

Abstract
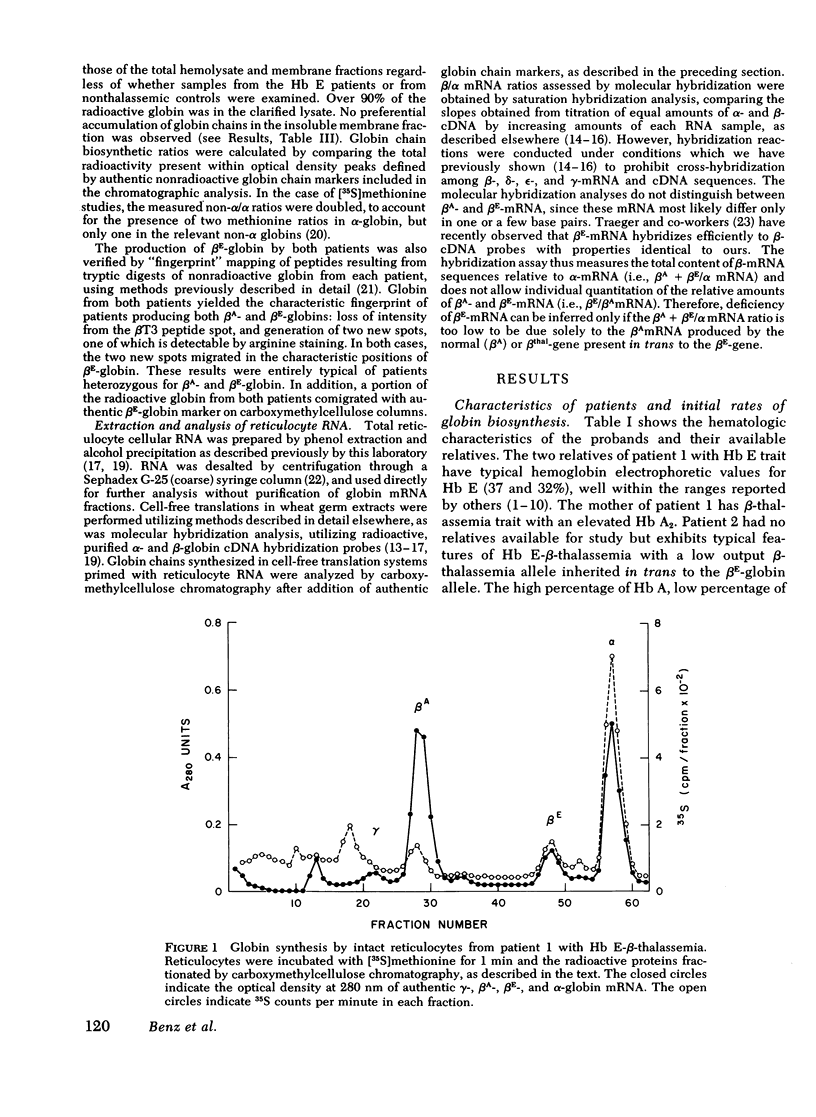
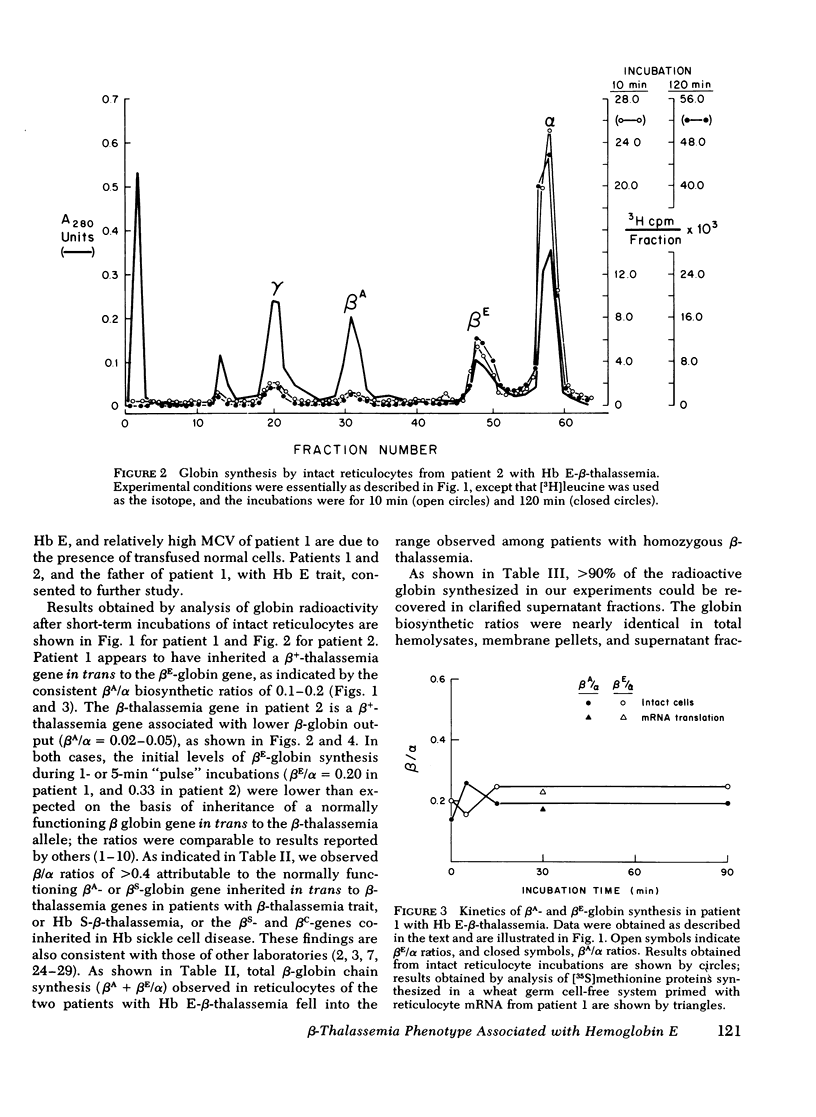
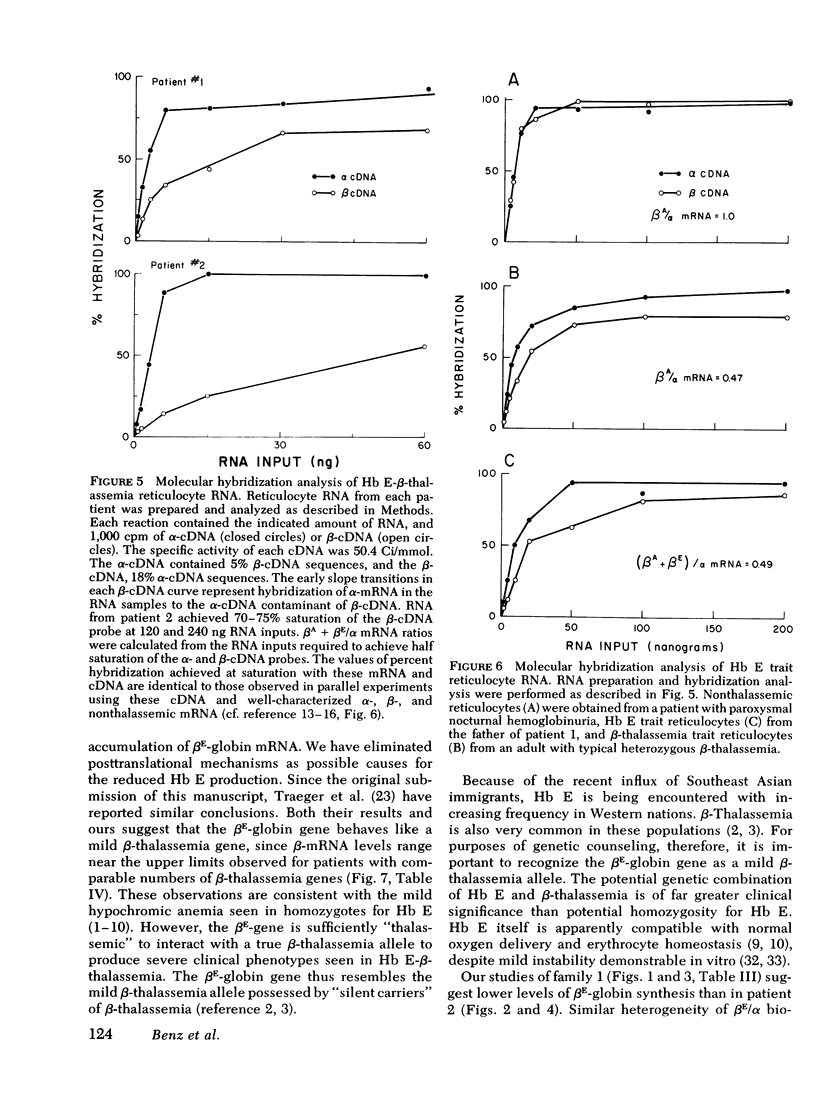
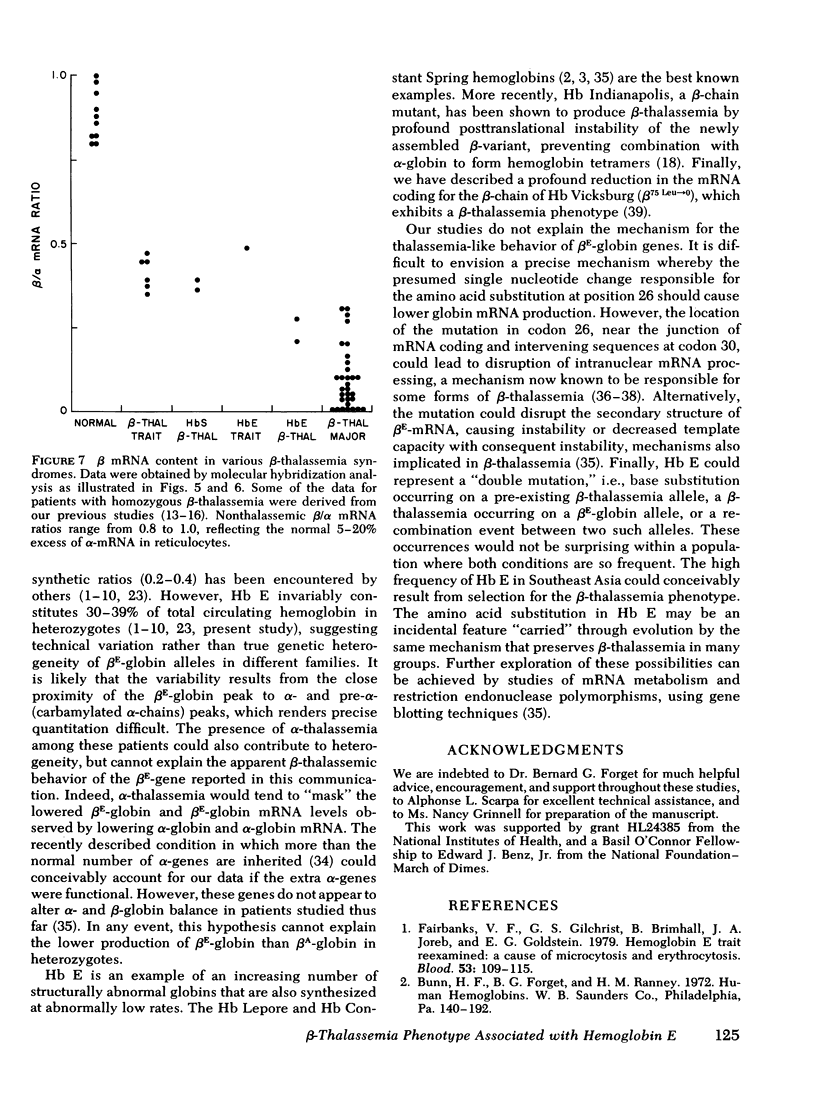
Inheritance of the gene for betaE-globin is associated with hypochromia and microcytosis, reminiscent of typical heterozygous beta-thalassemia. Patients with hemoglobin (Hb)E-beta-thalassemia exhibit clinical phenotypes of severe beta-thalassemia, a circumstance not encountered in other compound heterozygous states for structural beta-chain mutations and beta-thalassemia. We have analyzed the kinetics of globin synthesis and the levels of globin messenger (m) RNA accumulation in patients with Hb E-beta-thalassemia and Hb E trait. The initial rate of beta-globin synthesis (betaE/alpha=0.20-0.34) was less than expected on the basis of gene dosage, or comparable studies of other compound heterozygous states for beta-thalassemia and structurally abnormal beta-chains. betaE-globin synthesis was not only reduced during short-term incubations (1-5 min), but also remained relatively unchanged during long-term pulse or chase incubations up to 5h. Analysis of globin mRNA by cell-free translation and molecular hybridization confirmed that the unexpectedly low levels of betaE-globin synthesis were associated with comparable reduction in the levels of beta-globin mRNA. In Hb E-beta-thalassemia the betaA + betaE (alpha globin nRNA ratio observed were substantially lower than those obtained from reticulocytes of patients with heterozygous beta-thalassemia, or Hb S-betaO-thalassemia, while in Hb E trait, the betaA + betaE/alpha mRNA ratio was in the ranged observed for beta-thalassemia trait. The betaE-globin gene specifies reduced accumulation of betaE-globin mRNA, a property characteristic of other forms of beta-thalassemia. The beta-thalassemia phenotype associated with inheritance of Hb E is thus determined at the level of beta-globin mRNA metabolism.
Full text
PDF





Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Adams J. G., 3rd, Boxer L. A., Baehner R. L., Forget B. G., Tsistrakis G. A., Steinberg M. H. Hemoglobin Indianapolis (beta 112[G14] arginine). An unstable beta-chain variant producing the phenotype of severe beta-thalassemia. J Clin Invest. 1979 May;63(5):931–938. doi: 10.1172/JCI109393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J. G., 3rd, Steinberg M. H., Newman M. V., Morrison W. T., Benz E. J., Jr, Iyer R. beta-Thalassemia present in cis to a new beta-chain structural variant, Hb Vicksburg [beta 75 (E19)Leu leads to 0]. Proc Natl Acad Sci U S A. 1981 Jan;78(1):469–473. doi: 10.1073/pnas.78.1.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J. G., 3rd, Winter W. P., Tausk K., Heller P. Hemoglobin Rush (beta 101 (g3) glutamine): a new unstable hemoglobin causing mild hemolytic anemia. Blood. 1974 Feb;43(2):261–269. [PubMed] [Google Scholar]
- Ali M. A., Quinlan A., Wong S. C. Identification of hemoglobin E by the isopropanol solubility test. Clin Biochem. 1980 Aug;13(4):146–148. doi: 10.1016/s0009-9120(80)91014-0. [DOI] [PubMed] [Google Scholar]
- Benz E. J., Forget B. G., Hillman D. G., Cohen-Solal M., Pritchard J., Cavallesco C., Prensky W., Housman D. Variability in the amount of beta-globin mRNA in beta0 thalassemia. Cell. 1978 Jun;14(2):299–312. doi: 10.1016/0092-8674(78)90116-2. [DOI] [PubMed] [Google Scholar]
- Benz E. J., Geist C. E., Steggles A. W., Barker J. E., Nienhuis A. W. Hemoglobin switching in sheep and goats. Preparation and characterization of complementary DNAs specific for the alpha-, beta-, and gamma-globin messenger RNAs of sheep. J Biol Chem. 1977 Mar 25;252(6):1908–1916. [PubMed] [Google Scholar]
- Benz E. J., Jr, Forget B. G. Defect in messenger RNA for human hemoglobin synthesis in beta thalassemia. J Clin Invest. 1971 Dec;50(12):2755–2760. doi: 10.1172/JCI106778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz E. J., Jr, Forget B. G. Pathogenesis of the thalassemia syndromes. Pathobiol Annu. 1980;10:1–33. [PubMed] [Google Scholar]
- Benz E. J., Jr, Forget B. G. The biosynthesis of hemoglobin. Semin Hematol. 1974 Oct;11(4):463–523. [PubMed] [Google Scholar]
- Benz E. J., Jr, Swerdlow P. S., Forget B. G. Absence of functional messenger RNA activity for beta globin chain synthesis in beta 0-thalassemia. Blood. 1975 Jan;45(1):1–10. [PubMed] [Google Scholar]
- Benz E. J., Jr, Swerdlow P. S., Forget B. G. Globin messenger RNA in hemoglobin H disease. Blood. 1973 Dec;42(6):825–833. [PubMed] [Google Scholar]
- CHERNOFF A. I., MINNICH V., NANAKORN S., TUCHINDA S., KASHEMSANT C., BANGKOK, THAILAND, CHERNOFF R. R. Studies on hemoglobin E. I. The clinical, hematologic, and genetic characteristics of the hemoglobin E syndromes. J Lab Clin Med. 1956 Mar;47(3):455–489. [PubMed] [Google Scholar]
- Fairbanks V. F., Gilchrist G. S., Brimhall B., Jereb J. A., Goldston E. C. Hemoglobin E trait reexamined: a cause of microcytosis and erythrocytosis. Blood. 1979 Jan;53(1):109–115. [PubMed] [Google Scholar]
- Fairbanks V. F., Oliveros R., Brandabur J. H., Willis R. R., Fiester R. F. Homozygous hemoglobin E mimics beta-thalassemia minor without anemia or hemolysis: hematologic, functional, and biosynthetic studies of first North American cases. Am J Hematol. 1980;8(1):109–121. doi: 10.1002/ajh.2830080112. [DOI] [PubMed] [Google Scholar]
- Feldman R., Rieder R. F. The interaction of hemoglobin E with beta thalassemia: a study of hemoglobin synthesis in a family of mixed Burmese and Iranian origin. Blood. 1973 Nov;42(5):783–791. [PubMed] [Google Scholar]
- Forget B. G., Housman D., Benz E. J., Jr, McCaffrey R. P. Synthesis of DNA complementary to separated human alpha and beta globin messenger RNAs. Proc Natl Acad Sci U S A. 1975 Mar;72(3):984–988. doi: 10.1073/pnas.72.3.984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischer H., Bowman J. Hemoglobin E, an oxidatively unstable mutation. J Lab Clin Med. 1975 Apr;85(4):531–539. [PubMed] [Google Scholar]
- Higgs D. R., Old J. M., Pressley L., Clegg J. B., Weatherall D. J. A novel alpha-globin gene arrangement in man. Nature. 1980 Apr 17;284(5757):632–635. doi: 10.1038/284632a0. [DOI] [PubMed] [Google Scholar]
- Housman D., Forget B. G., Skoultchi A., Benz E. J., Jr Quantitative deficiency of chain-specific globin messenger ribonucleic acids in the thalassemia syndromes. Proc Natl Acad Sci U S A. 1973 Jun;70(6):1809–1813. doi: 10.1073/pnas.70.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor J. A., Turner P. H., Nienhuis A. W. Beta Thalassemia: mutations which affect processing of the beta-Globin mRNA precursor. Cell. 1980 Aug;21(1):149–157. doi: 10.1016/0092-8674(80)90122-1. [DOI] [PubMed] [Google Scholar]
- Kim H. C., Weierbach R. G., Friedman S., Schwartz E. Detection of sickle alpha- or beta0-thalassemia by studies of globin biosynthesis. Blood. 1977 May;49(5):785–792. [PubMed] [Google Scholar]
- Kim H. C., Weierbach R. G., Friedman S., Schwartz E. Globin biosynthesis in sickle cell, Hb SC, and Hb C diseases. J Pediatr. 1977 Jul;91(1):13–20. doi: 10.1016/s0022-3476(77)80434-4. [DOI] [PubMed] [Google Scholar]
- Maquat L. E., Kinniburgh A. J., Beach L. R., Honig G. R., Lazerson J., Ershler W. B., Ross J. Processing of human beta-globin mRNA precursor to mRNA is defective in three patients with beta+-thalassemia. Proc Natl Acad Sci U S A. 1980 Jul;77(7):4287–4291. doi: 10.1073/pnas.77.7.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan D. G., Lodish H. F., Kan Y. W., Housman D. Beta thalassemia and translation of globin messenger RNA. Proc Natl Acad Sci U S A. 1971 Oct;68(10):2514–2518. doi: 10.1073/pnas.68.10.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natta C., Banks J., Niazi G., Marks P. A., Bank A. Decreased beta globin mRNA activity in bone marrow cells in homozygous and heterozygous beta thalassaemia. Nat New Biol. 1973 Aug 29;244(139):280–281. doi: 10.1038/newbio244280a0. [DOI] [PubMed] [Google Scholar]
- Nienhuis A. W., Canfield P. H., Anderson W. F. Hemoglobin messenger RNA from human bone marrow. Isolation and translation in homozygous and heterozygous beta-thalassemia. J Clin Invest. 1973 Jul;52(7):1735–1745. doi: 10.1172/JCI107355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnier J., Wajcman H., Labie A. Defect in hemoglobin synthesis possibly due to a disturbed association. FEBS Lett. 1974 Sep 1;45(1):252–255. doi: 10.1016/0014-5793(74)80855-0. [DOI] [PubMed] [Google Scholar]
- Ruymann F. B., Popejoy L. A., Brouillard R. B. Splenic sequestration and ineffective erythropoiesis in hemoglobin E-beta-thalassemia disease. Pediatr Res. 1978 Oct;12(10):1020–1023. doi: 10.1203/00006450-197810000-00014. [DOI] [PubMed] [Google Scholar]
- STURGEON P., ITANO H. A., BERGREN W. R. Clinical manifestations of inherited abnormal hemoglobins. I. The interaction of hemoglobin-S with hemoglobin-D. Blood. 1955 May;10(5):389–404. [PubMed] [Google Scholar]
- Steinberg M. H., Dreiling B. J. Clinical, hematologic and biosynthetic studies in sickle cell-betao-thalassemia: a comparison with sickle cell anemia. Am J Hematol. 1976;1(1):35–44. doi: 10.1002/ajh.2830010105. [DOI] [PubMed] [Google Scholar]
- Testa U., Dubart A., Hinard N., Galacteros F., Vainchenker W., Rouyer-Fessard P., Beuzard Y., Rosa J. Beta O-thalassemia/Hb E association. Hemoglobin synthesis in blood reticulocytes and bone marrow cells fractionated by density gradient and in blood erythroid colonies in culture. Acta Haematol. 1980;64(1):42–52. doi: 10.1159/000207209. [DOI] [PubMed] [Google Scholar]
- Traeger J., Wood W. G., Clegg J. B., Weatherall D. J. Defective synthesis of HbE is due to reduced levels of beta E mRNA. Nature. 1980 Dec 4;288(5790):497–499. doi: 10.1038/288497a0. [DOI] [PubMed] [Google Scholar]
- Weatherall D. J., Clegg J. B., Na-Nakorn S., Wasi P. The pattern of disordered haemoglobin synthesis in homozygous and heterozygous beta-thalassaemia. Br J Haematol. 1969 Mar;16(3):251–267. doi: 10.1111/j.1365-2141.1969.tb00400.x. [DOI] [PubMed] [Google Scholar]
- White J. M. The synthesis of abnormal haemoglobins. Ser Haematol. 1971;4(3):116–132. [PubMed] [Google Scholar]