

Background: Mercaptopyruvate sulfurtransferase (MST) generates H2S, a signaling molecule.
Results: The detailed kinetics and crystal structure of human MST with bound substrate are reported.
Conclusion: Thioredoxin is the preferred persulfide acceptor from MST.
Significance: The structure provides molecular insights into activation and stabilization of MST reaction intermediates.
Keywords: Crystal Structure, Enzyme Kinetics, Hydrogen Sulfide, Redox, Sulfur
Abstract
Mercaptopyruvate sulfurtransferase (MST) is a source of endogenous H2S, a gaseous signaling molecule implicated in a wide range of physiological processes. The contribution of MST versus the other two H2S generators, cystathionine β-synthase and γ-cystathionase, has been difficult to evaluate because many studies on MST have been conducted at high pH and have used varied reaction conditions. In this study, we have expressed, purified, and crystallized human MST in the presence of the substrate 3-mercaptopyruvate (3-MP). The kinetics of H2S production by MST from 3-MP was studied at pH 7.4 in the presence of various physiological persulfide acceptors: cysteine, dihydrolipoic acid, glutathione, homocysteine, and thioredoxin, and in the presence of cyanide. The crystal structure of MST reveals a mixture of the product complex containing pyruvate and an active site cysteine persulfide (Cys248-SSH) and a nonproductive intermediate in which 3-MP is covalently linked via a disulfide bond to an active site cysteine. The crystal structure analysis allows us to propose a detailed mechanism for MST in which an Asp-His-Ser catalytic triad is positioned to activate the nucleophilic cysteine residue and participate in general acid-base chemistry, whereas our kinetic analysis indicates that thioredoxin is likely to be the major physiological persulfide acceptor for MST.
Introduction
Since the first report of the role of H2S as a possible endogenous neuromodulator (1), H2S has rapidly emerged as a signaling molecule impacting a broad range of physiological functions (2–6). The low steady-state concentration of H2S in tissues, estimated to be in the 10–30 nm range (7–9), is a product of both its biogenesis and clearance rates (8). H2S is produced by the transsulfuration pathway enzymes, cystathionine β-synthase and γ-cystathionase, which are cytosolic and by 3-mercaptopyruvate sulfurtransferase (MST),2 which is localized in the cytoplasm and the mitochondrion (10–13). Detailed steady-state (14, 15) and pre-steady (16, 17) kinetic studies on cystathione β-synthase and γ-cystathionase enzymes at physiological pH have provided insights into the efficiency of the various H2S-generating reactions that they catalyze. In contrast, studies on human MST have lagged behind. This, together with a number of earlier studies on MST from various mammalian sources that were conducted at high pH and varied reaction conditions, have made it difficult to compare the kinetics of H2S generation by MST versus the transsulfuration pathway enzymes.
MST is expressed predominantly in kidney cells, liver cells, cardiac cells, proximal tubular epithelium, pericentral hepatocytes, and neuroglial cells (13). Mutations in the MST gene lead to mercaptolactate-cysteine disulfiduria, an inborn error of metabolism (18). Recent studies support the role of MST as a potentially important generator of H2S in some organisms and tissues (19–22). In the MST-dependent route for H2S generation, cysteine is converted via the action of aspartate (or cysteine) aminotransferase, to 3-MP, which is acted upon by MST to give pyruvate and an enzyme-bound persulfide (Fig. 1, a and b). The MST-bound persulfide can be transferred to an acceptor such as cyanide to generate a less toxic product, thiocyanate (23). Alternatively, H2S can be released from the MST-bound persulfide in the presence of a reducing system. Examples of nonphysiological reductants that can liberate H2S include DTT (21, 24) and 2-mercaptoethanol (23). The potential for physiologically relevant small molecule thiols such as GSH, homocysteine, and cysteine, to couple with MST, has not been evaluated in a kinetically defined system. Thioredoxin in the presence of thioredoxin reductase and NADPH, or dihydrolipoic acid (DHLA), are examples of physiologically relevant reducing systems that can release H2S from MST-bound persulfide (19, 25). Trypanosomal variants of MST have a C-terminal thioredoxin-like domain, suggesting that thioredoxin might be a physiologically relevant partner of MST in other organisms as well (23, 26).
FIGURE 1.
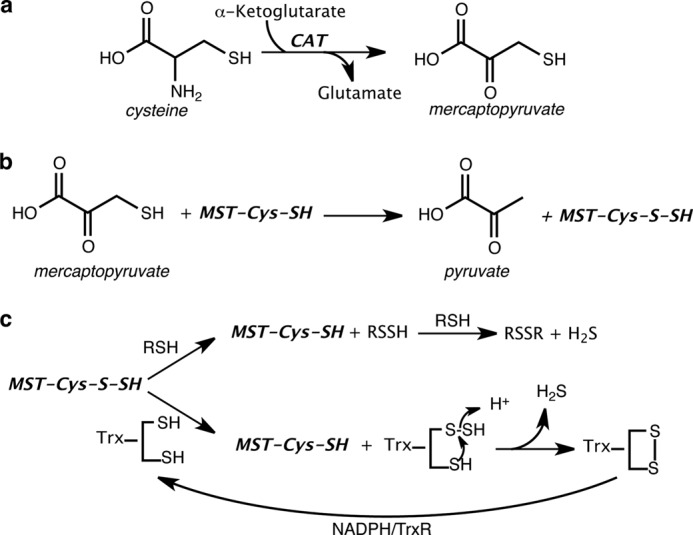
Reactions leading to H2S generation via the CAT/MST pathway. a, CAT catalyzes the transamination between cysteine and α-ketoglutarate to mercaptopyruvate and glutamate, respectively. b, MST catalyzes the sulfur transfer from 3-MP to an active site cysteine, giving pyruvate and MST-bound persulfide. c, MST-bound persulfide reacts with thiols or thioredoxin (in presence of NADPH/thioredoxin reductase) to generate H2S.
MST is a member of the sulfurtransferase family; it shares ∼60% sequence similarity with rhodanese (also known as thiosulfate sulfurtransferase), a prototypic member of this family. Both proteins have a conserved active site architecture with a catalytic cysteine residue, reflecting a common evolutionary origin (27). In rat MST, Cys247 is predicted to be the nucleophile that is persulfidated upon sulfur transfer from 3-MP (28). Two arginine residues are proposed to interact with the carbonyl oxygen and the carboxylate group of 3-MP and are important determinants of MST specificity for 3-MP versus thiosulfate (28).
In the present study, we report the purification of human MST and the kinetic analysis of H2S production in the presence of a range of physiologically relevant and artificial acceptors. We also report the first crystal structure of human MST that contains a mixture of a product complex (Cys248-SSH + pyruvate) and an intermediate, 3-MP linked to an active site cysteine via a disulfide bond. Our results allow us to propose a detailed reaction mechanism for MST in which a catalytic triad could be involved in activating the active site nucleophile (Cys248) and facilitating the reaction via general acid-base chemistry.
EXPERIMENTAL PROCEDURES
Materials
The sodium salt of 3-MP was purchased from Research Organics (Cleveland, OH). l-cysteine, dl-homocysteine, DTT, GSH, 2-mercaptoethanol, and NADPH were purchased from Sigma-Aldrich. DHLA was purchased from Santa Cruz Biotechnology (Dallas, TX).
Cloning of Human MST
Human MST cDNA cloned in the pOTB7 vector (ATCC number MGC-10492) was purchased from ATCC (Manassas, VA). The MST cDNA was PCR-amplified using Pfu ultra high fidelity DNA polymerase and the following primers: 5′-TACAGACATATGGTGTCGGCGCAATGG-3′ (forward) and 5′-CAACCGAAGCTTTCAGATGACATCCTCGGGC-3′ (reverse). The PCR product was subcloned into the pET28b Vector (Novagen) using NdeI and HindIII to generate the expression construct pET28b_hMST.
Expression and Purification of Human Thioredoxin and Thioredoxin Reductase
Expression constructs for human thioredoxin (cytoplasmic) and thioredoxin reductase were a generous gift from Dr. Vadim Gladyshev (Harvard Medical School). The cDNAs were expressed in Escherichia coli and purified as described previously (29, 30).
Expression and Purification of MST
The pET28b_hMST plasmid was co-transformed with the pTrc99A-EL plasmid harboring genes for the molecular chaperones GroES/GroEL, in E. coli BL21 (DE3). The transformed cells were grown overnight at 28 °C in 200 ml of Luria Bertani medium containing kanamycin (60 μg/ml) and ampicillin (100 μg/ml). The following day, six 1-liter volumes of Luria Bertani medium containing the same antibiotics were inoculated with the starter culture and grown at 28 °C (240 rpm). After 3 h, the temperature was reduced to 15 °C, and growth was continued until A600 reached 0.6. The cultures were induced with 100 μm isopropyl β-d-thiogalactopyranoside, and the cells were harvested 18 h later. The cells pellets were stored at −80 °C until use.
The cell pellets were suspended in 250 ml of 50 mm sodium phosphate buffer, pH 7.5, containing 500 mm NaCl, lysozyme (100 mg), MgCl2 (5 mm), DNase (10 mg), single protease inhibitor mixture tablet (Roche Applied Science), and 500 μm tris(2-carboxyethyl)phosphine. The cell suspension was stirred at 4 °C for 40 min and then sonicated (power setting = 7) for 10 min at 30-s intervals separated by 1-min cooling periods. The sonicate was centrifuged at 38,000 × g for 20 min, and the supernatant was loaded on to a nickel-nitrilotriacetic acid-agarose column (2.5 × 10 cm) pre-equilibrated with sodium phosphate buffer, pH 7.5, containing 500 mm NaCl and 20 mm imidazole. The column was washed with 300 ml of equilibration buffer containing 40 mm imidazole and eluted with 500 ml of a linear gradient ranging from 40 to 250 mm imidazole in 50 mm sodium phosphate buffer, pH 7.5, containing 500 mm NaCl. The fractions containing MST were identified following SDS-PAGE analysis, pooled, and concentrated to ∼10 ml and dialyzed overnight against 2 liters of 50 mm Tris buffer, pH 8.0. The dialyzed protein was loaded on to a DEAE-Sepharose column (2.5 × 15 cm) preequilibrated with 50 mm Tris buffer, pH 8.0. The column was washed with 200 ml of the buffer, and the protein was eluted with a 500-ml gradient ranging from 0 to 500 mm NaCl in the same buffer. The fractions of interest were concentrated and stored at −80 °C.
Estimation of the Molecular Mass of MST
Purified MST with an N-terminal His6 tag was separated on a prepacked Hiload 16/60 Superdex 200 column (GE Healthcare) with a flow rate of 0.5 ml min−1 in 50 mm Tris buffer, pH 8. The parent ion mass of MST (after purification by size exclusion chromatography) was determined by electrospray ionization mass spectrometry using a Q-Star XL Hybrid MS/MS mass spectrometer (Proteomics and Metabolomics Core Facility, Redox Biology Center, University of Nebraska, Lincoln).
MST Activity Assays
The following assays were used to determine MST activity. One unit of activity is defined as formation of 1 μmol of product min−1 mg−1 of protein at 37 °C. The Km and Vmax values were obtained by fitting the data sets to either the Michaelis-Menten or the Hill equation.
Activity Based on H2S Production
H2S formed by the reaction of 3-MP and reductants (2-mercaptoethanol, DTT, DHLA, l-cysteine, l-homocysteine, and GSH) was determined in the lead sulfide assay (15, 23). The reaction mixture containing 200 mm HEPES buffer, pH 7.4, 0.4 mm lead nitrate, BSA (100 μg/ml), 0.3 mm 3-MP, and 20 mm reductants was preincubated at 37 °C for 4 min, and the reaction was initiated by the addition of purified MST. Continuous formation of lead sulfide was monitored spectrophotometrically at 390 nm, and a molar extinction coefficient of 5,500 m−1 cm−1 for lead sulfide was used to determine its concentration (14). The Km values for the reductants were determined in the presence of 0.3 mm 3-MP and the following co-substrate concentrations: DTT (0–20 mm), DHLA (0–20 mm), l-cysteine (0–14 mm), l-homocysteine (0–70 mm), and GSH (0–100 mm). The Km for β-mercaptoethanol (0–300 mm) was determined in the presence of 0.5 mm 3-MP.
Activity Based on Thiocyanate Formation
Thiocyanate formation was monitored as described previously (23) with the following modifications. Briefly, 1 ml of the reaction mixture containing 200 mm HEPES buffer, pH 7.4, 3-MP (5 mm), KCN (25 mm), and bovine serum albumin (100 μg/ml) was preincubated at 37 °C for 4 min. The reaction was initiated by the addition of MST, and incubation was continued at 37 °C for 5 min. The reaction was stopped by the addition of 500 μl of formaldehyde, and 1.5 ml of the iron reagent (Fe(NO3)3, 50 g/liter; and 65% HNO3, 200 ml/liter) were added to detect FeSCN− formation at 460 nm. The Km for KCN was determined in the presence 5 mm 3-MP and 0–25 mm KCN. The Km for 3-MP was determined in the presence of 25 mm KCN and 0–2 mm 3-MP.
Thioredoxin-dependent Sulfurtransferase Activity
Oxidation of NADPH in a coupled MST-thioredoxin/thioredoxin reductase assay was measured as described previously (26). Briefly, a 1-ml reaction mixture containing 200 mm HEPES buffer, pH 7.4, 200 μm NADPH, 3.5 μm thioredoxin reductase, 20 μm thioredoxin, BSA (100 μg), and 3-MP (0–3 mm) was preincubated for 4 min at 37 °C. The reaction was started by addition of 0.3 μm MST, and the rate of NADPH oxidation was monitored at 340 nm using the extinction coefficient, 6,220 m−1 cm−1. Generation of H2S under these conditions (Fig. 1c) was monitored by the addition of 0.4 mm lead nitrate to the reaction mixture and detecting the increase in A390.
Determination of the pH Dependence of MST Activity
The pH dependence of MST was determined in the lead sulfide assay in the presence of 2-mercaptoethanol (50 mm) as described above. The following buffers were used: 200 mm citric acid-sodium citrate buffer (pH 4.5–6.5), HEPES buffer (pH 6.5–8.3), and boric acid-sodium borate buffer (pH 8.2–10.5). The data were fitted using a Gaussian function (Sigma-Plot 10).
Crystallization and Crystal Harvesting
Stock solutions of 9 mg/ml of truncated MST (t-MST, ΔN10, and ΔC8) in 50 mm HEPES, 100 mm NaCl, and 5% v/v glycerol were used for crystallization trials. Crystals of t-MST were obtained at 20 °C in 2 weeks by the vapor diffusion method from 1:1 mixtures of protein and reservoir solution in sitting drop plates. The reservoir solution contained 2 m ammonium sulfate, 0.1 m sodium acetate, pH 4.5, and 0.02 m betaine hydrochloride. Crystals of 3-MP bound to t-MST were obtained by soaking. Specifically, crystals of apo t-MST were transferred to a solution containing 2 m ammonium sulfate, 0.1 m sodium acetate, pH 4.5, 0.02 m betaine hydrochloride, 2 mm 3-MP, and 25% v/v glycerol. After 2 h of soaking, the crystals were harvested and were flash frozen in liquid N2. Crystals of 3-MP bound to t-MST were of space group C2 (a = 110.7 Å, b = 171.8 Å, c = 73.0 Å) with three molecules in the asymmetric unit (Matthews' coefficient VM = 3.1 Å3/Da for three molecules per asymmetric unit, and 60.5% solvent content).
Data Collection and Structure Determination
Diffraction data were collected at 100 K on beamline GM/CA-CAT 23-ID-B at the Advanced Photon Source, Argonne National Laboratory (Argonne, IL). The data were recorded on a Mar300 detector and processed with HKL2000 (31). Phases were obtained by molecular replacement using coordinates for the unpublished structure of human MST (Protein Data Bank code 3OLH) with the program PHASER (32). Initial simulated annealing refinement (torsional and cartesian) was performed for the model obtained from molecular replacement with phenix.refine (33) to remove potential model bias. Subsequently, restrained individual atomic refinement and restrained isotropic individual B-factor refinement with maximum likelihood targets using the Babinet model for bulk solvent scaling was performed using REFMAC5 (34) of the CCP4 suite (35). COOT (36) was used to manually correct the incorrectly modeled residues, and through successive iterative rounds of refinement and manual model building, the remaining residues were traced in the electron density to afford the structural model. In later rounds of refinement, electron density near the active site was assigned and modeled with 3-MP. The final 3-MP bound to t-MST model was refined to 2.15 Å resolution. Crystallographic information as well as refinement statistics are provided in Table 1. The geometric quality of the models was assessed with MolProbity (37). PyMOL (38) was used to create molecular images.
TABLE 1.
Data Collection and Refinement Statistics (Molecular Replacement)
| Protein | ΔMST |
|---|---|
| Data Collection | |
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 110.7, 171.9, 73.0 |
| α, β, γ (°) | 90, 117.2, 90 |
| Wavelength (Å) | 1.033 |
| Resolution (Å) | 50–2.16 (2.23–2.16) |
| Rsym (%) | 7.4 (50.3) |
| I/σI | 6.8 (1.7) |
| Completeness (%) | 96.1 (76.4) |
| Redundancy | 1.8 (1.7) |
| Refinement | |
| Resolution (Å) | 85.9–2.16 |
| No. reflections | 58794 |
| Rwork/Rfree | 0.185/0.220 |
| No. atoms | |
| Protein | 6620 |
| Water | 352 |
| Pyruvate | 18 |
| B-factors | |
| Protein | 32.4 |
| Water | 35.9 |
| Pyruvate | 31.4 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.011 |
| Bond angles (°) | 1.384 |
| Ramachandran plot (%) | |
| Favored/allowed/outliers | 97.0/3.0/0.0 |
| Clashscore, all atoms | 2.3 |
| MolProbity score | 1.29 |
| Protein Data Bank code | 4JGT |
RESULTS
Expression and Purification of MST
The human MST cDNA lacking 10 and 8 amino acids from the N and C termini, respectively, was cloned into the pET28b vector with an N-terminal six-histidine tag. The N-terminal residues were removed because they are predicted to represent the mitochondrial targeting sequence, whereas eight C-terminal residues were eliminated because they are predicted to be disordered. The yield of MST was ∼12 mg liter−1 of E. coli culture, and the protein was judged to be >95% pure based on SDS-PAGE analysis (Fig. 2a). This protein was used in the kinetic studies described below. MST migrated as a monomer by gel filtration chromatography on a Superdex 200 column and eluted with an apparent molecular mass of ∼34 kDa (not shown), consistent with the protein being a monomer. The exact molecular mass of the purified protein was determined by electrospray ionization mass spectrometry to be 33,455 Da (Fig. 2b) in good correspondence with the predicted molecular mass of 33,549 Da.
FIGURE 2.
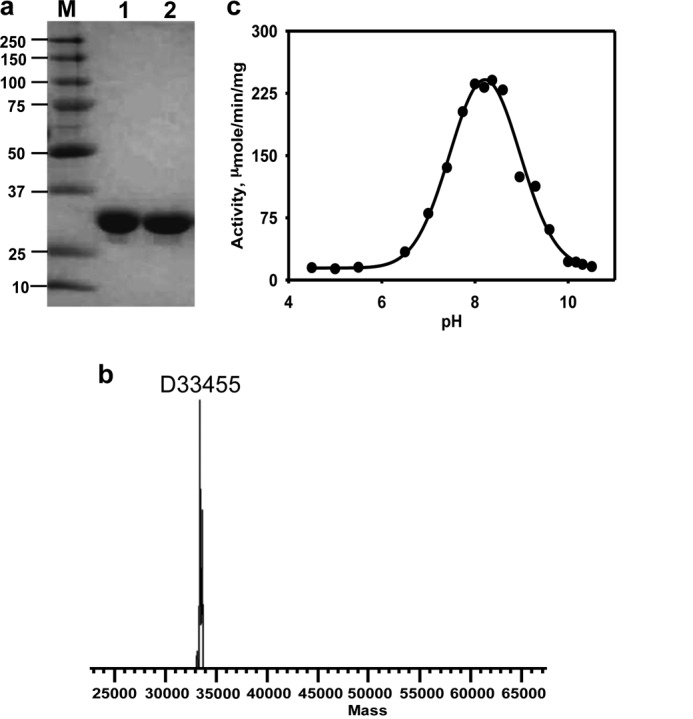
Characterization of MST. a, 10% SDS-polyacrylamide gel of purified recombinant human mercaptopyruvate sulfurtransferase. b, intact mass estimation of pure MST by electrospray ionization mass spectrometry. c, dependence of MST activity on pH. The buffers used in the different pH ranges were: citrate (pH 4.5–6.5), HEPES (pH 6.5–8.3), and borate (pH 8.2–10.5).
pH Dependence of MST Activity
In the literature, MST activity has often been measured at high pH (21, 39), making it difficult to evaluate the physiological significance of the reported kinetic data. The effect of pH on human MST activity was therefore determined in the lead sulfide assay in the presence of 2-mercaptoethanol as reductant and over a pH range of 4.5–10.5. MST exhibits a bell-shaped dependence on pH with a maximum at 8.21 ± 0.03 and inflection points at 7.2 and 9.2, respectively (Fig. 2c).
Kinetics of H2S Generation with Small Molecule Reductants
The kinetic parameters associated with H2S generation from 3-MP were determined in the presence of various small molecule acceptors including four physiologically relevant (DHLA, l-cysteine, l-homocysteine, and GSH) and two nonphysiological (2-mercaptoethanol and DTT) reductants (Table 2). The Km value for each reductant was separately determined at a saturating concentration of 3-MP (Table 2). Although a hyperbolic dependence of the reaction rate on the concentrations of DTT and DHLA was observed, a sigmoidal dependence was seen with the monothiols, 2-mercaptoethanol, cysteine, homocysteine, and GSH (Fig. 3). The Km values for DTT, DHLA, and cysteine, were ∼4 mm, and higher values were obtained for homocysteine (12.5 ± 1 mm), GSH (28 ± 2 mm), and 2-mercaptoethanol (108 ± 16 mm).
TABLE 2.
Summary of kinetic parameters of human MST
| Acceptor | Km Aa | Km 3-MPb | Vmax | kcat | kcat/Km A |
|---|---|---|---|---|---|
| mm | μm | μmol min−1 mg−1 | s−1 | mm−1 s−1 | |
| 2-Mercaptoethanol | 108 ± 16 | 130 ± 30 | 417 ± 38 | 229 | 2.1 |
| DTT | 4.6 ± 0.5 | 26 ± 5 | 11 ± 2 | 6.1 | 1.3 |
| Cyanide | 6 ± 1 | 350 ± 62 | 4.3 ± 0.3 | 2.4 | 0.4 |
| Dihydrolipoic acid | 4.4 ± 0.3 | 25 ± 6 | 3.1 ± 0.3 | 1.7 | 0.39 |
| l-Cysteine | 4.1 ± 0.6 | 22 ± 2 | 2.0 ± 0.2 | 1.1 | 0.27 |
| l-Homocysteine | 12.5 ± 1.6 | 30 ± 0.2 | 1.6 ± 0.2 | 0.8 | 0.06 |
| Glutathione | 28 ± 2 | 20 ± 0.4 | 0.61 ± 0.04 | 0.3 | 0.01 |
| Thioredoxin | 2.5 ± 0.4 × 10−3 | 350 ± 62 | 2.3 ± 0.2 | 1.3 | 520 |
a The Km values are for the acceptors (A) shown in the first column.
b The Km values were determined for mercaptopyruvate (3-MP) in the presence of the acceptors shown in the first column.
FIGURE 3.
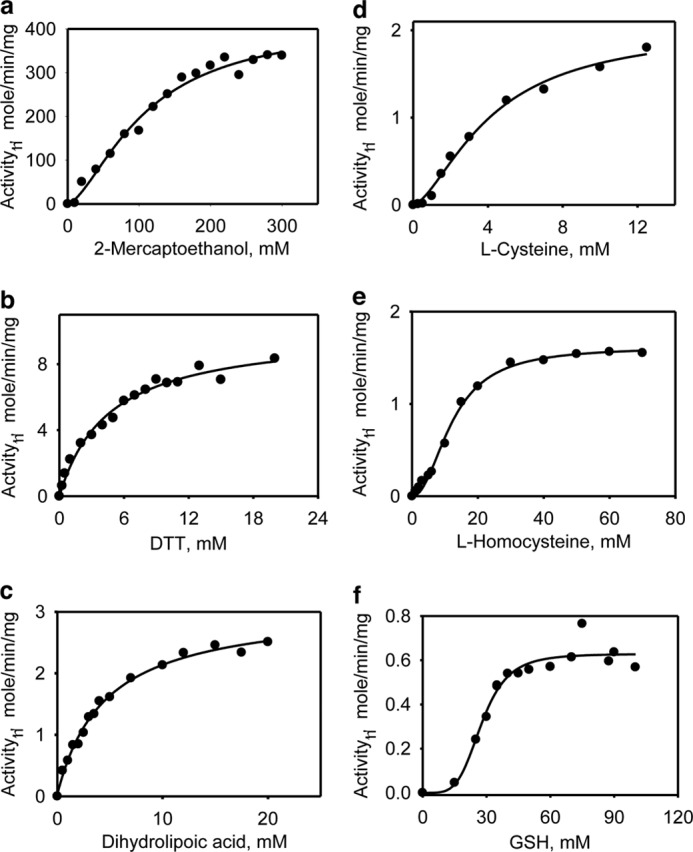
Kinetics of H2S generation by MST in the presence of varying concentrations of small molecule acceptors. The reaction mixtures contained 0–300 mm 2-mercaptoethanol (a), 0–20 mm dithiothreitol (b), 0–20 mm DHLA (c), 0–14 mm l-cysteine (d), 0–70 mm l-homocysteine (e), or 0–100 mm l-GSH (f) in 200 mm of HEPES, pH 7.4, 0.3 or 0.5 mm 3-MP, 0.1 mg/ml BSA, and 0.4 mm lead nitrate as described under “Experimental Procedures.” The data are representative of a minimum of two independent data sets.
The dependence of reaction rate on 3-MP concentration at saturating concentrations of the reductants revealed distinctly sigmoidal kinetics with homocysteine and cysteine (Fig. 4). The Km values for 3-MP were estimated at saturating concentrations of each acceptor and were similar for most (ranging from 20 to 30 μm) with the exception of the reaction with 2-mercaptoethanol (130 ± 30 μm), which yielded a significantly higher value. The Vmax for H2S generation clustered in the 0.61 ± 0.04 to 11 ± 2 μmol min−1 mg−1 protein range for all reductants but was considerably higher in the presence of 2-mercaptoethanol (417 ± 38 μmol min−1 mg−1 protein). The kcat/Km varied over a 200-fold range with 2-mercaptoethanol and DTT being the most proficient and GSH being the least proficient (Table 2).
FIGURE 4.
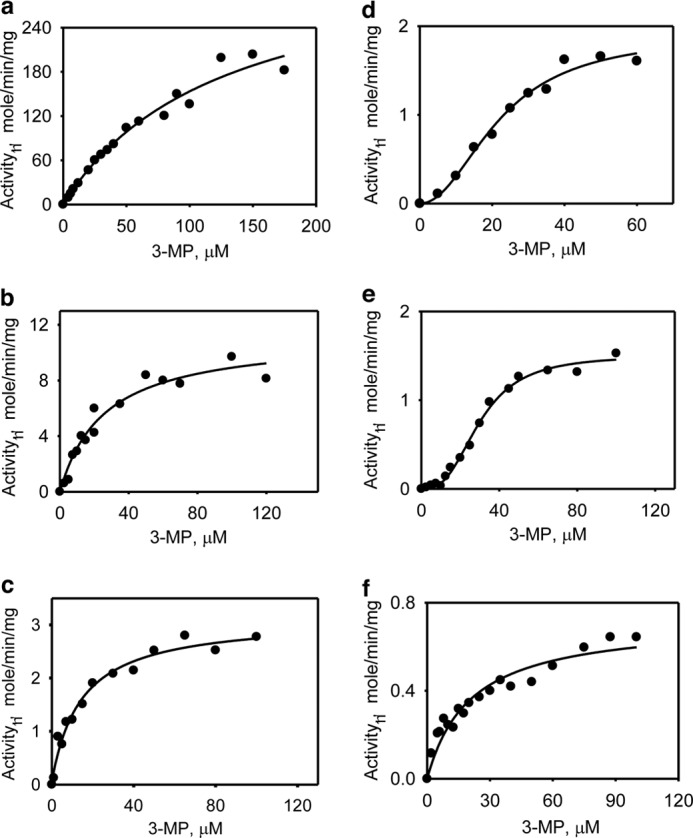
Kinetics of H2S generation by MST in the presence of varying concentrations of 3-MP. The reaction mixtures contained: 50 mm β-mercaptoethanol (a), 25 mm DTT (b), 25 mm DHLA (c), 20 mm l-cysteine (d), l-homocysteine (e), or 30 mm GSH (f) in 0–175 μm 3-MP and 200 mm of HEPES, pH 7.4, containing BSA (0.1 mg/ml) and lead nitrate (0.4 mm). Each panel is representative of a minimum of two independent data sets.
H2S Generation in the Presence of Thioredoxin
As shown in Fig. 1c, MST can be coupled to the thioredoxin/thioredoxin reductase/NADPH system for transfer and subsequent reduction of persulfide to H2S. We have determined the kinetic parameters for H2S generation by this system using the human proteins (Fig. 5). From the change in absorbance at 340 nm associated with oxidation of NADPH, the Km values for thioredoxin (Fig. 5a) and 3-MP (Fig. 5b) were estimated to be 2.5 ± 0.4 and 393 ± 26 μm, respectively (Table 2). The Vmax for NADPH oxidation was 2.3 ± 0.2 μmol min−1 mg−1 protein. To verify that H2S was indeed being generated in this reaction, its production was monitored directly in the lead acetate assay as described under “Experimental Procedures.” Under these conditions, the Vmax for H2S formation was 2.4 ± 0.2 μmol min−1 mg−1 protein. The kcat/Km for H2S production in the presence of thioredoxin (520 mm−1 s−1) was 400- and 250-fold higher than the most efficient nonphysiological reductants, DTT and β-mercaptoethanol, and 1,300–52,000-fold higher than the small molecule biological reductants (Table 2).
FIGURE 5.
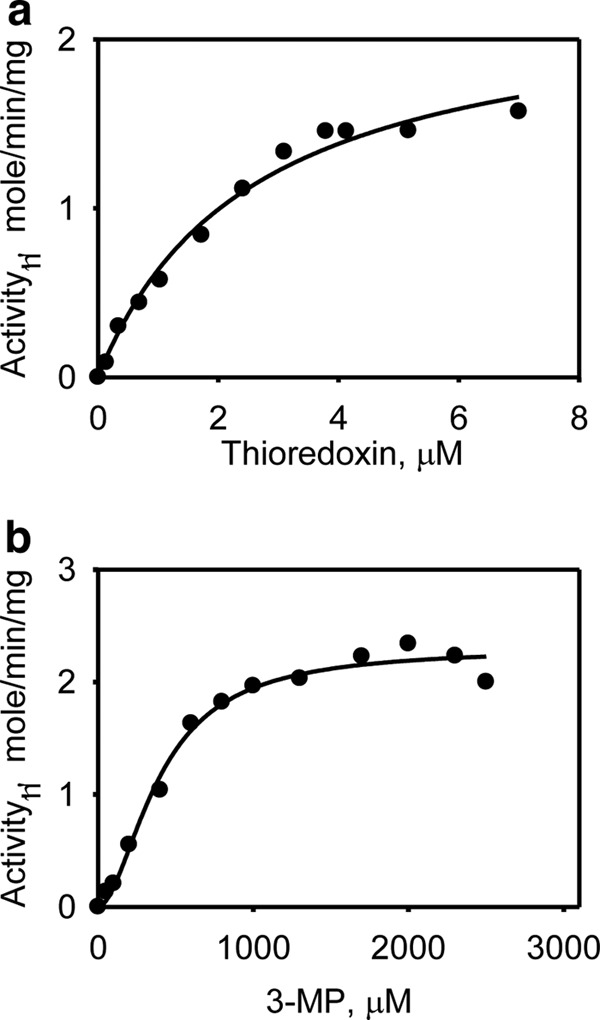
Kinetics of H2S generation by MST in the presence of thioredoxin. a, the dependence of the MST reaction on thioredoxin (0–7 μm) concentration was determined as described under “Experimental Procedures.” b, the dependence of the MST reaction on 3-MP in the presence of 3.5 μm thioredoxin reductase and 20 μm thioredoxin was determined as described under “Experimental Procedures.” The data are representative of a minimum of two independent data sets.
Kinetics of Cyanide Detoxification
One role long ascribed to MST is cyanide detoxification. The kinetic parameters for sulfur transfer to cyanide to form thiocyanate were assessed in the presence of 3-MP (Fig. 6). The estimated Vmax for this reaction was 4.3 ± 0.3 μmol min−1 mg−1 protein, and the Km values for KCN and 3-MP were 6 ± 1 mm and 350 ± 62 μm, respectively (Table 2).
FIGURE 6.
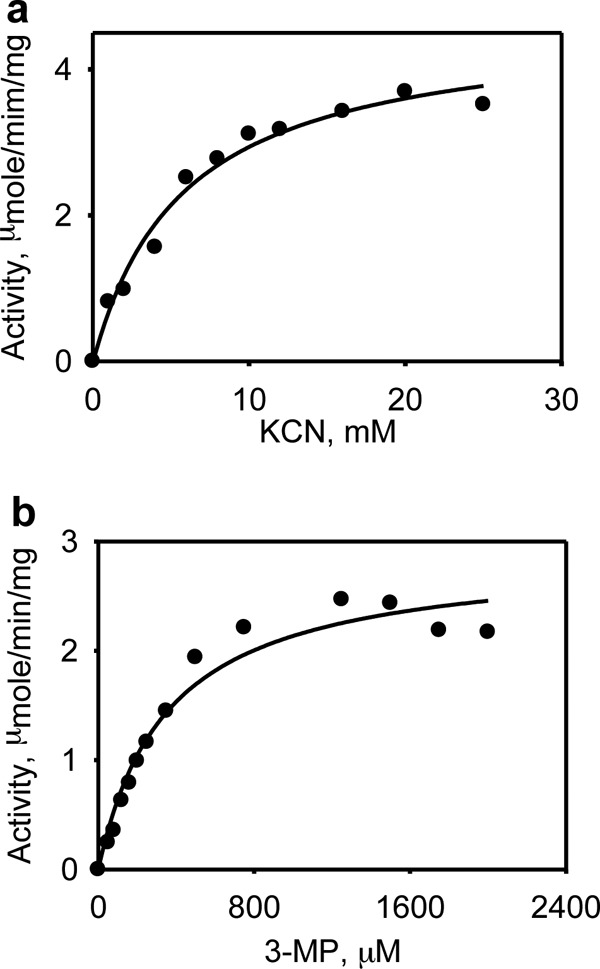
Kinetics of thiocyanate formation by MST. The dependence of the MST reaction on potassium cyanide concentration (a, 0–25 mm) and the dependence of the MST-catalyzed thiocyanate formation in the presence of 3-MP (b, 0–2 mm) were determined as described under “Experimental Procedures.” The data are representative of a minimum of two independent data sets.
Crystal Structure of the MST·3-MP Complex
The crystal structure of 3-MP bound to MST was solved at 2.15 Å resolution (Fig. 7a). The MST structure is comprised of two domains with a classical rhodanese-like fold: an N-terminal domain (residues 1–138, shown in blue in Fig. 7a), and a C-terminal domain (residues 165–285, shown in lavender), connected by a long linker (residues 139–164 shown in magenta) that wraps around and interacts tightly with both domains. A cleft located between the two rhodanese-like domains houses the active site (Fig. 7b). Both domains contribute residues that line the active site, and 3-MP binds in the cleft between them. The overall structure of 3-MP bound to MST is very similar to that of ligand-free MST (Protein Data Bank code 3OLH) (root mean square deviation of 0.38 Å based on all Cα atoms), indicating that 3-MP binding is not accompanied by large structural rearrangements (Fig. 7c).
FIGURE 7.
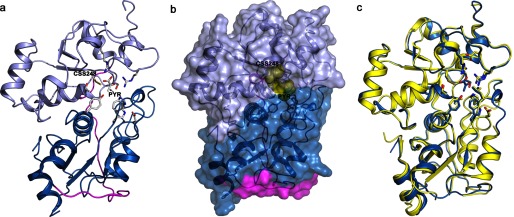
Structure of human MST. a, crystal structure of MST displayed in a ribbon drawing (N-terminal domain shown in blue; C-terminal domain shown in lavender; linker region between the two domains shown in magenta). The persulfide and pyruvate product as well as residues lining the active site are displayed as sticks. b, space filling model of MST with active site products persulfide and pyruvate shown as yellow spheres. c, superimposition of Cα atoms of the persulfide pyruvate-bound MST (blue) with ligand-free MST (Protein Data Bank code 3OLH; yellow). Active site residues are shown in stick display.
Interactions in the Active Site
Clear electron density is observed in the active site in the vicinity of Cys248 (Fig. 8a). We attempted to model two different sets of chemically plausible structures in the MST active site: a persulfide (Cys248-S-SH) plus pyruvate and a mixed disulfide between 3-MP and Cys248 (Cys248-S-SR). We were unable to unequivocally distinguish the two complexes from one another based on the active site electron density. Both MST·intermediate/product complexes were modeled and refined separately. The refinement statistics and fit to the electron density are slightly better for a persulfide (Cys248) plus pyruvate product complex (Fig. 8, b and c). Based on our results, we cannot exclude the possibility that we have a mixed population of both species in the active site. In both complexes, the positions of the carboxyl and carbonyl groups in 3-MP or pyruvate are indistinguishable. The main difference between the two models is in the position of the C3 methylene group of 3-MP or the equivalent C3 methyl group in pyruvate. We use the model of the persulfide plus pyruvate product complex (Fig. 8c) to describe the interactions in the active site in the next section.
FIGURE 8.

Close-up of the active site of human MST. a, unbiased experimental electron density in the MST active site based on a composite omit map. The green contours at 1 σ represent electron density at the active site prior to modeling and refinement of any substrates or products. The light blue contours at 1 σ represent unbiased electron density of selected active side residues. b, refined electron density (weighted 2Fo − Fc) with 3-MP in the active site contoured at 1 σ and shown in light blue. c, refined electron density (weighted 2Fo − Fc) with pyruvate and a persulfide in the active site contoured at 1 σ and shown in light blue. d, important active site residues and their interactions with pyruvate and persuldide are highlighted into two views of the active site.
An extensive hydrogen-bonding network is present between pyruvate and MST that includes Arg188 and Arg197, which hydrogen bond with the carboxyl group of pyruvate. Arg197 is also engaged in a hydrogen bond interaction with the carbonyl group of pyruvate. These two arginine residues were previously predicted to be important for substrate interactions (28, 40). In addition, Tyr108 and Thr253 form water-mediated contacts with an oxygen atom of the carboxylate group in pyruvate. Ser250 forms a hydrogen bonding interaction with the pyruvate carbonyl group. Previous work on MST from Leishmania major (40) suggested that MST contains a serine protease-like catalytic triad, predicted to comprise Ser250-His74-Asp63 in the human sequence. Our structure supports this prediction, with His74 hydrogen-bonded to Ser250 and Asp63 hydrogen-bonded to His74 (Fig. 8c). The C-terminal domain donates residues that make polar contacts with pyruvate such as Arg188, Arg197, Ser250, and Cys248. The methyl group of the pyruvate faces a region lined by Trp36 and Tyr108 that reside in the N-terminal domain of MST. The N-terminal domain also donates two of the catalytic triad residues, Asp63 and His74.
Structural Comparison of MST·3-MP and Rhodanese
MST is a member of the rhodanese/Cdc24 protein phosphatase superfamily. Superimposition of the substrate-bound MST (blue) and its closest structural homolog based on a Dali search, bovine rhodanese (Protein Data Bank code 1DP2; yellow), is shown in Fig. 9. The nucleophilic active site cysteine is conserved in rhodanese, whereas Arg197 and Ser250 in MST are replaced by an aspartate and a lysine, respectively, in rhodanese.
FIGURE 9.

Structural comparison of rhodanese and MST. a, superimposition of structures based on Cα traces of the 3-MP-bound MST and bovine rhodanese (Protein Data Bank code 1DP2). The structures are displayed as ribbon drawings with MST in blue and rhodanese shown in yellow. Active site residues are shown in stick display. b, close-up view of the active sites of MST and rhodanese.
DISCUSSION
3-MP, derived from cysteine via a transamination reaction, is an intermediate in the cysteine catabolism pathway (Fig. 1). The sulfur atom of 3-MP can be transferred via MST to various thiophilic acceptors. The presence of MST variants fused to thioredoxin in some protozoan parasites (23, 26) strongly suggests that thioredoxin is a physiological persulfide acceptor, and indeed, rat MST has been shown to couple to it (25). More recently, DHLA has been added to the list of potential physiological acceptors of MST, whereas cysteine and GSH were reportedly unable to couple to MST (19). In this study, we have determined the kinetic properties of human MST using highly pure enzyme and assessed its reactivity with a range of acceptors. Together with the first crystal structure of MST with bound substrate/product, our study provides insights into the reaction mechanism of this relatively poorly characterized H2S generator and lays the foundation for comparative analysis of the relative contributions of MST versus cystathione β-synthase and γ-cystathionase to H2S biogenesis.
Kinetic Analysis Human MST
MST can couple to both monothiols (2-mercaptoethanol, GSH, cysteine, and homocysteine) and dithiols (DTT, thioredoxin, and DHLA) for sulfur transfer from 3-MP (Figs. 3–5). In these reactions, H2S is eliminated following persulfide transfer as described by Reactions 1–3.
The steady-state kinetic analysis shows a hyperbolic dependence of the reaction rate of H2S production on dithiol acceptor concentration. In contrast, the kinetics are clearly sigmoidal in the presence of monothiols (Figs. 3 and 4). Although a single equivalent of dithiol is sufficient for the liberation of H2S from the transferred persulfide (Fig. 1c), two equivalents of monothiols are needed per transferred persulfide (Reactions 2 and 3). Based on kcat/Km values, a 52,000-fold range in efficacy of H2S production from 3-MP is observed in the presence of the various acceptors (Table 2). Of the physiological acceptors, thioredoxin is the most efficient (520 mm−1 s−1), and GSH is the least (0.01 mm−1 s−1). DHLA, cysteine, and homocysteine are intermediate, being ∼1,000–9,000-fold less efficient than thioredoxin.
A previous study with extracts from human lung adenocarcinoma cells (A549) failed to detect H2S production in the presence of 1 mm cysteine or GSH (19). This discrepancy between our results is likely due to the use in the previous study of crude cell extracts in which other enzymes that could compete for the utilization of the thiols were present combined with the high Km values for cysteine (4.1 ± 0.6 mm) and GSH (28 ± 2 mm) for MST-dependent H2S production (Table 2). The Km for DHLA is also high (4.4 ± 0.3 mm) compared with its reported physiological concentration (∼4–10 μm in murine tissues (41). However, much of the DHLA pool is covalently bound to proteins, and exogenous DHLA added to cell extracts would not have competed for these binding sites and therefore been available for the MST reaction in the cell extract assay. DHLA is a derivative of the hydrophobic octanoic acid and is unlikely to readily couple to soluble proteins like MST. In contrast to DHLA, the Km for human thioredoxin is low (2.5 ± 0.4 × 10−3 mm) and in the concentration range found in cells (∼20 μm) (42). On the other hand, the high Km for 3-MP in the thioredoxin-dependent reaction is high (350 ± 62 μm) relative to the Km values in the presence of other biotic thiol acceptors (20–30 μm) and raises questions about the relevance of thioredoxin-dependent H2S production by MST. The relative concentrations of each acceptor in the compartments where MST functions, together with their Km values, will determine their relative importance in coupling to MST for H2S biogenesis in the cellular milieu.
Proposed Reaction Mechanism for MST Based on the MST·3MP Structure
Our structure of MST with pyruvate or 3-MP bound provides rich insights into interactions between the substrate and active site residues. Two basic residues, Arg188 and Arg197 engage in electrostatic interactions with the carboxyl and carbonyl groups of pyruvate (Fig. 8). Mutations of the corresponding arginines in rat MST result in 10-fold (R196G) and 60-fold (R187G) increases in the Km for 3-MP (28). In the structures of the E. coli and L. major MSTs, a persulfide intermediate stabilized via hydrogen bonds with multiple backbone amides was seen (27, 40). The nucleophilic cysteine in MST needs to be activated to initiate the sulfur transfer reaction. Mutation of the corresponding cysteine residue in rat MST results in no detectable sulfurtransferase activity (28). A catalytic triad first observed in the L. major MST (40) comprising residues Ser250, His74, and Asp63 in human MST could be involved in activating Cys248. In serine proteases, the histidine and aspartate residues lower the pKa of the serine hydroxyl moiety, thereby activating it for covalent catalysis. We propose that in human MST, His74 could abstract a proton from Ser250, which in turn activates the Cys248 thiol for attack on the substrate thiol (Fig 10, step 1), leading to formation of an enzyme-bound persulfide and pyruvate anion (2). The resulting oxyanion intermediate could be stabilized via interactions with the hydroxyl group of Ser250, the side chain of Arg197, and by the positive electrostatic potential in the MST active site. Mutation of the corresponding serine residue in rat MST has a modest ∼10-fold effect on kcat without affecting the Km for 3-MP. Hence, other strategies for enhancing the reactivity of Cys248 must be deployed and could include orientation of α-helix dipoles (residues 253–264 and 276–283) in the MST active site (40). An alternative role has been postulated for the catalytic triad, i.e. in electrophilic catalysis via activation of the serine residue, which then polarizes the carbonyl group of 3-MP promoting attack by the nucleophilic cysteine (40).
FIGURE 10.

Postulated reaction mechanism of MST. Two arginine residues (Arg188 and Arg197) anchor the substrate carboxylate and carbonyl groups, whereas the catalytic triad (Asp63-His74-Ser250) in MST is postulated to activate Cys248 (step 1) for nucleophile attack on 3-MP (red) to give an enzyme bound persulfide. Release of the pyruvate anion following release and binding of the sulfur acceptor (step 2) set up the second half-reaction. Nucleophilic attack of the acceptor (a small molecule (di)thiol or thioredoxin) on the sulfane sulfur results in formation of the product persulfide, which is released (step 3) to complete the catalytic cycle. For clarity, not all interactions between the hydroxyl group of Ser250 and the substrate discussed in the text or the other steps in which the catalytic triad might participate in acid-base catalysis are shown.
Tautomerization and release of pyruvate followed by binding of an acceptor would initiate the second half-reaction, i.e. transfer of the sulfane sulfur and regeneration of the active site cysteine thiol (step 2). In this step, the His74–Ser250 dyad could be involved in acid-base catalysis, protonating the methylene group in the enolate tautomer of pyruvate prior to its departure from the active site. Our crystal structure with the Cys248 persulfide and pyruvate represents a product complex formed at the end of the first half-reaction. Transfer of the sulfane sulfur from the persulfide to an acceptor completes the second half-reaction (step 3). In the crystal structure of MST with pyruvate bound, the hydroxyl group of serine is too far from the Cys248 thiol to serve as a general base. It is possible that as the reaction progresses, the Ser250 hydroxyl is oriented away from Cys248 and toward the carbonyl group of 3-MP, stabilizing the nascent negative charge in the transition state.
Based on the proposed reaction mechanism, how can the presence of a mixed disulfide intermediate between Cys248 and 3-MP be explained? A mixed disulfide intermediate formed via oxidative addition of the active site cysteine and 3-MP has been proposed for MST (27). However, this mechanism seems unlikely because the MST reaction does not require a two-electron acceptor. Instead, formation of the mixed disulfide modeled in the crystal structure might be accounted for as follows. In the second half-reaction catalyzed by MST, the catalytic triad is postulated to activate the substrate thiol for nucleophilic attack on the sulfane sulfur on Cys248 (Fig. 11a). Consistent with this model, the hydroxyl group of Ser250 is closer to the sulfur on 3-MP (4.1 Å) than to the sulfur on Cys248 (5.5 Å). Thiol activation sets up a sulfur transfer reaction from the enzyme to the acceptor and regenerates the resting form of MST. An alternative and presumably side reaction occurs when the catalytic triad activates the MST-bound persulfide instead, setting up a nucleophilic attack on the C-3 methylene carbon of 3-MP and eliminating H2S (Fig. 11b). The disulfide could undergo a second round of sulfur transfer in the presence of a small molecule acceptor to regenerate resting enzyme. Controlling the orientation of the Ser250 hydroxyl group thus might be critical for ensuring the fidelity of the reaction in the first and second half-reactions. Mutation of Ser250 might lead to a change in the identity and distribution of products and warrants evaluation.
FIGURE 11.

Mechanism of sulfur transfer from the persulfide intermediate in MST and a side reaction with the sulfur acceptor. a, the catalytic triad is postulated to play a role in activating Ser250 during transfer of the MST-bound persulfide to an acceptor depicted here as 3-MP. b, the structure of the mixed disulfide seen in the crystal structure of 3-MST could arise from adventitious activation of the sulfane sulfur by Ser250, resulting in it attacking the C-3 methylene carbon of 3-MP. The products of this reaction are H2S and an enzyme-bound mixed disulfide between Cys248 and substrate.
It is possible that the low pH and temperature conditions used for crystallization promoted capture of the disulfide intermediate formed in a side reaction. Nevertheless, the structure provides valuable insights into interactions between pyruvate/3-MP and residues lining the MST active site. 3-MP is known to function as both a sulfur donor and an acceptor (10). Interestingly, attack of the cysteine thiolate on the methylene carbon of 3-cholorpyruvate, a stoichiometric inhibitor of MST, has been observed and generates a covalent adduct (43). This is similar to the postulated attack of the persulfide on the C-3 carbon of 3-MP to generate the observed mixed disulfide seen in the crystal structure (Fig. 11b) and further reinforces the importance of controlling reactivity and orientation of active site residues to promote the desired reaction while suppressing unwanted side reactions, exemplifying the principle of negative catalysis (44).
Acknowledgments
We thank Dr. Omer Kabil for helpful discussions and Dr. Nandakumar Madayiputhiya (University of Nebraska, Lincoln) for the mass spectrometric analysis of MST.
This work was supported, in whole or in part, by National Institutes of Health Grant HL58984.
The atomic coordinates and structure factors (code 4JGT) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- MST
- 3-mercaptopyruvate sulfurtransferase
- 3-MP
- 3-mercaptopyruvate
- DHLA
- dihydrolipoic acid.
REFERENCES
- 1. Abe K., Kimura H. (1996) The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 16, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yang G., Wu L., Jiang B., Yang W., Qi J., Cao K., Meng Q., Mustafa A. K., Mu W., Zhang S., Snyder S. H., Wang R. (2008) H2S as a physiologic vasorelaxant. Hypertension in mice with deletion of cystathionine γ-lyase. Science 322, 587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Elrod J. W., Calvert J. W., Morrison J., Doeller J. E., Kraus D. W., Tao L., Jiao X., Scalia R., Kiss L., Szabo C., Kimura H., Chow C. W., Lefer D. J. (2007) Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. U.S.A. 104, 15560–15565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li L., Bhatia M., Zhu Y. Z., Zhu Y. C., Ramnath R. D., Wang Z. J., Anuar F. B., Whiteman M., Salto-Tellez M., Moore P. K. (2005) Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 19, 1196–1198 [DOI] [PubMed] [Google Scholar]
- 5. Linden D. R., Levitt M. D., Farrugia G., Szurszewski J. H. (2010) Endogenous production of H2S in the gastrointestinal tract. Still in search of a physiologic function. Antioxid. Redox. Signal. 12, 1135–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Olson K. R., Whitfield N. L. (2010) Hydrogen sulfide and oxygen sensing in the cardiovascular system. Antioxid. Redox. Signal. 12, 1219–1234 [DOI] [PubMed] [Google Scholar]
- 7. Furne J., Saeed A., Levitt M. D. (2008) Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R1479–R1485 [DOI] [PubMed] [Google Scholar]
- 8. Vitvitsky V., Kabil O., Banerjee R. (2012) High turnover rates for hydrogen sulfide allow for rapid regulation of its tissue concentrations. Antioxid. Redox. Signal. 17, 22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Whitfield N. L., Kreimier E. L., Verdial F. C., Skovgaard N., Olson K. R. (2008) Reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1930–1937 [DOI] [PubMed] [Google Scholar]
- 10. Jarabak R., Westley J. (1978) Steady-state kinetics of 3-mercaptopyruvate sulfurtransferase from bovine kidney. Arch. Biochem. Biophys. 185, 458–465 [DOI] [PubMed] [Google Scholar]
- 11. Nagahara N., Ito T., Minami M. (1999) Mercaptopyruvate sulfurtransferase as a defense against cyanide toxication. Molecular properties and mode of detoxification. Histol. Histopathol. 14, 1277–1286 [DOI] [PubMed] [Google Scholar]
- 12. Nakamura T., Yamaguchi Y., Sano H. (2000) Plant mercaptopyruvate sulfurtransferases. Molecular cloning, subcellular localization and enzymatic activities. Eur. J. Biochem. 267, 5621–5630 [DOI] [PubMed] [Google Scholar]
- 13. Nagahara N., Ito T., Kitamura H., Nishino T. (1998) Tissue and subcellular distribution of mercaptopyruvate sulfurtransferase in the rat. Confocal laser fluorescence and immunoelectron microscopic studies combined with biochemical analysis. Histochem. Cell Biol. 110, 243–250 [DOI] [PubMed] [Google Scholar]
- 14. Chiku T., Padovani D., Zhu W., Singh S., Vitvitsky V., Banerjee R. (2009) H2S biogenesis by cystathionine γ-lyase leads to the novel sulfur metabolites, lanthionine and homolanthionine, and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 284, 11601–11612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Singh S., Padovani D., Leslie R. A., Chiku T., Banerjee R. (2009) Relative contributions of cystathionine β-synthase and γ-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem. 284, 22457–22466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Singh S., Ballou D. P., Banerjee R. (2011) Pre-steady-state kinetic analysis of enzyme-monitored turnover during cystathionine β-synthase-catalyzed H2S generation. Biochemistry 50, 419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yadav P. K., Banerjee R. (2012) Detection of reaction intermediates during human cystathionine β-synthase-monitored turnover and H2S production. J. Biol. Chem. 287, 43464–43471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crawhall J. C., Parker R., Sneddon W., Young E. P., Ampola M. G., Efron M. L., Bixby E. M. (1968) β Mercaptolactate-cysteine disulfide. Analog of cystine in the urine of a mentally retarded patient. Science 160, 419–420 [DOI] [PubMed] [Google Scholar]
- 19. Mikami Y., Shibuya N., Kimura Y., Nagahara N., Ogasawara Y., Kimura H. (2011) Thioredoxin and dihydrolipoic acid are required for 3-mercaptopyruvate sulfurtransferase to produce hydrogen sulfide. Biochem. J. 439, 479–485 [DOI] [PubMed] [Google Scholar]
- 20. Shatalin K., Shatalina E., Mironov A., Nudler E. (2011) H2S. A universal defense against antibiotics in bacteria. Science 334, 986–990 [DOI] [PubMed] [Google Scholar]
- 21. Shibuya N., Tanaka M., Yoshida M., Ogasawara Y., Togawa T., Ishii K., Kimura H. (2009) 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 11, 703–714 [DOI] [PubMed] [Google Scholar]
- 22. Modis K., Coletta C., Erdelyi K., Papapetropoulos A., Szabo C. (2012) Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 27, 601–611 [DOI] [PubMed] [Google Scholar]
- 23. Williams R. A., Kelly S. M., Mottram J. C., Coombs G. H. (2003) 3-Mercaptopyruvate sulfurtransferase of Leishmania contains an unusual C-terminal extension and is involved in thioredoxin and antioxidant metabolism. J. Biol. Chem. 278, 1480–1486 [DOI] [PubMed] [Google Scholar]
- 24. Kabil O., Banerjee R. (2010) The redox biochemistry of hydrogen sulfide. J. Biol. Chem. 285, 21903–21907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nagahara N., Katayama A. (2005) Post-translational regulation of mercaptopyruvate sulfurtransferase via a low redox potential cysteine-sulfenate in the maintenance of redox homeostasis. J. Biol. Chem. 280, 34569–34576 [DOI] [PubMed] [Google Scholar]
- 26. Westrop G. D., Georg I., Coombs G. H. (2009) The mercaptopyruvate sulfurtransferase of Trichomonas vaginalis links cysteine catabolism to the production of thioredoxin persulfide. J. Biol. Chem. 284, 33485–33494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Spallarossa A., Forlani F., Carpen A., Armirotti A., Pagani S., Bolognesi M., Bordo D. (2004) The “rhodanese” fold and catalytic mechanism of 3-mercaptopyruvate sulfurtransferases. Crystal structure of SseA from Escherichia coli. J. Mol. Biol. 335, 583–593 [DOI] [PubMed] [Google Scholar]
- 28. Nagahara N., Nishino T. (1996) Role of amino acid residues in the active site of rat liver mercaptopyruvate sulfurtransferase. J. Biol. Chem. 271, 27395–27401 [DOI] [PubMed] [Google Scholar]
- 29. Turanov A. A., Hatfield D. L., Gladyshev V. N. (2010) Characterization of protein targets of mammalian thioredoxin reductases. Methods Enzymol. 474, 245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Su D., Berndt C., Fomenko D. E., Holmgren A., Gladyshev V. N. (2007) A conserved cis-proline precludes metal binding by the active site thiolates in members of the thioredoxin family of proteins. Biochemistry 46, 6903–6910 [DOI] [PubMed] [Google Scholar]
- 31. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode, in Macromolecular Crystallography, Part A, pp. 307–326 [DOI] [PubMed] [Google Scholar]
- 32. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX. Building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 34. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 35.(1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 36. Emsley P., Cowtan K. (2004) COOT. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 37. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity. All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schrodinger L. L. C. (2010) The PyMOL Molecular Graphics System, Version 1.3r1 [Google Scholar]
- 39. Nagahara N., Okazaki T., Nishino T. (1995) Cytosolic mercaptopyruvate sulfurtransferase is evolutionarily related to mitochondrial rhodanese. Striking similarity in active site amino acid sequence and the increase in the mercaptopyruvate sulfurtransferase activity of rhodanese by site-directed mutagenesis. J. Biol. Chem. 270, 16230–16235 [DOI] [PubMed] [Google Scholar]
- 40. Alphey M. S., Williams R. A., Mottram J. C., Coombs G. H., Hunter W. N. (2003) The crystal structure of Leishmania major 3-mercaptopyruvate sulfurtransferase. A three-domain architecture with a serine protease-like triad at the active site. J. Biol. Chem. 278, 48219–48227 [DOI] [PubMed] [Google Scholar]
- 41. Kataoka H., Hirabayashi N., Makita M. (1993) Analysis of lipoic acid in biological samples by gas chromatography with flame photometric detection. J. Chromatogr. 615, 197–202 [DOI] [PubMed] [Google Scholar]
- 42. Holmgren A. (2008) The thioredoxin system. in Redox Biochemistry (Banerjee R., ed) John Wiley & Sons, Hoboken, NJ [Google Scholar]
- 43. Nagahara N., Sawada N., Nakagawa T. (2004) Affinity labeling of a catalytic site, cysteine (247), in rat mercaptopyruvate sulfurtransferase by chloropyruvate as an analog of a substrate. Biochimie 86, 723–729 [DOI] [PubMed] [Google Scholar]
- 44. Retey J. (1990) Enzymic reaction selectivity by negative catalysis or how do enzymes deal with highly reactive intermediates. Angew. Chem. Int. Ed. Engl. 29, 355–361 [Google Scholar]