

Abstract
It is established that efflux transporters of the ABC superfamily can affect the pharmacokinetics (PKs) of drugs through mechanisms pertaining to drug absorption, elimination and distribution. To characterize the role of multiple transporters in topotecan's PKs, total (lactone+carboxylate) and lactone forms were measured by LC/MS/MS in plasma, bile, urine and feces following IV administration at doses of 1 mg/kg and 4 mg/kg to eight mouse strains; C57BL/6 (WT), Abcb1−/−, Abcc2−/−, Abcc4−/−, Abcg2−/−, Abcc2;Abcb1−/−, Abcc2;Abcg2−/−, Abcc4;Abcg2−/−. Compared with WT mice and at both dose levels, the plasma areas under the curve (AUC) for topotecan lactone were not significantly different in the Abcc2−/−, Abcc4−/−, Abcb1−/− strains, whereas significant differences were found in Abcg2−/−, Abcc2;Abcb1−/− (only at the high dose), Abcc4;Abcg2−/− and Abcc2;Abcg2−/− mice and ranged from 2.1-fold to 3.3-fold higher. Consistent with these changes, the fecal and biliary excretion of topotecan was reduced, while renal elimination was elevated in Abcg2−/− based strains. Similarly, the Abcc2;Abcb1−/− strain also had elevated renal elimination and reduced fecal excretion of topotecan lactone, this was more pronounced at the 4 mg/kg dose level, suggesting possible saturation of Abcg2. The Abcc4 transporter was found not to be a major determinant of topotecan PKs. It is concluded that Abcg2 has the most significant effect on topotecan elimination, whereas both Abcb1 and Abcc2 have overlapping functions with Abcg2. As such it is relevant to examine how polymorphisms in these transporters influence topotecan activity in patients and whether coadministration of transport modulators could positively affect efficacy without increasing toxicity.
Introduction
Cancer chemotherapy involves concomitant administration of many drugs that could cause drug-drug interactions (DDIs) and serious adverse effects in patients. The deleterious effects of DDIs is further complicated when drugs are substrates for membranous ATP-binding cassette (ABC) multidrug efflux transporters due to competition and the nonlinear aspect of active transport. Efflux transporters, especially ABCB1 (P-glycoprotein, P-gp), ABCC2, ABCC3 and ABCC4 (also known as multidrug resistance proteins, MRP, 2, 3, 4), and ABCG2 (breast cancer resistance proteins, BCRP) can have an important impact on cancer chemotherapy, either at the systemic level where transporters, such as ABCB1, ABCC2 and ABCG2, are localized at apical membranes of important epithelial barriers in the kidney, intestine, and at the canalicular membrane of hepatocytes that determine both drug absorption and elimination, or locally at the tumor where transporters may be resistance factors limiting intracellular drug concentrations. Local effects of transporters are also seen at sanctuary sites, such as blood-brain, blood-placenta, and blood-testis barriers, where they exert an important protective function by restricting penetration of potentially harmful chemicals to the brain, fetus and testes, respectively. Evaluation of the effects of any single transporter has to be viewed in the context of their co-localization, overlapping substrate specificities and function (1, 2). It should also be appreciated that the membrane orientation of the transporters could offset unidirectional effects, and, possibly maintain a “balance”. For example, ABCC3 shares a similar substrate profile as that of ABCB1 and ABCC2 transporters; however, in contrast, it has a basolateral orientation in the hepatobiliary and intestinal axes and can have opposite effects on systemic availability. ABCC4 has a differential orientation; being apically localized in the renal epithelium and basolaterally localized in the hepatocytes (3, 4). The co-localization, redundancy and differential orientation of ABC-transporters provides not only functional flexibility but also limits the ability to predict their impact from in vitro permeability and in situ/ex vivo approaches. Therefore, the use of ABC transporter gene-disrupted mice offers a valuable means to characterize the role of each transporter on drug disposition, and was the approach utilized here.
Topotecan is a hydrophilic, semi-synthetic analog of the camptothecins plant alkaloid. It disrupts DNA formation in the S-phase of the cell cycle by inhibiting topoisomerase-I, which then prevents the religation of single strand DNA, thus affecting transcription and replication processes, eventually leading to cell death (5). Topotecan is an established treatment for certain types of lung cancer, ovarian cancer and cervical cancer and has shown favorable activity in the treatment of brain metastases (6, 7). Topotecan is a lactone species that coexists with an inactive open-ring or hydroxyacid form through a reversible pH-dependent equilibrium (Fig. 1). This chemical attribute has precluded the use of a single assay for measurement of topotecan in patient plasma samples and has allowed investigators the option to base their analyses on the lactone form or total topotecan, the sum of both the lactone and hydroxyacid forms (8, 9). The lactone form is rapidly and spontaneously converted to carboxylate form, which dominates at physiological pH (Fig. 1), yet the lactone form is essential for topoisomerase I binding, antitumor activity and toxicity (10, 11), and for that reason we focused on the lactone form in our pharmacokinetic [PK] studies.
Figure 1.
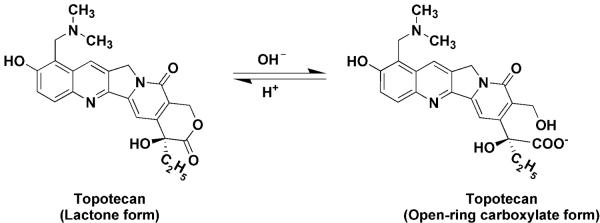
Chemical structure of the lactone and open-ring carboxylate forms of topotecan.
Topotecan is not metabolized to a significant degree, and thus, its elimination is dependent on transporters located in the hepatobiliary and renal axes. Definitive analyses of the role of ABC transporters on topotecan disposition are lacking at the systemic level, as well as those potentially related to tumor resistance. The magnitude of systemic and tumor cell ABC transport-mediated efflux could alter topotecan concentrations rendering it more or less effective within tumor cells. Studies report that topotecan is a substrate for ABCG2 and ABCB1, whereas conflicting reports are available for its interaction with ABCC4 transporters (12–16). Moreover, its interaction with other ABC transporters has not been reported in any detail, especially ABCC2, which like ABCG2, is localized in apical membranes in gut epithelium, and in liver canaliculi, and due to its high expression in these critical organs could affect topotecan PK. The present study was undertaken to characterize the role of all the relevant ABC transporters on the systemic disposition of topotecan using gene knockout mice. These studies included both single knockouts (i.e. Abcg2−/−, Abcb1−/−, Abcc2−/−, Abcc4−/−) and double knockout (i.e. Abcc2;Abcg2−/−, Abcc4;Abcg2−/−, Abcc2;Abcb1−/−) strains to not only clarify the role of each but to also assess their complimentary functions. PK studies of topotecan were conducted in each strain at dose levels (1 mg/kg and 4 mg/kg) that produce clinically relevant plasma concentrations (17) and could also indicate dose-dependent effects of the Abc –transporters given their capacity-limited function.
Materials and Methods
Reagents, antibodies and cell culture
Monoclonal antibodies (mAb) C-219 (to detect ABCB1/Abcb1) were acquired from Calbiochem, EMD Chemicals Inc. (San Diego, CA). Mouse mAB (IU2H10) to detect ABCC1/Abcc1 was obtained from Novus Biologicals (Littleton, CO). Rat mAb M41-80 to detect ABCC4/Abcc4 was bought from Alexis Biochemicals (Enzo Life Sciences, Farmingdale, NY). A rabbit polyclonal antibody (pAb) H300/sc-20766 against ABCC2/Abcc2, mouse mAb M3II-21/sc-59612 against ABCC3/Abcc3, rabbit pAb against ABCC10/Abcc10 (H-300/sc-67241), mouse mAb BXP-21/sc-58222 to detect ABCG2/Abcg2 were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Paclitaxel was obtained from LC labs (Woburn, MA). Mouse mAb β-Actin, infrared secondary antibodies IRDye 800 and 680-labeled anti-rabbit, anti-mouse, anti-rabbit and anti-rat, NewBlot PVDF stripping buffer and PVDF blocking buffer were purchased from Li-Cor Biosciences (Lincoln, NE). Mitoxantrone (MX), topotecan, ammonium formate, camptothecin, verapamil, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma Chemical Co. (St. Louis, MO). High performance liquid chromatography (HPLC)-grade acetonitrile and methanol were purchased from Fisher Scientific (Fair Lawn, New Jersey). Deionized water (Nanopure deionization system; Barnstead/Thermolyne, Dubuque, Iowa) was used to prepare all aqueous solutions. All other reagents and solvents were purchased from VWR (Bridgeport, NJ). HEK293/pcDNA3.1, HEK293/ABCB1, HEK293/ABCC1, HEK293/ABCG2-R2, and HEK293/MRP3 cells were previously established by transfecting HEK293 with either the empty pcDNA3.1 vector or pcDNA3.1 vector containing the full length ABCB1 (HEK293/ABCB1) or ABCC1 (HEK293/ABCC1) or ABCC3 (HEK293/MRP3), and were cultured in a medium with 2 mg/ml of G418 (18–20). MRP2-transfected LLC/PK1 cells (LLC/PK1-hMRP2) and parental plasmid-transfected cells (LLC/PK1-CMV) were described previously (21). The MRP4-transfected NIH-3T3 cell line (NIH-3T3/MRP4-3) and NIH-3T3 cells transfected with parental vector (NIH-3T3/pSRα) were described previously (22). In addition, non-small cell lung cancer (NSCLC) cells H460 were cultured with mitoxantrone up to 20 μmol/L to produce ABCG2 overexpressing H460/MX-20 cells. Similarly, NSCLC H460 cells were cultured with paclitaxel 50 nmol/L for up to 3 months to produce ABCB1 producing H460/TX-50 cells (18, 23). All of the cell lines were grown as adherent mono-layers in flasks with DMEM culture medium (Hyclone Co., UT) supplemented with 10% fetal bovine serum in a humidified incubator containing of 5% CO2 at 37°C. All the cell lines were authenticated and were separately tested periodically for mycoplasma or other biological contamination by CytoInspect™ Mycoplasma Detection Test Kits (Greiner Bio-One, Germany).
Cell cytotoxicity by MTT assay
The MTT colorimetric assay with slight modifications from that previously described (24) was used to detect the sensitivity of cells to topotecan. Cells were harvested with trypsin and re-suspended at a final concentration of 5 × 104 cells/ml for H460, 6 × 104 cells/ml for H460/MX-20 and H460/TX-50, 4 × 104 cells/ml for NIH-3T3/pSRα, 5 × 104 for NIH-3T3/MRP4-3 and 8 × 104 for all the other cell lines. Cells were seeded evenly into (180 μl/well) 96-well multiwell plates. Different concentrations of topotecan up to 10 μmol/L (20 μl/well) in triplicate were added into designated wells. After 72 h of incubation, 20 μl of MTT solution (4 mg/ml) was added to each well, and the plate was further incubated for 4 h, allowing viable cells to convert the yellow-colored MTT into dark-blue formazan crystals. Subsequently, the medium was discarded, and 100 μl of dimethylsulfoxide (DMSO) was added into each well to dissolve the formazan crystals. The absorbance was determined at 570 nm with a SpectraMax M3 multimode microplate reader (Molecular Devices, Sunnyvale, CA). The resistance-fold (RF) values were calculated by dividing the IC50 values (concentration required to inhibit growth by 50%) measured in the ABC transporter overexpressing cells by that measured in the parental sensitive cells. The SoftMax Pro Data Acquisition & Analysis software (Molecular Devices Sunnyvale, CA) was used for these analyses.
Western blot analysis
Crude membrane fractions from homogenized tissues were prepared and Western blots were carried out as previously described with minor modifications as noted (25, 26). Briefly, the samples were prepared by homogenizing approximately 40 mg liver or kidney in 0.5 ml of 10 mM Tris–Cl (pH 7.4), 150 mM NaCl, 0.1% (w/v) SDS, 0.5% (w/v) sodium deoxycholate, 1% (w/v) NP-40, 100 ug/ml phenylmethylsulfonyl fluoride (PMSF) and complete protease inhibitor cocktail mix (Roche Applied Sciences, Indianapolis, IN). Homogenized samples were incubated on ice for 30 min with occasional rocking for 15 min, which was followed by centrifugation at 15 000×g for 20 min. The supernatant was collected and total protein protein concentrations were determined using the BCA protein assay kit (Pierce Biotechnology, Inc., Rockford, IL). Approximately 50 μg of total protein per lane was resolved by electrophoresis on a NuPAGE 4 to 12% Bis-Tris gel (Invitrogen, Carlsbad, CA) and electrophoretically transferred onto polyvinylidene fluoride membranes. Equal protein loading was confirmed by comparing beta-actin expression. Samples were treated with one or more of the following primary antibodies to detect Abcc1 (IU2H10 - 1:100), Abcc2 (H-300 - 1:75), Abcc3 (M3II-21 - 1:150); Abcc4 (M41-80 - 1:100), Abcc10 (H300 – 1:200), Abcb1 (C-219 - 1:200); Abcg2 (BXP-21 - 1:200), and β-Actin (Licor92642212 - 1:2000). The following secondary antibodies were used: IRDye 680-labeled anti-rabbit (1:10,000), IRDye 800-labeled anti-mouse, anti-rabbit and anti-rat (1:5,000) and IRDye 680-labeled anti-mouse and anti-rat (1:5,000). Blotting and imaging were performed using the Odyssey imaging system according to the manufacturer's protocols (LI-COR, Lincoln, NE). Band intensity measurements were measured using Image J (NIH) software.
Animal handling, genotyping, blood chemistries, and hematology
Eight strains of male mice, C57BL/6 (wild type [WT]), Abcb1(a/b)−/−, Abcg2−/−, Abcc2−/−, Abcc4−/−, Abcb1(a/b);Abcc2−/−, Abcg2/Abcc2−/−, Abcg2/Abcc4−/−, (20 – 30 g, age 9 – 15 wk) were used in the study after confirmation of their genotype by regular polymerase chain reaction. Generation of the single gene knockout mouse strains are described elsewhere (27, 28). Abcb1(a/b);Abcc2−/−, Abcg2/Abcc2−/−, Abcg2/Abcc4−/−, mice with a C57BL/6 background were generated by crossbreeding two single knockout strains at Charles River Lab (Wilmington, MA). All animals were maintained on an alternating 12 hr light/dark cycle with free access to water and rodent chow ad libitum. The Institutional Animal Care and Use Committee at Mount Sinai Medical Center approved all animal procedures. Genotyping, blood chemistry and hematologic variables were determined at Charles River Research Animal Diagnostic Services (Wilmington, MA).
Pharmacokinetic, fecal and renal excretion studies
Each mouse under anesthesia had a cannula implanted in the left carotid artery that was exteriorized at the back of the neck to permit the collection of serial blood samples. The pharmacokinetic experiments were conducted on conscious, freely mobile mice placed in metabolic cages with an ample water supply (Nalgene; Braintree Scientific Inc., Braintree, MA) that allowed for the separate collection of urine and feces. Topotecan dissolved in saline was administered as an IV bolus via a tail vein to groups of mice at doses of 1 mg/kg and 4 mg/kg. Serial (n = 8–10) blood samples of 20 μL were collected through the left carotid artery cannula for up to 6 hours. The blood samples were centrifuged at 4 °C and the resultant plasma was separated into two parts (5 μL each). One part for measurement of total topotecan was stored at −80 °C immediately, whereas the other 5 μL aliquot designated for measurement of topotecan lactone had 4 parts of acetonitrile containing 80 ng/ml of the internal standard (camptothecin) added immediately followed by a 10 sec vortex and centrifugation for 10 min at 16,000 g. The resultant supernatant was stored at −80 °C to detect lactone form of topotecan by LC/MS/MS. To avoid blood volume depletion 20 μL of saline was replaced after each blood collection. Urine and feces of each animal were collected for 24 hours in pre-weighed tubes and were stored at −80 °C until analysis by LC/MS/MS.
Biliary topotecan excretion
A procedure to collect bile by gall bladder cannulation and bile duct ligation in male C57BL/6 WT, Abcb1(a/b)−/−, Abcg2−/−, Abcc2−/−, Abcc4−/−, Abcb1(a/b);Abcc2−/−, Abcg2/Abcc2−/−, Abcg2/Abcc4−/−, mice (n = 3–5) was slightly modified from that previously described (29). Briefly, mice were anesthetized with isoflorane anesthesia (VetEquip Inc., Pleasanton, CA) throughout the experiment. The common bile duct was ligated distally after opening the abdominal cavity and a durometer vinyl catheter (Scientific Commodities Inc, Lake Havasu City, AZ) with an inner diameter of 0.28 mm, was inserted into the incised gallbladder and fixed with two additional ligatures. After successful cannulation of the gall bladder, 4 mg/kg of topotecan was administered as an IV bolus through the lateral tail vein injection. Bile was collected at 10 min intervals for 1 hr. At the end of the 1 hr, mice were sacrificed by cervical dislocation. Several tissues were removed and homogenized in 4% BSA; intestinal contents were separated from intestinal tissues prior to homogenization and the plasma and organs were harvested on dry ice and stored at −80 °C until analyzed by LC/MS/MS.
Total and lactone form of topotecan determination by LC/MS/MS
Total topotecan (lactone and carboxylate forms) and topotecan lactone in plasma were quantified using an electrospray ionization interface LC/MS/MS system (API QTrap 5500; Applied Biosystems, Foster City, CA) operated in positive ion mode. Chromatography was performed using a Phenomenex Luna C18 (50 × 2.0 mm2) column with a gradient mobile phase of double-distilled water (pH ~ 6.5–7.0) and acetonitrile at a flow rate of 0.2 ml/min for topotecan lactone. For the determination of the lactone form the previously treated aliquots were vortexed and injected into the LC/MS/MS. In order to measure total (lactone+carboxylate) topotecan and ensure that topotecan was all in the lactone form (pH ~ 3–3.5), 5 μl plasma samples were acidified with 50% formic acid (0.5μl) followed by vortexing for 30 sec with 4-part of acetonitrile containing the internal standard camptothecin (80 ng/ml) and then processed as for the lactone fraction,. Chromatography was performed using Phenomenex Luna C18 (50 × 2.0 mm2) column with a gradient mobile phase of 5 mM ammonium formate (0.1% formic acid) and acetonitrile at a flow rate of 0.2 ml/min for topotecan. Mass transitions (m/z) monitored were 422.2 → 377.2 for topotecan and 349 → 305 for the internal standard, respectively. The lowest limit of quantitation was 1.0 ng/ml. Total topotecan and topotecan lactone concentrations in bile, urine, and feces were quantitated directly from the frozen samples using similar methods described above. In brief, each clean urine sample obtained by centrifugation was diluted 300 times and that of bile 500 times with double-distilled water followed by transfer of an aliquot (20 μl) that was deproteinized by a 2-fold volume of acetonitrile containing the internal standard camptothecin (80 ng/ml). After centrifugation (16,000 g, 10 min), the supernatant was injected into the LC/MS/MS system. Each sample of feces was first homogenized in a 2% (w/v) solution of double-distilled water. An aliquot (20 μl) of the resultant homogenate was deproteinized with a 2-fold volume of acetonitrile containing the internal standard camptothecin (80 ng/ml). After centrifugation (16,000 g, 10 min), the supernatant was injected into the LC/MS/MS system. Total (lactone+carboxylate) topotecan in urine, bile and feces were measured as described above after acidification with 50% formic acid (0.5μl) before deproteinization to convert to the lactone form (pH ~ 3–3.5).
Data analysis
Each individual subject's topotecan (total and lactone) plasma concentrations were analyzed by noncompartmental analysis using WinNonlin Professional Version 5.2 (Pharsight Co, Mountain View, CA) to obtain estimates of the pharmacokinetic parameters that included the volume of distribution (Vd), total clearance (CL), the elimination half-life (t1/2), and the area under the plasma concentration-time curve from 0 to infinity (AUCinf). Whereas, topotecan (total and lactone) elimination parameters i.e. biliary excretion rate (μg/hr), % dose in urine (fu, %) and % dose in feces (ff, %) were estimated separately. Comparison of PK variables and other elimination parameters between different strains at each dose level were completed using a one-way analysis of variance (ANOVA), followed by Tukey's multiple comparisons with significant differences based upon *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. In addition, intra-strain comparisons for CL, fu, % and ff, % were conducted based on the mean and standard deviation values from two independent groups (1mg/kg and 4 mg/kg) using unpaired Student's t tests, where P < 0.05 was considered statistically significant. Comparisons of RF values for parental and drug resistance cells were conducted based on the mean and standard deviation values from two independent groups using unpaired Student's t tests, where P < 0.05 was considered statistically significant. All in vitro experiments were repeated at least 3 times. All statistical analyses were completed using GraphPad Prism version 5.04.
Results
Topotecan interaction with ABC-transporters using in vitro systems
To generate an ABC transport profile for topotecan, in vitro cytotoxicity assays were completed in pairs of sensitive and drug-resistant or ABC transporter overexpressing cells (Fig. 2). The IC50 value ratio for each pair reported as a resistance factor (RF), was 3.7 to 12.1, and in low-to-high rank order was ABCC4 (Fig. 2F) = ABCB1 (transfected cells, Fig. 2A) < ABCC2 (Fig. 2E) < ABCB1 [paclitaxel-selected cells, Fig 2B) < ABCG2 (Fig. 2C and 2D). The findings that indicate topotecan may be a substrate for ABCB1 and ABCG2 are consistent with previous studies (12, 16). The interaction of topotecan with ABCC transporters family, whose role has only been recently implicated in drug resistance and drug disposition (4) indicated that ABCC2 (RF= 4.4, Fig. 2E) and ABCC4 (RF= 3.7, Fig. 2F) transporters conferred significant resistance to topotecan. RF values for other MRP members in the ABCC family, specifically, human ABCC1/MRP1, ABCC3/MRP3, ABCC5/MRP5, ABCC10/MRP7 and ABCC12/MRP9 were not significantly different for topotecan (Supplementary Table S1). In summary, based on cytotoxicity assays ABCB1, ABCG2, ABCC2 and ABCC4 conferred significant topotecan resistance in vitro and may be involved in its PK characteristics.
Figure 2.

Cell survival curves of multidrug resistant (MDR) cells and the corresponding parental cells towards topotecan are shown in (A) HEK293 and transfected HEK293/ABCB1 cells (B) NSCLC-H460 and paclitaxel selected ABCB1 overexpressing H460/TX-50 cells (C) HEK293 and transfected HEK293/ABCG2-R2 cells (D) NSCLC-H460 and mitoxantrone-selected ABCG2 overexpressing H460/MX-20 cells (E) Parental (LLC/PK1-CMV) and ABCC2-transfected LLC/PK1-hMRP2 cells (F) Parental (NIH-3T3/pSRα) and ABCC4-transfected NIH-3T3/MRP4-3 cells. Cell survival was determined by MTT assay as described in the Materials and Methods. Data points represent the means ± SD of triplicate determinations performed at least three independent times. The resistance fold (RF) was calculated by dividing the IC50 (concentrations required to inhibit growth by 50%) for the MDR cells by that of the parental sensitive cells. The statistical differences were determined using the Student's t-test with significant differences based upon *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with parental cells.
Overlapping functions of transporters in altering pharmacokinetic of topotecan
The in vitro findings guided us to select the relevant single and double ABC transporters knockout mouse models to study topotecan disposition. Each of the following strains received 1 mg/kg and 4 mg/kg of topotecan as an IV bolus, WT, Abcc2−/−, Abcc4−/−, Abcb1−/−, Abcg2−/−, Abcc2;Abcg2−/−, Abcc4;Abcg2−/− and Abcc2;Abcb1−/−, and both total topotecan (carboxylate + lactone) and topotecan lactone were measured by LC/MS/MS in plasma, urine, bile and feces (Supplementary Figures S1A–1H). At the 1 mg/kg dose, compared to WT mice, there were no significant changes in topotecan lactone AUC in Abcc2−/−, Abcc4−/− or Abcb1−/− strains; however higher topotecan lactone AUCs were observed in the order of 3.6-fold in Abcc2;Abcg2−/− > 3-fold in Abcg2−/− > 2.4-fold in Abcc4;Abcg2 > and 2.1-fold in Abcc2;Abcb1−/−. The changes in the AUC of topotecan lactone at the 4 mg/kg dose were analogous to those at the 1 mg/kg dose and those with more than a 2-fold change were; Abcc2;Abcg2−/− (3.3 fold) > Abcg2−/− (2.3 fold) > Abcc4;Abcg2−/− (2.2 fold) = Abcc2;Abcb1 (2.2 fold) (Table 1, Fig. 3A). A significantly increased AUC of topotecan lactone at 4 mg/kg dose was observed in the Abcc2;Abcb1−/− strain compared to WT, Abcb1−/− and Abcc2−/− strains, which was not obvious at 1 mg/kg, and suggests saturation of Abcg2 transporter at the higher dose.
Table 1.
Selected topotecan lactone pharmacokinetic parameters in ABC transporter KO strains at two i.v. dose levels.
| Parameters (TPT Lactone) | Mouse Strains | |||||||
|---|---|---|---|---|---|---|---|---|
| C57/BL6 | Abcc2−/− | Abcc4−/− | Abcb1−/− | Abcg2−/− | Abcc2; Abcg2−/− | Abcc4; Abcg2−/− | Abcc2; Abcb1−/− | |
|
| ||||||||
| (n=7) | (n=6) | (n=6) | (n=5) | (n=7) | (n=5) | (n=5) | (n=5) | |
| 1 mg | ||||||||
| Vd(l/kg) | 19.5 ± 5.6 | 17.9 ± 8.1 | 20.4 ± 5.5 | 14.0 ± 2.5 | 9.9 ± 4.3* | 7.1 ± 1.9**, ## | 9.1 ± 2.5*,‡‡ | 9.0 ± 2.7* |
| CL (l/hr/kg) | 12.8 ± 3.0 | 9.2 ± 3.9 | 14.2 ± 7.8 | 7.0 ± 2.2 | 4.9 ± 2.9** | 3.6 ± 1.0** | 5.1 ± 0.7‡ | 5.8 ± 0.8* |
| t1/2 (hr) | 1.1 ± 0.3 | 1.4 ± 0.2 | 1.3 ± 0.8 | 1.4 ± 0.2 | 1.5 ± 0.4 | 1.4 ± 0.3 | 1.2 ± 0.3 | 1.1 ± 0.4 |
| AUCInf (hr*ng/ml) | 81.9 ± 17.4 | 130.1 ± 65.0 | 91.2 ± 51.2 | 154.0 ± 46.7 | 242.4 ± 75.8*** | 291.5 ± 76.2***,### | 200.0 ± 25.5*,‡ | 175.7 ± 23.1 |
| Renal (fu, %) | 15.1 ± 9.3 | 21.8 ± 8.5 | 12.5 ± 6.7 | 24.0 ± 12.2 | 31.7 ± 9.1* | 37.9 ± 9.6** | 32.5 ± 8.1* | 25.4 ± 11.1 |
| Fecal (ff, %) | 35.7 ± 19.3 | 26.1 ± 12.5 | 44.7 ± 17.7 | 13.3 ± 11.4 | 19.1 ± 16.8 | 11.0 ± 4.9 | 14.3 ± 7.2 | 21.2 ± 5.6 |
|
| ||||||||
| 4 mg | (n=7) | (n=6) | (n=6) | (n=5) | (n=7) | (n=5) | (n=5) | (n=5) |
|
|
||||||||
| Vd(l/kg) | 23.6 ± 6.7 | 33.3 ± 10.2 | 21.7 ± 3.8 | 14.9 ± 5.4 | 10.5 ± 2.6* | 7.9 ± 3.8**,### | 9.5 ± 4.1* | 13.6 ± 12.5### |
| CL (l/hr/kg) | 14.0 ± 5.3 | 19.1 ± 7.9 | 15.9 ± 4.7 | 11.4 ± 2.8 | 5.9 ± 1.6* | 4.0 ± 0.6**,### | 6.5 ± 2.2‡ | 5.9 ± 0.5*,### |
| t1/2 (hr) | 1.3 ± 0.6 | 1.4 ± 0.9 | 1.0 ± 0.2 | 0.9 ± 0.2 | 1.2 ± 0.2 | 1.3 ± 0.4 | 1.0 ± 0.1 | 1.5 ± 1.2 |
| AUCInf (hr*ng/ml) | 314.3 ± 101.3 | 240.6 ± 94.3 | 271.3 ± 81.5 | 372.3 ± 105.4 | 735.6 ± 270.7*** | 1021.9 ± 151.2***,### | 684.0 ± 244.3**,‡‡ | 676.4 ± 55.8**,## |
| Renal (fu, %) | 18.0 ± 11.8 | 16.5 ± 9.9 | 18.8 ± 11.9 | 14.7 ± 7.8 | 32.6 ± 18.1 | 34.7 ± 5.1 | 30.7 ± 11.9 | 35.7 ± 11.3 |
| Fecal (ff, %) | 25.5 ± 10.4 | 18.0 ± 9.7 | 24.2 ± 16.5 | 12.1 ± 8.8 | 12.0 ± 9.8 | 4.9 ± 2.7* | 12.5 ± 6.9 | 5.9 ± 3.3* |
Topotecan lactone pharmacokinetic parameters - volume of distribution (Vd), systemic clearance (CL), elimination half life (t1/2) area under the plasma curve at infinity (AUCinf), renal fraction of dose (fu, %), fecal fraction of dose (ff, %) in male C57BL/6 (WT), Abcc2−/−, Abcc4−/−, Abcb1−/−, Abcg2−/−, Abcc2;Abcg2, Abcc4;Abcg2−/− and Abcc2;Abcb1−/− mice, after IV administration of 1 mg/kg and 4 mg/kg topotecan. The pharmacokinetic parameters were analyzed by noncompartmental analysis using WinNonlin Professional Version 5.2. The statistical differences were determined using the One-Way ANOVA with Tukey's multiple comparison (GraphPad Prism version 5.04) tests to compare inter-strain differences. Data are expressed as mean values of the Vd, CL, AUC, t1/2, fu, %, ff, %±SD (n = 5–7;
P < 0.05
P < 0.01
P < 0.001 compared with wild-type
P < 0.05
P < 0.01
P < 0.001 compared with Abcc2−/−
P < 0.05
P < 0.01
P < 0.001 compared with Abcc4−/−).
Figure 3.

Topotecan lactone pharmacokinetic variables (A) Area under the plasma concentration-time curve (AUCinf) (B) Systemic clearance (CL) and (C) Volume of distribution (Vd) in male C57BL/6 WT, Abcc2−/−, Abcc4−/−, Abcb1−/−, Abcg2−/−, Abcc2;Abcg2, Abcc4;Abcg2−/− and Abcc2;Abcb1−/− mice, after IV administration of 1 mg/kg and 4 mg/kg topotecan. The pharmacokinetic parameters were estimated by noncompartmental analysis using WinNonlin Professional Version 5.2. The statistical differences were determined using the One-Way ANOVA with Tukey's multiple comparison (GraphPad Prism version 5.04) tests to compare inter-strain differences. Data are expressed as mean values of the AUC, CL or Vd ± SD (n = 5–9; *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with wild-type; #, P < 0.05; ##, P < 0.01; ###, P < 0.001 compared with Abcc2−/−).
Comparisons within the different gene knockout strains allowed the overlapping function of the transporters to be discerned. The mean topotecan AUC in the Abcc2;Abcg2−/− strain was 2.2-fold (1 mg/kg) and 4.2-fold (4 mg/kg) greater than the single knockout Abcc2−/− strain, and 1.2-fold (1 mg/kg) and 1.5-fold (4 mg/kg) greater than the Abcg2−/− strains, and attests to the dominant effect of Abcg2, as well as to a contributing effect of Abcc2 on topotecan elimination. Abcc4 did not have a significant effect on topotecan elimination based on small changes in the AUC obtained in the Abcc4;Abcg2−/− mouse relative to the Abcg2−/− mouse. To decipher the role of Abcb1, AUC comparisons between the Abcc2;Abcb1−/− and both the Abcc2−/−/ and Abcb1−/− strains indicate complimentary functions with the greatest fold changes, 2.8 and 1.8, obtained for the double knockout group at the 4 mg/kg dose level relative to the Abcc2−/− and Abcb1−/− groups, respectively (Table 1, Fig. 3). These changes seem to emanate from reduced biliary excretion as only 5.9% of the dose was found in feces in the Abcc2;Abcb1−/− strain at the high dose level.
The total clearance values of topotecan lactone at both dose levels (Fig. 3B) follow the same pattern as the AUC values, yet in the opposite direction as required, since the CL is the inverse of the total AUC (i.e. CL = dose/AUC). The apparent volume of distribution (Vd) is dependent on drug-plasma protein and drug-tissue binding, yet in the absence of changes in these parameters in each strain, the Vd values reflect changes in total clearance and the terminal elimination rate constant (i.e. Vd = CL/λn), and therefore the significant differences in Vd between strains (Table 1) are likely due to primary changes in clearance; transport-mediated effects on biliary and renal excretion. For completeness, the pharmacokinetic variables obtained from total topotecan measurements are provided (Supplementary Table S1 and Supplementary Figures S2 and S3) and were found to follow the same trends amongst different strains as those obtained for topotecan lactone.
Role of transporters in renal, hepatic and biliary elimination of topotecan
Measurement of topotecan in urine, bile and feces can delineate the contribution of biliary and renal elimination pathways to total clearance. In the preclinical setting, the use of “metabolism” cages permits the separate collection of urine, needed for renal clearance, and feces; however drug contained in feces represents that eliminated in bile and by intestinal extrusion following IV administration. A direct means to determine biliary excretion can be achieved through the combined use of bile duct ligation and gall bladder cannulation, yet due to the intricacies of this approach and the difficulty to maintain the patency of the exteriorized cannula limit the duration of the experiment to one hour. Therefore, two experimental approaches were used; one in which individual mice in each strain were placed in metabolism cages to obtain serial blood samples, urine and fecal samples, and two, separate groups of mice at the 4 mg/kg dose only underwent the bile duct surgical procedure to provide bile samples to assess biliary excretion. In this manner, a thorough analysis of the routes of topotecan elimination and contribution of each transporter could be ascertained.
Figures 4A and 4B show the topotecan lactone excreted by renal and fecal excretion, respectively, at both dose levels. At 1 mg/kg (Fig. 4A), topotecan lactone was excreted in urine from a low of 12.5 % to a high of 37.9 % of the dose over all the strains with the rank order of Abcc2;Abcg2−/− > Abcc4;Abcg2−/− > Abcg2−/− > Abcc2;Abcb1−/− > Abcb1−/− > Abcc2−/− > WT > Abcc4−/−; the 3 strains based on Abcg2 deletion being significantly greater than the WT strain with the differences on the order of over 2-fold. Of some interest, renal excretion of topotecan lactone was also increased close to 2-fold in Abcc2;Abcb1−/− (Fig. 4A). A quite similar pattern in renal disposition was seen at the higher 4 mg/kg topotecan dose, yet none of the differences reached significance, that could either reflect experimental variability or saturation of compensatory renal clearance mechanisms. The compensatory increases in renal excretion of topotecan in the knockout strains, particularly in the Abcg2−/− strains, are partially attributed to enhanced Abcc4-mediated renal excretion - supported by increased protein expression - yet may be considered paradoxical since even the Abcc4;Abcg2−/− strain had elevated renal excretion. Similar patterns of renal excretion were observed for total topotecan at both dose levels in WT and KO mice (Supplementary Fig. S3). Overall, it can be concluded that the absence of Abcg2 has the most profound effect on topotecan biliary excretion that is compensated for by elevated renal excretion; to a lesser extent Abcb1 and Abcc2 function in a similar manner.
Figure 4.

Urinary (A) and fecal (B) excretion of topotecan lactone in male C57BL/6 (WT), Abcc2−/−, Abcc4−/−, Abcb1−/−, Abcg2−/−, Abcc2;Abcg2, Abcc4;Abcg2−/− and Abcc2;Abcb1−/− mice, over 24 h after IV administration of 1 mg/kg and 4 mg/kg topotecan. The statistical differences were determined using the One-Way ANOVA with Tukey's multiple comparison (GraphPad Prism version 5.04) tests to compare inter-strain differences. Data are expressed as percentage of dose ± SD (n = 5–9; *, P < 0.05; **, P < 0.01; compared with wild-type).
The patterns of changes in fecal elimination expressed as the % dose are essentially inverse to those for renal excretion for each strain. The mean % dose eliminated in the feces at both the 1 mg/kg and 4 mg/kg ranged from about 5% to a high of 45%, with the Abcg2−/− based strains having the lowest fractional fecal excretion at about 5%. Fecal excretion of topotecan lactone was also decreased in the absence of both Abcc2 and Abcb1 at both dose levels, but not to the extent observed in the absence of Abcg2 (Fig. 4B). Intrastrain dose comparisons showed a significant reduction of fecal elimination in the Abcc2;Abcb1−/− strain at 4 mg/kg compared to 1 mg/kg that is consistent with saturation of Abcg2 transporter at high dose levels. Fecal elimination of total topotecan followed a similar trend in all the mouse strains as that of topotecan lactone (Supplementary Fig. S3B and Supplementary Table S2).
Topotecan biliary excretion rates were obtained in bile duct ligated, gall-bladder cannulated mice at the 4mg/kg IV dose (Fig. 5). Compared to the WT strain, significantly decreased biliary excretion of topotecan lactone was found for Abcc2;Abcb1−/− and for all the Abcg2−/− based strains, with a high of a 19-fold reduction in the Abcc2;Abcg2−/− strain. Within strain comparisons indicated significantly decreased biliary elimination of topotecan lactone in the Abcc2;Abcb1−/− strain compared to Abcc2−/− and Abcb1−/− strains, and in Abcc2; Abcg2−/− strain compared to both the Abcc2−/− and Abcg2−/−strains. These excretion rate data, (amount excreted/hour) take into account the changes in bile flow rate that relative to WT mice decreased in the order of Abcc2−/− = Abcc2;Abcg2−/− < Abcc2;Abcb1−/− < Abcg2−/− = Abcc4;Abcg2−/− (Supplementary Fig. S4A), which except for the Abcc2−/− strain coincided with the excretion rate. This suggests that bile flow may partially depend on the functional capacity of the biliary pumps for topotecan. Total topotecan biliary excretion pattern were similar to that of topotecan lactone in all strains (Supplementary Fig. S4B). In summary, the changes in fecal excretion of topotecan are primarily attributed to changes in its biliary excretion afforded by Abcg2, Abcb1 and Abcc2.
Figure 5.

Biliary excretion rate (μg/hr) of topotecan lactone in male C57BL/6 (WT), Abcc2−/−, Abcc4−/−, Abcb1−/−, Abcg2−/−, Abcc2;Abcg2, Abcc4;Abcg2−/− and Abcc2;Abcb1−/− mice, over 1 h after IV administration of 4 mg/kg topotecan. The statistical differences were determined using the One-Way ANOVA with Tukey's multiple comparison tests to compare inter-strain differences. Each bar represents the mean ± SD (n = 3–5; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 compared with wild-type; P < 0.001 compared with wild-type; #, P < 0.05; ##, P < 0.01; ###, P < 0.001 compared with Abcc2−/−; ˙, P < 0.05; compared with Abcb1−/−).
Transporters expression analysis and clinical chemistry in the ABC KO models
It is possible that elimination of one or more transporters could result in altered expression of the remaining transporters to compensate for the loss of function. Examination of protein expression of the ABC transporters in kidney and liver, prior to any drug treatment, indicated (Figures 6A and 6B) there were insignificant differences in the expression in the different gene knockout strains compared to the WT strain except in a few organ specific cases. For example, in liver Abcc4 is basolaterally localized and was significantly elevated in the Abcc2−/−, Abcb1−/−, Abcc2;Abcg2−/− and Abcc2;Abcb1−/− strains compared to WT (Fig. 6A). This elevated expression of Abcc4 could have contributed to higher topotecan plasma concentrations in addition to the loss in biliary excretion potential in the absence of Abcc2, Abcb1 and Abcg2. In kidney, the most pronounced change occurred with Abcc3, a basolateral transporter was increased about 1.6-fold, in Abcc2;Abcb1−/− mouse, while Abcc4, an apical transporter, was significantly up-regulated in the Abcc2−/−, Abcc2;Abcg2−/− and Abcc2;Abcb1−/− mouse strains compared to WT (Fig. 6B) and certainly may have had a role in increased renal excretion in these strains.
Figure 6.

Representative Western blots of Abcb1, Abcg2, Abcc1, Abcc2, Abcc3 and Abcc4 in (A) liver and (B) kidney of male C57BL/6 WT, Abcc2−/−, Abcc4−/−, Abcb1−/−, Abcg2−/−, Abcc2;Abcg2, Abcc4;Abcg2−/− and Abcc2;Abcb1−/− mice. A densitometric analysis using Image J software was done to compare the relative change in the expression of Abcb1, Abcg2, Abcc1, Abcc2, Abcc3 and Abcc4 in (A) liver and (B) kidney of male Abcc2−/−, Abcc4−/−, Abcb1−/−, Abcg2−/−, Abcc2;Abcg2, Abcc4;Abcg2−/− and Abcc2;Abcb1−/− mice compared to male C57BL/6 WT. The statistical differences were determined using the Student's t-test with significant differences based upon P < 0.05. The color format chosen for expression analysis were – green (no change), orange (slightly up-regulated but less than being significant at P < 0.05), red (P < 0.05), blue (slightly down-regulated but less than being significant at P < 0.05), purple (undetectable in these samples) and black (deleted gene – no detection).
All the mouse strains used in this study were healthy and had no obvious phenotypic abnormalities. The mice gained normal weight and had no abnormality in their fertility or survival (data not shown). Consistent with previous findings total bilirubin levels were increased in the Abcc2−/− and Abcc2;Abcg2−/− strains, whereas higher levels of triglycerides and cholesterol were seen in Abcc2−/− mice. There were no other significant changes in the clinical chemistry panels in any of the strains compared to WT mice (Supplementary Table S3).
Discussion
The analysis of how ABC transporters function in drug disposition has become more complex as their overlapping roles and substrate profiles are delineated. The pivotal locations of ABC transporters in the gastrointestinal tract, liver and kidneys make their contribution to drug absorption and elimination register as changes in macro-PK parameters such as oral bioavailability and clearance. Their presence in regional sites, such as the BBB, can also have profound influences on drug distribution, but may not be realized without tissue analyses of drug concentrations. The net effects, whether they emanate on a systemic or regional tissue scale can curtail or enhance the effectiveness of drugs depending on target site drug concentrations, and further, contribute to toxicity in non-target organs. Coupled to the ABC transporter somewhat ubiquitous presence, overlapping function and their capacity to efflux diverse chemical structures is their ability to operate in a nonlinear and saturable manner, subject to competitive inhibition, making predictions of their influence difficult without detailed PK investigations. The current study was designed to characterize the role of ABC transporters on the systemic PKs of topotecan, a highly used anticancer drug.
Topotecan is a known substrate for the ABCG2 transporter and confirmed here, while its interaction with ABCB1 and ABCC4 uncertain, and details of other ABCC family members lacking. Moreover, there have been few studies to address the overlapping function of different transporters that limit an understanding of the role of single ABC transporters in topotecan PKs. The benefits of using double knockout mice are known (30, 31), and can resolve conflicting interpretations of PK data obtained in single ABC transporter knockout mouse models, when there is functional redundancy of at least two transporters. In the present study we have dissected the overlapping function of ABC transporters as it pertains to topotecan systemic pharmacokinetic properties through the use of gene knockout mouse models that provide a realistic means to predict the consequences of the transporters in humans.
The PKs of topotecan in cancer patients appear to be dose-proportional and multicompartmental in nature with an elimination t ½ of 2–3 hrs (10, 32). Renal elimination and biliary excretion are the major routes of elimination for topotecan, with 26–80% of the dose recovered in urine, 17.9 ± 3.6% in feces and 18% of the dose eliminated in the bile (33). Comparing these general characteristics to the WT mice in our study, a similar dose-proportional relationship in topotecan lactone AUC was observed at 1 mg/kg to 4 mg/kg doses (Table 1). dose eliminated by each route for the two doses was a mean of 15–18 % for renal, 26–36 % for fecal (Table 1), and about 20% by biliary excretion based on the 1 hour colleciton (data not shown). Thus, compared to normal mice, patients seem to have a slightly greater propensity to eliminate topotecan by renal excretion, yet the variability is high and could represent disease states and polymorphic changes in transporter genes (34, 35).
Topotecan is not extensively metabolized (36–38) with only low concentrations of three topotecan metabolites (N-desmethyl topotecan, TPT-O-glucuronide and N-desmethyl TPT-O-glucuronide) identified in blood in patients. Other than the ABC transporters the involvement of other membrane transporters to topotecan elimination appear limited, with a recent finding that OAT3, a member of the SLC22 family, may contribute to topotecan secretion in the renal tubules of the rats and humans (39). Thus, there is general agreement in the clearance mechanisms involved in topotecan elimination in mice and humans, and that these processes are intimately connected to active transporters of the ABC family.
Through an extensive set of pharmacokinetic investigations that utilized eight mouse strains, two dose levels, and the collection of urine, feces, bile (one dose level) and serial blood samples, we determined the most important ABC transporters and their complimentary functions in the disposition of topotecan. These analyses were facilitated by a sensitive LC/MS/MS method to measure topotecan, both total and lactone forms, in as little as 5 μl of plasma that permitted a serial blood sampling protocol in mice. There were similar trends in the PK variables, renal, fecal and hepatobiliary excretion at both dose levels for both lactone and total forms of topotecan suggesting that either form is sufficient to assess its disposition in mice.
Abcg2 had the most pronounced influence on topotecan elimination via biliary excretion, yet Abcc2 and Abcb1 also contributed to this process that is revealed at the high dose level in the double knockout strains (Table 1). Indeed, the Abcc2;Abcb1−/− and Abcc2;Abcg2−/− strains had elevated renal, reduced fecal and biliary excretion, particularly prominent at the 4 mg/kg dose level. It was through the use of the double knockout strains and two dose levels that a role of Abcc2 and its overlapping function with Abcg2 and Abcb1 could be ascertained. There is some conflicting data on the interaction between ABCC4 or Abcc4 and topotecan. Previously, Leggas et al (14) have shown that topotecan is a substrate of ABCC4 and in the Abcc4−/− mouse showed significant accumulation of topotecan in brain tissues and cerebrospinal fluid compared to WT mice. Somewhat counter findings were reported by de Vries and colleagues (12), where topotecan concentrations in brain homogenates of Abcc4−/− were not significantly different from that of WT mice. Although our study was not designed to evaluate how Abcc4−/− affected brain distribution of topotecan, we found an approximate 4-fold change in the RF in cells that overexpressed ABCC4 that supports a topotecan-Abcc4 interaction. However, sole deletion of Abcc4 did not significantly affect topotecan clearance even though it would be anticipated to contribute to renal elimination based on its apical orientation in renal tubules (40); apparently topotecan either has a low affinity for Abcc4 in renal tubules or other transporters compensate for this function since the % dose (Table 1) eliminated in urine was not significantly different than in WT mice. In fact, in the absence of both Abcc4 and Abcg2 renal excretion increased relative to WT mice, although paradoxical, could be indicative of another renal transport mechanism. Nonetheless, examination of the renal expression of Abcc4 amongst the different strains showed appreciable upregulation in the Abcc2−/−, Abcc2−/−;Abcg2−/− and Abcc2−/−;Abcb1−/− strains, which except for the Abcc2−/−/ strain, and elevated renal excretion that supports a contributory role of Abcc4 in the renal excretion of topotecan. Therefore, although the role of Abcc4 in the renal elimination of topotecan is not compelling there is support for a partial role.
The therapeutic index of topotecan like many anticancer drugs is narrow, which presents a greater risk of toxicity and of therapeutic failure due to small variations in exposure (i.e. AUC) (41). For such drugs that are also substrates for the ABC transporters the task of maintaining therapeutic concentrations is compounded by the saturable nature of transport function, the potential for drug-drug interactions and gene polymorphisms that may influence transporter activity. With respect to the latter, a pilot study indicated that the presence of single nucleotide polymorphisms (SNP) in ABCG2 Q141K (ABCG2 421C>A) in a cohort of cancer patients significantly altered topotecan bioavailability and increased plasma AUC by 1.35-fold (34). This mutation also significantly altered imatinib PK (35), and was associated with a significantly high risk of diarrhea in patients treated with oral gefitinib (42). Therefore, the PK information obtained in Abc gene knockout mouse models can anchor our assessment of their roles, and support examination of how pharmacogenetic variations in these genes in patients may alter the PKs of a drug.
In summary, we found that either topotecan lactone form or total topotecan can reliably be used to determine topotecan disposition. It is clear that Abcg2 is the primary transporter involved in topotecan disposition; however, in its absence or under saturating conditions, Abcb1 and Abcc2 can partially assume its role via biliary excretion. Assuming these transporters function similarly in humans with regard to topotecan elimination, their induction, inhibition or polymorphic changes that alter their activity warrant scrutiny as to the ultimate impact on topotecan's PK characteristics. However, it is precisely this overlapping function that may minimize the deleterious impact on patients by maintaining therapeutic drug exposures and limiting toxicities.
Supplementary Material
Acknowledgements
This work was supported by the National Institute of Health grant CA114574 to J. M. Gallo. The authors thank Dr. A. Schinkel (Netherlands Cancer Institute, Netherlands) for providing the detailed protocol for gall bladder cannulation and bile duct ligation.
Abbreviations
- ABC
ATP-binding cassette
- ABCB1/P-gp
ABC sub-family B member 1/Phosphoglycoprotein
- ABCC/MRP
ABC sub-family C member/multiple resistance protein
- ABCG2/BCRP
ABC sub-family G member 2/Breast Cancer Resistance Protein
- ANOVA
analysis of variance
- AUC
area under the plasma concentration-time curve
- CL
total systemic clearance
- ff
fecal fraction of dose
- fu
renal fraction of dose
- IV
intravenous
- LC/MS/MS
liquid chromatography/tandem mass spectrometry
- MRP
multidrug resistance proteins
- PK
pharmacokinetic
- WT
wild type
Footnotes
CONFLICT: The authors have no conflicts of interest to declare.
Authors' Contributions Conception and design: A.K. Tiwari and J.M. Gallo
Development of methodology: A.K. Tiwari and R. Zhang
Acquisition of data (provided animals, acquired and managed animals, provided facilities, etc.): A.K. Tiwari, R. Zhang and J.M. Gallo
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): A.K. Tiwari and J.M. Gallo
Writing, review, and/or revision of the manuscript: A.K. Tiwari and J.M. Gallo
Study supervision: J.M. Gallo
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71:537–92. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- 2.Chan LM, Lowes S, Hirst BH. The ABCs of drug transport in intestine and liver: efflux proteins limiting drug absorption and bioavailability. Eur J Pharm Sci. 2004;21:25–51. doi: 10.1016/j.ejps.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Kruh GD, Belinsky MG, Gallo JM, Lee K. Physiological and pharmacological functions of Mrp2, Mrp3 and Mrp4 as determined from recent studies on gene-disrupted mice. Cancer Metastasis Rev. 2007;26:5–14. doi: 10.1007/s10555-007-9039-1. [DOI] [PubMed] [Google Scholar]
- 4.Chen ZS, Tiwari AK. Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. FEBS J. 2011;278:3226–45. doi: 10.1111/j.1742-4658.2011.08235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Jr., Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci U S A. 2002;99:15387–92. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruce JN, Fine RL, Canoll P, Yun J, Kennedy BC, Rosenfeld SS, et al. Regression of recurrent malignant gliomas with convection-enhanced delivery of topotecan. Neurosurgery. 2011;69:1272–9. doi: 10.1227/NEU.0b013e3182233e24. discussion 9–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong ET, Berkenblit A. The role of topotecan in the treatment of brain metastases. Oncologist. 2004;9:68–79. doi: 10.1634/theoncologist.9-1-68. [DOI] [PubMed] [Google Scholar]
- 8.Gallo JM, Laub PB, Rowinsky EK, Grochow LB, Baker SD. Population pharmacokinetic model for topotecan derived from phase I clinical trials. J Clin Oncol. 2000;18:2459–67. doi: 10.1200/JCO.2000.18.12.2459. [DOI] [PubMed] [Google Scholar]
- 9.Blaney SM, Cole DE, Balis FM, Godwin K, Poplack DG. Plasma and cerebrospinal fluid pharmacokinetic study of topotecan in nonhuman primates. Cancer Res. 1993;53:725–7. [PubMed] [Google Scholar]
- 10.Herben VM, ten Bokkel Huinink WW, Beijnen JH. Clinical pharmacokinetics of topotecan. Clin Pharmacokinet. 1996;31:85–102. doi: 10.2165/00003088-199631020-00001. [DOI] [PubMed] [Google Scholar]
- 11.Hertzberg RP, Caranfa MJ, Holden KG, Jakas DR, Gallagher G, Mattern MR, et al. Modification of the hydroxy lactone ring of camptothecin: inhibition of mammalian topoisomerase I and biological activity. J Med Chem. 1989;32:715–20. doi: 10.1021/jm00123a038. [DOI] [PubMed] [Google Scholar]
- 12.de Vries NA, Zhao J, Kroon E, Buckle T, Beijnen JH, van Tellingen O. P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res. 2007;13:6440–9. doi: 10.1158/1078-0432.CCR-07-1335. [DOI] [PubMed] [Google Scholar]
- 13.Tian Q, Zhang J, Tan TM, Chan E, Duan W, Chan SY, et al. Human multidrug resistance associated protein 4 confers resistance to camptothecins. Pharm Res. 2005;22:1837–53. doi: 10.1007/s11095-005-7595-z. [DOI] [PubMed] [Google Scholar]
- 14.Leggas M, Adachi M, Scheffer GL, Sun D, Wielinga P, Du G, et al. Mrp4 confers resistance to topotecan and protects the brain from chemotherapy. Mol Cell Biol. 2004;24:7612–21. doi: 10.1128/MCB.24.17.7612-7621.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen J, Carcaboso AM, Hubbard KE, Tagen M, Wynn HG, Panetta JC, et al. Compartment-specific roles of ATP-binding cassette transporters define differential topotecan distribution in brain parenchyma and cerebrospinal fluid. Cancer Res. 2009;69:5885–92. doi: 10.1158/0008-5472.CAN-09-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jonker JW, Smit JW, Brinkhuis RF, Maliepaard M, Beijnen JH, Schellens JH, et al. Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J Natl Cancer Inst. 2000;92:1651–6. doi: 10.1093/jnci/92.20.1651. [DOI] [PubMed] [Google Scholar]
- 17.Verweij J, Lund B, Beijnen J, Planting A, de Boer-Dennert M, Koier I, et al. Phase I and pharmacokinetics study of topotecan, a new topoisomerase I inhibitor. Ann Oncol. 1993;4:673–8. doi: 10.1093/oxfordjournals.annonc.a058623. [DOI] [PubMed] [Google Scholar]
- 18.Robey RW, Lin B, Qiu J, Chan LL, Bates SE. Rapid detection of ABC transporter interaction: potential utility in pharmacology. J Pharmacol Toxicol Methods. 2011;63:217–22. doi: 10.1016/j.vascn.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shukla S, Skoumbourdis AP, Walsh MJ, Hartz AM, Fung KL, Wu CP, et al. Synthesis and characterization of a BODIPY conjugate of the BCR-ABL kinase inhibitor Tasigna (nilotinib): evidence for transport of Tasigna and its fluorescent derivative by ABC drug transporters. Mol Pharm. 2011;8:1292–302. doi: 10.1021/mp2001022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tiwari AK, Sodani K, Wang SR, Kuang YH, Ashby CR, Jr., Chen X, et al. Nilotinib (AMN107, Tasigna) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem Pharmacol. 2009;78:153–61. doi: 10.1016/j.bcp.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Kawabe T, Chen ZS, Wada M, Uchiumi T, Ono M, Akiyama S, et al. Enhanced transport of anticancer agents and leukotriene C4 by the human canalicular multispecific organic anion transporter (cMOAT/MRP2) FEBS Lett. 1999;456:327–31. doi: 10.1016/s0014-5793(99)00979-5. [DOI] [PubMed] [Google Scholar]
- 22.Lee K, Klein-Szanto AJ, Kruh GD. Analysis of the MRP4 drug resistance profile in transfected NIH3T3 cells. J Natl Cancer Inst. 2000;92:1934–40. doi: 10.1093/jnci/92.23.1934. [DOI] [PubMed] [Google Scholar]
- 23.Tiwari AK, Sodani K, Dai CL, Abuznait AH, Singh S, Xiao ZJ, et al. Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett. 2013;328:307–17. doi: 10.1016/j.canlet.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res. 1987;47:936–42. [PubMed] [Google Scholar]
- 25.Ogihara H, Saito H, Shin BC, Terado T, Takenoshita S, Nagamachi Y, et al. Immuno-localization of H+/peptide cotransporter in rat digestive tract. Biochem Biophys Res Commun. 1996;220:848–52. doi: 10.1006/bbrc.1996.0493. [DOI] [PubMed] [Google Scholar]
- 26.Peng KC, Cluzeaud F, Bens M, Duong Van Huyen JP, Wioland MA, Lacave R, et al. Tissue and cell distribution of the multidrug resistance-associated protein (MRP) in mouse intestine and kidney. J Histochem Cytochem. 1999;47:757–68. doi: 10.1177/002215549904700605. [DOI] [PubMed] [Google Scholar]
- 27.Belinsky MG, Guo P, Lee K, Zhou F, Kotova E, Grinberg A, et al. Multidrug resistance protein 4 protects bone marrow, thymus, spleen, and intestine from nucleotide analogue-induced damage. Cancer Res. 2007;67:262–8. doi: 10.1158/0008-5472.CAN-06-2680. [DOI] [PubMed] [Google Scholar]
- 28.Wang Z, Zhou Q, Kruh GD, Gallo JM. Dose-dependent disposition of methotrexate in Abcc2 and Abcc3 gene knockout murine models. Drug Metab Dispos. 2011;39:2155–61. doi: 10.1124/dmd.111.041228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Herwaarden AE, Jonker JW, Wagenaar E, Brinkhuis RF, Schellens JH, Beijnen JH, et al. The breast cancer resistance protein (Bcrp1/Abcg2) restricts exposure to the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. Cancer Res. 2003;63:6447–52. [PubMed] [Google Scholar]
- 30.Lagas JS, Vlaming ML, Schinkel AH. Pharmacokinetic assessment of multiple ATP-binding cassette transporters: the power of combination knockout mice. Mol Interv. 2009;9:136–45. doi: 10.1124/mi.9.3.7. [DOI] [PubMed] [Google Scholar]
- 31.Tiwari AK, Sodani K, Dai CL, Ashby CR, Jr., Chen ZS. Revisiting the ABCs of multidrug resistance in cancer chemotherapy. Curr Pharm Biotechnol. 2011;12:570–94. doi: 10.2174/138920111795164048. [DOI] [PubMed] [Google Scholar]
- 32.Blaney SM, Balis FM, Cole DE, Craig C, Reid JM, Ames MM, et al. Pediatric phase I trial and pharmacokinetic study of topotecan administered as a 24-hour continuous infusion. Cancer Res. 1993;53:1032–6. [PubMed] [Google Scholar]
- 33.Shah DK, Balthasar JP. Physiologically based pharmacokinetic model for topotecan in mice. J Pharmacokinet Pharmacodyn. 2011;38:121–42. doi: 10.1007/s10928-010-9181-1. [DOI] [PubMed] [Google Scholar]
- 34.Sparreboom A, Loos WJ, Burger H, Sissung TM, Verweij J, Figg WD, et al. Effect of ABCG2 genotype on the oral bioavailability of topotecan. Cancer Biol Ther. 2005;4:650–8. doi: 10.4161/cbt.4.6.1731. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi N, Miura M, Scott SA, Kagaya H, Kameoka Y, Tagawa H, et al. Influence of CYP3A5 and drug transporter polymorphisms on imatinib trough concentration and clinical response among patients with chronic phase chronic myeloid leukemia. J Hum Genet. 2010;55:731–7. doi: 10.1038/jhg.2010.98. [DOI] [PubMed] [Google Scholar]
- 36.Sung C, Blaney SM, Cole DE, Balis FM, Dedrick RL. A pharmacokinetic model of topotecan clearance from plasma and cerebrospinal fluid. Cancer Res. 1994;54:5118–22. [PubMed] [Google Scholar]
- 37.Herben VM, Schoemaker n E, Rosing H, van Zomeren DM, ten Bokkel Huinink WW, Dubbelman R, et al. Urinary and fecal excretion of topotecan in patients with malignant solid tumours. Cancer Chemother Pharmacol. 2002;50:59–64. doi: 10.1007/s00280-002-0454-2. [DOI] [PubMed] [Google Scholar]
- 38.Rosing H, van Zomeren DM, Doyle E, Bult A, Beijnen JH. O-glucuronidation, a newly identified metabolic pathway for topotecan and N-desmethyl topotecan. Anticancer Drugs. 1998;9:587–92. doi: 10.1097/00001813-199808000-00002. [DOI] [PubMed] [Google Scholar]
- 39.Matsumoto S, Yoshida K, Ishiguro N, Maeda T, Tamai I. Involvement of rat and human organic anion transporter 3 in the renal tubular secretion of topotecan [(S)-9-dimethylaminomethyl-10-hydroxy-camptothecin hydrochloride] J Pharmacol Exp Ther. 2007;322:1246–52. doi: 10.1124/jpet.107.123323. [DOI] [PubMed] [Google Scholar]
- 40.Chen C, Slitt AL, Dieter MZ, Tanaka Y, Scheffer GL, Klaassen CD. Up-regulation of Mrp4 expression in kidney of Mrp2-deficient TR- rats. Biochem Pharmacol. 2005;70:1088–95. doi: 10.1016/j.bcp.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 41.Gerrits CJ, Schellens JH, Burris H, Eckardt JR, Planting AS, van der Burg ME, et al. A comparison of clinical pharmacodynamics of different administration schedules of oral topotecan (Hycamtin) Clin Cancer Res. 1999;5:69–75. [PubMed] [Google Scholar]
- 42.Cusatis G, Gregorc V, Li J, Spreafico A, Ingersoll RG, Verweij J, et al. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst. 2006;98:1739–42. doi: 10.1093/jnci/djj469. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.