the averages from cell populations.
FIGURE 8.

H3.3 regulates transcription at the CMV promoter-regulated transgene array. A, strand-specific qRT-PCR analysis of total RNA collected from U2OS 2-6-3/YFP-MS2/rtTA cells expressing full-length FLAG-tagged H3.3 constructs 0 and 3 h after activation with doxycycline (Dox) using a primer pair in rabbit β-globin exon 3. Results are the average of at least three independent experiments. S.D. values, in the form of error bars, and p values, calculated using unpaired t test, are presented in the graphs. Western blot analysis of FLAG-tagged H3.3 constructs using the FLAG antibody and γ-tubulin as a loading control. B, strand-specific qRT-PCR analysis of total RNA collected from U2OS 2-6-3/YFP-MS2/rtTA, expressing control pLKO.1 and H3.3 shRNA constructs, 0 and 3 h after activation with doxycycline. C, qRT-PCR analysis of hH3F3A and hH3F3B mRNA levels in U2OS 2-6-3/YFP-MS2/rtTA cells after knockdown with gene-specific shRNAs. Shown is Western blot analysis of H3.3 levels in control and knockdown cells using an H3.3-specific antibody and γ-tubulin as a loading control.
To evaluate the effects of depleting H3.3 on transcription, we measured S and AS RNA levels after knocking down the endogenous genes, hH3F3A and hH3F3B, using shRNAs (Fig. 8B). Depletion of both the mRNA and protein was confirmed using qRT-PCR and immunoblotting, respectively (Fig. 8C). Remarkably, H3.3 knockdown induced an ∼5-fold increase in both S and AS RNA levels in transcriptionally activated cells (Fig. 8B). Additionally, an ∼6-fold increase in AS RNA was detected in inactive cells, suggesting that H3.3 represses AS transcription at silent chromatin. The increase in S and AS RNA after H3.3 knockdown suggests that H3.3 inhibits transcription at Daxx and ATRX-regulated RI incorporation sites. It also suggests that the RNA recruitment signal is attenuated by nucleosomal deposition and amplified when the Daxx and ATRX pathway is inhibited.
DISCUSSION
Although RI H3.3 chromatin assembly is highly conserved in eukaryotes and H3.3 incorporation patterns indicate that it regulates a wide range of sites, its essential function in genome regulation is still not well understood. Disruption of RI H3.3 chromatin assembly negatively impacts nuclear reprogramming (53, 54), gametogenesis (55–57), and fertilization (30, 58), indicating a critical mode of action in epigenetic regulation. Here, we provide evidence that H3.3 is recruited to a RI incorporation site (an array of a CMV promoter-regulated transgene) through an RNA-mediated chaperone-independent mechanism. When RI H3.3 chromatin assembly is blocked, H3.3 accumulates with S and AS RNA at the activated site (Fig. 9A). S and AS RNA transcripts also increase after H3.3 knockdown, suggesting that H3.3 regulates their expression and that nucleosomal deposition attenuates the RNA recruitment signal. Taken together, these results indicate that RI H3.3 chromatin assembly is intimately connected with transcriptional events, which could provide important new insight into RNA-mediated epigenetic regulation.
FIGURE 9.

Models of RNA-mediated H3.3 recruitment. A, model of Daxx- and ATRX-mediated transcriptional repression of the CMV promoter-regulated transcription site. When both Daxx and ATRX are present, H3.3 is incorporated into the nucleosomes at the array, and the transcriptional activator is unable to access its binding sites and recruit RNA polymerase II to the site. In the ATRX-null U2OS cell line, the activator is able to accumulate at the transcription site and induce transcription. Under these conditions, S and AS RNA accumulate at the activated site. Sequences in the N-terminal tail and αN helix mediate H3.3 recruitment to the activated transgene array. H3.3 colocalizes with RNA at the activated site but is not incorporated into the chromatin. B, RI H3.3 chromatin assembly can be divided into recruitment and chromatin assembly steps. In step 1, an epigenetic transcriptional regulatory event triggers H3.3 recruitment to its site of incorporation. N-terminal tail and αN helix sequences, which are 100% conserved between H3.1, H3.2, and H3.3, mediate recruitment, which means that all three variants can be recruited. In step 2, the H3.3-specific chaperones present at the incorporation site specifically utilize H3.3 for chromatin assembly.
This analysis was done using an experimental system that permits the single-cell visualization of a chromatinized CMV promoter-regulated transgene array. The Daxx and ATRX pathway is required for transcriptional repression and H3.3 nucleosomal deposition at this site (9). The integration of this transgene into the ATRX-null U2OS cell line provides the opportunity to analyze RI H3.3 chromatin assembly in a genetic background in which this pathway is blocked. In U2OS cells, transcription is rapidly induced from the array; within minutes, the coding strand is transcribed and subsequently processed, exported, and translated (33, 34). In contrast to the transcriptional activator (Cherry-tTA-ER), H3.3 does not begin to accumulate until ∼1 h postinduction, which suggests that additional regulatory events are first required. The accumulation of H3.3 with both S and AS RNA suggests that its recruitment may be regulated by AS transcription. It is likely that we can clearly detect these RNA products at the transcription site because their synthesis is amplified in the ATRX-null background. Therefore, this system provides a unique opportunity to decipher regulatory events upstream of nucleosomal deposition.
Our experiments also suggest that RI H3.3 chromatin assembly can be divided into recruitment and nucleosomal deposition steps and that chromatin assembly factors are only required for the latter (Fig. 9B). Residues in the H3.3 N-terminal tail and αN helix (amino acids 36–63) comprise the minimal region required for recruitment. Because it is 100% conserved in H3.1, H3.2, and H3.3, they accumulate similarly when chromatin assembly is blocked. However, only H3.3 is specifically deposited into nucleosomes by Daxx and ATRX (9) (Fig. 9B). Although Daxx and ATRX do not utilize H3.1 and H3.2, our demonstration that they can be recruited to RI chromatin assembly sites suggests that they could have a regulatory effect on them. It is possible that their effects are limited to S phase when the canonical histones are expressed. It is also possible that the chromatin assembly machinery can efficiently exclude H3.1 and H3.2 if low levels of these variants are available throughout the cell cycle. However, as discussed below, because H3.1 and H3.2 can be recruited to RI assembly sites, mutations in them that interfere with events upstream of nucleosomal deposition could deleteriously affect cellular function.
We also provide several lines of evidence that H3.3 recruitment is chaperone-independent. In addition to the finding that H3.1 and H3.2, which do not interact with Daxx (12), accumulate at the activated transgene array, we also show that H3.3 is recruited after Daxx knockdown in the ATRX-null U2OS cell line, indicating that neither Daxx nor ATRX is required. Analysis of H3.3 deletion constructs also indicates that helix α2, which includes the Daxx interaction domain, is not sufficient for transcription site targeting. Finally, H3.3 accumulates at the activated array in cells expressing ICP0, which removes both Daxx and ATRX from the site, further indicating that they are not required for its recruitment (9).
It is not understood how heterochromatin and euchromatin are functionally united by H3.3 deposition. However, because both types of chromatin are regulated by transcription (59–62), an RNA-mediated H3.3 recruitment step could provide the mechanistic link. The colocalization of H3.3 with S and AS RNA at the transcription site when chromatin assembly is blocked suggests that dsRNA could be its recruitment signal. In support of this hypothesis, we show that the region of H3.3 that mediates recruitment also interacts with dsRNA in vitro. Its lower affinity for dsRNA compared with the canonical ADAR1 dsRNA-binding domain could be mechanistically important if H3.3 interacts transiently with dsRNA en route to nucleosomal deposition. Interestingly, histone/RNA interactions and a role for RNA in chromatin assembly have been reported previously (63, 64). Mutations in the αN helix of S. cerevisiae H3 also disrupt sporulation but not mitotic growth, indicating that this region plays a critical role in epigenetic regulation (65, 66), which could include RNA-mediated H3 recruitment.
Our recent report showing that Daxx and ATRX are enriched at the CMV promoter of the inducible transgene suggests that promoter sequences recruit RI H3.3 chromatin assembly factors (9). This model is also consistent with the finding that H3.3 chaperones regulate mutually exclusive sites (11, 50). Furthermore, RI chromatin assembly machineries seem to have regulatory effects on transcription (Fig. 9B). For example, telomeres, pericentromeres, and the CMV promoter, which utilize Daxx and ATRX, are transcriptionally silent (9–12). HIRA-regulated sites, which include single copy housekeeping genes (11), the genomes of senescent cells (13, 14), and male pronuclei (15–17), are both active and silent. It is possible that their transcriptional states are determined by the chromatin remodeling factors with which HIRA functions. Thus, our data suggest a model in which a universal RNA signal recruits H3.3 to sites of RI incorporation, and sequence-specifically targeted chromatin assembly factors function downstream to regulate transcriptional activity and epigenetic inheritance.
Surprisingly, the gain-of-function brain cancer mutations significantly affect the interaction of H3.3 with both the activated array and dsRNA, which suggests that the disease etiology could be due in part to the misregulation of RNA regulatory events upstream of nucleosomal deposition. In support of this model is the finding that the G34R mutation, which increases the affinity of H3.3 for dsRNA, is only found in tumors that also harbor a mutation in DAXX or ATRX (31). The fact that G34R only induces malignancy when RI chromatin assembly is blocked suggests that it misregulates a step(s) upstream of nucleosomal deposition. Support for the K27M mutation also interfering with events upstream of nucleosomal deposition comes from the finding that it is also found in the replication-dependent variant, H3.1 (HIST3H1B) (32), which we show can be recruited to the CMV promoter-regulated array but which is not utilized for RI nucleosomal deposition. H3 variants containing the K27M mutation have recently been shown to reduce overall levels of H3 Lys-27 trimethylation (67, 68). The K27M mutation inhibits the activity of the H3 Lys-27 methyltransferase, EZH2, through a direct interaction (67). Our results suggest the possibility that this dominant gain-of-function interaction occurs at RI chromatin assembly sites prior to nucleosomal deposition.
Chromatin immunoprecipitation analyses indicate that H3.3 is enriched at the 5′- and 3′-ends of genes and that its incorporation levels correlate with transcriptional activity (11, 14, 18, 21–26). Interestingly, genome-wide RNA sequencing has identified AS RNAs corresponding to the 5′- and 3′-ends of genes (69–72). Because they could be a source of dsRNA, it is possible that their expression is linked to H3.3 recruitment. Further evidence for a connection between RNA and H3.3 deposition comes from studies in S. cerevisiae showing that H3 Lys-36 methylation suppresses both histone exchange and S and AS transcription from cryptic promoters (73–75). It is also possible that the misregulation of telomeric and pericentromeric transcription seen after ATRX and Daxx knockout and knockdown (10, 11, 30) reflects the deregulation of an H3.3 RNA recruitment signal. Double-stranded RNA is a good candidate to be a universal H3.3 recruitment signal because it is structural and can be formed at any site. However, the differential affinity of the K27M and G34R mutants for it suggests that there may also be sequence specificity to this interaction. Because both mRNA and heterochromatic RNA transcripts are polyadenylated (76–78), H3.3 binding could be related to sequences that regulate this untemplated RNA synthesis event.
At this time, we do not know the mechanism regulating AS transcription at the CMV promoter-regulated array or whether synthesis is DNA- or RNA-templated. Canonical RNA-dependent RNA polymerases are components of plant, nematode, and S. pombe RNA interference (RNAi) pathways (79), but they are thought to have been lost in the ancestor of animals (80). RNA exosome sequencing suggests, however, that RNA-dependent RNA polymerase activity is also present in mammals because AS transcripts, which cross splice junctions and contain non-encoded 5′ stretches of polyuridine, have been detected (71, 81). TERT, the protein subunit of telomerase, is reported to function as an RNA-dependent RNA polymerase (82). However, TERT is not expressed in the ALT-positive U2OS cell line (83, 84), suggesting that it is not responsible for the AS transcription at the CMV promoter-regulated array.
Our discovery of a link between RNA and Daxx- and ATRX-mediated RI H3.3 deposition may also contribute to our understanding of RNA-mediated gene silencing in mammals. In invertebrates and plants, RNAi plays a significant role in heterochromatin regulation and is the primary defense against invasive foreign nucleic acids (85–87). In contrast, RNAi is thought to have regressed in mammals with the emergence of the interferon response and adaptive immunity, making it possible that its role in heterochromatin regulation also regressed (88). Interestingly, Daxx is metazoan-specific (50) and a component of PML nuclear bodies (PML-NBs). Many viruses degrade and/or disorganize PML-NB proteins, including Daxx and PML, in their efforts to escape intrinsic immunity (89). In support of a connection between PML-NBs, RI H3.3 chromatin assembly, and genome defense, we found that PML is also enriched at the CMV promoter of the inducible transgene (9). Others have also reported the colocalization of Daxx and H3.3 in PML-NBs (10, 90). Therefore, the finding that RI H3.3 chromatin assembly is connected to RNA regulatory events could provide important new insight into RNA-mediated gene silencing and genome defense in mammals.
Supplementary Material
Acknowledgments
We thank Kazuko Nishikura, Peter Adams, Roger Everett, Steve Henikoff, and Kami Ahmad for reagents. We also thank Michael Showe, Dario Altieri, and Aswathy Rai for critical discussions and comments on the manuscript; Sylvie Shaffer for artistic contributions to the models; and Laurence Bell and Elizabeth Strumfels for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM 093000-02 and P30 CA10815 (Wistar Cancer Center Core Grant Facilities: Genomics, Bioinformatics, Protein Expression and Library Screening, Imaging, and Research Supply). This work was also supported by start-up funding from the Wistar Institute and grants from the Arnold and Mabel Beckman Foundation, the Mallinckrodt Foundation, and the Emerald Foundation, the March of Dimes Basil O'Connor Award.

This article contains supplemental Movie 1.
- RI
- replication-independent
- ALT
- alternative lengthening of telomeres
- dsRBD
- double-stranded RNA binding domain
- CFP
- cyan fluorescent protein
- FISH
- fluorescence in situ hybridization
- S
- sense
- AS
- antisense
- qRT-PCR
- quantitative RT-PCR
- tTA
- tetracycline transcriptional activator
- rtTA
- reverse tTA
- ER
- endoplasmic reticulum
- HTS
- high throughput sequencing
- PML
- promyelocytic leukemia
- NB
- nuclear body.
REFERENCES
- 1. Talbert P. B., Henikoff S. (2010) Histone variants. Ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 11, 264–275 [DOI] [PubMed] [Google Scholar]
- 2. Santaguida S., Musacchio A. (2009) The life and miracles of kinetochores. EMBO J. 28, 2511–2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Waterborg J. H. (2012) Evolution of histone H3. Emergence of variants and conservation of post-translational modification sites. Biochem. Cell Biol. 90, 79–95 [DOI] [PubMed] [Google Scholar]
- 4. Ahmad K., Henikoff S. (2002) The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 9, 1191–1200 [DOI] [PubMed] [Google Scholar]
- 5. Schwartz B. E., Ahmad K. (2005) Transcriptional activation triggers deposition and removal of the histone variant H3.3. Genes Dev. 19, 804–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Szenker E., Ray-Gallet D., Almouzni G. (2011) The double face of the histone variant H3.3. Cell Res. 21, 421–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Campos E. I., Reinberg D. (2010) New chaps in the histone chaperone arena. Genes Dev. 24, 1334–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elsaesser S. J., Goldberg A. D., Allis C. D. (2010) New functions for an old variant. No substitute for histone H3.3. Curr. Opin. Genet. Dev. 20, 110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Newhart A., Rafalska-Metcalf I. U., Yang T., Negorev D. G., Janicki S. M. (2012) Single cell analysis of Daxx and ATRX-dependent transcriptional repression. J. Cell Sci. 125, 5489–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Drané P., Ouararhni K., Depaux A., Shuaib M., Hamiche A. (2010) The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 24, 1253–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goldberg A. D., Banaszynski L. A., Noh K. M., Lewis P. W., Elsaesser S. J., Stadler S., Dewell S., Law M., Guo X., Li X., Wen D., Chapgier A., DeKelver R. C., Miller J. C., Lee Y. L., Boydston E. A., Holmes M. C., Gregory P. D., Greally J. M., Rafii S., Yang C., Scambler P. J., Garrick D., Gibbons R. J., Higgs D. R., Cristea I. M., Urnov F. D., Zheng D., Allis C. D. (2010) Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140, 678–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lewis P. W., Elsaesser S. J., Noh K. M., Stadler S. C., Allis C. D. (2010) Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. U.S.A. 107, 14075–14080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang R., Poustovoitov M. V., Ye X., Santos H. A., Chen W., Daganzo S. M., Erzberger J. P., Serebriiskii I. G., Canutescu A. A., Dunbrack R. L., Pehrson J. R., Berger J. M., Kaufman P. D., Adams P. D. (2005) Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 8, 19–30 [DOI] [PubMed] [Google Scholar]
- 14. Rai T. S., Puri A., McBryan T., Hoffman J., Tang Y., Pchelintsev N. A., van Tuyn J., Marmorstein R., Schultz D. C., Adams P. D. (2011) Human CABIN1 is a functional member of the human HIRA/UBN1/ASF1a histone H3.3 chaperone complex. Mol. Cell Biol. 31, 4107–4118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bonnefoy E., Orsi G. A., Couble P., Loppin B. (2007) The essential role of Drosophila HIRA for de novo assembly of paternal chromatin at fertilization. PLoS Genet. 3, 1991–2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Loppin B., Bonnefoy E., Anselme C., Laurençon A., Karr T. L., Couble P. (2005) The histone H3.3 chaperone HIRA is essential for chromatin assembly in the male pronucleus. Nature 437, 1386–1390 [DOI] [PubMed] [Google Scholar]
- 17. Konev A. Y., Tribus M., Park S. Y., Podhraski V., Lim C. Y., Emelyanov A. V., Vershilova E., Pirrotta V., Kadonaga J. T., Lusser A., Fyodorov D. V. (2007) CHD1 motor protein is required for deposition of histone variant H3.3 into chromatin in vivo. Science 317, 1087–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harada A., Okada S., Konno D., Odawara J., Yoshimi T., Yoshimura S., Kumamaru H., Saiwai H., Tsubota T., Kurumizaka H., Akashi K., Tachibana T., Imbalzano A. N., Ohkawa Y. (2012) Chd2 interacts with H3.3 to determine myogenic cell fate. EMBO J. 31, 2994–3007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mito Y., Henikoff J. G., Henikoff S. (2005) Genome-scale profiling of histone H3.3 replacement patterns. Nat. Genet. 37, 1090–1097 [DOI] [PubMed] [Google Scholar]
- 20. Wirbelauer C., Bell O., Schübeler D. (2005) Variant histone H3.3 is deposited at sites of nucleosomal displacement throughout transcribed genes while active histone modifications show a promoter-proximal bias. Genes Dev. 19, 1761–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wollmann H., Holec S., Alden K., Clarke N. D., Jacques P. É., Berger F. (2012) Dynamic deposition of histone variant H3.3 accompanies developmental remodeling of the Arabidopsis transcriptome. PLoS Genet. 8, e1002658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choi E. S., Shin J. A., Kim H. S., Jang Y. K. (2005) Dynamic regulation of replication independent deposition of histone H3 in fission yeast. Nucleic Acids Res. 33, 7102–7110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jin C., Zang C., Wei G., Cui K., Peng W., Zhao K., Felsenfeld G. (2009) H3.3/H2A.Z double variant-containing nucleosomes mark “nucleosome-free regions” of active promoters and other regulatory regions. Nat. Genet. 41, 941–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tamura T., Smith M., Kanno T., Dasenbrock H., Nishiyama A., Ozato K. (2009) Inducible deposition of the histone variant H3.3 in interferon-stimulated genes. J. Biol. Chem. 284, 12217–12225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ooi S. L., Henikoff J. G., Henikoff S. (2010) A native chromatin purification system for epigenomic profiling in Caenorhabditis elegans. Nucleic Acids Res. 38, e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stroud H., Otero S., Desvoyes B., Ramírez-Parra E., Jacobsen S. E., Gutierrez C. (2012) Genome-wide analysis of histone H3.1 and H3.3 variants in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 109, 5370–5375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heaphy C. M., de Wilde R. F., Jiao Y., Klein A. P., Edil B. H., Shi C., Bettegowda C., Rodriguez F. J., Eberhart C. G., Hebbar S., Offerhaus G. J., McLendon R., Rasheed B. A., He Y., Yan H., Bigner D. D., Oba-Shinjo S. M., Marie S. K., Riggins G. J., Kinzler K. W., Vogelstein B., Hruban R. H., Maitra A., Papadopoulos N., Meeker A. K. (2011) Altered telomeres in tumors with ATRX and DAXX mutations. Science 333, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lovejoy C. A., Li W., Reisenweber S., Thongthip S., Bruno J., de Lange T., De S., Petrini J. H., Sung P. A., Jasin M., Rosenbluh J., Zwang Y., Weir B. A., Hatton C., Ivanova E., Macconaill L., Hanna M., Hahn W. C., Lue N. F., Reddel R. R., Jiao Y., Kinzler K., Vogelstein B., Papadopoulos N., Meeker A. K. (2012) Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 8, e1002772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bower K., Napier C. E., Cole S. L., Dagg R. A., Lau L. M., Duncan E. L., Moy E. L., Reddel R. R. (2012) Loss of wild-type ATRX expression in somatic cell hybrids segregates with activation of alternative lengthening of telomeres. PLoS One 7, e50062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Santenard A., Ziegler-Birling C., Koch M., Tora L., Bannister A. J., Torres-Padilla M. E. (2010) Heterochromatin formation in the mouse embryo requires critical residues of the histone variant H3.3. Nat. Cell Biol. 12, 853–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwartzentruber J., Korshunov A., Liu X. Y., Jones D. T., Pfaff E., Jacob K., Sturm D., Fontebasso A. M., Quang D. A., Tönjes M., Hovestadt V., Albrecht S., Kool M., Nantel A., Konermann C., Lindroth A., Jäger N., Rausch T., Ryzhova M., Korbel J. O., Hielscher T., Hauser P., Garami M., Klekner A., Bognar L., Ebinger M., Schuhmann M. U., Scheurlen W., Pekrun A., Frühwald M. C., Roggendorf W., Kramm C., Dürken M., Atkinson J., Lepage P., Montpetit A., Zakrzewska M., Zakrzewski K., Liberski P. P., Dong Z., Siegel P., Kulozik A. E., Zapatka M., Guha A., Malkin D., Felsberg J., Reifenberger G., von Deimling A., Ichimura K., Collins V. P., Witt H., Milde T., Witt O., Zhang C., Castelo-Branco P., Lichter P., Faury D., Tabori U., Plass C., Majewski J., Pfister S. M., Jabado N. (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 [DOI] [PubMed] [Google Scholar]
- 32. Wu G., Broniscer A., McEachron T. A., Lu C., Paugh B. S., Becksfort J., Qu C., Ding L., Huether R., Parker M., Zhang J., Gajjar A., Dyer M. A., Mullighan C. G., Gilbertson R. J., Mardis E. R., Wilson R. K., Downing J. R., Ellison D. W., Zhang J., Baker S. J. (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 44, 251–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Janicki S. M., Tsukamoto T., Salghetti S. E., Tansey W. P., Sachidanandam R., Prasanth K. V., Ried T., Shav-Tal Y., Bertrand E., Singer R. H., Spector D. L. (2004) From silencing to gene expression. Real-time analysis in single cells. Cell 116, 683–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rafalska-Metcalf I. U., Powers S. L., Joo L. M., LeRoy G., Janicki S. M. (2010) Single cell analysis of transcriptional activation dynamics. PLoS One 5, e10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ahmad K., Henikoff S. (2002) Histone H3 variants specify modes of chromatin assembly. Proc. Natl. Acad. Sci. U.S.A. 99, 16477–16484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Raitskin O., Cho D. S., Sperling J., Nishikura K., Sperling R. (2001) RNA editing activity is associated with splicing factors in lnRNP particles. The nuclear pre-mRNA processing machinery. Proc. Natl. Acad. Sci. U.S.A. 98, 6571–6576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Newhart A., Negorev D. G., Rafalska-Metcalf I. U., Yang T., Maul G. G., Janicki S. M. (2013) Sp100A promotes chromatin decondensation at a CMV-promoter regulated transcription site. Mol. Biol. Cell 24, 1454–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Femino A. M., Fogarty K., Lifshitz L. M., Carrington W., Singer R. H. (2003) Visualization of single molecules of mRNA in situ. Methods Enzymol. 361, 245–304 [DOI] [PubMed] [Google Scholar]
- 39. Zhang R., Chen W., Adams P. D. (2007) Molecular dissection of formation of senescence-associated heterochromatin foci. Mol. Cell Biol. 27, 2343–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Everett R. D., Parada C., Gripon P., Sirma H., Orr A. (2008) Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J. Virol. 82, 2661–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wagner R. W., Nishikura K. (1988) Cell cycle expression of RNA duplex unwindase activity in mammalian cells. Mol. Cell Biol. 8, 770–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lukashchuk V., Everett R. D. (2010) Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J. Virol. 84, 4026–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim H. D., O'Shea E. K. (2008) A quantitative model of transcription factor-activated gene expression. Nat. Struct. Mol. Biol. 15, 1192–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zaslavsky E., Hershberg U., Seto J., Pham A. M., Marquez S., Duke J. L., Wetmur J. G., Tenoever B. R., Sealfon S. C., Kleinstein S. H. (2010) Antiviral response dictated by choreographed cascade of transcription factors. J. Immunol. 184, 2908–2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bintu L., Buchler N. E., Garcia H. G., Gerland U., Hwa T., Kondev J., Phillips R. (2005) Transcriptional regulation by the numbers. Models. Curr. Opin. Genet. Dev. 15, 116–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mirny L. A. (2010) Nucleosome-mediated cooperativity between transcription factors. Proc. Natl. Acad. Sci. U.S.A. 107, 22534–22539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Polach K. J., Widom J. (1996) A model for the cooperative binding of eukaryotic regulatory proteins to nucleosomal target sites. J. Mol. Biol. 258, 800–812 [DOI] [PubMed] [Google Scholar]
- 48. Proudfoot N. J. (1989) How RNA polymerase II terminates transcription in higher eukaryotes. Trends Biochem. Sci. 14, 105–110 [DOI] [PubMed] [Google Scholar]
- 49. Rohrbaugh M. L., Johnson J. E., 3rd, James M. D., Hardison R. C. (1985) Transcription unit of the rabbit β1 globin gene. Mol. Cell Biol. 5, 147–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Elsaesser S. J., Allis C. D. (2010) HIRA and Daxx constitute two independent histone H3.3-containing predeposition complexes. Cold Spring Harb. Symp. Quant. Biol. 75, 27–34 [DOI] [PubMed] [Google Scholar]
- 51. Wu B., Davey C. A. (2010) Using soft x-rays for a detailed picture of divalent metal binding in the nucleosome. J. Mol. Biol. 398, 633–640 [DOI] [PubMed] [Google Scholar]
- 52. Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 53. Jullien J., Astrand C., Szenker E., Garrett N., Almouzni G., Gurdon J. B. (2012) HIRA dependent H3.3 deposition is required for transcriptional reprogramming following nuclear transfer to Xenopus oocytes. Epigenetics Chromatin 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ng R. K., Gurdon J. B. (2008) Epigenetic memory of an active gene state depends on histone H3.3 incorporation into chromatin in the absence of transcription. Nat. Cell Biol. 10, 102–109 [DOI] [PubMed] [Google Scholar]
- 55. Cui B., Liu Y., Gorovsky M. A. (2006) Deposition and function of histone H3 variants in Tetrahymena thermophila. Mol. Cell Biol. 26, 7719–7730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sakai A., Schwartz B. E., Goldstein S., Ahmad K. (2009) Transcriptional and developmental functions of the H3.3 histone variant in Drosophila. Curr. Biol. 19, 1816–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. van der Heijden G. W., Derijck A. A., Pósfai E., Giele M., Pelczar P., Ramos L., Wansink D. G., van der Vlag J., Peters A. H., de Boer P. (2007) Chromosome-wide nucleosome replacement and H3.3 incorporation during mammalian meiotic sex chromosome inactivation. Nat. Genet. 39, 251–258 [DOI] [PubMed] [Google Scholar]
- 58. Torres-Padilla M. E., Bannister A. J., Hurd P. J., Kouzarides T., Zernicka-Goetz M. (2006) Dynamic distribution of the replacement histone variant H3.3 in the mouse oocyte and preimplantation embryos. Int. J. Dev. Biol. 50, 455–461 [DOI] [PubMed] [Google Scholar]
- 59. Eymery A., Callanan M., Vourc'h C. (2009) The secret message of heterochromatin. New insights into the mechanisms and function of centromeric and pericentric repeat sequence transcription. Int. J. Dev. Biol. 53, 259–268 [DOI] [PubMed] [Google Scholar]
- 60. Maison C., Bailly D., Peters A. H., Quivy J. P., Roche D., Taddei A., Lachner M., Jenuwein T., Almouzni G. (2002) Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat. Genet. 30, 329–334 [DOI] [PubMed] [Google Scholar]
- 61. Muchardt C., Guilleme M., Seeler J. S., Trouche D., Dejean A., Yaniv M. (2002) Coordinated methyl and RNA binding is required for heterochromatin localization of mammalian HP1α. EMBO Rep. 3, 975–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Grewal S. I., Elgin S. C. (2007) Transcription and RNA interference in the formation of heterochromatin. Nature 447, 399–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Huang R. C., Bonner J. (1965) Histone-bound RNA, a component of native nucleohistone. Proc. Natl. Acad. Sci. U.S.A. 54, 960–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nelson T., Wiegand R., Brutlag D. (1981) Ribonucleic acid and other polyanions facilitate chromatin assembly in vitro. Biochemistry 20, 2594–2601 [DOI] [PubMed] [Google Scholar]
- 65. Nakanishi S., Sanderson B. W., Delventhal K. M., Bradford W. D., Staehling-Hampton K., Shilatifard A. (2008) A comprehensive library of histone mutants identifies nucleosomal residues required for H3K4 methylation. Nat. Struct. Mol. Biol. 15, 881–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Govin J., Dorsey J., Gaucher J., Rousseaux S., Khochbin S., Berger S. L. (2010) Systematic screen reveals new functional dynamics of histones H3 and H4 during gametogenesis. Genes Dev. 24, 1772–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lewis P. W., Müller M. M., Koletsky M. S., Cordero F., Lin S., Banaszynski L. A., Garcia B. A., Muir T. W., Becher O. J., Allis C. D. (2013) Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chan K. M., Fang D., Gan H., Hashizume R., Yu C., Schroeder M., Gupta N., Mueller S., James C. D., Jenkins R., Sarkaria J., Zhang Z. (2013) The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 27, 985–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kapranov P., Cheng J., Dike S., Nix D. A., Duttagupta R., Willingham A. T., Stadler P. F., Hertel J., Hackermüller J., Hofacker I. L., Bell I., Cheung E., Drenkow J., Dumais E., Patel S., Helt G., Ganesh M., Ghosh S., Piccolboni A., Sementchenko V., Tammana H., Gingeras T. R. (2007) RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316, 1484–1488 [DOI] [PubMed] [Google Scholar]
- 70. He Y., Vogelstein B., Velculescu V. E., Papadopoulos N., Kinzler K. W. (2008) The antisense transcriptomes of human cells. Science 322, 1855–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kapranov P., Ozsolak F., Kim S. W., Foissac S., Lipson D., Hart C., Roels S., Borel C., Antonarakis S. E., Monaghan A. P., John B., Milos P. M. (2010) New class of gene-termini-associated human RNAs suggests a novel RNA copying mechanism. Nature 466, 642–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Affymetrix ENCODE Transcriptome Project, Cold Spring Harbor Laboratory ENCODE Transcriptome Project (2009) Post-transcriptional processing generates a diversity of 5′-modified long and short RNAs. Nature 457, 1028–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Venkatesh S., Smolle M., Li H., Gogol M. M., Saint M., Kumar S., Natarajan K., Workman J. L. (2012) Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature 489, 452–455 [DOI] [PubMed] [Google Scholar]
- 74. Lin L. J., Minard L. V., Johnston G. C., Singer R. A., Schultz M. C. (2010) Asf1 can promote trimethylation of H3 K36 by Set2. Mol. Cell Biol. 30, 1116–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Carrozza M. J., Li B., Florens L., Suganuma T., Swanson S. K., Lee K. K., Shia W. J., Anderson S., Yates J., Washburn M. P., Workman J. L. (2005) Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123, 581–592 [DOI] [PubMed] [Google Scholar]
- 76. Zaratiegui M., Castel S. E., Irvine D. V., Kloc A., Ren J., Li F., de Castro E., Marín L., Chang A. Y., Goto D., Cande W. Z., Antequera F., Arcangioli B., Martienssen R. A. (2011) RNAi promotes heterochromatic silencing through replication-coupled release of RNA Pol II. Nature 479, 135–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Porro A., Feuerhahn S., Reichenbach P., Lingner J. (2010) Molecular dissection of telomeric repeat-containing RNA biogenesis unveils the presence of distinct and multiple regulatory pathways. Mol. Cell Biol. 30, 4808–4817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Luke B., Panza A., Redon S., Iglesias N., Li Z., Lingner J. (2008) The Rat1p 5′ to 3′ exonuclease degrades telomeric repeat-containing RNA and promotes telomere elongation in Saccharomyces cerevisiae. Mol. Cell 32, 465–477 [DOI] [PubMed] [Google Scholar]
- 79. Pak J., Fire A. (2007) Distinct populations of primary and secondary effectors during RNAi in C. elegans. Science 315, 241–244 [DOI] [PubMed] [Google Scholar]
- 80. Zong J., Yao X., Yin J., Zhang D., Ma H. (2009) Evolution of the RNA-dependent RNA polymerase (RdRP) genes. Duplications and possible losses before and after the divergence of major eukaryotic groups. Gene 447, 29–39 [DOI] [PubMed] [Google Scholar]
- 81. Carninci P., Sandelin A., Lenhard B., Katayama S., Shimokawa K., Ponjavic J., Semple C. A., Taylor M. S., Engström P. G., Frith M. C., Forrest A. R., Alkema W. B., Tan S. L., Plessy C., Kodzius R., Ravasi T., Kasukawa T., Fukuda S., Kanamori-Katayama M., Kitazume Y., Kawaji H., Kai C., Nakamura M., Konno H., Nakano K., Mottagui-Tabar S., Arner P., Chesi A., Gustincich S., Persichetti F., Suzuki H., Grimmond S. M., Wells C. A., Orlando V., Wahlestedt C., Liu E. T., Harbers M., Kawai J., Bajic V. B., Hume D. A., Hayashizaki Y. (2006) Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 38, 626–635 [DOI] [PubMed] [Google Scholar]
- 82. Maida Y., Yasukawa M., Furuuchi M., Lassmann T., Possemato R., Okamoto N., Kasim V., Hayashizaki Y., Hahn W. C., Masutomi K. (2009) An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature 461, 230–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhu Y., Tomlinson R. L., Lukowiak A. A., Terns R. M., Terns M. P. (2004) Telomerase RNA accumulates in Cajal bodies in human cancer cells. Mol. Biol. Cell 15, 81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bryan T. M., Englezou A., Dalla-Pozza L., Dunham M. A., Reddel R. R. (1997) Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 3, 1271–1274 [DOI] [PubMed] [Google Scholar]
- 85. Grewal S. I. (2010) RNAi-dependent formation of heterochromatin and its diverse functions. Curr. Opin. Genet. Dev. 20, 134–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bivalkar-Mehla S., Vakharia J., Mehla R., Abreha M., Kanwar J. R., Tikoo A., Chauhan A. (2011) Viral RNA silencing suppressors (RSS). Novel strategy of viruses to ablate the host RNA interference (RNAi) defense system. Virus Res. 155, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Obbard D. J., Gordon K. H., Buck A. H., Jiggins F. M. (2009) The evolution of RNAi as a defence against viruses and transposable elements. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 99–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sioud M. (2007) RNA interference and innate immunity. Adv. Drug Deliv. Rev. 59, 153–163 [DOI] [PubMed] [Google Scholar]
- 89. Geoffroy M. C., Chelbi-Alix M. K. (2011) Role of promyelocytic leukemia protein in host antiviral defense. J. Interferon Cytokine Res. 31, 145–158 [DOI] [PubMed] [Google Scholar]
- 90. Delbarre E., Ivanauskiene K., Küntziger T., Collas P. (2013) DAXX-dependent supply of soluble (H3.3-H4) dimers into PML bodies pending deposition into chromatin. Genome Res. 23, 440–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.