

Abstract
Glioblastoma multiforme (GBM) is the most common and prognostically unfavorable form of brain tumor. The aggressive and highly invasive phenotype of these tumors makes them among the most anatomically damaging human cancers with a median survival of less than one year. Although canonical WNT pathway activation in cancers has been historically linked to the presence of mutations involving key components of the pathway (APC, β-CATENIN or AXIN proteins), an increasing number of studies suggest that elevated WNT signaling in GBM is initiated by several alternative mechanisms that are involved in different steps of the disease. Therefore, inhibition of WNT signaling may represent a therapeutically relevant approach for GBM treatment. After the selection of a GBM cell model responsive to WNT inhibition, we set out to develop a screening approach for the identification of compounds capable of modulating canonical WNT signaling and associated proliferative responses in GBM cells. Here we show that the small molecule SEN461 inhibits the canonical WNT signaling pathway in GBM cells, with relevant effects at both molecular and phenotypic levels in vitro and in vivo. These include SEN461-induced AXIN stabilization, increased β-CATENIN phosphorylation/degradation, and inhibition of anchorage-independent growth of human GBM cell lines and patient-derived primary tumor cells in vitro. Moreover, in vivo administration of SEN461 antagonized WNT signaling in Xenopus embryos and reduced tumor growth in a GBM xenograft model. These data represent the first demonstration that small molecule-mediated inhibition of WNT signaling may be a potential approach for GBM therapeutics.
Keywords: Canonical WNT signaling, glioma, small molecule, inhibitor, pathway screening
INTRODUCTION
The WNT signaling pathways, the best studied of which is the canonical (β-CATENIN dependent) branch, are amongst the most evolutionarily conserved and universally important signaling cascades in metazoans, with key roles in cellular proliferation, differentiation, development and function (1, 2). Dysfunctional WNT signaling has been associated with a variety of human pathologies (3) affecting different cell types and tissues including several types of cancer, bone diseases, and diseases of the central nervous system. An increasing number of studies suggest that aberrant WNT signaling can be initiated by several mechanisms affecting key elements of the pathway (4–11). For instance, mutations (inactivating mutations on APC or AXIN1 tumor suppressor genes or activating mutations on the β-CATENIN oncogene), autocrine activation (increased expression of pathway components including WNT ligands, FRIZZLED (FZD) receptors and DISHEVELLED (DVL) family members) and epigenetic phenomena (e.g. promoter hypermethylation) in negative modulators of the WNT pathway which act homeostatically (e.g. SFRPs, DKKs and NKDs genes). Although studied in multiple diseases, the role and importance of the WNT signaling pathway has not been extensively described in GBM. Recent literature data supports the role of WNT/β-CATENIN signaling in glioma initiation, proliferation and invasion (12–18). The protooncogene PLAG2, amplified in GBM, imparts stem-cell properties to glioma cells by regulating WNT signaling (12). The interaction between the transcription factor FORKHEAD BOX M1 (FOXM1) and β-CATENIN, is a mechanism for controlling canonical WNT signaling and is required for glioma formation (13). RNAi mediated depletion of the scaffold protein DVL affects proliferation and promotes differentiation of GBM cells in vitro and in vivo (16). To explore further the relevance of the WNT pathway in GBM and to provide evidence that small molecule inhibition of WNT signaling has therapeutic potential in this CNS tumor, we demonstrate that both genetic and pharmacological WNT inhibition results in modulation of pathway activity at both the biochemical and functional level, and in decreased proliferative capacity both in vitro and in vivo.
We also report the identification and initial characterization of SEN461, a novel, potent small molecule inhibitor of canonical WNT signaling which acts through AXIN stabilization through a mechanism which is not entirely dependent on TANKYRASES, and possesses strong in vitro and in vivo anti-tumour activity in GBM settings.
MATERIALS AND METHODS
Cell lines and human GBMs
The cell lines HEK293, A172, LN229, U87MG, U251 and T98G were obtained from the American Type Culture Collection (ATCC). DBTRG-05-MG was purchased from ICLC (Genoa, Italy), identification and authentication was performed by CELL ID™ System (Promega). All cell lines were cultured according to the supplier’s recommendations. Primary glioma cells (GBMR9, GBMR11, GBMR16 and BTR1) were obtained from patients undergone to surgery at IRCCS Besta Hospital (Milan, Italy), and cultured in RPMI 1640 medium supplemented with 10% FBS. Mouse Wnt3a containing conditioned media (Wnt3a-CM), and control conditioned media (CTR-CM) from mouse L cells, were harvested according to ATCC protocol.
Plasmids, lentiviral vectors and protein production
For the generation of TCF-Luciferase reporter, three copies of a 4x TCF responsive element were cloned into the pcDNA3.1/Zeo vector (Invitrogen) after deletion of the constitutive CMV promoter and the insertion of the Firefly Luciferase ph-FL-TK (Promega). For the TA-Renilla reporter, pcDNA3.1/Hygro (Invitrogen) and ph-RL-TK (Promega) vectors, were digested with restriction enzymes Mlu1 and BamH1 and ligated by T4-Ligase to form the final construct. Human AXIN1 and WNT3A were purchased from Origene as “transfection ready” plasmids. Dominant negative TCF4 cDNA was purchased from Upstate. Human LRP6 and WNT1 have been cloned into pcDNA3.1/Zeo (Invitrogen) by PCR amplification of human cDNA (Clontech).β-CATENIN siRNA was purchased from Ambion. Lentiviral vectors for inducible dominant negative TCF4 (rLV-EF1-tTS, rLV-EF1-rtTA and rLV.TRE-CMV.HA-TCF4DN) were purchased from Vectalys. To generate GST fusion proteins, the PARP domain of human TNKS1 and TNKS2 (934-1166) were synthesized (GenScript) with EcoRI and SalI sites at the 5′ and 3′ ends of the constructs to allow in-frame subcloning into the expression vector pGEX-6P-1.
Primary screening
A structurally diverse, low molecular weight library of 16,000 compounds was screened in stable transfected DBTRG cells containing TCF-Luciferase. For single concentration testing, 6500 cells/well, plated in 96 well-plates were incubated with compounds at 10μM (0.5% DMSO (v/v)) 36 hours after plating. Each compound was tested in duplicate on two different copy cell plates. Luciferase signal was detected using Luclite Luminescence Reporter Gene Assay System 10000 (Perkin Elmer). Data were expressed as % of negative control (DMSO) and the activity threshold was set to 50% reduction. For IC50 determination, stable transfected DBTRG cells (plated at the same density used for the single concentration testing) containing TCF-Luciferase and TA-Renilla were incubated with 8-points dilutions (from 60μM to 0.185μM) compound 36 hours after plating. Each compound was tested in triplicate in a single plate. Luciferase detection was done with Dual-Luciferase Reporter Assay System (Promega). For IC50 calculation, the data were expressed as % of negative control (DMSO) for Firefly and Renilla luciferase independently. Values were calculated using XLFit version 4.2, with a four parameters sigmoid model (XLFit model 205). A luciferase biochemical assay enabled the identification of compounds acting directly on the enzyme rather than true inhibitors. Quantilum recombinant Luciferase (Promega) was employed to test compounds at single concentration (10μM). Data were expressed as % of negative control (DMSO).
Auto-PARsylation reactions
To assess the effect of SEN461 and XAV939 on auto-PARsylation of TANKYRASES, reactions were carried out in 40μl volumes in the presence of the compounds (concentration varying from 0.006 to 100μM, 2.5% DMSO), 20nM GST-TNKS1/2 and 250μM NAD+ (Sigma). Reactions were incubated at room temperature for 2 hours and then quenched by adding 10μl of 20% formic acid. Then, 100μl of acetonitrile was added and the samples were centrifuged for 30 min at 3,500 rpm, 4ºC. The supernatant was transferred to a new plate and subjected to the LC/MS analysis, to detect the formation of nicotinamide (a by-product of the PARsylation reaction).
AXIN ubiquitination assay
For the ubiquitination assay DBTRG cells were pre-treated with 10μM SEN461 for 4 hours and subsequently treated with 25μM of the proteasome inhibitor MG-132 (Sigma) in combination with 10μM of SEN461 overnight. Proteins were extracted with Ripa buffer (50mM Tris-HCl ph 7.4, 150mM NaCl, 1% NP40, 0.5% Na-deoxycholate, 0.1% SDS, 1mM EDTA) supplemented with 5mM NEM, to block the activities of deubiquitinase. To immunoprecipitate AXIN2, 1mg of total lysate was incubated 2 hours at 4°C with 3μg of specific antibody and subsequently the immunocomplexes were incubated with Dynabeads Protein-A-conjugated magnetic beads (Dynal) at 4°C overnight. Samples were analyzed by western blotting with anti-multi UBIQUITIN (MBL) antibody.
Immunoblotting and antibodies
Total cells lysates were prepared in RIPA buffer (50mM Tris-HCl ph 7.4, 150mM NaCl, 0.1% SDS, 1% NP-40, 1mM EDTA, 0.5% Na- deoxycholate) containing fresh protease (Sigma) and phosphatase (Upstate) inhibitors cocktail. Cytosolic lysates were prepared using a cell fractionation Kit (Thermo Scientific). Commercial antibodies used in this study include anti-AXIN1, anti-AXIN2, anti-β-CATENIN, anti-P-β CATENIN Ser33/Ser37/Thr41 and anti-HA from Cell Signaling Technologies, anti-TANKYRASES (Abcam), anti-TUBULIN (Calbiochem), anti-GAPDH (Sigma) and anti-multi UBIQUITIN (MBL).
qRT-PCR
RNA was extracted from cultured cells using TRIzol reagent (Gibco) followed by isopropanol-alcohol precipitation (RNeasy Plus Mini Kit Qiagen) before quantification. Transcript levels were assessed using the Bio-Rad iQ5 (Kit iQ™ SYBR Green Supermix) machine, according to the manufacturer’s instructions, and each experiment was repeated three times using independent RNAs samples. Gene expression analysis was performed using the human housekeeping genes, GAPDH and RPL13a. Primers for the hAXIN2 were the following: forward: 5′-CAAGGGCCAGGTCACCAA-3′; reverse: 5′-CCCCCAACCCATCTTCGT-3′.
Transfections, infections and reporter assays
Plasmids and siRNA transfections were carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Inducible lentiviral expression of dominant negative TCF4 was carried out following Vectalys instructions. For reporter assays, luciferase activities were measured with the Dual Luciferase Assay Kit (Promega) according to the manufacturer’s instructions, twenty-four hours after transient transfections or lentiviral infections. In HEK293 cells WNT pathway was activated by ectopic expression of WNT1 and WNT3A or through addiction of Wnt3a condition medium.
FACS analysis
Cell cycle distribution after silencing (siRNA) of β-CATENIN was determined by measuring the amount of cellular DNA using propidium iodide staining. For G0/G1 synchronization, confluent cells were maintained for 24h in complete medium followed by 24h in serum free medium. Cells were then collected by centrifugation and fixed with 50% ethanol overnight. Following fixation, cells were washed with PBS, treated with 100μg/ml RNAse for 15min, and then incubated with 50μg/ml propidium iodide for additional 15min. DNA content was determined using a flow cytometer (FACScalibur, BD Biosciences Immunocytometry System) by measuring propidium iodide emission at 580nm. Cell cycle distribution was analyzed using BD CellQuest™ Pro software (BD Biosciences Immunocytometry System).
Soft agar assay
In the soft agar assay, a suspension of 2.5 × 103 cells per well, containing 0.36% (wt/vol) agar, was mixed with various concentrations of compounds, SiRNA or inducible lentiviral vectors prior to setting. The cell layer was overlaid onto a layer of culture medium containing 0.6% (wt/vol) agar in 24-well plates. Subsequently, the plates were kept in culture (37°C and 5% CO2) for 14 or 21 days depending on the cell line. At the end of the incubation, colonies were stained overnight with 5mg/ml of MTT (Sigma) and counted using Oxford Optronix GelCount® instrument.
Xenopus experiments and in vivo tumor growth assay
Xenopus injections were performed as previously described (19). For the glioma xenograft model, 5 × 106 DBTRG cells were injected subcutaneously into the right flank of athymic female nude mice (CD-1 nu/nu, Charles Rivers, Calco, Italy). All mice were maintained in a conventional-specific pathogen-free facility (according to the NIH guidelines). Twice a week tumor growth and body weight were evaluated and recorded. Dimensions of the tumors were measured by a digital caliper, and tumor masses were calculated using the following formula: length (mm) × width2 (mm) × d/2, assuming density d= 1 mg/mm3 for tumor tissue. When measurable tumors were established in the majority of mice, animals were randomly assigned into treatment groups (10 mice/group). Animals received the compound by oral gavage at three dose levels and at the dose volume of 10 mL/kg. SEN461 was administered at the following doses and schedules: 30 mg/kg twice daily for 14 consecutive days, 100 mg/kg once daily for 14 consecutive days, 500 mg/kg once weekly for two consecutive weeks. Mice were sacrificed when the tumors reached a volume around 10% of total body weight.
Statistical analysis
Statistical analysis for soft agar, reporter and qPCR assays was performed with one way Anova test followed by Tukey’s test for multiple comparisons. Auto-PARsylation data were analyzed by non-linear regression. Significant difference of embryos developing second axis was tested with Fischer’s exact test, and a t-test was performed on proportions to assess good reproducibility of results across the independent experiments. For the glioma xenograft model, a mixed-effects ANOVA model was performed at the end of the study on body weight and tumor mass data considering “treatment” and “time” as main effects. Statistical analysis was performed using GraphPad Prism and Matlab statistical software.
Compounds
IWR2 and XAV939 molecules were purchased from Asinex and Maybridge respectively. SEN461 [6-Methoxy-3-{4-[4-(2-methoxy-acetyl)-piperazine-1-carbonyl]-cyclohexylmethyl}-1-methyl-1H-quinazoline-2,4-dione] and SEN973 [3-[4-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-cyclohexylmethyl]-6,7-dimethoxy-1H-quinazoline-2,4-dione] were designed and synthesized at Siena Biotech. The chemical structures are reported in Figure 1A. Synthetic details for SEN461 are reported in the patent application WO 2011/042145 (compound 66). All compounds tested for the in vitro assays were dissolved in DMSO. SEN461 was formulated in 0.5% methocel for the in vivo studies.
Figure 1. SEN461 inhibits WNT-induced transcriptional activity and suppresses anchorage independent growth of DBTRG cells.

DBTRG cells stably transfected for TCF-Luciferase and TA-Renilla were exposed to different amount of SEN461 or SEN973 and reporter activity was measured 24h later (A). Black and grey lines represent TCF-luciferase and TA-Renilla respectively. The half-maximal inhibitory concentration (IC50) for DBTRG cancer cells after SEN461 and SEN973 is shown (B), determined from the soft agar assay.
RESULTS
Effect of WNT pathway modulation and pathway screening approach to identify small molecule WNT inhibitors in glioma cells
To investigate the consequences of WNT signaling inhibition in glioblastoma, we used the DBTRG-05MG (DBTRG) cell line (20). The cell line, originally derived from a human recurrent GBM, harbours mutations in PTEN, CDKN2A, and BRAF but has a wild-type TP53 gene (Wellcome Trust Sanger Institute: http://www.sanger.ac.uk/genetics/CGP/CellLines/). No mutations involving APC, AXIN and/or β-CATENIN genes have been reported for DBTRG cells, which are considered to have an intact canonical WNT pathway cascade (Wellcome Trust Sanger Institute Database). We characterized this cellular system for WNT pathway activity and relevance by specific biological and biochemical tools at the molecular and phenotypic level. In the canonical pathway, WNT signaling activity is controlled by the intracellular β-CATENIN level through its phosphorylation-dependent degradation. Upon stimulation by an appropriate WNT signal, accumulating β-CATENIN translocates to the nucleus where it binds TCF (T cell factor) transcription factor (also known as lymphoid enhancer-binding factor-l, LEF1), serving as a co-activator of TCF/LEF-induced transcription and leading to increased expression of WNT target genes (21, 22). β-CATENIN therefore represents a key intracellular effector of the genomic response of the cell to an incoming WNT signal. The phenotypic effects of transient β-CATENIN knock-down via an siRNA included a decrease of a WNT target gene CYCLIN D1 (Supplemental Figure S1B) and a reduction in the ability of GBM cancer cells to grow in an anchorage-independent fashion (Supplemental Figure S1A), and a substantial change in the cell cycle profile with a G0/G1 cell cycle arrest and an S phase reduction (Supplemental Figure S1C), demonstrating that cell growth is WNT/β-CATENIN dependent in this glioblastoma cell line. DBTRG cells therefore represent a suitable model for the initial identification of small molecule modulators of WNT signaling with relevance for GBM.
Having selected DBTRG as a GBM cell model responsive to WNT inhibition, we set out to develop a screening approach for the identification of compounds capable of modulating canonical WNT signaling and associated proliferative responses in GBM cells. The screening cascade to identify WNT signaling pathway inhibitors included cellular and biochemical-based assays. A WNT-responsive Luciferase (TCF-Luciferase (Firefly)) and a (WNT-independent) constitutive promoter-driven Renilla Luciferase (TA-Renilla) reporter plasmid (alone and in combination) were stably transfected in DBTRG cells and constituted our primary screening assay. As an additional validation step for this readout, we employed the dominant-negative TCF4 (dnTCF4), which cannot bind to β-CATENIN (23, 24). As result, the output of the Luciferase-based reporter system was strongly inhibited (Supplemental Figure S1D) in a concentration dependent fashion indicating that the reporter cell line is sensitive to genetic WNT inhibition. A random set of 16000 small molecules from Siena Biotech internal compounds collection was tested at single concentration (10μM) in stably transfected DBTRG-TCF-Luciferase cells. Compounds showing ≥ 50% inhibition were then tested in a concentration response manner on DBTRG cells stably transfected with TCF-Luciferase and TA-Renilla plasmids, in order to select the compounds displaying potency associated with minimal signs of cellular toxicity. A luciferase biochemical assay applied before hit selection enabled the identification of true inhibitors of the WNT pathway and the elimination of compounds acting directly on the enzyme, such as luciferase modulators and/or quenchers. Several structurally distinct hit series were identified and validated. The lead compound SEN461 inhibited WNT reporter activity in the DBTRG cell line (Figure 1A) with an IC50 of 1.3μM, and affected their ability to grow in anchorage independent fashion (Figure 1B); no effect either in the reporter (Figure 1A) or in the growth inhibition assay (Figure 1B) was showed by SEN973 (Figure 2), a structural analogue of SEN461 (Figure 2).
Figure 2. Chemical structures.
Chemical structures of different AXIN stabilizers and SEN973 an inactive structural analogue of SEN461.
SEN461 increases AXIN and decreases β-CATENIN levels in DBTRG cells
In order to link inhibition of WNT signaling and anchorage independent growth in glioblastoma cells we started to analyze the effect of SEN461 treatment on key components of the canonical WNT pathway at the protein level. The effect of SEN461 on AXIN steady-state protein levels was compared to that of XAV939 and IWR2 molecules (Figure 2); two previously published AXIN stabilizers (25, 26). DBTRG cells treated overnight with two different concentrations (3μM and 10μM) of SEN461 showed an increase of phosphorylated β-CATENIN (a prerequisite for proteasome-mediated degradation of β-CATENIN) in the cytoplasmic fraction, which correlated with a concomitant decrease in the total amount of β-CATENIN, and a simultaneous accumulation of AXIN1 and AXIN2 compared to vehicle (DMSO) treated cells (Figure 3A). By contrast, the inactive structural analogue SEN973 didn’t produce any such effects (data not shown). As presented in figure 3B, all three small molecules demonstrated comparable effects on the accumulation of both AXIN1 and AXIN2 in DBTRG cells. The increase in AXIN protein levels after compound treatment could be explained by protein stabilization as reported for these recently identified inhibitors of the TANKYRASE (25–27), which acts through AXIN destabilization. It can be hypothesized that SEN461 treatment protected AXIN from proteosomal degradation, because co-treatment of SEN461 and the reversible proteasome inhibitor MG-132 almost completely blocked the ubiquitination of AXIN2 (Figure 3C). TANKYRASES, TNKS1 and TNKS2, are enzymes of the PARP family mediating the PARsylation of substrate proteins, a fundamental step in ubiquitin-mediated protein degradation. To test whether the negative modulation of WNT activity induced by SEN461 was the consequence of the inhibition of the PARP catalytic activity of TNKS, we performed biochemical assays for TNKS1 and TNKS2. As shown in the Supplemental Figure S2A, SEN461 showed much weaker activity than XAV939 (from three hundred to almost two thousand folds) in auto-PARsylation of TNKS1 and TNKS2 (IC50 of 18μM and 2.9μM respectively). Moreover, we also tested whether SEN461 was able to stabilize TNKS1 and TNKS2 protein levels as demonstrated for XAV939 and IWR2 (26). There was no sign of TNKS stabilization after SEN461 treatment in DBTRG cells (regardless the accumulation of AXIN1), while both IWR2 and XAV939 induced significant TNKS stabilization (Figure 3D), as previously reported (26). A weak TNKS stabilization was observable only following very high (100μM) exposure to SEN461 (Supplemental Figure S2B). These results suggest that AXIN stabilization induced by SEN461 is accompanied by minimal TNKS stabilization, implying that AXIN stabilization by SEN461 occurs via a mechanism distinct from that by known TNKS inhibitors. The identification of SEN461 as a structurally novel small molecule inhibitor of the WNT pathway acting at the level of AXIN stabilization further supports the modulation of AXIN levels as a pharmacological approach in WNT inhibition. The comparable activity of SEN461, IWR2 and XAV939 in inhibition of TCF-luciferase activity and GBM cell growth in vitro (Supplemental Figure S3) suggests the relevance of such approach for the development of GBM therapeutics.
Figure 3. Effects of different compound AXIN stabilizers on key protein components of the WNT pathway in DBTRG cells.

Western blotting analysis from cytoplasmic DBTRG lysates demonstrating that SEN461 treatment stabilizes AXIN1 and AXIN2, and increases phosphorylated β-CATENIN (Ser33/Ser37/Thr41) with a concomitant decrease of total β-CATENIN (A). GAPDH was used as loading control. Western blotting from cytoplasmic DBTRG lysates after treatment with two different amounts of SEN461, XAV939 and IWR2 molecules demonstrated comparable stabilization of AXIN1 and AXIN2 proteins (B). DBTRG cells were exposed overnight with 25μM of the proteasome inhibitor MG-132 alone or in combination with 10μM of SEN461. Lysates were immunoprecipitated with anti-AXIN2 and immunoblotted with anti-UBIQUITIN. Total cell lysate (TCL) was analyzed by Western blotting with anti-AXIN1, anti-AXIN2 and anti-GAPDH (C). DBTRG cells were treated overnight with 10μM of different WNT inhibitor molecules. Lysates were then analyzed by Western blotting with anti-TNKS (26), anti-AXIN1 and anti-TUBULIN as loading control (D). The asterisk represents a background band, clearly evident in the DMSO lane, migrating below the TNKS1 band.
In vitro and in vivo characterization of WNT signaling inhibition by SEN461
To further characterize the effects of SEN461 on canonical WNT signaling in a different, non GBM cellular background, we investigated the compound in a non-tumorigenic, immortalized cell line widely employed for WNT studies, namely HEK293 cells, where individual WNT ligands can be efficiently expressed and downstream responses studied (28, 29). In order to study the effects of SEN461 on pathway stimulation by selected WNTs, we transiently co-transfected the Luciferase and Renilla-based reporter plasmids (already employed in the screening campaign) in HEK293 cells transiently overexpressing the canonical WNT pathway ligands WNT1 (Figure 4A) or WNT3A (Figure 4B) alone or in combination with the co-receptor LRP6 (Supplemental Figure S4). WNT1 and WNT3A were chosen because they represent the members of the WNT family with the strongest association with stimulation of the canonical pathway, and because of their relevance to tumour biology (30–33). The results indicated that SEN461 inhibited with comparable potency either WNT1 or WNT3A-mediated luciferase activity in a concentration-dependent manner, without affecting WNT-independent, constitutive TA-Renilla activity. Stimulation of HEK293 cells with Wnt3a exogenously provided in conditioned medium (CM) produced an increase in the amount of total β-CATENIN protein levels as expected after Wnt3a stimulation. SEN461 reversed the effects of Wnt3a by inducing a reduction of total β-CATENIN and an increment in the phosphorylated β-CATENIN fraction (Figure 4C), demonstrating that the WNT inhibitory effect of SEN461 is not mediated through inhibition of ligand expression/secretion (as recently reported for a Porcupine inhibitor (25)). Consistent with the reporter data, the mRNA levels for the WNT/β-CATENIN target gene AXIN2, induced by Wnt3a CM stimulation, was inhibited by SEN461 treatment (Figure 4D). The Xenopus axis duplication assay represents a valuable and sensitive way to test the in vivo efficacy and specificity of WNT signaling modulators (34). Injecton of 10pg of XWnt8 mRNA into the ventral regions of a four-cell stage Xenopus embryo produced ectopic axis formation in almost 80% of the injected embryos (Figure 5A). In contrast, co-injection of XWnt8 mRNA with 1 pmol SEN461, produced a 56% reduction of axis duplication compared to DMSO treated embryos (Figure 5A). These results support the specific and selective WNT canonical inhibitory activity in both cellular based assays and in vivo.
Figure 4. SEN461 affects canonical WNT ligands mediated transcription and modulates molecular markers of the pathway.

HEK293 cells, transiently transfected with TCF-Luciferase and TA-Renilla and different combination of WNT1 (A) and WNT3A (B) expression plasmids were either treated with DMSO (vehicle) or different amounts of SEN461. The data showed potent concentration-dependent inhibition of WNT transcriptional activity either in WNT1 or WNT3A mediated luciferase activity, without affecting WNT-independent TA-Renilla activity. Data represent means ± SEMs. HEK293 cells were stimulated overnight with Wnt3a conditioned medium or control conditioned medium, alone or in combination with SEN461. Lysates were analyzed by Western blotting with total and phosphorylated anti-β-CATENIN and anti-AXIN1 (C). The effect of SEN461 treatment after Wnt3a CM stimulation on the WNT target gene AXIN2 (mRNA) was measured by quantitative RT-PCR (D). Data represent means ± SEM. *P<0.001 relative to Wnt3a stimulated cells (Tukey’s Multiple Comparison Test).
Figure 5. SEN461 inhibits XWnt8-induced axis duplication in Xenopus embryos.
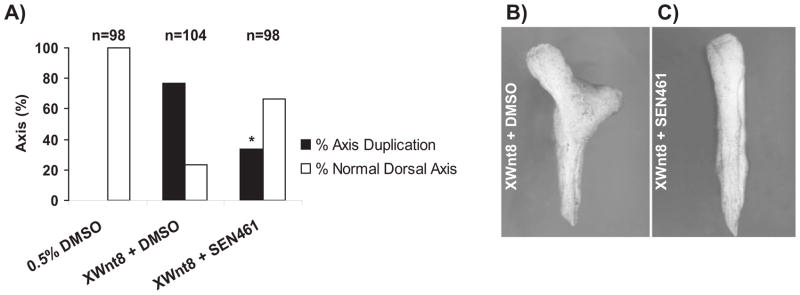
Injection of 10pg of XWnt8 mRNA induced axis duplication, which was inhibited by co-injection of SEN461 (1pmol/embryo). The histogram (A) shows the percent of embryos with normal (white bars) or duplicated axes (black bars); n= number of embryos examined for each group. Data collected from three independent experiments showed that SEN461 significantly decreased the proportion of embryos with duplicated axes (* p<0.001). Representative images of embryos with duplicated axis (B) and normal development after SEN461 co-injection (C) are shown.
In-vitro antitumor activity of SEN461 in glioblastoma cell lines
To explore the pharmacological effects of WNT signaling inhibition on glioblastoma cell viability, and to extend the observations obtained in DBTRG cells, we examined the consequences of SEN461 treatment in a set of 9 additional glioma cell lines, either commercially available or primary tumour, patient-derived. As shown in Figure 6A, soft agar assay results demonstrated a wide range of sensitivities, from an IC50 of 0.5μM in sensitive T98 cells to >20μM in some cell lines. Overall SEN461 showed significant in vitro activity across the panel of GBM cells tested, with most of the lines (7 out of 10) showing IC50 in the low μM range (from 0.5 to 3.5μM). To provide additional evidence that WNT signaling activity was indeed responsible for growth inhibition, we transduced T98G (sensitive to genetic WNT inhibition as showed in Figure 6B) glioblastoma cell line with a doxycyclin (Doxy) inducible dominant negative TCF4 (dnTCF4) lentivirus. As a consequence, we observed a strong decrease in anchorage-independent growth ability (Supplemental Figures S5A). We next examined the effect of SEN461 treatment on β-CATENIN and AXIN protein levels in T98G cells (Figure 6C), where we observed a pattern resembling the one already obtained in DBTRG cells: increased phosphorylation of β-CATENIN and stabilized AXIN1 levels, with a concomitant decrease in the cytoplasmic fraction of total β-CATENIN. We then asked whether over-expression of AXIN1 would affect the phenotypic behaviour of the GBM cell lines examined. Indeed this was the case; AXIN1 over-expression showed a profound effect on T98G (Supplemental Figure S5B) as well as on DBTRG (Supplemental Figure S1E) anchorage-independent growth ability, phenocopying the pharmacological effects of SEN461 at a morphological and molecular level.
Figure 6. The response of a panel of glioblastoma cells to SEN461 in vitro.

The half- maximal inhibitory concentration (IC50) for 10 GBM cancer cells is shown (A), determined from the soft agar assay, and ranked from lowest to highest. (#= primary patient derived GBMs). Inhibition of canonical WNT signaling by transient transfection with dnTCF4 produced a strong concentration-dependent reduction in the WNT transcriptional activity (B). Data represent means ± SEM. *P<0.001 relative to control cells (Tukey’s Multiple Comparison Test). Lysates from cells treated with SEN461 for different length of time were analyzed by Western blotting with anti-P-β-CATENIN, anti total β-CATENIN and anti AXIN1 (C).
SEN461 affects tumor growth of DBTRG xenograft model
In order to investigate the relevance of SEN461 antiproliferative capacity in an in vivo setting, a subcutaneous xenograft model was used, to confirm the in vitro observation that WNT/β-CATENIN signaling pathway inhibition by SEN461 has an effect on tumour growth. Due to the very poor blood brain barrier (BBB) penetration index of the compound (data not shown) a subcutaneous model was used instead of an orthotopic one. DBTRG cells were subcutaneously injected into CD-1 nude mice on day 0 and dosing was initiated when tumours reached a mean tumour volume of 200 mm3. SEN461 was administered orally using three different schedules: 30 mg/kg twice daily (BID) for 14 consecutive days (from Day 28 to 41), 100 mg/kg daily for 14 consecutive days (from Day 28 to 41) and 500 mg/Kg once weekly for 2 consecutive weeks (on Days 28 and 34). Figure 7 shows the effect of SEN461 on tumour volume inhibition over time until Day 79 (more than a month beyond treatment and 51 days after the start of treatment). All schedules were well tolerated with no observable gross toxicities and minimal difference in body weights (Supplemental Figure S6) between control and SEN461 treated animals. Analysis on tumor mass showed a very significant effect on treatment (p≪ 0.01), and additional pair wise comparison between treatment groups showed all treatments to be significantly different from vehicle. Significant treatment × day effect (p ≪ 0.01) was observed due to a treatment-specific increase of tumor mass over time: vehicle group tumor mass significantly increased starting from day 58 with respect to day 28; on the contrary, tumor mass re-growth on SEN461 treatment is observed only after day 69. At the end of the study, all SEN461 treatments were found to be significantly different from vehicle. The antitumor activity observed for SEN461 at 30 mg/Kg BID shows the highest efficacy level (54% tumor growth inhibition with respect to control group) at the final endpoint (37 days after the end of the treatment).
Figure 7. Antitumor activity of SEN461 in a DBTRG xenograft tumor model in vivo.

DBTRG cells were injected s.c. into CD-1 nude mice on day 0 and SEN461 p.o. dosing started on day 28. Treatment groups (10 mice per group) were 30 mg/kg twice a day from day 28 to 41, 100 mg/kg/day daily from day 28 to 41 and 500 mg/kg/day once weekly on day 28 and 34. Tumor volume was followed over time until day 79 (37 days after the end of the treatment).
DISCUSSION
Despite the efforts to characterize at the molecular level targets and pathways that drive glioma tumorigenesis, the prognosis for patients with such kind of brain tumors remains poor (35). The complexity of the tumor is increased by its extreme heterogeneity, supported by a vast array of genetic changes and signaling pathways cross-talk (36, 37), which supply and sustain its high proliferation rate and its ability to infiltrate the surrounding normal parenchyma, representing the major driving force behind tumor recurrence. It has recently been reported that aberrant WNT signaling represents a critical mechanism for the genesis, proliferation and invasion of glioma (12–14), suggesting that inhibition of WNT signaling provides an important approach to cancer therapies. Our data support and reinforce the involvement of the WNT signaling pathway in glioblastoma, using either commercially available cell lines or primary patient-derived tumors. Here we show for the first time that WNT pharmacological modulation in glioblastoma, mediated by a small molecule, can affect therapeutically relevant phenotypes. We demonstrate that the WNT inhibitory compound SEN461 selectively affects canonical WNT signaling, negatively regulates WNT transcriptional activity in glioblastoma cell lines, inhibits XWnt8-mediated axis duplication in Xenopus embryos, and attenuates in vitro growth and in vivo tumorigenicity of GBM cells. The pharmacological inhibition of the WNT pathway by SEN461 is likely to be mediated through stabilization of AXIN, which is a key negative modulator of the pathway and represents a concentration-limiting component of the β-CATENIN destruction complex. As a consequence, the pool of β-CATENIN (the phosphorylated fraction) committed to degradation increases. A critical negative role of AXIN in GBM proliferation was also confirmed in DBTRG and T98G cells, where its over-expression phenocopies the pharmacological activity of SEN461. In recent years, multiple reports (either based on mechanistic studies or pharmacological tools) fueled interest around AXIN as potential pharmacological target. AXIN levels were in fact reported to inversely correlate with the grades of human astrocytoma, and its over-expression in astrocytoma cells induced cell death and reduced cell proliferation (38). The precise mechanisms regulating the degradation of AXIN are at the moment however only partially understood, and its PARsylation by TANKYRASE, its sumoylation and its stability regulated by the Ubiquitin specific protease USP34 or by Smurf2 have recently been shown to control its ubiquitin-dependent degradation (26, 39, 40, 41). As an additional selectivity step, SEN461 activity was also biochemically tested against a panel of forty-eight kinases (ExpresS Diversity Kinase Profile, Cerep), where it did not show any significant activity (data not shown). Based on chemical structure diversity and biochemical and biological activity data, TNKS may not be the primary and/or direct pharmacological target of SEN461, which we are trying to identify. In conclusion, the data presented here support the WNT canonical signaling as a valid therapeutic opportunity to treat glioblastoma.
Supplementary Material
Acknowledgments
Financial support:
M.S. acknowledges support by Monte Dei Paschi Foundation. S-M. C. acknowledges support in part by the National Research Foundation of Korea (NFR-2012R1A6A3A03039818). X. H. acknowledges support by NIH (RO1 GM074241) and by the Boston Children’s Hospital Intellectual and Developmental Disabilities Research Center (P30 HD-18655).
We thank Letizia Magnoni and Elisa Mori for the statistical support and Giuseppe Pollio and Daniela Diamanti for the assistance with the reporter plasmids.
Footnotes
Conflict of Interest: The authors declare no financial conflicts of interest
References
- 1.Nusse R, Varmus HE. Wnt genes. Cell. 1992;69:1073–1087. doi: 10.1016/0092-8674(92)90630-u. [DOI] [PubMed] [Google Scholar]
- 2.Nusse R. Wnt signaling in disease and in development. Cell Research. 2005;15:28–32. doi: 10.1038/sj.cr.7290260. [DOI] [PubMed] [Google Scholar]
- 3.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discovery. 2006;5:997–1014. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- 5.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–90. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 6.Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nature Genetics. 2000;24:245–250. doi: 10.1038/73448. [DOI] [PubMed] [Google Scholar]
- 7.Benhaj K, Akcali KC, Ozturk M. Redundant expression of canonical Wnt ligands in human breast cancer cell lines. Oncol Rep. 2006;15:701–707. [PubMed] [Google Scholar]
- 8.Yang L, Wu X, Wang Y, Zhang K, Wu J, Yuan YC, et al. Fzd7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene. 2011;30:4437–4446. doi: 10.1038/onc.2011.145. [DOI] [PubMed] [Google Scholar]
- 9.Uematsu K, He B, You L, Xu Z, McCormick F, Jablons DM. Activation of the Wnt pathway in non small cell lung cancer: evidence of dishevelled overexpression. Oncogene. 2003;22:7218–7221. doi: 10.1038/sj.onc.1206817. [DOI] [PubMed] [Google Scholar]
- 10.Bafico A, Liu G, Goldin L, Harris V, Aaronson SA. An autocrine mechanism for constitutive Wnt pathway activation in human cancer cells. Cancer Cell. 2004;6:497–506. doi: 10.1016/j.ccr.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 11.Rajan N, Burn J, Langtry J, Sieber-Blum M, Lord CJ, Ashworth A. Transition from cylindroma to spiradenoma in CYLD-defective tumours is associated with reduced DKK2 expression. J Pathol. 2011;224:309–321. doi: 10.1002/path.2896. [DOI] [PubMed] [Google Scholar]
- 12.Zheng H, Ying H, Wiedemeyer R, Yan H, Quayle SN, Ivanova EV, et al. PLAGL2 regulates Wnt signaling to impede differentiation in neural stem cells and gliomas. Cancer Cell. 2010;17:497–509. doi: 10.1016/j.ccr.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H, et al. FoxM1 promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20:427–442. doi: 10.1016/j.ccr.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Augustin I, Goidts V, Bongers A, Kerr G, Vollert G, Radlwimmer B, et al. The wnt secretion protein Evi/Gpr177 promotes glioma tumourigenesis. EMBO Mol Med. 2011;4:38–51. doi: 10.1002/emmm.201100186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pu P, Zhang Z, Kang C, Jiang R, Jia Z, Wang G, et al. Downregulation of Wnt2 and beta-catenin by siRNA suppresses malignant glioma cell growth. Cancer Gene Ther. 2009;16:351–61. doi: 10.1038/cgt.2008.78. [DOI] [PubMed] [Google Scholar]
- 16.Pulvirenti T, Van Der Heijden M, Droms LA, Huse JT, Tabar V, Hall A. Dishevelled 2 signaling promotes self-renewal and tumorigenicity in human gliomas. Cancer Res. 2011;71:7280–90. doi: 10.1158/0008-5472.CAN-11-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lambiv WL, Vassallo I, Delorenzi M, Shay T, Diserens AC, Misra A, et al. The Wnt inhibitory factor 1 (WIF1) is targeted in glioblastoma and has a tumor suppressing function potentially by induction of senescence. Neuro Oncology. 2011;13:736–747. doi: 10.1093/neuonc/nor036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin X, Jeon HY, Joo KM, Kim JK, Jin J, Kim SH, et al. Frizzled 4 regulates stemness and invasiveness of migrating glioma cells established by serial intracranial transplantation. Cancer Res. 2011;71:3066–3075. doi: 10.1158/0008-5472.CAN-10-1495. [DOI] [PubMed] [Google Scholar]
- 19.Semënov MV, Tamai K, Brott BK, Kühl M, Sokol S, He X. Head inducer Dickkopf-1 is a ligand for Wnt coreceptor LRP6. Curr Biol. 2001;11:951–961. doi: 10.1016/s0960-9822(01)00290-1. [DOI] [PubMed] [Google Scholar]
- 20.Kruse CA, Mitchell DH, Kleinschmidt-DeMasters BK, Franklin WA, Morse HG, Spector EB, et al. Characterization of a continuous human glioma cell line DBTRG-05MG: growth kinetics, karyotype, receptor expression, and tumor suppressor gene analyses. In Vivo Cell Dev Biol. 1992;28A:609–614. doi: 10.1007/BF02631035. [DOI] [PubMed] [Google Scholar]
- 21.Clevers H. Wnt beta-catenin/signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 22.Moon RT, Kohn AD, De Ferrari GV, Kaykas A. Wnt and β-catenin signaling: diseases and therapies. Nat Rev Genet. 2004;5:691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- 23.Van De Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–250. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 24.Kolligs FT, Nieman MT, Winer I, Hu G, Van Mater D, Feng Y, et al. ITF-2, a downstream target of the Wnt/TCF pathway, is activated in human cancers with β-catenin defects and promotes neoplastic transformation. Cancer Cell. 2002;1:145–155. doi: 10.1016/s1535-6108(02)00035-1. [DOI] [PubMed] [Google Scholar]
- 25.Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW, et al. Small molecule–mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009;5:100–107. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang A, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signaling. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 27.Waaler J, Machon O, Tumova L, Dinh H, Korinek V, Wilson SR, et al. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012;72:2822–2832. doi: 10.1158/0008-5472.CAN-11-3336. [DOI] [PubMed] [Google Scholar]
- 28.Semënov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;29:26770–26775. doi: 10.1074/jbc.M504308200. [DOI] [PubMed] [Google Scholar]
- 29.Oloumi A, Syam S, Dedhar S. Modulation of Wnt3a-mediated nuclear beta-catenin accumulation and activation by integrin-linked kinase in mammalian cells. Oncogene. 2006;25:7747–7757. doi: 10.1038/sj.onc.1209752. [DOI] [PubMed] [Google Scholar]
- 30.Kumar R, Balasenthil S, Pakala SB, Rayala SK, Sahin AA, Ohshiro K. Metastasis-associated protein 1 short form stimulates Wnt1 pathway in mammary epithelial and cancer cells. Cancer Res. 2010;70:6598–608. doi: 10.1158/0008-5472.CAN-10-0907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakashima N, Huang CL, Liu D, Ueno M, Yokomise H. Intratumoral Wnt1 expression affects survivin gene expression in non-small cell lung cancer. Int J Oncol. 2010;37:687–94. doi: 10.3892/ijo_00000718. [DOI] [PubMed] [Google Scholar]
- 32.Verras M, Brown J, Li X, Nusse R, Sun Z. Wnt3a growth factor induces androgen receptor-mediated transcription and enhances cell growth in human prostate cancer cells. Cancer Res. 2004;64:8860–8866. doi: 10.1158/0008-5472.CAN-04-2370. [DOI] [PubMed] [Google Scholar]
- 33.Katoh M. Regulation of WNT3 and WNT3A mRNAs in human cancer cell lines NT2, MCF-7, and MKN45. Int J Oncol. 2002;2:373–7. [PubMed] [Google Scholar]
- 34.Harland R, Gerhart J. Formation and function of Spermann’s organizer. Annu Rev Cell Dev Biol. 1997;13:611–67. doi: 10.1146/annurev.cellbio.13.1.611. [DOI] [PubMed] [Google Scholar]
- 35.Zhu Y, Parada LF. The molecular and genetic basis of neurological tumours. Nature Rev Cancer. 2002;2:616–626. doi: 10.1038/nrc866. [DOI] [PubMed] [Google Scholar]
- 36.Holland EC. Glioblastoma multiforme: the terminator. Proc Natl Acad Sci. 2000;97:6242–6244. doi: 10.1073/pnas.97.12.6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang LY, Ye J, Zhang F, Li FF, Li H, Gu Y, et al. Axin induces cell death and reduces cell proliferation in astrocytoma by activating the p53 pathway. Int J of Cancer. 2009;35:25–32. doi: 10.3892/ijo_00000309. [DOI] [PubMed] [Google Scholar]
- 39.Kim MJ, Chia IV, Costantini F. SUMOylation target sites at the C terminus protect Axin from ubiquitination and confer protein stability. Faseb J. 2008;22:3785–94. doi: 10.1096/fj.08-113910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lui TT, Lacroix C, Ahmed SM, Goldenberg SJ, Leach CA, Daulat AM, et al. The Ubiquitin specific protease USP34 regulates Axin stability and Wnt/β-catenin signaling. Mol Cell Biol. 2011;10:2053–2065. doi: 10.1128/MCB.01094-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim S, Jho EH. The Protein Stability of Axin, a Negative Regulator of Wnt Signaling, Is Regulated by Smad Ubiquitination Regulatory Factor 2 (Smurf2) J of Biol Chem. 2010;285:36420–36426. doi: 10.1074/jbc.M110.137471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.