

Abstract
SIRT1 is a purported central regulator of skeletal muscle mitochondrial biogenesis. Herein we discuss our recent work utilizing conditional mouse models, which highlight the complexities of SIRT1 biology in vivo, and question its role in regulating mitochondrial function and mitochondrial adaptions to endurance exercise. Further, we discuss the possible contribution of proposed SIRT1 substrates to muscle mitochondrial biogenesis.
Keywords: Exercise, sirtuins, acetylation, transcription, adaptation
INTRODUCTION
The seminal studies of Dr. John Holloszy were the first to definitively show that aerobic exercise training increases mitochondrial biogenesis and oxidative capacity in skeletal muscle (13). Since these studies, much research has focused on elucidating the molecular mechanisms linking muscle contraction to mitochondrial plasticity. There are numerous allosteric factors that change in concentration during contraction (e.g. Ca2+, AMP, NAD+, Pi), and thus provide a logical starting point for connecting muscle contraction to the adaptive response. Because of their sensitivity to changes in cellular NAD+ levels, and their ability to deacetylate histone and protein targets (and therefore regulate their function), over the past decade much research has focused on the sirtuin family of proteins, and particularly, sirtuin 1 (SIRT1), as a potential signaling node that connects exercise to mitochondrial biogenesis (33)
SIRT1, in particular is known to regulate the activity of more than 40 protein targets (21), a number of which, as described below, are potentially important regulators of mitochondrial biogenesis. Because an extensive discussion on the multifaceted roles of SIRT1 is beyond the scope of this article, readers are encouraged to read other comprehensive reviews on SIRT1 (21), alternative sirtuin family members (12), and the role of NAD+ as a signaling entity in skeletal muscle (33).
SIRT1 SIGNALING IN SKELETAL MUSCLE – MUSCLE SPECIFIC SUBSTRATES AND INTERACTING PROTEINS
The rapid expansion of SIRT1 as an important modulator of muscle metabolism over the past decade has been due, in part, to the identification that SIRT1, through its deacetylase activity, regulates the activity of four proposed central regulators of intermediary metabolism: the AMP activated protein kinase (AMPK), the transcriptional co-activator, peroxisome proliferator activated receptor-gamma coactivator-1α (PGC-1α) and the transcription factors, p53 and cyclic AMP response element binding protein (CREB). Thus, SIRT1 represents a fine-tuned metabolic sensing node that translates fluctuations in peripheral and cellular substrate availability (primarily through its sensitivity to perturbations in lactate, pyruvate and NAD+) to a cellular adaptive response (Figure 1).
Figure 1. Proposed SIRT1-regulated signaling pathways in skeletal muscle.
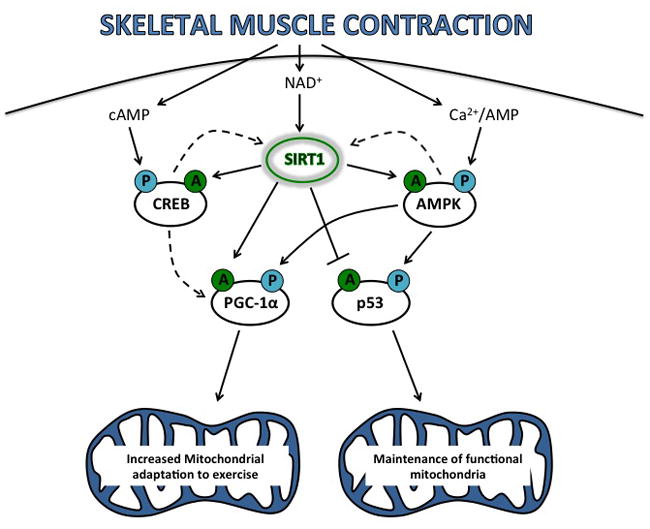
Skeletal muscle contraction results in increased cellular cAMP, NAD+, Ca2+ and AMP, which leads to the increased activity of SIRT1, CREB and AMPK. Through a coordinated interplay between phosphorylation (P) and deacetylation (A), SIRT1, CREB and AMPK promote adaptation of the mitochondrial reticulum through their downstream targets, PGC-1α and p53. Filled arrows represent activation, flat line represents inactivation, broken arrows represent suggested feedback loops.
Below we examine findings identifying AMPK, CREB, p53 and PGC-1α as SIRT1 targets, and discuss the relevance of these proteins in the context of mitochondrial adaptation, particularly to endurance exercise, in skeletal muscle. Our hope is to encourage discussion on whether or not these targets are, (i) specific SIRT1 substrates, and (ii) important metabolic regulators of skeletal muscle mitochondrial function, in vivo. Based on our research (23, 29), and that of others (3, 11, 20), we suggest that SIRT1 is not required for basal maintenance, or exercise-induced mitochondrial biogenesis in skeletal muscle, and that in fact, in certain scenarios, elevated SIRT1 may even impair skeletal muscle mitochondrial function. Further, our research introduces the hypothesis that exercise-mediated modulation of acetyltransferase activity may be an important regulator of mitochondrial adaptation to exercise (23).
SIRT1 AND AMPK: ARE THEY (DIS)CONNECTED IN SKELETAL MUSCLE?
AMPK is a key energy sensor in skeletal muscle that is activated by an increase in cellular AMP levels, and therefore, is activated by cellular energy stress (5). Considering that AMPK and SIRT1 are sensitive to changes in cellular nucleotides, it is not unexpected that much research has focused on an interconnectivity and overlap between these two proteins. For example, both AMPK and SIRT1 can initiate signaling cascades that promote the utilization of lipid stores, whilst blunting anabolic processes such as protein synthesis (5). Recent debate has however centered on the hierarchy of this process and whether SIRT1 and AMPK directly interact. Lan and coworkers (16) were amongst the first to suggest that AMPK activity may be under the direct regulation of SIRT1. Specifically, theses authors demonstrated that SIRT1 deacetylated the AMPK-kinase, LKB1, thereby regulating AMPK activity (16). Using both in vitro (HEK293T cells) and in vivo (mouse adipose and rat liver) approaches, the authors identified that mutation of lysine 48 of AMPK to arginine (to mimic deacetylation), increased AMPK activity, increased the cytoplasmic/nuclear ratio of LKB1, and increased the association of LKB1 with STRAD, its activating protein (16). In parallel, shRNA mediated knockdown of SIRT1 reduced phosphorylation of AMPK and the AMPK-related kinase MARK1, indicating that LKB1 activity is directly regulated by SIRT1 (16). Finally, hepatic LKB1 deacetylation was increased by 60% following 48h starvation in rats, which resulted in modest activation of LKB1 and AMPK (16).
Extending this data in skeletal muscle, Price et al. (25) recently reported that functional SIRT1 is required for the activation of AMPK in response to treatment with the polyphenol, resveratrol. Using a whole-body, inducible, SIRT1 knockout mouse model, these authors demonstrated that at low doses of resveratrol (25 μM in cells, or 25mg/kg/day in mice), AMPK activation and mitochondrial biogenesis induced by resveratrol was dependent on SIRT1 (25). Interestingly, higher doses of resveratrol increased AMPK activity in a SIRT1-independent manner (25), indicating that resveratrol might act via a number of molecular targets, as has previously been reported (8). Further, Price et al. (25) suggested a direct role for SIRT1 to activate AMPK, as muscle from SIRT1 over-expressing mice (SIRT1-Tg) displayed increased levels of AMPK phosphorylation. In a similar mechanism to that proposed by Lan et al. (16), the SIRT1-Tg mice had elevated deacetylation of LKB1, which may explain the increased AMPK activity observed. Collectively these studies indicate that AMPK activity is reliant upon a functional and active SIRT1 protein both in the liver and skeletal muscle.
Contrary to these two studies, there have been a number of reports suggesting that SIRT1 activity is dependent upon AMPK, and not vice-versa. Fulco et al. (9) identified that AMPK can increase SIRT1 activity in vitro through modulation of the NAD+ biosynthetic enzyme, nicotinamide phosphoribosyltransferase (Nampt). Following glucose restriction, Nampt activity was increased in an AMPK-dependent process, which in turn increased the NAD+:NADH ratio, decreased nicotinamide and activated SIRT1 (9). Interestingly, in the context of this study, the activation of SIRT1 led to a decrease in cell myogenesis, indicating that SIRT1 may regulate aspects of skeletal muscle growth/development, however this function is yet to be tested in skeletal muscle, in vivo. Canto and colleagues (6) extended this study by examining the biological effect of the AMPK activator, AICAR, in C2C12 cells. Specifically, these authors reported that AICAR treatment results in deacetylation of PGC-1α through a SIRT1-dependent mechanism (6). In agreement to the work from Fulco et al. (9), AICAR-mediated activation of AMPK increased cellular levels of NAD+, which in turn led to the activation of SIRT1 (6). In an interesting caveat, Canto et al. (6) also reported that dual phosphorylation (AMPK) and deacetylation (SIRT1) was required to fully activate PGC-1α, thus suggesting interdependence and coordinated dual regulation of PGC-1α.
To further probe the specific function of SIRT1 on AMPK activation, we studied mice with muscle-specific knockout of SIRT1 (termed hereafter as SIRT1 mKO) (23, 29), in which exon 4 of the murine SIRT1 gene is excised, leaving a truncated, deacetylase inactive SIRT1 protein (7). Importantly, SIRT1 activity is lost in skeletal muscle of SIRT1 mKO mice, as evidenced by reduced deacetylation of the SIRT1 target, p53, in response to physiological stimuli such as calorie restriction (CR) and endurance exercise (23, 29). In addition, we did not observe any compensation from other sirtuins in skeletal muscle to offset the lack of SIRT1 deacetylase activity, as the protein abundance of SIRT3 and SIRT6, as well as the gene expression of SIRT2-7 were unchanged in mKO compared to WT muscle (White A.T., Philp A. and Schenk S., unpublished observations). Following acute endurance exercise we observed parallel increases in AMPK phosphorylation and the phosphorylation of the AMPK targets, histone deacetylase 5 (HDAC5) and acetyl CoA carboxylase (ACC)-β in SIRT1 mKO and WT mice, indicating that AMPK activity is not affected in vivo by loss of SIRT1 (23). Further, in ex-vivo studies, activation of AMPK in extensor digitorum longus (EDL) muscles (29) and glucose transport in EDL (29) and soleus (Schenk S. and Philp A., unpublished observations) in response to AICAR was equivalent in SIRT1 mKO and WT mice (Figure 2). Collectively, there appears to be considerable disconnect between in vitro and in vivo studies, such that in vitro studies suggest that either, AMPK activation requires SIRT1 (16), or that SIRT1 activation requires AMPK (6), whilst in vivo studies suggest that activation of AMPK does not require SIRT1 deacetylase activity (23, 29).
Figure 2. Activation of AMPK by acute exercise and AICAR does not require SIRT1 deacetylase activity.
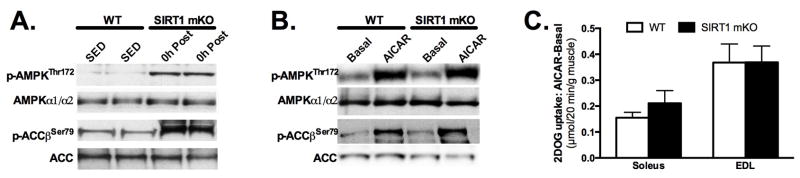
(A) Western blot analysis of phosphorylated AMPKThr172 (p-AMPKThr172) and phosphorylated acetyl CoA carboxylase (p-AccβSer79), and respective totals, in gastrocnemius muscle from wildtype (WT) mice or mice with skeletal muscle-specific knockout of SIRT1 deacetylase activity (mKO). Muscle was taken after a 4 h fast (Sedentary) or immediately after 60 min of variable-intensity endurance exercise (0h Post). The data demonstrates that SIRT1 deacetylase activity is not required for exercise-induced activation of AMPK. (Reprinted from (23). Copyright 2011 © the American Society for Biochemistry and Molecular Biology. Used with permission.) (B–C) Paired extensor digitorum longus (EDL) or soleus muscles from fasted (4 h) WT and SIRT1 mKO mice were incubated ex vivo without (Basal) or with AICAR (AICAR; 2mM). (B) Western blot analysis of p-AMPKThr172 and p-AccβSer79 in EDL muscles demonstrates normal AICAR-induced activation of AMPK in SIRT1 mKO muscle (29). (Reprinted from (29) Copyright 2011 © American Society for Clinical Investigation. Used with permission.) (C) AICAR-stimulated (Basal minus AICAR) 3H-2-deoxyglucose (2DOG) uptake in soleus and EDL muscles demonstrates that AICAR-induced glucose uptake is SIRT1 deacetylase independent. Data presented as mean±SEM (n=6/group). [Adapted from (29) Copyright 2011 © American Society for Clinical Investigation. Used with permission.]
SIRT1 AND PGC-1α: IS PGC-1α ACTIVITY REGULATED BY SIRT1 DURING ENDURANCE EXERCISE?
The first clear mechanistic link between SIRT1 and mitochondrial adaptation was provided by Nemoto and colleagues (20), who demonstrated functional interaction between SIRT1 and the transcriptional co-activator, PGC-1α. Specifically, these authors provided evidence that SIRT1 and PGC-1α form a complex, that SIRT1 deacetylates PGC-1α in an NAD+-dependent manner, and that SIRT1-mediated deacetylation of PGC-1α may be an important factor linking altered cellular energy status to increased mitochondrial gene transcription (20). Whilst the results provided by Nemoto et al. (20) were instrumental in driving SIRT1-PGC-1α research forward, there are two often overlooked observations from their study, which suggest that in certain situations, SIRT1 may negatively regulate PGC-1α and mitochondrial function. First, they reported that overexpression of SIRT1 in PC12 cells reduced (not increased) cellular oxygen consumption (by 25%) and the protein abundance of COXII (by ~45%), which is indicative of reduced mitochondrial function (20). Secondly, these authors demonstrated that SIRT1 over-expression led to an approximate 50% reduction in intrinsic PGC-1α transcriptional activity, as assessed via a GAL4-PGC-1α reporter assay (20). Intriguingly, the authors also reported unpublished data to suggest that SIRT1 knockdown also impaired cellular respiration, indicating a complex relationship between SIRT1, PGC-1α and mitochondrial function (20).
At a similar time, Rodgers and colleagues (27) described the first functional assessment of SIRT1 in the liver in response to fasting and re-feeding. These authors observed that both SIRT1 and PGC-1α protein content were rapidly up-regulated following fasting, and returned to basal conditions upon re-feeding (27), mirroring increased hepatic pyruvate and NAD+ concentrations. Further, Rodgers et al. (27) observed that PGC-1α was deacetylated following fasting, with in vitro analysis suggesting that this process was dependent on functional SIRT1 and NAD+. Tandom mass- spectrometry analysis on purified PGC-1α identified 13 lysine residues spanning the length of the PGC-1α protein that underwent reversible acetylation (27). Performing lysine to arginine mutation on five residues (K183R, K253R, K320R, K346R, K441R) significantly reduced nicotinamide-mediated repression of PGC-1α by ~50% compared to WT control (27). Repression was further rescued by an additional 25% with the mutation of a further five residues (K450R, K778R, K77R, K412R, K757R) (27). It is important to note that neither the relevance of each of these lysine residues for PGC-1α function, or the effect of a single lysine residue on PGC-1α function, is known. Interestingly, in contrast to Nemoto’s (20) previous findings, Rodgers et al. (27) also demonstrated that SIRT1-mediated deacetylation of PGC-1α increased co-activator activity. Specifically this interaction triggered an increase in hepatic genes involved in gluconeogenesis, while suppressing glycolytic genes (27). In addition, SIRT1 had limited effects on PGC-1α-mediated adaptations in mitochondrial genes in the liver, indicating that SIRT1 deacetylation of PGC-1α could serve to specify transcriptional targets for the co-activator, in this case directing PGC-1α to initiate a program of gluconeogenesis, and not mitochondrial biogenesis (27).
The notion that SIRT1 specifically directs PGC-1α-mediated gene transcription was further supported by Gerhart-Hines et al. (10), who demonstrated that SIRT1 localizes at PGC-1α target gene promoters to presumably synergize PGC-1α action on these target genes. Importantly, SIRT1 abundance was regulated by PGC-1α activity, as ectopic overexpression of PGC-1α increased SIRT1-associated promoter abundance by 7–10 fold (10). This study was also important, as it was the first evidence for a direct effect of SIRT1 on mitochondrial adaptation in skeletal muscle, albeit predominantly in cell culture experiments. In contrast to the previous work of Rodgers et al (27), the study from Gerhart-Hines et al. (10) also demonstrated that SIRT1 may regulate the expression of genes involved in mitochondrial respiration and fatty acid utilization, as shRNA mediated knockdown of SIRT1 in both C2C12 and mouse primary myotubes reduced the expression of cytochrome-c, isocitrate dehydrogenase-3α (IDH-3α), cytochrome-c oxidase subunit IV (COXIV), medium-chain acyl-CoA dehydrogenase (MCAD), carnitine palmitoyltransferase-1 (CPT-1) and pyruvate dehydrogenase kinase-4 (PDK4) (10). Functionally, SIRT1 knockdown also reduced citrate synthase (CS) activity in primary mouse myotubes in parallel to reductions in estrogen-related receptor-α (ERRα), PGC-1α and mitochondrial transcription factor-A (mtTFA) gene expression (10). In addition, SIRT1 overexpression in SIRT1−/− mouse embryonic fibroblasts (SIRT1−/− MEFs) increased the expression of cytochrome-c, IDH-3α, MCAD, ERRα and PDK4 in a dose-response manner (10). Finally, the authors demonstrated that increased mitochondrial metabolic activity in vitro in response to glucose reduction was dependent on SIRT1 as both SIRT1−/− MEFS and SIRT1 shRNA knockdown blocked the increase in mitochondrial gene expression and fatty acid utilization observed in wild-type cells (10). Collectively, this study provided mechanistic data to support the concept that SIRT1 is a key signaling node in skeletal muscle linking alterations in cellular energy status to mitochondrial adaptation and β-oxidation.
Whilst work from the Puigserver lab had strongly suggested that SIRT1 mediated deacetylation of PGC-1α can increase its co-activator activity toward target gene promoters, these studies did not address whether SIRT1 can regulate PGC-1α gene transcription. This issue appeared to be resolved, in part, following the work of Amat and coworkers (2) who reported that SIRT1, via interaction with myogenic determining factor (MyoD) promotes a positive autoregulatory PGC-1α expression loop in vitro. In their model, the authors propose that SIRT1 deacetylates PGC-1α, which increases PGC-1α activity on its own promoter through interaction with MyoD (2).
Putting these studies together, a paradigm was set such that SIRT1, through its actions on PGC-1α, could drive mitochondrial metabolic adaptation in response to transient fluctuation in cellular NAD+ levels. Interestingly, the proposed pro-metabolic effects of SIRT1 in skeletal muscle are primarily based on studies performed in vitro, with very little data established from intact skeletal muscle studies. Based on these studies, using our SIRT1 mKO mouse model, we hypothesized that loss of SIRT1 deacetylase activity in skeletal muscle would impair basal mitochondrial function, and block mitochondrial adaptation in response to endurance exercise (23). Surprisingly, however, we found that loss of SIRT1 activity did not lead to the widespread decline in skeletal muscle function that we anticipated (23). Functionally, mKO mice had similar skeletal muscle force characteristics (tetanic stress and time-to-fatigue) compared to wild-type (WT) controls and showed no decrements during voluntary wheel running (VWR) (23). At the cellular level, loss of SIRT1 function had no detrimental effect on mitochondrial respiration or mitochondrial gene expression, protein content or enzyme activity in skeletal muscle of SIRT1 mKO mice (23). Collectively, this data would therefore suggest that loss of SIRT1 function in skeletal muscle does not have detrimental effects muscle function.
To test whether mKO mice adapt to exercise, we performed both acute (treadmill running) and chronic (20 d of VWR) exercise and measured alterations in mitochondrial content and function, PGC-1α signaling, and protein transduction pathways thought to be central in mediating mitochondrial adaptation (23). All in all, we found that the ability of SIRT1 mKO muscle to adapt to acute and chronic exercise training was identical to that of WT mice, including the deacetylation and activation of PGC-1α (23). In fact, in response to an acute bout of endurance exercise, PGC-1α gene expression increased to a greater extent in the mKO mice compared to WT (~10 fold vs. ~6 fold induction in SIRT1 mKO and WT mice, respectively) (23). Although notably, induction of the PGC-1α targets mitofusin-2, cytochrome-c and PDK4 was increased similarly after exercise in both groups (23). Why the elevated increase in PGC-1α gene expression did not result in differential target gene expression is not apparent. It could potentially be explained by the use of only a single time-point post exercise, or potentially that a certain level of PGC-1α induction is required, beyond which further activity does not initiate additional target gene induction. Regardless, what was clear was that in contrast to the in vitro data from Amat et al. (2), PGC-1α gene expression is not dependent upon SIRT1 activity, in vivo.
To assess the in vivo relevance of PGC-1α acetylation, we next examined the acetylation status of PGC-1α at 0h and 3h post exercise and the interaction between PGC-1α and the acetyltransferase, general control of amino-acid synthesis (GCN5), which had previously been identified by the Puigserver group as a predominant acetyltransferase regulating PGC-1α activity (10, 17). We demonstrated that endogenous PGC-1α and GCN5 interact in skeletal muscle, and that acute exercise leads to a dissociation of PGC-1α from GCN5, which results in a reduction in nuclear GCN5 (23). Thus, we believe that exercise-induced deacetylation of PGC1-1α occurs not because of increased SIRT1 deacetylase activity, but due to reduced GCN5 activity on PGC1-1α. The net results being that exercise-induced deacetylation of PGC-1α occurs independent of functional SIRT1 (23).
Whilst our data in the SIRT1 mKO mouse is clearly at odds with cell-based studies, published in vivo studies do support the hypothesis that SIRT1 is not as pivotal in mediating mitochondrial adaptation as previously proposed. While this has been reviewed in detail elsewhere (33), SIRT1 protein content displays a poor correlation with skeletal muscle oxidative capacity (11) and transient overexpression of SIRT1 actually leads to a decrease in skeletal muscle mitochondrial respiration, enzyme activity and PGC-1α protein content (11). In addition, muscle from mice with whole-body knockout of SIRT1 do not show any decrements in respiratory capacity (3). To address some of the findings from Gurd et al. (11), Price and colleagues (25) recently generated a whole-body SIRT1 transgenic mouse, in which SIRT1 protein in skeletal muscle is increased ~5–10-fold. In this model, state 3 mitochondrial respiration and the mtDNA:nDNA ratio are increased by ~50% as is the mtDNA:nDNA ratio, indicative of increased mitochondrial number and function. In addition, the expression of multiple mitochondrial genes was increased, as were the expression of PGC-1α, PGC-1β, mtTFA, nuclear respiratory factor-1 (NRF-1) and mitochondrial transcription factor-b2 (TFB2M) (25). At present, the reasons for the differences between the rat data from Gurd et al. (11) and the mouse data from Price et al., (25), are not readily apparent, given that similar levels of SIRT1 over-expression were achieved by the two approaches. One possible reason could be that the mouse model from Price et al. (25) overexpresses SIRT1 in all tissues, and so skeletal muscle mitochondrial adaptation might be as a result of systemic as well as local effects. Thus, using a similar approach to Price et al. (25), but in a skeletal muscle-specific model would perhaps help to resolve the differences between the two data sets and help clarify the role of SIRT1 in skeletal muscle.
SIRT1 AND p53: NOT A STRAIGHTFORWARD RELATIONSHIP IN SKELETAL MUSCLE
One of the most characterized SIRT1 substrates is the oncoprotein and transcription factor p53 (12). Vaziri et al. (31) initially reported that SIRT1-mediated deacetylation of p53 in the C-terminus at lysine 382, reduced p53 activity and down-regulated p53 target genes involved in growth arrest and apoptosis. This important observation linked SIRT1 activation to cellular senescence and DNA repair (12). In vivo support for this mechanism was provided in work utilizing a SIRT1-deficient mouse model (7), in which p53 was observed to be hyperacetylated when SIRT1 activity was ablated. Thus, there is clear evidence, both in cell and rodent models that SIRT1-mediated deacetylation of p53 is a robust post-translational modification to down-regulate p53 activity (12). Whilst the described SIRT1-p53 interaction appears to make sense with regard to DNA damage and toxic stress, this process in skeletal muscle, in relation to mitochondrial adaptation is not so straightforward. The main reason for this is that p53 activity in skeletal muscle has been suggested to be an important factor in the maintenance of mitochondrial integrity and oxidative function (28). Saleem et al. (28) reported that p53 null mice have significant reductions in mitochondrial content, COX activity and PGC-1α protein content. Further, respiration measurements in isolated mitochondria revealed that state 3 respiration was impaired in the inter-myofibrillar (IMF) pool in parallel with increased reactive oxygen species (ROS) production (28). Notably, however, p53 null mice were able to increase mitochondrial biogenesis in response to chronic endurance training (8 weeks of VWR) to a similar extent as wild-type controls, despite a significant reduction in the total distance run per week. Thus, these data suggest that p53 is dispensable for exercise-induced mitochondrial adaptations, but may play a role in regulating ‘steady state’ mitochondrial mass and function (28).
Surprisingly, little is known about how pro-mitochondrial signals, such as endurance exercise may regulate p53 activity. After acute exercise, p53 phosphorylation at serine 15 was increased following acute exercise, as were the purported kinases for this specific residue, AMPK and the mitogen activated protein kinase, p38 (28). This observation has also been reported in humans, suggesting that phosphorylation of p53 at serine 15 is a conserved process in response to exercise. It is known that phosphorylation increases p53 activity, and so could potentially provide a mechanism as to how exercise alters p53 signaling. However, SIRT1 is also reported to be activated by acute endurance exercise in both rodent and human models (33), which would result in suppressed p53 activity, and be counter-active to phosphorylation. If p53 is indeed an important exercise-induced regulator of mitochondrial activity, then clearly more research should be conducted to understand the interplay between acetylation and phosphorylation in the regulation of p53 biological function after exercise. Some recently published data from our SIRT1 mKO mouse might help to address the role of SIRT1-mediated p53 deacetylation. That is, following acute exercise we observed a robust increase in nuclear p53 abundance in WT but not SIRT1 mKO mice, indicating that SIRT1-mediated deacetylation might alter the cellular location of p53 in response to exercise (Figure 3).
Figure 3. Exercise-induced nuclear translocation of p53 requires SIRT1 deacetylase activity.
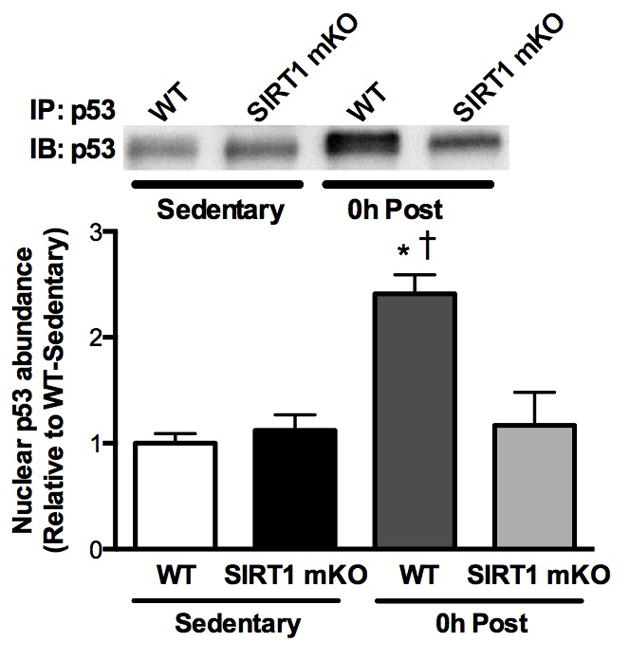
Gastrocnemius (GA) muscle was collected from wildtype (WT) mice or mice with skeletal muscle-specific knockout of SIRT1 deacetylase activity after a 4 h fast (Sedentary) or immediately after 60 min of variable-intensity of endurance exercise (0h Post). Subsequently, nuclear fractions were isolated from the GA, p53 was immunoprecipitated (IP) and IPs were immunoblotted for p53. *, within genotype; †, within 0h Post, P<0.05. Data presented as mean±SEM (n=4/group). [Adapted from (23). Copyright 2011 © the American Society for Biochemistry and Molecular Biology. Used with permission.]
This observation should be verified in a SIRT1 gain-of-function model, and obviously does not give any insight into the role of acetylation on p53 function. Nevertheless, this result does suggest that nuclear translocation of p53 is not required for exercise-induced gene transcription (23), which assimilates with the finding that mitochondrial biogenesis is not impaired after VWR studies in p53 null and SIRT1 mKO mice (23, 28). One interesting caveat to this issue is the report from Kamel et al. (15) who demonstrated that whilst SIRT1 interacts with p53, SIRT1 mediated deacetylation did not effect the expression of a number of known p53 target genes, indicating maintained p53 activity. Thus, it may be that SIRT1 deacetylation of p53 in skeletal muscle targets specific genes unrelated to mitochondrial adaptation, in a similar model to that proposed by Puigserver’s group for hepatic vs. skeletal muscle PGC-1α (10). Specific analysis of p53 promoter binding on target genes in response to exercise would help to address this issue.
SIRT1 AND CREB: ANOTHER LEVEL OF ENERGY SENSING?
Domenico Accili’s group (26) recently reported that the CREB is deacetylated at lysine 136 by SIRT1, which in turns blunts CREB activity by preventing cAMP dependent phosphorylation at Ser133 in the liver. In vitro expression of a constitutively acetylated CREB (K136Q) mimicked CREB activation and the activation of gluconeogenic genes (26). Thus, this data provides a fine-tuned association between SIRT1 and CREB to integrate fasting-induced gene expression. In contrast, data from Jeong et al. (14) suggests that SIRT1 may exert a positive effect on CREB activity in the brain, but in this case not through direct deacetylation of CREB, but via deacetylation of the CREB-related transcription coactivator 1 (TORC1). Under normal conditions, SIRT1 deacetylates and activates TORC1 by promoting its dephosphorylation and interaction with CREB (14). To add an additional layer of complexity, Johan Auwerx’s group recently reported that CREB transcriptionally regulates SIRT1 (22). In response to fasting or glucose deprivation, SIRT1 was found to be rapidly upregulated by a CREB dependent mechanism (22). Thus, it would appear that SIRT1 and CREB interact in a feedback loop that is sensitive to transient alterations in glucose availability, which in turn modulate cAMP abundance. How this interaction is regulated in skeletal muscle is currently unknown. However, CREB is thought to play an important role in exercise-induced adaptation in skeletal muscle via interaction with AMPK (30), and the fact that CREB activity is required for PGC-1α promoter activity following contraction in vivo (1) would suggest that SIRT1 deacetylation of CREB in skeletal muscle would either directly increase CREB activity, or promote CREB Ser133 phosphorylation. However, as yet this process has not been examined in response to exercise.
SIRT1 AND MEF2: DISCORDANCE BETWEEN PROPOSED REGULATORS OF OXIDATIVE METABOLISM
An intriguing extension to the CREB model is the reported interaction between SIRT1 and the myogenic enhancing factor-2 (MEF2) transcription factor. Similarly to CREB, MEF2 is required for PGC-1α promoter activity following muscle contraction (1), and has been extensively studied with regard to muscle development and oxidative capacity (24). Interestingly, Zhao et al. (35) were the first to report that MEF2 activity is regulated by lysine acetylation. Previous work had shown that MEF2 is negatively regulated by the class II deacetylase HDAC4, which represses MEF2 transcriptional activity by binding to MEF2 (REF). Further, Zhao et al. (35) showed that HDAC4 also represses MEF2 activity via sumoylation, most likely via the action of a SUMO E3 ligase. This targeted sumoylation of MEF2 is important, as the lysine residue in which it takes place is also a target for acetylation by the acetyltransferase, CREB binding protein (CBP). The authors observed that MEF2 acetylation correlates with activity and so disruption of this process via sumoylation interfered with the MEF2-CBP interaction and resulted in reduced acetylation and subsequent repression of MEF2 activity (35). The most surprising aspect of this process was the observation that SIRT1 potently deacetylates MEF2 and represses (not enhances) MEF2 transcriptional activity (35).
In vivo support for the positive role of acetylation in MEF2 activity was recently provided by Yamamoto et al. (34), who reported that increased acetylation of the MEF2-D isoform increased its binding to the promoter region of its target genes, glucose transporter type 4 (GLUT4) and muscle creatine kinase (MCK) (34). Whilst there is limited data relating to SIRT1 and MEF2 interactions, particularly in skeletal muscle, it is somewhat paradoxical to think that such a pro-mitochondrial adaptive mechanism such as SIRT1 deacetylation, might also inhibit the activity of MEF2, a well-described positive regulator of transcription factors that enhance oxidative metabolism (24). Clearly SIRT1-MEF2 interactions require further investigation. A further question that should also be addressed is whether the effect of SIRT1 on MEF2 are direct, as suggested by Zhao et al. (35), or occur indirectly through SIRT1 interaction with acetyltransferases such as p300 or CBP (4). Finally this data also highlights the need to understand tissue-dependent functions of SIRT1 and emphasizes how different skeletal muscle signaling can be compared to cardiac and liver signaling pathways.
WHY IS THERE SO MUCH DISPARITY IN THE LITERATURE?
Collectively, these studies suggest that the in vivo regulation of skeletal muscle mitochondrial biogenesis is far more complex than can be studied solely in vitro, and supports the notion that the acetylation status of a given protein is governed by a balance between the deacetylases and acetyltransferases that regulate its acetylation. For example, our data and others suggests that exercise is capable of dually modulating the acetylation status of protein targets such as PGC-1α by increasing the deacetylase activity of SIRT1 and/or reducing the acetyltransferase activity of GCN5.
A likely underlying reason for the disparity between many of the findings on SIRT1 and its role in regulating skeletal muscle mitochondrial biogenesis relates to the ‘system’ used to study its role. For instance, many of the mechanistic studies on SIRT1 have been performed in vitro, and as such, were commonly performed in non-muscle cell lines. As is the case in many fields, extrapolating findings from the in vitro to in vivo setting can be problematic, particularly with a tissue as unique as skeletal muscle. In addition, even when studies are conducted in muscles cells (be it myoblasts or myotubes), the cellular milieu in which cell culture experiments are conducted is important to consider. For instance, cells are routinely cultured in chronic hyperglycemic conditions (25mM glucose, approximately 5 times euglycemia), along with an abundance of growth factors, amino acids and serum, which cumulatively in vivo would most likely lead to chronic hyperinsulinemia and significant modulation of NAD and NADH levels. Whether or not cell culture experiments produce the same results when conducted in the physiological range is unknown. However, given the cells reliance upon high glucose concentrations, it is not surprising that such widespread metabolic adaptation is observed in vitro when cells are shifted from high to low glucose, as is typically used to study the action of SIRT1. That is not to say that cell culture experiments cannot be used as a useful tool to study molecular regulation of skeletal muscle mitochondrial biogenesis. However, it is important to be careful not to over-extrapolate in vitro findings to an in vivo setting. In addition, with respect to mitochondrial biogenesis, many studies rely on changes in gene expression as their outcome marker of ‘biogenesis’. Whilst an increase in gene expression is obviously an important initial step in the adaptative process, complete and functional mitochondrial biogenesis represents an increase in transcriptional activity, coupled with an increase in mitochondrial enzyme activity, respiratory chain protein content, and ultimately an increase in mitochondrial protein synthesis (19). Interestingly, when reviewing the SIRT1 literature, on the whole, in vivo studies that have provided contradicting results to in vitro models tend to perform far more rigorous assessment of mitochondrial biogenesis, potentially suggesting that reports solely relying on readouts of gene expression changes might be overlooking important physiological data relating to mitochondrial function.
CONCLUSION
Clearly there are many unanswered questions relating to the role of SIRT1 in regulating skeletal muscle mitochondrial biogenesis. Whilst our SIRT1 mKO mouse model has allowed us to explore SIRT1-specific functions in skeletal muscle, to truly test this question, suitable muscle-specific gain-of-function models are required to fully verify skeletal muscle specific targets in skeletal muscle. Ultimately the use of inducible transgenic models, as have been developed for PGC-1α (32) might also assist in the study of transient bursts of SIRT1, or bypass developmental issues that may arise in germline models. Beyond mouse models, there are very few detailed human studies that have examined the role of lysine acetylation on skeletal muscle function. Certainly, with the development of proteomic approaches to skeletal muscle (18), measuring multi-site acetylation appears to be a technique that will shed considerable new light on the acetylation field. Given the sensitivity of SIRT1 to fluctuations in cellular substrate stores, it should be possible to design well-controlled human studies that can examine the effect of manipulating SIRT1 activity, on specific cellular adaptation. Once this synergy between experimental approaches and whole system physiology is reached, then it is likely that we can confirm or dismiss currently proposed functions for SIRT1, and discover many new roles for SIRT1 in skeletal muscle, in the context of athletic performance, health and disease.
Summary.
Endurance exercise-induced increases in skeletal muscle mitochondrial content are proposed to work through SIRT1 and its downstream substrates.
Acknowledgments
Funding sources: This publication was supported in part by National Institutes of Health grants, R01 AG043120, R24 HD050837 and P30 AR058878.
Footnotes
Conflict of interest: There are no conflicts of interest to declare for both authors.
References
- 1.Akimoto T, Sorg BS, Yan Z. Real-time imaging of peroxisome proliferator-activated receptor-gamma coactivator-1alpha promoter activity in skeletal muscles of living mice. Am J Physiol Cell Physiol. 2004;287(3):C790–6. doi: 10.1152/ajpcell.00425.2003. [DOI] [PubMed] [Google Scholar]
- 2.Amat R, Planavila A, Chen SL, Iglesias R, Giralt M, Villarroya F. SIRT1 controls the transcription of the peroxisome proliferator-activated receptor-gamma Co-activator-1alpha (PGC-1alpha) gene in skeletal muscle through the PGC-1alpha autoregulatory loop and interaction with MyoD. J Biol Chem. 2009;284(33):21872–80. doi: 10.1074/jbc.M109.022749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boily G, Seifert EL, Bevilacqua L, et al. SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS One. 2008;3(3):e1759. doi: 10.1371/journal.pone.0001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bouras T, Fu M, Sauve AA, et al. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J Biol Chem. 2005;280(11):10264–76. doi: 10.1074/jbc.M408748200. [DOI] [PubMed] [Google Scholar]
- 5.Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20(2):98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canto C, Gerhart-Hines Z, Feige JN, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458(7241):1056–60. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng HL, Mostoslavsky R, Saito S, et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100(19):10794–9. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung JH, Manganiello V, Dyck JR. Resveratrol as a calorie restriction mimetic: therapeutic implications. Trends Cell Biol. 2012;22(10):546–54. doi: 10.1016/j.tcb.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fulco M, Cen Y, Zhao P, et al. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14(5):661–73. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerhart-Hines Z, Rodgers JT, Bare O, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26(7):1913–23. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gurd BJ, Yoshida Y, Lally J, Holloway GP, Bonen A. The deacetylase enzyme SIRT1 is not associated with oxidative capacity in rat heart and skeletal muscle and its overexpression reduces mitochondrial biogenesis. J Physiol. 2009;587(Pt 8):1817–28. doi: 10.1113/jphysiol.2008.168096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–95. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holloszy JO. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem. 1967;242(9):2278–82. [PubMed] [Google Scholar]
- 14.Jeong H, Cohen DE, Cui L, et al. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat Med. 2012;18(1):159–65. doi: 10.1038/nm.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamel C, Abrol M, Jardine K, He X, McBurney MW. SirT1 fails to affect p53-mediated biological functions. Aging Cell. 2006;5(1):81–8. doi: 10.1111/j.1474-9726.2006.00191.x. [DOI] [PubMed] [Google Scholar]
- 16.Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283(41):27628–35. doi: 10.1074/jbc.M805711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 2006;3(6):429–38. doi: 10.1016/j.cmet.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 18.Lundby A, Lage K, Weinert BT, et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012;2(2):419–31. doi: 10.1016/j.celrep.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller BF, Hamilton KL. A perspective on the determination of mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2012;302(5):E496–9. doi: 10.1152/ajpendo.00578.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280(16):16456–60. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 21.Nogueiras R, Habegger KM, Chaudhary N, et al. Sirtuin 1 and sirtuin 3: physiological modulators of metabolism. Physiol Rev. 2012;92(3):1479–514. doi: 10.1152/physrev.00022.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noriega LG, Feige JN, Canto C, et al. CREB and ChREBP oppositely regulate SIRT1 expression in response to energy availability. EMBO Rep. 2011;12(10):1069–76. doi: 10.1038/embor.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Philp A, Chen A, Lan D, et al. Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation following endurance exercise. J Biol Chem. 2011;286(35):30561–70. doi: 10.1074/jbc.M111.261685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Potthoff MJ, Wu H, Arnold MA, et al. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J Clin Invest. 2007;117(9):2459–67. doi: 10.1172/JCI31960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price NL, Gomes AP, Ling AJ, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012;15(5):675–90. doi: 10.1016/j.cmet.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qiang L, Lin HV, Kim-Muller JY, Welch CL, Gu W, Accili D. Proatherogenic abnormalities of lipid metabolism in SirT1 transgenic mice are mediated through Creb deacetylation. Cell Metab. 2011;14(6):758–67. doi: 10.1016/j.cmet.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434(7029):113–8. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 28.Saleem A, Adhihetty PJ, Hood DA. Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol Genomics. 2009;37(1):58–66. doi: 10.1152/physiolgenomics.90346.2008. [DOI] [PubMed] [Google Scholar]
- 29.Schenk S, McCurdy CE, Philp A, et al. Sirt1 enhances skeletal muscle insulin sensitivity in mice during caloric restriction. J Clin Invest. 2011;121(11):4281–8. doi: 10.1172/JCI58554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomson DM, Herway ST, Fillmore N, et al. AMP-activated protein kinase phosphorylates transcription factors of the CREB family. J Appl Physiol. 2008;104(2):429–38. doi: 10.1152/japplphysiol.00900.2007. [DOI] [PubMed] [Google Scholar]
- 31.Vaziri H, Dessain SK, Ng Eaton E, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107(2):149–59. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 32.Wende AR, Schaeffer PJ, Parker GJ, et al. A role for the transcriptional coactivator PGC-1alpha in muscle refueling. J Biol Chem. 2007;282(50):36642–51. doi: 10.1074/jbc.M707006200. [DOI] [PubMed] [Google Scholar]
- 33.White AT, Schenk S. NAD(+)/NADH and skeletal muscle mitochondrial adaptations to exercise. Am J Physiol Endocrinol Metab. 2012;303(3):E308–21. doi: 10.1152/ajpendo.00054.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto H, Williams EG, Mouchiroud L, et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell. 2011;147(4):827–39. doi: 10.1016/j.cell.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao X, Sternsdorf T, Bolger TA, Evans RM, Yao TP. Regulation of MEF2 by histone deacetylase 4- and SIRT1 deacetylase-mediated lysine modifications. Mol Cell Biol. 2005;25(19):8456–64. doi: 10.1128/MCB.25.19.8456-8464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]