

Abstract
Background
22q13 deletion syndrome, also known as Phelan-McDermid syndrome, is a neurodevelopmental disorder characterized by intellectual disability, hypotonia, delayed or absent speech, and autistic features. SHANK3 has been identified as the critical gene in the neurological and behavioral aspects of this syndrome. The phenotype of SHANK3 deficiency has been described primarily from case studies, with limited evaluation of behavioral and cognitive deficits. The present study used a prospective design and inter-disciplinary clinical evaluations to assess patients with SHANK3 deficiency, with the goal of providing a comprehensive picture of the medical and behavioral profile of the syndrome.
Methods
A serially ascertained sample of patients with SHANK3 deficiency (n = 32) was evaluated by a team of child psychiatrists, neurologists, clinical geneticists, molecular geneticists and psychologists. Patients were evaluated for autism spectrum disorder using the Autism Diagnostic Interview-Revised and the Autism Diagnostic Observation Schedule-G.
Results
Thirty participants with 22q13.3 deletions ranging in size from 101 kb to 8.45 Mb and two participants with de novo SHANK3 mutations were included. The sample was characterized by high rates of autism spectrum disorder: 27 (84%) met criteria for autism spectrum disorder and 24 (75%) for autistic disorder. Most patients (77%) exhibited severe to profound intellectual disability and only five (19%) used some words spontaneously to communicate. Dysmorphic features, hypotonia, gait disturbance, recurring upper respiratory tract infections, gastroesophageal reflux and seizures were also common. Analysis of genotype-phenotype correlations indicated that larger deletions were associated with increased levels of dysmorphic features, medical comorbidities and social communication impairments related to autism. Analyses of individuals with small deletions or point mutations identified features related to SHANK3 haploinsufficiency, including ASD, seizures and abnormal EEG, hypotonia, sleep disturbances, abnormal brain MRI, gastroesophageal reflux, and certain dysmorphic features.
Conclusions
This study supports findings from previous research on the severity of intellectual, motor, and speech impairments seen in SHANK3 deficiency, and highlights the prominence of autism spectrum disorder in the syndrome. Limitations of existing evaluation tools are discussed, along with the need for natural history studies to inform clinical monitoring and treatment development in SHANK3 deficiency.
Keywords: 22q13 deletion syndrome, Autism, Microarrays, Mutation, Phelan-McDermid syndrome, SHANK3
Background
22q13 deletion syndrome, also known as Phelan-McDermid syndrome, is a genetic disorder characterized by global developmental delay, hypotonia, delayed or absent speech, and autistic behaviors [1]. SHANK3 is the critical gene for the core neurological and behavioral symptoms in this syndrome, as the loss of one copy (haploinsufficiency) of SHANK3, occurring through intragenic deletion or point mutation, is sufficient to cause the neurobehavioral manifestations of Phelan-McDermid syndrome [2-5]. SHANK3 codes for a master scaffolding protein that forms a key framework in the postsynaptic density of glutamatergic synapses and plays a critical role in synaptic function, learning and memory [6].
SHANK3 deletion or mutation (which we will refer to together as SHANK3 deficiency) is found in about 0.5% of patients ascertained for autism spectrum disorder (ASD), including 0.2% with a SHANK3 mutation identified by sequencing [2-4,7,8] and 0.3% with a SHANK3 deletion, as shown by microarray analyses of over 7,000 individuals with ASD([3,9-15], Autism Genome Project, unpublished). Analysis of a very large cohort of patients with intellectual disability (ID) also indicates that about 0.3% of such patients have SHANK3 deletions [16], with some studies reporting rates above 1% [17]. The rate of SHANK3 mutation in ID is still being determined, but the first two studies estimate it at approximately 1% [17,18]. These findings suggest that SHANK3 deficiency is one of the more common monogenic causes of ASD and ID. Furthermore, recent evidence suggests that disruption of the SHANK3 and glutamate signaling pathway is common to multiple forms of ASD, including Fragile X syndrome and tuberous sclerosis [19,20]; dissecting this pathway may therefore represent an important opportunity to improve understanding of the biological pathways associated with ASD and ID not involving haploinsufficiency of SHANK3.
More than 150 affected individuals with SHANK3 deficiency have been described in published case studies since the first case report by Nesslinger and colleagues in 1994 [21]. These cases suggest a common underlying phenotype that includes global developmental delay, severe expressive language delay, hypotonia, autistic features and minor dysmorphic features [21-30]. However, clinical and genetic methodology varied across studies, as did estimates of the nature and prevalence of ASD, relying in the majority of cases on parental reports or questionnaires. These reports highlight a broad and clinically heterogeneous phenotype. Dysmorphic features are commonly described and include dysplastic toenails; dysplastic ears; large, fleshy hands; long eyelashes; dolichocephaly; pointed chin; and bulbous nose [21-26,28,29]. Medical conditions associated with the syndrome are less well defined but have been reported to include seizures, renal abnormalities, cardiac defects, hearing loss, gastroesophageal reflux and lymphedema [23-25,28].
The first explicit association between ASD and SHANK3 deficiency was published in 2000 by Prasad and colleagues, who described three cases of individuals with pervasive developmental disorder and terminal 22q13 deletion [31]. Among the case series published since then, several have specifically evaluated the presence of ASD using a variety of measures such as medical record review [24,26], developmental questionnaires [23] and various standardized diagnostic instruments [22,32]. Likely because of the diverse approaches, estimates of rates of ASD vary dramatically across studies. A review of 107 cases previously described in the literature reported a rate of ‘autistic behaviors’ of 44% [33]. Studies that prospectively evaluate ASD in patients with 22q13 deletions and utilized standardized assessments (for example, Childhood Autism Rating Scale, Social Communication Questionnaire) suggest rates of 60% to 94% [22,25,28]. Philippe et al. [27] administered the Autism Diagnostic Interview-Revised (ADI-R, [34]) in eight participants and found that, although none met criteria for an autism classification, five (63%) met cutoff scores in the social and communication domains and were within one point of cutoff scores on the repetitive behavior domain, suggesting a level of autistic features consistent with Diagnostic and Statistical Manual of Mental Disorders - Fourth Edition (DSM-IV) diagnoses of pervasive developmental disorder. To date, published investigations have not evaluated ASD in SHANK3 deficiency using best practice recommendations, which include combining information from clinician evaluations, structured observation, and an autism-focused, structured developmental history [35]. As such, the nature and prevalence of ASD among patients with SHANK3 deficiency remains an important area of study.
Evaluations of medical comorbidities associated with SHANK3 deficiency have been likewise limited to primarily retrospective reports. Electroencephalography (EEG) recordings have never been prospectively collected in published studies of 22q13 deletion syndrome, although five case series report on the prevalence of seizure disorders using retrospective review and parent survey methods [22,23,25,27,28]. These prior prevalence estimates of seizure disorders range from 0% (out of 8) [27] to 31% (4 out of 13) [28], depending on the study.
Among the published case series, 31 patients have had brain magnetic resonance imaging (MRI) results described, either through prospective assessment [27,28,36] or medical record review [24]. Brain abnormalities were evident in 22 out of 31 (71%) cases, ranging from 2 out of 4 (50%) [24] to 10 out of 10 (100%) [36]. Changes in the corpus callosum were described in 16 out of 31 patients, including thinning or hypoplasia; 14 had white matter changes, including delayed myelination, generalized white matter atrophy and nonspecific white matter hyperintensities; 12 had ventricular dilatation; and 3 had interventricular, cerebellar or temporal sylvian arachnoid cysts. The first case series in SHANK3 deficiency reported that four out of seven patients had ventricular dilatation [21] although methods were not specified. The most recent study specifically examined cerebellar malformations in 10 patients with 22q13 deletions using MRI and found evidence of cerebellar vermis hypoplasia in six patients, including an enlarged cisterna magna in five [36].
The nature and prevalence of other clinical features (for example, cognitive impairment, language impairment and motor deficits) have also not been systematically evaluated in some of the previous studies. Phillipe et al. [27] conducted prospective neuropsychological evaluations of eight children with 22q13 deletions and present a rich clinical picture of neurocognitive features, particularly in the motor domain. The authors reported mild delays in acquisition of early motor milestones and two patterns of motor disabilities: orthopedic disorders (including hypotonia of lower limbs) and impairments in motor control (for example, praxis, pyramidal signs and postural abnormalities).
The present study seeks to expand upon the existing literature by contributing results from systematic evaluations of phenotypic domains in patients with SHANK3 deficiency using a prospective design and standardized evaluation methods. Detailed clinical and molecular analyses represent an important opportunity to improve the understanding of underlying biological pathways associated with ASD, and neurodevelopmental disorders more broadly. In addition, the knowledge gained from comprehensive evaluations may help to establish a neurobiological signature of the disorder and serve to identify important phenotypic targets for treatment intervention or candidate biomarkers of treatment response. Finally, results from SHANK3 sequencing and high-resolution chromosomal microarray analysis (CMA) may also provide insight into the relationship between genotype and phenotype in this complex syndrome.
Methods
Participants
Participants were enrolled into an institutional review board-approved project with parents providing informed consent for participation. Over an 18-month period, 32 children and adults (18 males and 14 females) were seen. Referral sources included the Phelan-McDermid Syndrome Foundation, on-going research studies and word-of mouth. Patients ranged in age from 1.6 to 45.4 years (mean 8.8, SD 9.2). All patients were of Caucasian descent. Phenotypic data were available on all participants with rates of 90% to 100% completion on diagnostic, cognitive and clinical genetics evaluations. Half of the participants received structured neurological examinations by a neurologist, but all patients were assessed for neurologic abnormalities during the clinical genetics evaluation.
Clinical evaluation
An inter-disciplinary team evaluated patients using the following clinical tools:
1. Psychiatric evaluations using the DSM-IV [37] were conducted by board-certified child and adolescent psychiatrists at the first patient visit and focused on the assessment of pervasive developmental disorders.
2. Clinical genetics evaluations and dysmorphology examinations were performed by clinical geneticists to assess growth, pubertal development, head size, craniofacial features, digits, extremities, chest, spine, skin, organ malformations (such as congenital heart or renal defects) and neurological abnormalities.
3. Neurological examinations were conducted by a pediatric neurologist to evaluate gross motor skills and gait, fine motor coordination, cranial nerves and deep tendon reflexes.
4. Autism Diagnostic Observation Schedule-G (ADOS-G [38]), a direct semi-structured assessment, was used to assess the presence of the three main domains of autism (communication, reciprocal social interaction and repetitive, restricted behaviors). ADOS-G, Module 1, intended for minimally verbal individuals, was administered by a trained clinician to all patients in this sample.
5. ADI-R[34], an investigator-based, semi-structured instrument, was used to differentiate autistic disorder from non-autistic ID in individuals with a developmental age greater than 18 months. It was administered by a trained interviewer to parents or caregivers, making use of an algorithm that incorporates the World Health Organization International Classification of Diseases-10 and DSM-IV criteria for diagnosis.
6. Cognitive testing was conducted by licensed clinical psychologists or doctoral students to provide estimates of cognitive functioning. Tests were selected based on age of child and developmental level. The Mullen Scales of Early Learning [39] were used on 27 participants, including children with chronological ages older than the standardization sample, to allow for flexible administration of items. The Stanford-Binet Intelligence Scales, Fifth Edition [40] were used for one school-aged child who used single words on a regular basis, and the Leiter International Performance Scale-Revised [41] was used on two adult patients. Ratio intelligence quotients (IQs) were calculated using mental age estimates from the cognitive tests and used to provide an estimate of nonverbal IQ.
7. The Vineland Adaptive Behavior Scales, Second Edition[42] were used to evaluate independence in daily life skills, including communication, socialization and motor skills.
8. Medical record reviews were conducted to ascertain information on medical, neurological and physical manifestations not directly assessed in the clinical evaluations listed above. Results from EEG and brain MRI were collected from record review when available.
Genetic testing
CMA and multiplex ligation-dependent probe amplification (MLPA) were used to confirm previous clinical genetic testing in 29 out of 30 patients with 22q13 deletions. DNA from one participant (SH27) was not available for in-house validation, so the clinical laboratory CMA report using the Affymetrix 6.0 platform was reviewed to confirm loss of SHANK3. Two patients (SH29 and SH32) had deleterious de novo point mutations in SHANK3, and Sanger sequencing was carried out to confirm the mutation (see below).
MLPA probe set SALSA P188 MLPA kit 22q13 from MRC-Holland (Amsterdam, the Netherlands) was used according to the manufacturer’s protocols. A total of 37 probes are on 22q13 and four of them on the SHANK3 gene. Briefly, 200 ng genomic DNA in 5 μl water was denatured, mixed with the probe set and high salt MLPA buffer. The mixture was first hybridized at 60°C for 16 h, followed by a ligation reaction; 5 μl of the ligation reaction product was used for PCR. PCR products were subjected to capillary electrophoresis on an ABI 3130 genetic analyzer (Applied Biosystems, Foster City, CA, USA). Raw traces from the electrophoresis were imported into GeneMarker software (SoftGenetics, State College, PA, USA) for MLPA analysis. After population normalization, data were compared with a synthetic control sample, which represents the median of all normal samples in the experiment. A threshold of dosage change <0.75 was used to identify deletions, and a threshold >1.30 was used to identify duplications.
Twenty-nine samples were genotyped on Illumina Omni 2.5-8 v1 array (Illumina, San Diego, CA, USA) containing a total of 2,379,855 markers. Samples were processed according to the manufacturer’s protocol and normalization and genotype calling were performed using Illumina Genome Studio software V2011.1. All samples had an array call rate above 99%, SD for log R ratio values in the autosomes <0.28 and SD for B allele frequency values <0.06. Copy number variants (CNVs) were called using PennCNV algorithm [43]. Fragmented CNVs were merged and the breakpoints were inspected by visual investigation of log R ratio and B allele frequency values within and in the vicinity of the called CNVs. The exact deletion breakpoints in the 22q13 region were confirmed using the moving average of the number of markers with B allele frequency between 0.45 and 0.55, in addition to the visual investigation of the PennCNV call. Genomic locations are based on GRCh37 (hg 19).
SHANK3 point mutations were identified in two patients, SH32 had a nonsense mutation (c.1527G>A, p.W509*) in exon 12 and SH29 had a frameshift single base deletion (c.2497delG, p.P834Rfs*59) in exon 21 of the SHANK3 gene (NM_033517.1). As we reported previously [44], because the current version of reference human genome assembly GRCh37 misses the beginning of exon 11, the cDNA and amino acid positions here have been corrected according to the most updated SHANK3 mRNA and protein sequence (NM_033517.1 and NP_277052.1) in RefSeq. PCRs were designed to amplify the corresponding target fragments from 20 ng genomic DNA of each family member using AccuPrime Pfx (Invitrogen, Carlsbad, CA, USA) in a 10 μl total PCR reaction volume. Two DNA fragments were amplified: a 389-bp fragment covering exon 12 (forward primer 5′-CGAGCCCATCTGTTCCTTT-3′, reverse primer 5′-TCCATGTGGATTTAGCACCA-3′), and a 264-bp exonic fragment covering exon 21 (forward primer 5′-GAGCACCTCGATGCAAGAC-3′, reverse primer 5′-TTCTGCCGCTCGGGATAC-3′). The amplified DNA fragments were then purified and sequenced on an ABI 3730 DNA analyzer using the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems).
Data analysis
Descriptive statistics were calculated across all measures and organized into phenotypic domains: medical comorbidities, dysmorphic features, behavioral and ASD features, cognitive ability, receptive and expressive language, and motor skills. For genotype-phenotype analyses, the deletion size for the two patients with point mutations was entered as the size of the SHANK3 gene, 58,572 bp. Spearman rank order correlations (SROC) were used to explore associations between deletion size and continuous phenotyping variables (for example, number of medical comorbidities, number of dysmorphic features, IQ). Pairwise deletion of missing cases was used in correlation analysis to address missing data in the SROC calculations. P-values were calculated by bootstrapping 95% confidence intervals around the bootstrap-estimated SROC, with bias corrected and accelerated confidence interval reported below. Mann–Whitney U tests were conducted to explore potential associations between each medical comorbidity and dysmorphic features (dichotomous variables) with the continuous but skewed measure of deletion size.
Results
We carried out comprehensive, prospective clinical evaluations of 32 patients with a 22q13 deletion (n = 30), SHANK3 mutation (n = 1), or ascertained with ASD and subsequently identified by whole exome sequencing as having a de novo SHANK3 mutation (n = 1).
Genetic testing
As shown in Table 1, all 32 participants had confirmed SHANK3 deficiency secondary to mutation or deletion. Two participants had single base-pair changes leading to a premature stop, either due to a nonsense change (c.1527G>A) or a frameshift (c.2497delG). Among the 30 patients with deletions, six had ring chromosome 22, two had unbalanced translocations with loss of distal 22q, and one had an interstitial deletion that included SHANK3; the remaining 21 participants (66%) had simple terminal deletions of 22q. Deletion sizes ranged from 101 kb to 8.45 Mb (mean ±SD, 4.21 ±2.75 Mb) (Figure 1). Deletions and mutations were confirmed to be de novo in 25 parent dyads. Samples from three fathers and three mothers from an additional six participants were studied and did not carry deletions of SHANK3. All parents were healthy, suggesting a de novo origin for those remaining. In one patient for whom blood could not be obtained for further molecular characterization (SH27), the affected child had a 22q terminal deletion based on a clinical laboratory report. The patient’s mother has a balanced translocation, 46,XX, t(11;22)(q23;q11.2); the child’s karyotype was normal.
Table 1.
Description of the genetic changes in 32 participants with 22q13 deletions or SHANK3 mutations
| Patient | Gender | Ascertainment method | Rearrangement | Del 22q13, array coordinates (hg19) | Del 22q13 size | Validation | Inheritance | Additional genomic findings |
|---|---|---|---|---|---|---|---|---|
| SH1 |
Male |
Karyotype, FISH |
Ring 22 |
48927548-51224208 |
2,296,661 |
MLPA |
Mother negative |
None |
| SH2 |
Male |
Karyotype, aCGH |
Ring 22 |
48444959-51224208 |
2,779,250 |
MLPA |
De novo |
None |
| SH3 |
Male |
Karyotype, SNP array |
Ring 22 |
49114430-51224208 |
2,109,779 |
MLPA |
De novo |
None |
| SH4 |
Female |
aCGH |
Terminal deletion |
44321641-51224208 |
6,902,568 |
MLPA |
De novo |
None |
| SH5 |
Male |
aCGH |
Terminal deletion |
46143471-51224208 |
5,080,738 |
MLPA |
De novo |
None |
| SH6 |
Female |
aCGH, FISH |
Terminal deletion |
44427703-51224208 |
6,796,506 |
MLPA |
De novo |
None |
| SH7 |
Female |
Karyotype, FISH |
Ring 22 |
46905533-51224208 |
4,318,676 |
MLPA |
Mother negative |
None |
| SH8 |
Female |
aCGH, and FISH |
Terminal deletion |
49574124-51224208 |
1,650,085 |
MLPA |
De novo |
None |
| SH9 |
Female |
aCGH, FISH |
Terminal deletion |
49028732-51224208 |
2,195,477 |
MLPA |
De novo |
None |
| SH10 |
Male |
SNP microarray |
Terminal deletion |
51122946-51224208 |
101,263 |
MLPA |
De novo |
None |
| SH11 |
Female |
aCGH, FISH |
Terminal deletion |
49028732-51224208 |
2,195,477 |
MLPA |
De novo |
None |
| SH12 |
Male |
aCGH |
Terminal deletion |
42773732-51224208 |
8,450,477 |
MLPA |
De novo |
None |
| SH13 |
Female |
Karyotype |
Terminal deletion |
43745129-51224208 |
7,479,080 |
MLPA |
De novo |
None |
| SH14 |
Female |
aCGH, FISH |
Unbalanced translocation |
50267252-51224208 |
956,957 |
MLPA |
De novo |
None |
| SH15 |
Female |
SNP microarray |
Terminal deletion |
45902119-51224208 |
5,322,090 |
MLPA |
De novo |
None |
| SH16 |
Female |
CMA, FISH |
Terminal deletion |
42918711-51224208 |
8,305,498 |
MLPA |
Father negative |
None |
| SH17 |
Female |
SNP microarray |
Terminal deletion |
45583935-51224208 |
5,640,274 |
MLPA |
De novo |
None |
| SH18 |
Female |
aCGH, FISH |
Terminal deletion |
50077362-51224208 |
1,146,847 |
MLPA |
De novo |
None |
| SH19 |
Male |
FISH |
Interstitial deletion |
48551989-51206201 |
2,654,212 |
MLPA |
Father negative |
None |
| SH20 |
Male |
aCGH |
Terminal deletion |
51083118-51224208 |
141,091 |
MLPA |
De novo |
None |
| SH21 |
Male |
aCGH |
Terminal deletion |
45428606-51224208 |
5,795,603 |
MLPA |
De novo |
None |
| SH22 |
Male |
FISH |
Terminal deletion |
44800014-51224208 |
6,424,195 |
MLPA |
De novo |
None |
| SH23 |
Male |
Karyotype, FISH |
Unbalanced translocation |
44023173-51224208 |
7,201,036 |
MLPA |
De novo |
None |
| SH24 |
Male |
Karyotype, aCGH |
Ring 22 |
43218614-51224208 |
8,005,595 |
MLPA |
De novo |
None |
| SH25 |
Male |
aCGH, FISH |
Terminal deletion |
46787434-51224208 |
4,436,775 |
MLPA |
De novo |
None |
| SH26 |
Male |
Karyotype, FISH |
Ring 22 |
49460840-51224208 |
1,763,369 |
MLPA |
De novo deletion; maternal balanced translocation, 46,XX, t(1;6)(p13.3;q22,2) |
None |
| SH27a |
Male |
SNP microarray |
Terminal deletion |
51115526-51234443 |
118,917 |
no DNA, validated in a clinical lab |
De novo deletion; maternal balanced translocation, 46,XX, t(11;22)(q23;q11.2) |
None |
| SH28 |
Female |
aCGH, FISH |
Terminal deletion |
45705241-51224208 |
5,518,968 |
MLPA |
De novo |
None |
| SH29 |
Male |
Sequencing |
SHANK3 frameshift mutation (c.2497delG) |
No 22q13 deletion detected |
|
Sanger sequencing |
De novo |
None |
| SH30 |
Female |
FISH |
Terminal deletion |
49004395-51224208 |
2,219,814 |
MLPA |
Father negative |
None |
| SH31 |
Male |
aCGH |
Terminal deletion |
42822943-51224208 |
8,401,266 |
MLPA |
Mother negative |
None |
| SH32 | Male | Sequencing | SHANK3 nonsense mutation (c.1527G>A) | No 22q13 deletion detected | Sanger sequencing | De novo | De novo 17q12 microduplication (chr17:34815184–36249059)b |
aNo blood was available for participant SH27 or his parents so we present the findings of the clinical report, which used the Affymetrix 6.0 platform. bReported previously [45]. aCGH, array comparative genomic hybridization; CMA, chromosomal microarray analysis; FISH, fluorescent in situ hybridization; hg, human genome version; MLPA, multiplex ligation-dependent probe amplification; SNP, single nucleotide polymorphism.
Figure 1.

Deletion sizes at 22q13.2-11q13.3 in 32 patients. Genes are indicated across the top in green, the red triangles represent MLPA probes. The two patients with SHANK3 mutations are shown in red and the patient with an interstitial deletion is shown in pink. Genomic coordinates correspond to the hg19 genome assembly (Build 37).
Autism spectrum disorder evaluation
We used standardized methods including the ADI-R, ADOS-G and DSM-IV diagnosis based on psychiatric evaluation to evaluate for ASD symptoms in the sample (Table 2). Classifications of ‘autism’, ‘autism spectrum’ and ‘not ASD’ were used in evaluating the diagnostic data. Patients classified as ‘autism’ met DSM-IV criteria for autistic disorder on psychiatric intake, met autism spectrum or autism cutoffs on ADOS-G, Module 1 revised algorithm, and met cutoffs for autism on at least two of three domains on the ADI-R. Patients classified as ‘autism spectrum’ met DSM-IV criteria for pervasive developmental disorder not otherwise specified, autism spectrum cutoffs on the ADOS-G, and met cutoffs on at least two of three domains of the ADI-R. Twenty-four patients (75%) met full criteria for an autism classification based on these criteria. An additional three patients (9.3%) met criteria for autism spectrum. Five patients (15.6%) did not meet study criteria for an ASD (Additional file 1: Table S1). In approximately 50% of cases, the ADOS-G, ADI-R and DSM-IV were in full agreement. The ADOS-G and DSM-IV diagnosis, which considered all sources of information, were in agreement in all but one case where the participant (SH2) met DSM-IV criteria for autistic disorder but only autism spectrum criteria on the ADOS-G. By contrast, the ADI-R yielded ‘autism’ classifications in four out of five ‘not ASD’ patients (that is, patients who did not meet DSM-IV or ADOS-G criteria for an ASD). In nine ‘autism’ cases, the ADI-R was below threshold on one out of three domains, typically the repetitive behavior/restricted interests domain (Domain C).
Table 2.
Autism spectrum disorder and intelligence quotient diagnostic classifications
| N | % | |
|---|---|---|
|
Consensus ASD diagnosis (n = 32) |
|
|
| Autism |
24 |
75 |
| Autism spectrum |
3 |
9.4 |
| Not ASD |
5 |
15.6 |
|
Nonverbal IQ classification (n = 30) |
|
|
| Average (IQ 100–110) |
1 |
3.3 |
| Mild intellectual disability (IQ 50–55 to 70) |
3 |
10 |
| Moderate intellectual disability (IQ 35–40 to 50–55) |
3 |
10 |
| Severe intellectual disability (IQ 20–25 to 35–40) |
7 |
23.3 |
| Profound intellectual disability (IQ <20-25) | 16 | 53.3 |
ASD, autism spectrum disorder; IQ, intelligence quotient.
Results from the ADOS-G indicated that 23 participants met criteria for autism and four for autism spectrum (Additional file 1: Table S1). Five participants (age range 2.9 to 25.9 years old) were below thresholds for an autism spectrum classification on the ADOS-G. We also calculated calibrated severity scores [46] in the current sample to provide an estimate of ASD severity compared to reports of severity scores in children with ASD and ID in prior research. The mean calibrated severity score for patients classified as ASD or autism in our sample was 7.07 (SD 1.82). Severity scores between 4 and 6 represent ‘autism spectrum’ classification, and between 6 and 10 correspond to ‘autism’ classifications. Minimally verbal, school-aged children in a previous study presented with scores in the more severe end of this range [46].
The ADI-R sample included 30 out of 32 participants. Two of the youngest participants (<25 months) were not administered the ADI-R as it has been shown to have strongest validity in individuals with chronological and mental ages over 18 months. Strict interpretation of the ADI-R indicates that 18 out of 30 participants (60%) met full criteria on the ADI-R. An additional 11 participants met criteria in two of three domains. Eight of these 11 participants had subthreshold scores on the repetitive behaviors domain (that is, 1 to 2 points below). Figure 2 presents the average scores of each domain and subdomain of the ADI-R. The data reflect deficits across ASD symptom domains, including several social communication skills such social reciprocity, engagement, and play skills.
Figure 2.

Mean scores on Autism Diagnostic Interview - Revised. (A) ADI-R domains and (B) subdomains and repetitive behaviors factors (n = 29). Data represent mean ± standard deviation. Cutoff scores for domains are as follows: Social = 10, Communication (nonverbal) = 7, Repetitive behaviors and restricted interests = 3. ADI, Autism Diagnostic Interview.
A separate analysis of the ADI-R repetitive behavior domain was conducted to allow comparison with previous research suggesting low rates of repetitive behaviors in 22q13 deletion syndrome [27]. Figure 2 includes data from an analysis of two repetitive behavior factors identified in research on the ADI-R: insistence on sameness, and repetitive sensory and motor behaviors [47]. Interpretation of the ADI-R algorithm alone indicates many children presented with subthreshold levels of repetitive behaviors. However, analysis of all items suggests repetitive and sensory-motor behaviors are reported in all but one participant and represent a range of behaviors seen in idiopathic autism. The item analysis is also consistent with reports from the psychiatric evaluation (see below), which indicated the presence of repetitive behaviors or restricted interests in most participants. The most common repetitive behaviors or restricted interests reported on the ADI-R included unusual sensory interests (n = 21), repetitive use of objects (n = 19), whole body or complex motor mannerisms (n = 15), hand and finger motor mannerisms (n = 13), circumscribed interests (n = 11), unusual sensory sensitivities (n = 11), and negative reactions to changes in personal routines (n = 10). Few reported their children as presenting with unusual preoccupations, having negative reactions to changes in environment, or showing unusual attachment to objects.
Psychiatric evaluation
A psychiatric review of systems was performed on all participants, and focused primarily on DSM-IV criteria for pervasive developmental disorders. Associated psychiatric and behavioral manifestations were assessed through parent interview, direct child assessment and an autism-focused mental status examination [48] during psychiatric evaluation. On the psychiatric evaluation, the most commonly reported repetitive behaviors included chewing and mouthing objects; hand flapping; jumping; spinning objects; opening and closing doors, drawers and lights; sniffing; breath holding; forced expirations; stereotypic vocalizations; and teeth grinding. Sixteen participants (50%) exhibited current manifestations of hyperactivity and 14 (44%) had episodes of aggression or self-injury. No participants reported current or past motor or vocal tics. Sleep disturbance was also reported in 13 participants (41%).
Behavioral and emotional problems reported in the psychiatric evaluation were also supported by parent ratings on the Vineland Adaptive Behavior Scales. Vineland reports indicated high rates of internalizing behaviors (18.47 ± 2.10), and externalizing behaviors (16.3 ± 2.03). However, item analysis suggests the Vineland does not represent an accurate evaluation of associated symptoms for individuals with SHANK3 deficiency. The most commonly endorsed ‘internalizing symptoms’ on the Vineland included eating difficulties, sleeping difficulties and poor eye contact, symptoms more likely representing developmental disability rather than psychiatric comorbidity. Items more likely to represent internalizing symptoms consistent with psychiatric illness, such as sadness (n = 3), lethargy (n = 1) and anxiety (n = 4), were endorsed at a low rate in our sample.
We also used information from the psychiatric evaluation and ADI-R to evaluate the presence of skill regression, a developmental pattern commonly reported by parents. Regression was reported to occur as early as 15 months and up to 17 years old. Parents of nine participants (28%) reported skill regression during the psychiatric evaluation, most commonly in the language domain. In at least five of these patients, regression was reported to be associated with onset of seizures. The patient who experienced regression at 17 years of age (SH25) did so after being placed in a residential program, and the regression included loss of language and toileting skills. The ADI-R requires presence of functional, daily spontaneous words prior to loss to capture language regression. Using this more stringent criterion, no participants met criteria for language loss on the ADI-R because of their limited verbal abilities. However, 7 out of 30 (23%) reported loss of skills in other domains, including hand movements (n = 5), other motor skills (n = 3), self help skills (n = 3), play skills (n = 2) and social engagement (n = 4).
Cognitive and language testing
Results of cognitive testing (Table 2) indicate that the majority of the sample had ID, with only one child scoring in the average range for the nonverbal IQ estimate. Most patients (77%) scored in the severe to profound range of ID. Language was evaluated by clinical intake evaluation, Vineland subscales and, in a subset of the sample, the language domains of the Mullen (Table 3). Five patients (19%) used some words spontaneously to communicate on a regular basis, but none were using spontaneous phrase speech consistently. Standardized testing of language abilities based on Mullen and Vineland scores suggest that the majority of the patients were functioning 2 to 3 SDs below the mean. Figure 3 depicts a scatterplot of receptive and expressive language V scores from the Vineland. Two patients scored in the average range for receptive language abilities, and one patient scored in the average range for expressive language abilities. The latter patient is one of the youngest in the sample (under 2 years old) and as such his score is interpreted with care, given the instability of standardized test scores in very young children with developmental disabilities.
Table 3.
Language and motor age equivalence scores
| N | Age equivalence months (mean ± SD) | |
|---|---|---|
|
Language measures |
|
|
|
Receptive language |
|
|
| Mullen receptive language |
27 |
11.63±6.25 |
| Vineland receptive language |
28 |
12.40±8.07 |
|
Expressive language |
|
|
| Mullen expressive language |
27 |
7.52±4.72 |
| Vineland expressive language |
29 |
9.95±5.98 |
|
Motor ability measures |
|
|
|
Fine motor skills |
|
|
| Mullen fine motor skills |
27 |
14.89±6.07 |
| Vineland fine motor skills |
29 |
19.62±9.48 |
|
Gross motor skills |
|
|
| Mullen gross motor skills |
19 |
17.95±8.52 |
| Vineland gross motor skills | 29 | 21.03±9.02 |
Figure 3.
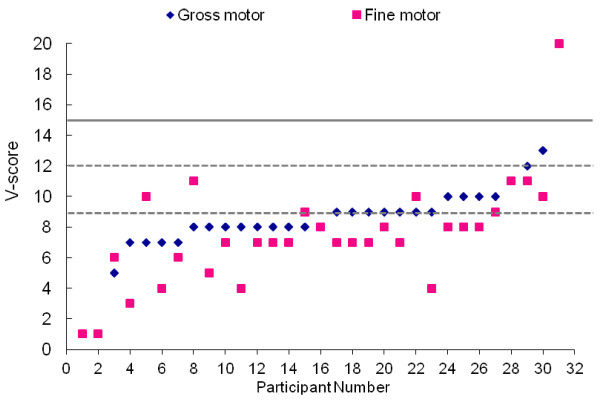
Motor functioning based on Vineland Adaptive Behavior Scales (n = 32) represented by V scores. Population mean (15) and standard deviation (3) are represented by solid and dashed lines, respectively.
Neurological examination
A detailed neurological examination was performed on 16 participants ranging in age from 20 months to 45 years old. The major neurological findings were hypotonia, gait disturbance, and motor planning and coordination abnormalities. Signs often began in early infancy, most commonly manifesting as feeding abnormalities, reported in 7 out of 16 patients (44%). Hypotonia involving limbs and trunk was present in 16 out of 16 patients (100%). At the time of the examination, hypotonia was rated as mild in seven patients and moderate in nine. Signs of upper motor neuron dysfunction, including increased ankle tone, clonus, toe-walking, extensor (Babinski) signs, abnormal posturing of upper extremities and ‘cortical thumbs’, were present in 9 out of 16 patients (56%) evaluated.
Gait was abnormal in 15 out of 16 patients (94%). Four out of 16 patients were not able to walk independently; three were young children with developmental delay who had not yet walked independently and the fourth patient was an adult who was unable to ambulate after a pelvic fracture and spinal surgery. Gait abnormalities included wide-based stance (n = 5), in-toeing (n = 5), toe-walking (n = 2), steppage (n = 3), feet dragging (n = 2), circumduction (n = 2), knee flexion (n = 4) and upper extremity posturing (n = 2). Motor coordination was found to be grossly abnormal in 14 out 16 patients (88%). In the remaining two patients, gross motor coordination abnormalities were not present, but precise testing was difficult because of cognitive and behavioral limitations. Results of the neurological examination were supported by standardized assessments of gross and fine motor abilities. Scores indicated that patients performed in the 14 to 19 month range on average, regardless of chronological age, on both direct observation (Mullen) and parent report (Vineland) measures of fine and gross motor abilities (Table 3 and Figure 4). While these tests provide insight into the potential extent of the delay, specific patterns of motor deficits have not clearly emerged.
Figure 4.

Language functioning based on Vineland Adaptive Behavior Scales (n = 32) represented by V scores. Population mean (15) and standard deviation (3) are represented by solid and dashed lines, respectively.
Dysmorphology evaluation
Thirty-one patients had comprehensive clinical genetic and dysmorphology examinations at Mount Sinai; for one patient we relied on medical record review of an examination at another clinical genetics program. A total of 32 dysmorphic features were evaluated and all patients had at least one abnormal feature (Table 4, Additional file 2: Table S2). The most common dysmorphic feature was the appearance of large fleshy hands, present in 53% of patients, followed by bulbous nose in 47%, long eyelashes in 44%, and minor ear anomalies in 41%. Other frequent features included hypoplastic or dysplastic nails in 34% and full lips in 31%. Accelerated growth has been reported in previous studies [21,22,33,49]. However, only one patient in our study was above the 95th percentile in height. Six children of the 29 participants below the age of 18 had height above the 85th percentile and four children had short stature with height below the 5th percentile. None of the participants with short stature had ring chromosome 22, although a relationship between the two has been previously suggested [23].
Table 4.
Dysmorphic features identified in the clinical genetic evaluation (N = 32)
| Dysmorphic features | N | % | Estimated frequency from previous reports [50] |
|---|---|---|---|
| Large, fleshy hands |
17 |
53 |
>50% |
| Bulbous nose |
15 |
47 |
>50% |
| Long eyelashes |
14 |
44 |
>50% |
| Ear anomalies |
13 |
41 |
>50% |
| Hypoplastic/dysplastic nails |
11 |
34 |
>50% |
| Full lips |
10 |
31 |
|
| Epicanthal folds |
10 |
31 |
>25% |
| Macrocephalya |
10 |
31 |
|
| Dolichocephaly |
8 |
25 |
>50% |
| High arched palate |
8 |
25 |
>25% |
| Hyperextensibility |
8 |
25 |
|
| Full cheeks |
8 |
25 |
>50% |
| Periorbital fullness |
8 |
25 |
>50% |
| Pointed chin |
7 |
22 |
>50% |
| Abnormal spine curvature |
7 |
22 |
|
| Wide nasal bridge |
5 |
16 |
>50% |
| Long philtrum |
5 |
16 |
>25% |
| Sparse hair/abnormal whorl |
5 |
16 |
|
| Malocclusion/widely spaced teeth |
6 |
19 |
>25% |
| Micrognathia |
4 |
13 |
|
| Hypertelorism |
4 |
13 |
|
| Short statureb |
4 |
13 |
|
| Sacral dimple |
4 |
13 |
>50% |
| Syndactyly of toes 2 and 3 |
3 |
9 |
|
| Malar hypoplasia |
3 |
9 |
|
| Fifth finger clinodactyly |
3 |
9 |
<14% |
| Microcephalyc |
2 |
6 |
|
| Deep set eyes |
2 |
6 |
>50% |
| Accelerated growthd |
1 |
3 |
|
| Flat midface |
1 |
3 |
>50% |
| Ptosis |
1 |
3 |
>25% |
| Low set ears | 1 | 3 |
a defined as head circumference >97th percentile; b defined as height <3rd percentile; c defined as head circumference <3rd percentile; d defined as height >95th percentile.
Medical comorbidities
Comprehensive medical record review was performed with records provided by families. EEG was performed in 21 out of 32 cases. Thirteen of 32 participants (41%) had clinical seizures reported by parents, including seven with febrile seizures only (22%; four of these had EEG recordings interpreted as normal; one was interpreted as abnormal but the report was not available for review; two did not receive an EEG), four participants (13%) had non-febrile seizures and two (6%) had both febrile and non-febrile seizures. Among patients with non-febrile seizures, five out of six (83%) had generalized seizures and one (17%) had partial complex seizures; all patients with non-febrile seizures had evidence of EEG abnormalities. In one patient, the febrile seizures were complex seizures and one patient required temporal lobectomy due to uncontrolled seizure disorder. Four patients (13%) had abnormal EEG recordings without reported clinical seizures.
Among other comorbid medical conditions (Table 5), increased pain tolerance and hypotonia were the most common, occurring in 88% and 75% of the patients, respectively. In addition, 17 patients (53%) had a history of recurring infection of the upper respiratory tract, most commonly associated with ear infections in 14 patients (44%) . Two patients also had recurring staphylococcus skin infections with a history of cellulitis. Other common conditions were gastroesophageal reflux disease (44%), sleep disturbance (41%), constipation and/or diarrhea (38%), renal abnormalities (38%), lymphedema (22%), seasonal allergies (19%) and food allergies (16%). Specific renal abnormalities on medical record review included vesicoureteral reflux (n = 4), hydronephrosis (n = 4), renal agenesis (n = 2), extrarenal pelvis (n = 1), dysplastic kidney (n = 1), and bilateral horseshoe kidneys and pyelectasis (n = 1). Only one patient presented with a cardiac defect, aortic regurgitation. Two patients had strabismus and the following conditions affected a single patient each: hypothyroidism, hypertrichosis, vitiligo, intestinal lymphangiectasia, celiac disease, eczema, and esophageal yeast overgrowth with esophagitis.
Table 5.
Medical comorbidities identified from clinical interviews and medical record reviews (n = 32)
| Medical comorbidity | N | % | Estimated frequency from previous reports [50] |
|---|---|---|---|
| Increased pain tolerance |
28 |
88 |
>50% |
| Hypotonia |
24 |
75 |
>75% |
| Recurring upper respiratory tract infections |
17 |
53 |
|
| Gastroesophageal reflux |
14 |
44 |
>25% |
| Sleep disturbance |
13 |
41 |
|
| Seizures (febrile and/or non-febrile) |
13 |
41 |
>25% |
| Constipation and/or diarrhea |
12 |
38 |
|
| Renal abnormalities |
12 |
38 |
>25% |
| Lymphedema |
7 |
22 |
>25% |
| Seasonal allergies |
6 |
19 |
|
| Food allergies |
5 |
16 |
|
| Asthma |
3 |
9 |
|
| Strabismus |
2 |
6 |
>25% |
| Cardiac abnormalities |
1 |
3 |
>25% |
| Hypothyroidism |
1 |
3 |
5% |
| Hypertrichosis |
1 |
3 |
|
| Vitiligo | 1 | 3 |
Neuroimaging
Medical record review indicated that 28 out of 32 participants (88%) had MRIs performed and 21 out of 28 (75%) had abnormal findings. Reported abnormalities were consistent with those reported in previous studies [24,27,28,36] and included thinning or hypoplasia of the corpus callosum (n = 5), white matter changes (for example, delayed myelination, generalized white matter atrophy, and nonspecific white matter hyperintensities or gliosis) (n = 9), ventricular dilation (n = 7) and arachnoid cysts (n = 5).
Genotype-phenotype correlations
First, broad domains were used in the analysis of the association between deletion size and clinical features (Table 6). The domains included the total number of dysmorphic features identified by clinical examination, number of medical comorbidities, nonverbal IQ, Vineland language domain V scores, Vineland motor domain V scores and ADI subdomain scores to capture autism symptomatology. The results of the SROC analysis indicated significant associations between larger deletion size and the following variables: number of dysmorphic features (rs = 0.474), number of medical comorbidities (rs = 0.402), ADI Social (rs = 0.466) and ADI Communication (rs = 0.386) domain scores. In addition, SROC estimates for gross motor skills approached statistical significance (rs = −0.402). Descriptive review also suggests a possible relationship between deletion size and diagnostic classification based on consensus diagnosis. Mean deletion sizes for non-ASD, autism spectrum and autism groups were 2.28 ± 3.52 Mb, 3.57 ± 2.31 Mb and 4.35 ± 2.76 Mb, respectively. While the number of patients in the non-ASD and spectrum categories is small, the progressive increase in deletion size associated with autism diagnosis may warrant further research in larger samples.
Table 6.
Association between deletion size and phenotypic variables
| Phenotypic variable | N | Deletion size |
BCa confidence intervala |
|
|---|---|---|---|---|
| Lower | Upper | |||
| Number of dysmorphic features |
32 |
0.474b |
0.145 |
0.738 |
| Number of medical comorbidities |
32 |
0.386b |
0.022 |
0.640 |
| Nonverbal IQ estimate |
29 |
−0.332 |
−0.640 |
0.112 |
| Gross motor skills (Vineland) |
31 |
−0.402c |
−0.728 |
0.036 |
| Fine motor skills (Vineland) |
31 |
−0.123 |
−0.473 |
0.254 |
| Expressive language skills (Vineland) |
32 |
−0.184 |
−0.531 |
0.199 |
| Receptive language skills (Vineland) |
32 |
−0.231 |
−0.553 |
0.154 |
| Qualitative abnormalities in reciprocal social interactions (ADI-R) |
30 |
0.466b |
0.073 |
0.723 |
| Qualitative abnormalities in communication (ADI-R) |
30 |
0.498b |
0.091 |
0.740 |
| Restricted, repetitive, and stereotyped patterns of behavior (ADI-R) | 30 | −0.229 | −0.592 | 0.214 |
aBooststrap estimates and bias corrected and accelerated confidence intervals for Spearman rank order correlations; bsignificant at 0.05 level (two-tailed test) as confidence interval does not contain 0; capproached significance at 0.05 level (two-tailed test). ADI-R, Autism Diagnostic Interview-Revised; BCa, bias corrected and accelerated confidence intervals; IQ, intelligence quotient.
The association patterns found for medical comorbidities and dysmorphic features were explored further through analysis of each individual feature with deletion size using Mann–Whitney U tests. Significant differences between groups with and without specified features were found for seizures, renal abnormalities, lymphedema, pointed chin and bulbous nose (MWU = 36.00, x2 = 5.513, degrees of freedom = 1, P = 0.019); however, differences were not significant when correcting for multiple comparisons. Note that patient SH32 carried both a point mutation in SHANK3 and a 1.4 Mb duplication at 17q12 harboring the HNF1B gene previously reported [45], so analyses were run with and without this patient, with similar findings.
Second, we looked at the relationship between major medical findings seen in at least four patients and deletion size to begin to understand whether certain genes are associated with specific phenotypes outside the core phenotypes (see Additional file 3: Table S3). Seizures and abnormal EEG were present in many patients, including those with the smallest deletion or just a point mutation in SHANK3. This was also true for gastroesophageal reflux disease, hypotonia, sleep disturbances, abnormal brain MRI and certain dysmorphic features (for example, dolichocephaly, long eyelashes, dysplastic nails, large fleshy hands and full lips). For this reason, it is most parsimonious to attribute these phenotypes to SHANK3. By contrast, lymphedema, asthma, cardiac abnormality and renal abnormalities were only observed in larger deletions, consistent with the potential involvement of additional genes in these manifestations.
Sex differences
The relatively equivalent male:female ratios in SHANK3 deficiency allowed for analysis of sex differences on phenotyping variables. No differences were found between males (n = 18) and females (n = 14) on deletion size, IQ, autism severity, autism diagnosis, adaptive behavior, number of dysmorphic features or number of medical features.
Discussion
The aim of this study was to evaluate a serially ascertained sample of children and adults with SHANK3 deletion or mutation and provide an overview of diagnostic, medical and psychological presentations to guide future research on this syndrome. The results present a picture of SHANK3 deficiency as a disorder characterized by ID, ASD, predominance of gross motor impairments including gait disturbance and hypotonia, and severe speech delays. Approximately half of the sample also reported a history of abnormal EEG findings and gastroesophageal reflux disease on medical record review.
Patterns of dysmorphic features were not observed in our sample. While all patients presented with at least one dysmorphic feature, the most common feature, large fleshy hands, was only present in 53%. It is also important to note that hand size was within height expectations for the sample and, as such, this feature may represent appearance rather than actual size differences. Other commonly observed dysmorphic features included bulbous nose, long eyelashes and ear anomalies. Our sample also did not present with a pattern of accelerated growth reported in previous studies [21,22,33,49]. These results are consistent with a recent study suggesting that the majority of children with SHANK3 deficiency have normal growth [51].
The integration of careful clinical evaluation, caregiver report and direct structured observations used in autism research has been shown to increase the validity of ASD diagnoses [52]. To date, published studies have not utilized standard research methods for assessing the presence of ASD in SHANK3 deficiency. Using the recommended diagnostic methodology, our study found rates of approximately 84% for all ASDs, with rates of 75% for strictly defined autistic disorder and 9.3% for subthreshold symptoms consistent with ASD on the DSM-IV. Rates of autism in our sample were higher than studies relying only on clinical judgment or parent report, which report rates ranging from 26% [29] to 54% [24]. By contrast, studies utilizing at least one standardized assessment tool report rates comparable to the present study. Using the Childhood Autism Rating Scale [53], an investigator rated instrument, Phelan et al. [22] found 94% of their sample met criteria for an ASD and Jeffries and colleagues [25] found a rate of ASD of 85% using the Social Communication Questionnaire [32], a caregiver report screening measure.
Autism-specific diagnostic evaluation tools have been reported to possess limited specificity in individuals with severe ID [35,46]. Our findings suggest discrepancies between ADOS-G and ADI-R results for 13 out of 32 participants. In 4 out of 13 patients, the ADI-R yielded an autism classification when both ADOS-G and clinical diagnosis did not support the presence of ASD. In 8 out of 13 cases, the ADI-R did not yield an autism classification as a result of subthreshold levels of repetitive behaviors. Overall, the ADOS-G and DSM-IV clinical diagnoses likely provide a more representative picture of the social communication and repetitive behavior profiles presenting in children with SHANK3 deficiency and ASD. As previously mentioned, the ADI-R has less validity in populations with chronological and mental ages below 18 months. In our sample, the mean language age equivalence scores were below 12 months and motor age equivalence scores were below 21 months, suggesting a level of ID which may present diagnostic challenges when using the ADI-R.
Importantly, social communication impairments, and repetitive behaviors or restricted interests were observed in this sample of individuals with SHANK3 deficiency. The most common social communication impairments included social engagement, reciprocity and play. Our results also indicated a significant association between deletion size and scores on social and communication domains of the ADI-R. This finding suggests that the severity of the behavioral phenotype is associated with larger deletion sizes and runs in contrast to results from Sarasua and colleagues [29], who found that individuals with smaller deletion sizes were more likely to have an ASD. However, the presence of ASD in that study [29] was based on parental report and only 26% of the sample was identified as having ASD.
Previous research had suggested a low rate of repetitive behaviors in individuals with 22q13 deletion syndrome [27]. In the repetitive behavior and restricted interest domain of the ADI-R, over half of our sample displayed intense sensory interests and repetitive toy or object play, which are commonly found in individuals with ID alone. However, approximately one-third of the sample also reported compulsive behaviors such as adhering to rigid routines. Further evaluation of repetitive behaviors and restricted interest domains may be an important direction for future research, particularly given the discrepancy between repetitive behaviors reported on the ADI-R and clinical evaluations, as well as the high rates of sensory-related behaviors observed in this sample of individuals with SHANK3 deficiency. Increasing sample size and improving specificity of measurements of the repetitive behaviors will be important in reconciling published reports.
The severity of the phenotype in SHANK3 deficiency is highlighted by the results of the standardized evaluations of cognitive, language and functional skills in this study. No participants used phrase speech on a daily basis, and only five used single words to communicate consistently. Scores from direct testing of language and cognitive skills, as well as parent reports on the Vineland, reflect the severe language disability found in most individuals with SHANK3 deficiency. One of 32 participants had a nonverbal IQ estimate in the average range, with approximately three-quarters of participants performing in the severe to profound range of ID.
The floor effects observed on standardized testing did not allow for comparison of differences within domains (for example, receptive or expressive language). While standardized tests allow comparison of the phenotype relative to general population norms, available tools are hampered by a restricted range in individuals with ID. As such, traditional methods are likely insufficient for the purposes of treatment development or progress evaluations. Development of evaluation tools designed to capture precursors of language and early cognitive signals, such as auditory discrimination and visual attention, are needed in future investigations of SHANK3 deficiency.
One significant pattern of results relates to the prevalence of motor impairments found across evaluation methods. Motor disabilities were identified in the earliest clinical profiles of SHANK3 deficiency and their predominance is supported by findings in our sample. The comprehensive clinical evaluation suggests hypotonia as a dominant, early presenting clinical feature, present in 100% of the 16 children who underwent neurological examinations in our sample and described by parents in 75% of the total 32 patients. Motor impairments due to hypotonia may result in feeding difficulties, which are commonly reported as one of the first signs noticed by families in infancy. The neurological evaluation in this study also found high rates of gait abnormalities. Gross motor impairments were moderately associated with deletion size, albeit slightly below the threshold of significance.
Other common clinical features in our sample included gastroesophageal reflux disease, seizures, sleep disturbance, renal abnormalities, and constipation and/or diarrhea. The presence of recurring ear and upper respiratory tract infections was notable and may reflect poor airway protection and sputum clearance due to low muscle tone in SHANK3 deficiency. It is also possible that SHANK3 may play an important role in immune function. Immunocytochemical analyses in rats have verified the presence of Shank3 proteins in thymic tissue and suggest a role of Shank3 in the coordination of immune cell signal transduction [54]. Previous case reports have also suggested a link between immune dysfunction and SHANK3 deficiency [55,56]. Our findings suggest an increased prevalence of recurring infections in our sample of patients with SHANK3 deficiency and its relationship to SHANK3 and neighboring genes is an area that should be more carefully explored in the future.
Twenty-eight of our patients had MRIs performed and, among them, 75% had abnormal findings based on clinical reports. The prevalence of structural brain abnormalities in our sample highlights the paucity of data on the abnormal neural systems underlying this syndrome. Understanding how brain structure is affected in SHANK3 deficiency will be important for future studies. Given that little is known about the neurobiology associated with SHANK3 deficiency, identification of specific brain abnormalities will aid in more thorough characterization and may provide a critical link between SHANK3 deficiency and associated behavior.
Significant regression in motor and social skills has also been reported among patients in our sample and in the literature [2,5,30,57,58]. Isolated reports have emerged recently describing significant medical complications of SHANK3 deficiency in the context of severe cognitive and behavioral regression, including seizure-induced aspiration [2], renal failure [5] and pneumonia [58]. In addition, recent reports of psychiatric comorbidity in individuals with SHANK3 deficiency suggest that, as patients age, they may be at increased risk for bipolar disorder [30,57,59]. Our results suggest that regression may occur at various ages, across multiple domains, and is often associated with the onset of seizures. Future studies examining the natural history of SHANK3 deficiency should clarify the nature and extent of regression.
Several studies have examined genotype-phenotype correlations in SHANK3 deficiency and results are inconsistent [21,23,25,26,28,29,60]. The genotype-phenotype correlations in this study are consistent with at least two previous reports [28,29], indicating a correlation between deletion size and more severe phenotypes. In our sample, larger deletion sizes were associated with a greater number of dysmorphic features and medical comorbidities. In addition, a correlation between larger deletion sizes and social communication impairments associated with autism was found in the present study using standardized research administrations of the ADI-R. Nonverbal IQ, fine motor skills, repetitive behaviors and language abilities did not show an association with size of deletion in the 22q13 region. The associations identified in our analysis are limited by several factors, including sample size. However, should a clear pattern of genotype-phenotype correlations emerge in larger samples, it may delineate a role for other genes and pathways in SHANK3 deficiency. Replication of potential associations between deletion size and medical comorbidity, social impairments, and possibly motor skills, would also provide valuable information for medical monitoring and treatment planning.
Conclusions
This study provides additional evidence of the severity of intellectual, motor and speech impairments seen in SHANK3 deficiency, and highlights the prevalence of ASD symptoms in the syndrome. The findings are presented to guide future investigations seeking to comprehensively map the phenotype of SHANK3 deficiency. Although our results provide support for the use of a standardized, prospective methodology for evaluating language, psychiatric, cognitive and medical features of SHANK3 deficiency, they also highlight limitations of available methods and emphasize the need for novel measurement tools in subsequent investigations. Finally, in light of recent reports on adolescence or adult onset psychiatric disturbances [30,57,59] and significant cognitive and behavioral regression [2,5,30,57,58], the need for natural history studies that systematically collect health information is apparent. These studies will facilitate our knowledge of disease progression, facilitate selection of treatment targets, improve medical monitoring, and ultimately improve clinical care across the lifespan of people with SHANK3 deficiency.
Abbreviations
ADI-R: Autism Diagnostic Interview – Revised; ADOS-G: Autism Diagnostic Observation Schedule-G; ASD: Autism spectrum disorder; CMA: Chromosomal microarray analysis; CNV: Copy number variant; DSM-IV: Diagnostic and Statistical Manual of Mental Disorders – Fourth Edition; EEG: Electroencephalography; FISH: Fluorescent in situ hybridization; Hg: Human genome; ID: Intellectual disability; IQ: Intelligence quotient; Kb: Kilobase; Mb: Megabase; MLPA: Multiplex ligation-dependent probe amplification; MRI: Magnetic resonance imaging; PCR: Polymerase chain reaction; SD: Standard deviation; SNP: Single nucleotide polymorphism; SROC: Spearman rank order correlations.
Competing interests
JDB has submitted a patent on IGF1 treatment for SHANK3 deficiency. All other authors declare that they have no competing interests.
Authors’ contributions
LSo participated in study design, data collection, data analysis and manuscript preparation. AK participated in study design, data collection, data analysis and manuscript preparation. JZ participated in data collection and analysis. TL participated in data analysis. DY participated in data analysis. LSc participated in data collection and analysis. YF performed neurological evaluations. ATW participated in study design, data collection, data analysis and manuscript preparation. CG participated in molecular genetic analysis. EP participated in molecular genetic analysis. DH participated in data collection. DG participated in data collection. BA participated in data collection. JPW performed clinical genetics evaluations. AY performed clinical genetics evaluations. RC participated in study design. WC performed statistical analyses. CB assisted with data interpretation and manuscript preparation. JDB assisted with study design, data interpretation and manuscript preparation. All authors read and approved the final manuscript.
Supplementary Material
Descriptive and diagnostic data by patient: nonverbal IQ, Vineland Adaptive Behavior Scales, ADI-R, ADOS-G, DSM-IV and consensus diagnosis.
Dysmorphic features organized by deletion size.
Clinical features and medical comorbidities organized by deletion size.
Contributor Information
Latha Soorya, Email: latha_soorya@rush.edu.
Alexander Kolevzon, Email: alexander.kolevzon@mssm.edu.
Jessica Zweifach, Email: jessica.zweifach@gmail.com.
Teresa Lim, Email: teresa.lim@mssm.edu.
Yuriy Dobry, Email: yuriy.dobry@mssm.edu.
Lily Schwartz, Email: lily.schwartz@mssm.edu.
Yitzchak Frank, Email: yitzchak.frank@mssm.edu.
A Ting Wang, Email: ting.wang@mssm.edu.
Guiqing Cai, Email: guiqing.cai@mssm.edu.
Elena Parkhomenko, Email: elena.parkhomenko@mssm.edu.
Danielle Halpern, Email: danielle.halpern@mssm.edu.
David Grodberg, Email: david.grodberg@mssm.edu.
Benjamin Angarita, Email: benjamin.angarita@mssm.edu.
Judith P Willner, Email: judith.willner@mssm.edu.
Amy Yang, Email: amy.yang@mssm.edu.
Roberto Canitano, Email: r.canitano@ao-siena.toscana.it.
William Chaplin, Email: chaplinw@stjohns.edu.
Catalina Betancur, Email: Catalina.Betancur@inserm.fr.
Joseph D Buxbaum, Email: joseph.buxbaum@mssm.edu.
Acknowledgements
This work was supported by grants from the Beatrice and Samuel A. Seaver Foundation and the National Institutes of Health (NIMH grant MH089025 to JDB). We would like to thank the many families that work closely with us to better understand neurodevelopmental disability.
References
- Phelan K, Rogers C. Phelan-McDermid Syndrome. GeneReviews. 2011. [ http://wwwncbinlmnihgov/books/NBK1198/]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H, Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni MC, de Mas P, Bieth E, Roge B, Heron D, Burglen L, Gillberg C, Leboyer M, Bourgeron T. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, Zwaigenbaum L, Fernandez B, Roberts W, Szatmari P, Scherer SW. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007;81:1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J, Spiegelman D, Piton A, Lafreniere RG, Laurent S, St-Onge J, Lapointe L, Hamdan FF, Cossette P, Mottron L, Fombonne E, Joober R, Marineau C, Drapeau P, Rouleau GA. Novel de novo SHANK3 mutation in autistic patients. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:421–424. doi: 10.1002/ajmg.b.30822. [DOI] [PubMed] [Google Scholar]
- Bonaglia MC, Giorda R, Beri S, De Agostini C, Novara F, Fichera M, Grillo L, Galesi O, Vetro A, Ciccone R, Bonati MT, Giglio S, Guerrini R, Osimani S, Marelli S, Zucca C, Grasso R, Borgatti R, Mani E, Motta C, Molteni M, Romano C, Greco D, Reitano S, Baroncini A, Lapi E, Cecconi A, Arrigo G, Patricelli MG, Pantaleoni C. et al. Molecular mechanisms generating and stabilizing terminal 22q13 deletions in 44 subjects with Phelan/McDermid syndrome. PLoS Genet. 2011;7:e1002173. doi: 10.1371/journal.pgen.1002173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckers TM. The postsynaptic density. Cell Tissue Res. 2006;326:409–422. doi: 10.1007/s00441-006-0274-5. [DOI] [PubMed] [Google Scholar]
- Schaaf CP, Sabo A, Sakai Y, Crosby J, Muzny D, Hawes A, Lewis L, Akbar H, Varghese R, Boerwinkle E, Gibbs RA, Zoghbi HY. Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum Mol Genet. 2011;20:3366–3375. doi: 10.1093/hmg/ddr243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccuto L, Lauri M, Sarasua SM, Skinner CD, Buccella D, Dwivedi A, Orteschi D, Collins JS, Zollino M, Visconti P, Dupont B, Tiziano D, Schroer RJ, Neri G, Stevenson RE, Gurrieri F, Schwartz CE. Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur J Hum Genet. 2013;21:310–316. doi: 10.1038/ejhg.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimaki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King MC, Skuse D, Geschwind DH, Gilliam TC. et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilmatre A, Dubourg C, Mosca AL, Legallic S, Goldenberg A, Drouin-Garraud V, Layet V, Rosier A, Briault S, Bonnet-Brilhault F, Laumonnier F, Odent S, Le Vacon G, Joly-Helas G, David V, Bendavid C, Pinoit JM, Henry C, Impallomeni C, Germano E, Tortorella G, Di Rosa G, Barthelemy C, Andres C, Faivre L, Frebourg T, Saugier Veber P, Campion D. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry. 2009;66:947–956. doi: 10.1001/archgenpsychiatry.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bolte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Bryson SE, Carson AR, Casallo G, Casey J, Chung BH, Cochrane L, Corsello C. et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Ballif BC, Torchia BS, Sahoo T, Ravnan JB, Schultz R, Lamb A, Bejjani BA, Shaffer LG. Copy number variations associated with autism spectrum disorders contribute to a spectrum of neurodevelopmental disorders. Genet Med. 2010;12:694–702. doi: 10.1097/GIM.0b013e3181f0c5f3. [DOI] [PubMed] [Google Scholar]
- Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB, Miller KJ, Frazier JA, Silverstein I, Picker J, Weissman L, Raffalli P, Jeste S, Demmer LA, Peters HK, Brewster SJ, Kowalczyk SJ, Rosen-Sheidley B, McGowan C, Duda AW 3rd, Lincoln SA, Lowe KR, Schonwald A, Robbins M, Hisama F, Wolff R, Becker R, Nasir R, Urion DK, Milunsky JM. et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125:e727–735. doi: 10.1542/peds.2009-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer A, Giacobini M, Eriksson M, Gustavsson P, Nordin V, Fernell E, Gillberg C, Nordgren A, Uppstromer A, Anderlid BM, Nordenskjold M, Schoumans J. Copy number variation characteristics in subpopulations of patients with autism spectrum disorders. Am J Med Genet B Neuropsychiatr Genet. 2011;156:115–124. doi: 10.1002/ajmg.b.31142. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, Mason CE, Bilguvar K, Celestino-Soper PB, Choi M, Crawford EL, Davis L, Wright NR, Dhodapkar RM, DiCola M, DiLullo NM, Fernandez TV, Fielding-Singh V, Fishman DO, Frahm S, Garagaloyan R, Goh GS, Kammela S, Klei L, Lowe JK, Lund SC. et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, Abdel-Hamid H, Bader P, McCracken E, Niyazov D, Leppig K, Thiese H, Hummel M, Alexander N, Gorski J, Kussmann J, Shashi V, Johnson K, Rehder C, Ballif BC, Shaffer LG, Eichler EE. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X, Jiang YW, Zhang X, An Y, Zhang J, Wu Y, Wang J, Sun Y, Liu Y, Gao X, Shen Y, Wu X, Qiu Z, Jin L, Wu BL, Wang H. High proportion of 22q13 deletions and SHANK3 mutations in Chinese patients with intellectual disability. PLoS One. 2012;7:e34739. doi: 10.1371/journal.pone.0034739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Araki Y, Lin DT, Yoshizawa Y, Higashi K, Park AR, Spiegelman D, Dobrzeniecka S, Piton A, Tomitori H, Daoud H, Massicotte C, Henrion E, Diallo O, Shekarabi M, Marineau C, Shevell M, Maranda B, Mitchell G, Nadeau A, D'Anjou G, Vanasse M, Srour M, Lafreniere RG, Drapeau P, Lacaille JC, Kim E, Lee JR, Igarashi K. et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet. 2011;88:306–316. doi: 10.1016/j.ajhg.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai Y, Shaw CA, Dawson BC, Dugas DV, Al-Mohtaseb Z, Hill DE, Zoghbi HY. Protein interactome reveals converging molecular pathways among autism disorders. Sci Transl Med. 2011;3:86ra49. doi: 10.1126/scitranslmed.3002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesslinger NJ, Gorski JL, Kurczynski TW, Shapira SK, Siegel-Bartelt J, Dumanski JP, Cullen RF Jr, French BN, McDermid HE. Clinical, cytogenetic, and molecular characterization of seven patients with deletions of chromosome 22q13.3. Am J Hum Genet. 1994;54:464–472. [PMC free article] [PubMed] [Google Scholar]
- Phelan MC, Rogers RC, Saul RA, Stapleton GA, Sweet K, McDermid H, Shaw SR, Claytor J, Willis J, Kelly DP. 22q13 deletion syndrome. Am J Med Genet. 2001;101:91–99. doi: 10.1002/1096-8628(20010615)101:2<91::AID-AJMG1340>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Luciani JJ, de Mas P, Depetris D, Mignon-Ravix C, Bottani A, Prieur M, Jonveaux P, Philippe A, Bourrouillou G, de Martinville B, Delobel B, Vallee L, Croquette MF, Mattei MG. Telomeric 22q13 deletions resulting from rings, simple deletions, and translocations: cytogenetic, molecular, and clinical analyses of 32 new observations. J Med Genet. 2003;40:690–696. doi: 10.1136/jmg.40.9.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning MA, Cassidy SB, Clericuzio C, Cherry AM, Schwartz S, Hudgins L, Enns GM, Hoyme HE. Terminal 22q deletion syndrome: a newly recognized cause of speech and language disability in the autism spectrum. Pediatrics. 2004;114:451–457. doi: 10.1542/peds.114.2.451. [DOI] [PubMed] [Google Scholar]
- Jeffries AR, Curran S, Elmslie F, Sharma A, Wenger S, Hummel M, Powell J. Molecular and phenotypic characterization of ring chromosome 22. Am J Med Genet A. 2005;137:139–147. doi: 10.1002/ajmg.a.30780. [DOI] [PubMed] [Google Scholar]
- Koolen DA, Reardon W, Rosser EM, Lacombe D, Hurst JA, Law CJ, Bongers EM, van Ravenswaaij-Arts CM, Leisink MA, van Kessel AG, Veltman JA, de Vries BB. Molecular characterisation of patients with subtelomeric 22q abnormalities using chromosome specific array-based comparative genomic hybridisation. Eur J Hum Genet. 2005;13:1019–1024. doi: 10.1038/sj.ejhg.5201456. [DOI] [PubMed] [Google Scholar]
- Philippe A, Boddaert N, Vaivre-Douret L, Robel L, Danon-Boileau L, Malan V, De Blois MC, Heron D, Colleaux L, Golse B, Zilbovicius M, Munnich A. Neurobehavioral profile and brain imaging study of the 22q13.3 deletion syndrome in childhood. Pediatrics. 2008;122:e376–382. doi: 10.1542/peds.2007-2584. [DOI] [PubMed] [Google Scholar]
- Dhar SU, del Gaudio D, German JR, Peters SU, Ou Z, Bader PI, Berg JS, Blazo M, Brown CW, Graham BH, Grebe TA, Lalani S, Irons M, Sparagana S, Williams M, Phillips JA 3rd, Beaudet AL, Stankiewicz P, Patel A, Cheung SW, Sahoo T. 22q13.3 deletion syndrome: clinical and molecular analysis using array CGH. Am J Med Genet A. 2010;152A:573–581. doi: 10.1002/ajmg.a.33253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarasua SM, Dwivedi A, Boccuto L, Rollins JD, Chen CF, Rogers RC, Phelan K, DuPont BR, Collins JS. Association between deletion size and important phenotypes expands the genomic region of interest in Phelan-McDermid syndrome (22q13 deletion syndrome) J Med Genet. 2011;48:761–766. doi: 10.1136/jmedgenet-2011-100225. [DOI] [PubMed] [Google Scholar]
- Denayer A, Van Esch H, de Ravel T, Frijns JP, Van Buggenhout G, Vogels A, Devriendt K, Geutjens J, Thiry P, Swillen A. Neuropsychopathology in 7 patients with the 22q13 deletion syndrome: presence of bipolar disorder and progressive loss of skills. Mol Syndromol. 2012;3:14–20. doi: 10.1159/000339119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad C, Prasad AN, Chodirker BN, Lee C, Dawson AK, Jocelyn LJ, Chudley AE. Genetic evaluation of pervasive developmental disorders: the terminal 22q13 deletion syndrome may represent a recognizable phenotype. Clin Genet. 2000;57:103–109. doi: 10.1034/j.1399-0004.2000.570203.x. [DOI] [PubMed] [Google Scholar]
- Berument SK, Rutter M, Lord C, Pickles A, Bailey A. Autism screening questionnaire: diagnostic validity. Br J Psychiatry. 1999;175:444–451. doi: 10.1192/bjp.175.5.444. [DOI] [PubMed] [Google Scholar]
- Cusmano-Ozog K, Manning MA, Hoyme HE. 22q13.3 deletion syndrome: a recognizable malformation syndrome associated with marked speech and language delay. Am J Med Genet C Semin Med Genet. 2007;145C:393–398. doi: 10.1002/ajmg.c.30155. [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- Gotham K, Bishop SL, Lord C. In: Autism Spectrum Disorders. Amaral D, Geschwind D, Dawson G, editor. New York: Oxford University Press; 2011. Diagnosis of Autism Spectrum Disorders; pp. 30–43. [Google Scholar]
- Aldinger KA, Kogan J, Kimonis V, Fernandez B, Horn D, Klopocki E, Chung B, Toutain A, Weksberg R, Millen KJ, Barkovich AJ, Dobyns WB. Cerebellar and posterior fossa malformations in patients with autism-associated chromosome 22q13 terminal deletion. Am J Med Genet A. 2013;161A:131–136. doi: 10.1002/ajmg.a.35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- Lord C, Risi S, Lambrecht L, Cook EH Jr, Leventhal BL, DiLavore PC, Pickles A, Rutter M. The Autism Diagnostic Observation Schedule-Generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord. 2000;30:205–223. doi: 10.1023/A:1005592401947. [DOI] [PubMed] [Google Scholar]
- Mullen EM. Mullen Scales of Early Learning. Circle Pines, MN: American Guidance Service; 1995. [Google Scholar]
- Roid GH. Stanford-Binet Intelligence Scales. 5. Itasca, IL: Riverside; 2003. [Google Scholar]
- Roid GH, Miller LJ. Leiter International Performance Scale-Revised. Wood Dale, IL: Stoelting Co.; 1997. [Google Scholar]
- Sparrow SS, Cicchetti DV, Balla DA. Vineland-II Survey Forms Manual (Vineland Adaptive Behavior Scales) 2. Minneapolis, MN: AGS Publishing; 2005. [Google Scholar]
- Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolevzon A, Cai G, Soorya L, Takahashi N, Grodberg D, Kajiwara Y, Willner JP, Tryfon A, Buxbaum JD. Analysis of a purported SHANK3 mutation in a boy with autism: clinical impact of rare variant research in neurodevelopmental disabilities. Brain Res. 2011;1380:98–105. doi: 10.1016/j.brainres.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Brandt T, Desai K, Grodberg D, Mehta L, Cohen N, Tryfon A, Kolevzon A, Soorya L, Buxbaum JD, Edelmann L. Complex autism spectrum disorder in a patient with a 17q12 microduplication. Am J Med Genet A. 2012;158A:1170–1177. doi: 10.1002/ajmg.a.35267. [DOI] [PubMed] [Google Scholar]
- Gotham K, Pickles A, Lord C. Standardizing ADOS scores for a measure of severity in autism spectrum disorders. J Autism Dev Disord. 2009;39:693–705. doi: 10.1007/s10803-008-0674-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatmari P, Georgiades S, Bryson S, Zwaigenbaum L, Roberts W, Mahoney W, Goldberg J, Tuff L. Investigating the structure of the restricted, repetitive behaviours and interests domain of autism. J Child Psychol Psychiatry. 2006;47:582–590. doi: 10.1111/j.1469-7610.2005.01537.x. [DOI] [PubMed] [Google Scholar]
- Grodberg D, Weinger PM, Kolevzon A, Soorya L, Buxbaum JD. Brief report: the Autism Mental Status Examination: development of a brief autism-focused exam. J Autism Dev Disord. 2012;42:455–459. doi: 10.1007/s10803-011-1255-4. [DOI] [PubMed] [Google Scholar]
- Phelan MC. Deletion 22q13.3 syndrome. Orphanet J Rare Dis. 2008;3:14. doi: 10.1186/1750-1172-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan K, McDermid HE. The 22q13.3 deletion syndrome (Phelan-McDermid syndrome) Mol Syndromol. 2012;2:186–201. doi: 10.1159/000334260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins JD, Sarasua SM, Phelan K, DuPont BR, Rogers RC, Collins JS. Growth in Phelan-McDermid syndrome. Am J Med Genet A. 2011;155A:2324–2326. doi: 10.1002/ajmg.a.34158. [DOI] [PubMed] [Google Scholar]
- Risi S, Lord C, Gotham K, Corsello C, Chrysler C, Szatmari P, Cook EH Jr, Leventhal BL, Pickles A. Combining information from multiple sources in the diagnosis of autism spectrum disorders. J Am Acad Child Adolesc Psychiatry. 2006;45:1094–1103. doi: 10.1097/01.chi.0000227880.42780.0e. [DOI] [PubMed] [Google Scholar]
- Schopler E, Reichler RJ, Renner BR. The Childhood Autism Rating Scale. Los Angeles: Western Psychological Services; 1988. [Google Scholar]
- Redecker P, Bockmann J, Bockers TM. Expression of postsynaptic density proteins of the ProSAP/Shank family in the thymus. Histochem Cell Biol. 2006;126:679–685. doi: 10.1007/s00418-006-0199-9. [DOI] [PubMed] [Google Scholar]
- Chen CP, Lin SP, Chern SR, Tsai FJ, Wu PC, Lee CC, Chen YT, Chen WL, Wang W. A de novo 7.9 Mb deletion in 22q13.2-qter in a boy with autistic features, epilepsy, developmental delay, atopic dermatitis and abnormal immunological findings. Eur J Med Genet. 2010;53:329–332. doi: 10.1016/j.ejmg.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Tufano M, Della Corte C, Cirillo F, Spagnuolo MI, Candusso M, Melis D, Torre G, Iorio R. Fulminant autoimmune hepatitis in a girl with 22q13 deletion syndrome: a previously unreported association. Eur J Pediatr. 2009;168:225–227. doi: 10.1007/s00431-008-0732-z. [DOI] [PubMed] [Google Scholar]
- Vucurovic K, Landais E, Delahaigue C, Eutrope J, Schneider A, Leroy C, Kabbaj H, Motte J, Gaillard D, Rolland AC, Doco-Fenzy M. Bipolar affective disorder and early dementia onset in a male patient with SHANK3 deletion. Eur J Med Genet. 2012;55:625–629. doi: 10.1016/j.ejmg.2012.07.009. [DOI] [PubMed] [Google Scholar]
- Willemsen MH, Rensen JH, Van Schrojenstein-Lantman De Valk HM, Hamel BC, Kleefstra T. Adult phenotypes in Angelman- and Rett-like syndromes. Mol Syndromol. 2012;2:217–234. doi: 10.1159/000335661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven WM, Egger JI, Willemsen MH, de Leijer GJ, Kleefstra T. Phelan-McDermid syndrome in two adult brothers: atypical bipolar disorder as its psychopathological phenotype? Neuropsychiatr Dis Treat. 2012;8:175–179. doi: 10.2147/NDT.S30506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson HL, Wong AC, Shaw SR, Tse WY, Stapleton GA, Phelan MC, Hu S, Marshall J, McDermid HE. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet. 2003;40:575–584. doi: 10.1136/jmg.40.8.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Descriptive and diagnostic data by patient: nonverbal IQ, Vineland Adaptive Behavior Scales, ADI-R, ADOS-G, DSM-IV and consensus diagnosis.
Dysmorphic features organized by deletion size.
Clinical features and medical comorbidities organized by deletion size.