

Abstract
Propionic acidemia (PA) is a recessive genetic disease that results in an inability to metabolize certain amino acids and odd-chain fatty acids. Current treatment involves restricting consumption of these substrates or liver transplantation. Deletion of the Pcca gene in mice mimics the most severe forms of the human disease. Pcca− mice die within 36 hours of birth, making it difficult to test intravenous systemic therapies in them. We generated an adult hypomorphic model of PA in Pcca− mice using a transgene bearing an A138T mutant of the human PCCA protein. Pcca−/−(A138T) mice have 2% of wild-type PCC activity, survive to adulthood, and have elevations in propionyl-carnitine, methylcitrate, glycine, alanine, lysine, ammonia, and markers associated with cardiomyopathy similar to those in patients with PA. This adult model allowed gene therapy testing by intravenous injection with adenovirus serotype 5 (Ad5) and adeno-associated virus 2/8 (AAV8) vectors. Ad5-mediated more rapid increases in PCCA protein and propionyl-CoA carboxylase (PCC) activity in the liver than AAV8 and both vectors reduced propionylcarnitine and methylcitrate levels. Phenotypic correction was transient with first generation Ad whereas AAV8-mediated long-lasting effects. These data suggest that this PA model may be a useful platform for optimizing systemic intravenous therapies for PA.
Introduction
Propionic acidemia (PA) is an inborn error of metabolism affecting ~1 in 100,000 live births in the United States and up to 1 in 1,000 births in high-risk populations.1 PA results from defects in the mitochondrial enzyme propionyl-CoA carboxylase (PCC). The α and β subunits of the PCC enzyme are encoded by the nuclear PCCA and PCCB genes, respectively.2,3,4 Missense, nonsense, or splicing mutations can occur in either gene.5,6,7,8 Approximately 49% of all observed mutations are missense mutations that can produce hypomorphic proteins with reduced, but not completely ablated enzyme activity (http://cbs.lf1.cuni.cz/pcc/pccmain.htm).9 Loss of PCC function leads to an inability to metabolize odd-chain fatty acids and the amino acids valine, isoleucine, methionine, and threonine (reviewed in ref. 10). These metabolic effects produce elevated levels of propionylcarnitine (C3), methyl citrate (MeCit), and glycine in the blood, allowing PA to be detected during newborn screening.11,12,13,14
Clinical presentation of PA usually occurs within the first week of life and includes poor feeding, vomiting, lethargy, and metabolic acidosis.15,16 Because only certain amino acids and odd-chain fatty acids are substrates for PCC, standard treatment focuses on restricting consumption of protein.17 Patients under active protein restriction may still undergo metabolic crises, explained in part by difficulty adhering to these life-long dietary interventions.17 Even when careful dietary control is maintained, patients can still undergo metabolic decompensation, particularly in response to infection, trauma, or stress.18 Recurrence of these metabolic crises can cause cognitive damage and sometimes death.19 Grunert et al. showed a significant correlation between reduced intelligence quotient and the number of metabolic crises experienced by patients.20 Although the frequency of metabolic decompensation generally decreases with age, long-term survival is still reduced to 67%.19 In addition, PA can lead to cardiac abnormalities including cardiomyopathy and arrhythmia.21,22,23,24,25
There is no cure for PA. Elective liver transplantation is being increasingly used to reduce metabolic crises and temper some of the most severe symptoms of the disease, including hyperammonemia.26,27 Although liver transplantation has positive effects, it is invasive, has high morbidity, requires long-term immunosuppression, and rejection of the transplant is possible. Because of these drawbacks, it is important to develop alternative and safer therapies for PA.
A mouse model of PA was generated previously by disrupting the murine Pcca gene.28 These Pcca−/− mice lack Pcca protein, have drastic increases in C3 and MeCit and die within 36 hours of birth.28 Proof of principle for liver transplantation and liver gene therapy in this model was demonstrated by transgenesis with a liver-specific PCCA transgene. Pcca−/− mice with this covering transgene maintained 50% of wild-type enzyme activity and survived for many months. In contrast, mice with <20% of wild-type activity died within 3 weeks.
Pcca−/− mice have been used to test the feasibility of gene therapy for PA. In a first study, treatment of newborn mice with helper-dependent adenovirus serotype 5 (HD-Ad5) or adeno-associated virus 2/8 (AAV8) vectors expressing the human PCCA cDNA were able to statistically extend lifespan; however, the increases were modest.29 Subsequently, AAV8-hPCCA treatment of newborn Pcca−/− mice extended the lifespan of 50% of animals up to 150 days.30
Although Pcca−/− mice have provided proof of principle for gene therapy of PA, death of Pcca null animals within 36 hours makes treatment of this model challenging. In particular, the need to inject newborn mice within hours of birth makes intravenous injection strategies difficult to test. Given the need to test systemic therapies for application to humans, in this work, we have developed a hypomorphic model of PA in mice that allows mice to survive to adulthood. These mice express a mutant human PCCA protein that partially replaces deletion in the original Pcca- mice. We show that this model accurately recapitulates many aspects of PA in humans, particularly in the 50% of patients with PA who have hypomorphic enzyme activity. We also show that this adult model now allows testing of gene therapy approaches by intravenous administration.
Results
A hypomporphic mouse model of PA
To avoid the difficulties in testing systemic gene therapy in newborn Pcca−/− mice used in previous studies, a hypomorphic model of PA was engineered using a cDNA for human PCCA harboring a nucleotide mutation that results in an A138T mutation. This mutation was previously identified in PA patients with mild to moderate symptoms.6,7,31 This mutation in exon 5 results in reduced PCCA protein levels in patient cells. Transfection of PCCA-deficient human fibroblasts with A138T cDNA produces a protein with reduced mitochondrial stability and only 9.4% of wild-type PCC activity.9
The A138T human cDNA was generated and cloned into a construct under control of a ubiquitously active CAG promoter followed by an internal ribosome entry site–enhanced green fluorescent protein (IRES-EGFP) to aid in phenotyping (Figure 1). This cassette was microinjected into mouse embryos for random insertion into the genome. Six A138T founder mice were identified by PCR and three of them had detectable GFP fluorescence. A138T founders were crossed to Pcca+/− mice to identify Pcca−/− mice with covering transgenes. Pcca−/−(A138T) mice were rescued and ultimately bred from one GFP-positive founder and used for subsequent studies.
Figure 1.

Strategy for generating PCCA hypomorph mice. Depiction of the insertion cassette microinjected into FVB mouse fertilized eggs (top). Two separate hypomorph constructs were injected, one with an A138T mutation and another with an A75P mutation. IRES-GFP DNA sequence was included to aid in genotyping founder mice. (bottom) Breeding strategies of Pcca−/− and Pcca−/−(A138T) mice. The A138T transcript increases survival well beyond the 36 hours observed in full knockout mice. CAG, cytomegalovirus early enhancer coupled with chicken β-actin promoter; EGFP, enhanced green fluorescent protein; IRES, internal ribosome entry site.
Copy number analysis of Pcca−/−(A138T) mice by Taqman assay demonstrated a single copy of transgene, which was transmitted in a Mendelian pattern of inheritance (data not shown). Pcca+/−(A138T) mice were phenotypically normal. Crossing Pcca+/−(A138T) to Pcca+/−(A138T) to produce Pcca−/−(A138T) mice was feasible, but produced only 2 to 3 Pcca−/−(A138T) pups per litter. Pcca−/−(A138T) mice produced from Pcca+/−(A138T) dams were generally indistinguishable from wild-type littermates until after weaning at which point their phenotype became more evident as they exhibited significantly delayed growth (Figure 2a). Pcca−/−(A138T) mice with two copies of the A138T transgene could also be bred to each other, generating litters with 5 to 9 Pcca−/−(A138T) pups. Unlike the phenotypically normal Pcca−/−(A138T) pups produced from Pcca+/−(A138T) dams, mice born to Pcca−/−(A138T) mothers were drastically smaller and experienced significantly delayed growth throughout their neonatal development (Figure 2b,c). These data suggested that low PCC activity in the dams affected the growth and development of their mutant pups. As these mice survived and were produced in larger numbers per litter, subsequent studies were performed with mice from Pcca−/−(A138T) parents. These Pcca−/−(A138T) mice had marginally decreased survival over 3 months after birth (Figure 2d). Although survival was reduced, it was still markedly longer than the 36-hour survival of the Pcca−/− mice.29
Figure 2.

Growth of PCCA−/−(A138T) hypomorph mice. (a) Growth curve of Pcca−/−(A138T) mice versus wild-type animals with intact endogenous mouse Pcca (n ≥ 7 for all data points). (b) Growth curve of pups born from indicated dams. Pups from litters with ≥5 pups were analyzed (n = 14). (c) Photo of Pcca−/−(A138T) mice birthed by a Pcca+/− dam (left) alongside a mouse birthed by a Pcca−/−(A138T) dam (right), picture was taken when both mice were weaned at 3 weeks of age. (d) Kaplan–Meier survival curve depicting death rate of Pcca−/−(A138T) mice (n = 73) versus wild-type (n = 30). Error bars in the line graph depict SEM. ****P < 0.0001; **P < 0.01.
Liver PCC activity in Pcca−/−(A138T) mice
The A138T human cDNA produced 9.4% activity in transfected fibroblasts.9 To test PCC activity in Pcca−/−(A138T) mice, livers from wild-type and Pcca−/−(A138T) mice were analyzed for PCC activity by incorporation of labeled CO2 (Figure 3a). This assay demonstrated that Pcca−/−(A138T) mouse livers had 2.2% of wild-type PCC activity (P < 0.0001 by two-tailed t-test).
Figure 3.

Biomarker concentrations in PCCA hypomorphs. (a) Determination of PCC enzyme activity in liver of Pcca−/−(A138T) mice (n = 10) as compared with wild-type mice (n = 3). (b) Ratio of propionylcarnitine (C3) to acetylcarnitine (C2) in dried blood spots. Pcca−/−(A138T) mice exhibit ~18-fold higher C3/C2 than wild-type animals (n ≥ 12 for all data points). (c) Levels of methyl citrate (MeCit) in the same samples (n ≥ 12 for all data points). (d) Comparison of indicated amino acid levels in plasma of wild-type or Pcca−/−(A138T) mice. Error bars in line and bar graphs depict SEM. ****P < 0.0001; **P < 0.01.
Elevated PA-associated biomarkers in Pcca−/−(A138T) mice
Elevated propionylcarnitine (C3) and MeCit are observed in patients with PA during newborn screening. Blood spots were collected on filter paper from the animals in a manner similar to the method used for newborn screening to assess these biomarkers. C3 levels were normalized to acetylcarnitine (C2) to control for variations in sample recovery from blotted blood. Measurements of Pcca−/−(A138T) mice beginning at 4 weeks of age showed significantly increased levels of C3/C2 and MeCit (Figure 3b,c). On average, there was a 18-fold increase in both C3/C2 ratios and in MeCit levels when compared with wild-type mice (P < 0.0001 for both by one-way analysis of variance (ANOVA)). Analysis of amino acid levels also demonstrated significant increases in glycine, alanine, and lysine in the Pcca−/−(A138T) mice (Figure 3d).
Hyperammonemia and cardiac perturbations in Pcca−/−(A138T) mice
Hyperammonemia and cardiomyopathy are significant life-threatening symptoms in patients with PA. Ammonia levels in humans are below 35 µmol/l in humans32,33 and are similar in wild-type mice (Figure 4a). In patients with PA, ammonia levels are usually elevated from 50 to 150 µmol/l with some being >400 µmol/l.32 Significant elevations in plasma ammonia levels similar to those in humans were observed in 8-month-old Pcca−/−(A138T) mice when compared with age-matched wild-type animals (Figure 4a). Hearts from these 8-month-old mice were also analyzed and mass measurements revealed that those from Pcca−/−(A138T) mice were significantly larger than those in wild-type mice (Figure 4b). Elevations in brain natriuretic peptide (BNP) has been proposed as a potential marker for cardiomyopathy and cardiac dysfunction.34 When BNP mRNA levels in the hearts of these animals were compared, message levels were significantly higher in the Pcca−/−(A138T) mice (Figure 4c).
Figure 4.
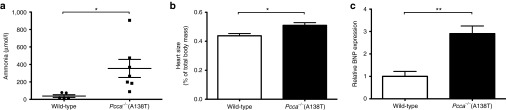
Phenotypic markers. At 8 months of age, mice were killed and their body mass was recorded. (a) Blood was drawn to determine concentration of ammonia in plasma samples. (b) Hearts were removed from the animals and their mass recorded and was normalized to total body mass. (c) Level of BNP mRNA obtained from mouse heart tissue was also determined. **P < 0.01; *P < 0.05.
Gene therapy in adult Pcca−/−(A138T) mice
To test gene therapy in the new mouse model, a codon-optimized human PCCA cDNA (hPCCAco) was inserted into a first generation Ad5 (FG-Ad5) vector and into a single-stranded AAV2/8 vector. Adult Pcca−/−(A138T) mice of 10 weeks old were injected with optimal doses of either vector: 5 × 1011 vg of AAV2/8 CMV-hPCCAco or 5 × 1010 vp of Ad5 CMV-hPCCAco by tail vein and C3/C2 ratios and MeCit levels were assayed after 1 week (Figure 5). Treatment with either vector drove C3/C2 and MeCit below the levels observed in untreated mice, but the Ad5 vector mediated more significant reductions 1 week after injection (Figure 5, P < 0.0001 by one-way ANOVA). When metabolite levels were tracked over several weeks, phenotypic correction by the two vectors was quite different (Figure 6a,b). FG-Ad5 treated mice had a rapid reduction in C3/C2 ratios within 1 week of treatment, but this effect was transient and rebounded within 8 weeks (Figure 6a). In contrast, AAV8 induced gradual reductions in C3/C2 ratios over 8 weeks and these remained low throughout the experiment. MeCit levels generally mimicked changes in C3/C2 ratios, but with somewhat different kinetics (Figure 6b).
Figure 5.

Biomarker response to treatment. (a) C3/C2 ratio assayed 1 week after administration of 5 × 1011 vg AAV2/8-hPCCAco at either 5 or 10 weeks of age along with 5 × 1010 vp Ad-hPCCAco at either 5 or 10 weeks of age. (b) The same samples were analyzed for MeCit levels. ****P < 0.0001; **P < 0.01; *P < 0.05; and asterisks also represent statistical significance compared with untreated Pcca-/- A138T mice. Error bars in graphs depict SEM.
Figure 6.

Therapeutic response to viral vector treatment. (a) C3/C2 ratio assayed at time points up to 13 weeks after administration of control AAV2/8-GFPLuc at 5 weeks old (n = 5), AAV2/8-hPCCAco (n = 10 at 5 weeks old, n = 9 at 10 weeks old) or Ad-hPCCAco (n = 5 at 5 weeks old, n = 7 at 10 weeks old) as in Figure 4. (b) The same samples were analyzed for MeCit levels. (c) Body mass response 1 week after vector administration. Mass was recorded at 5 and 6 weeks of age for wild-type and Pcca−/−(A138T) mice born from a Pcca−/−(A138T) dam. At the time of the 5-week measurement, Pcca−/−(A138T) mice were injected with control AAV2/8-GFPLuc (n = 5), Ad5-GFPLuc (n = 5), Ad5-hPCCAco (n = 5), or AAV2/8-hPCCAco (n = 10). Five-week mass measurements are represented in the left column and 6-week measurements are represented in the right column for each treatment data set. Error bars in line and bar graphs depict SEM. NS, not significance.***P < 0.001.
Gene therapy in young Pcca−/−(A138T) mice
FG-Ad5 and AAV8 treatment of adult mice (age 10 weeks) had detectable therapeutic effects. Given that FG-Ad5 effects were short-lived presumably due to anti-Ad immune responses, both vectors were tested again in younger 5-week-old mice (Figures 5 and 6). AAV8 and FG-Ad5 treatment of 5-week-old mice drastically reduced C3/C2 ratios and MeCit levels within one weak of treatment when compared with untreated Pcca−/−(A138T) mice (P < 0.0001 for both vectors for both metabolite levels by one-way ANOVA). These metabolite levels were not significantly different than wild-type animals by one-way ANOVA. C3/C2 ratios declined over time in mice treated with AAV8 at 5 weeks, but increased over time when mice were treated with Ad at 5 weeks. MeCit levels remained low in both groups at least through 8 weeks after treatment. These results indicate that earlier administration of both AAV and FG-Ad vector greatly increased their efficacy in this mouse model. Ad and AAV treatment also significantly increased body mass in the young animals (Figure 6c). Mice injected with Ad-hPCCAco and AAV2/8-hPCCAco gained 34% and 15% body mass respectively within just the first week as compared with a 7% increase observed in mice injected with control AAV2/8-GFPLuc (AAV8-GL) and a 6% increase in mice injected with control Ad.
Effects of gene therapy on PCCA protein and activity levels
To determine whether the two gene therapy vectors produced different protein levels, we assayed liver PCCA protein levels in SDS-PAGE gels using Neutravidin and PCCA antibody. PCCA is one of four proteins in mammalian cells that are covalently biotinylated by holocarboxylase synthetase.35 Neutravidin can detect all four. Therefore, whereas PCCA antibody is specific for our protein of interest, Neutravidin not only detects PCCA, but also detects three other proteins that serve as an internal loading control.
Using this system, we probed PCCA protein levels in the livers of wild-type mice and Pcca−/−(A138T) mice by western blot (Figure 7a). PCCA band intensities from the livers of wild-type mice were visually stronger than that from Pcca−/−(A138T) livers. The signal observed in the Pcca−/−(A138T) lane may be from residual mouse Pcca, human PCCA(A138T), or overlapping signal with methylcrotonoyl CoA carboxylase when probed with Neutravidin. When blots were probed with PCCA antibody that recognizes both human and murine PCCA, no PCCA protein band was detected in untreated Pcca−/−(A138T) mice (Figure 7a) consistent with the reduced protein stability of the A138T protein.9
Figure 7.

Endogenous and treatment protein levels. Pcca−/−(A138T) mice were treated with 5 × 1011 vg AAV2/8-hPCCAco or 5 × 1010 vp Ad-hPCCAco at 5 weeks of age. Livers were removed either 3 or 10 days after vector administration. (a) 25 μg of total liver protein was run on SDS-PAGE gels and blotted onto PVDF membrane. Blots were probed with either Neutravidin or anti-PCCA antibody. The upper band is consistent with pyruvate carboxylase and served as a loading control. (b) Pieces of the same livers were analyzed for PCC enzyme activity. ****P < 0.0001.
PCCA protein levels increased drastically in both AAV and Ad-treated mice as assessed by both Neutravidin and anti-PCCA antibody, reaching levels much higher than that seen in wild-type mice (Figure 7a).
When PCC enzymatic activity was measured from the livers of the same animals, activity trended in a similar manner to that for protein levels (Figure 7a,b). Ad5-hPCCAco–induced PCC activity reached maximal levels within 3 days of treatment whereas AAV8-hPCCAco–induced levels were higher at 10 days post-treatment. However, although the vectors produced PCCA protein levels higher than were observed wild-type mice (Figure 7a), PCC enzyme activity reached only 30% of wild-type levels (Figure 7b).
Discussion
This work was motivated by a need for an adult small animal model of PA where systemic therapies could readily be tested by intravenous injection. Full knockout of mouse Pcca generated a model wherein mice die within 36 hours of birth28 making this a challenging system with which to test intravenous delivery methods and vectors.
In the original mouse model study by Miyazaki's group, Pcca knockout was rescued by transgenesis with the mouse Pccac DNA that was expressed in a largely liver-specific fashion using the serum amyloid protein (SAP) promoter. This transgene's ability to rescue Pcca deficiency provided excellent proof of principle for liver-directed gene therapy or stem cell therapy,28 but did not recapitulate human disease where PCC deficiency is cell autonomous and affects every cell in the body.
To recapitulate systemic disease and generate a less stringent mouse model, we introduced human PCCA cDNA containing a well-described hypomorphic A138T mutation under control of the promiscuous CAG promoter. Under these conditions, we were able to obtain A138T transgenic mice on the Pcca−/− background. In these mice, the A138T human transgene partially rescued the mouse Pcca deficiency resulting in 2.2% of wild-type liver PCC activity in the mice. This enzymatic activity is interestingly similar to the 4% activity that has been directly observed in fibroblasts of a human patient homozygous for the A138T mutation.9,31
Numerous aspects of the Pcca−/−(A138T) mice were probed to determine how closely their phenotype mirrors that of human patients. The 2.2% level of PCC activity translated to 18-fold increases in C3/C2 ratios and MeCit levels that mimicked levels observed in humans with PA. Unlike Pcca−/− mice that die within 36 hours, Pcca−/−(A138T) mice survive through the neonatal period with >75% survival beyond 90 days. Pcca−/−(A138T) mice were sufficiently healthy to allow breeding as homozygote mutants. However, PCC deficiency in the Pcca−/−(A138T) dams clearly affected the health of their progeny as demonstrated by the markedly reduced size of their pups when compared with genetically identical pups that were produced from Pcca+/−(A138T) mothers.
Analysis of plasma amino acid levels indicated that the Pcca−/−(A138T) animals exhibit elevations in glycine similar to that seen in humans.36 In addition, increases in alanine are a consistent correlate of lactic acidosis, which is often observed in patient samples.10,37 Elevations in lysine are also frequently observed in patients with PA and positively correlate with hyperammonemia.10,32,38 We observed hyperammonemia in the mice similar to the levels observed in patients with PA.32,33 Hyperammonemia likely causes some degree of neurological perturbation in addition to contributing to premature death in some of the animals. The relative increases in heart mass and in BNP mRNA in the hearts of Pcca−/−(A138T) mice also suggest some degree of cardiac involvement. Although these data are suggestive, additional studies will be needed to determine how well this mouse model recaptulates cardiac involvement observed in patients with PA.
These data indicate that the A138T model mimics many of the phenotypes observed in patients with PA. To investigate usefulness of this model for testing systemic therapies, Ad and AAV vectors were injected intravenously into 5- and 10-week-old Pcca−/−(A138T) mice. Adenoviral vectors expressing codon-optimized human PCCA produced significant increases in growth of animals and partial correction of C3/C2 and MeCit levels. However, these effects were transient and were only observed for 4 weeks after vector administration. These results were not surprising, because first generation adenoviral vectors are known to be attenuated by immune-mediated clearance of Ad-transduced cells.39 Future work will test if helper-dependent Ad vectors lacking all Ad genes mediate a more persistent phenotypic correction in this model.
AAV8 vectors expressing codon-optimized human PCCA produced more persistent and robust correction of disease marker levels. In contrast to the Ad vector, mice treated with AAV8 underwent a rapid drop in C3/C2 and MeCit that was maintained for the duration of the 13-week study. Of particular interest is the differential effect of both vectors when they were administered to mice that were 5 weeks of age rather than 10 weeks. Animals of 5 weeks old receiving either vector had markedly stronger effects on metabolite levels as exhibited by lower C3/C2 and MeCit levels. This could be due to modification of a higher fraction of the liver by the vectors in the slightly smaller mice or be due to reduced therapeutic effects in older animals that have suffered longer from metabolic damage.
Comparison of PCCA protein levels and PCC enzyme activity levels also revealed an interesting discordance in gene therapy efficacy. Although both vectors produced PCCA protein levels that actually exceeded normal levels, this high level expression did not translate into PCC enzyme activities at or above wild-type activities. This discrepancy could be due to two intrinsic features of the PCC enzyme. First, PCC consists of a dodecamer of two protein subunits: PCCA as well as PCCB. Therefore, the failure to reconstitute 100% PCC activity may be due to imbalanced PCCA and PCCB protein expression. If so, better results may occur if both cDNAs are delivered at the same time. Dual cDNA delivery would also be appealing given that ~50% of patients with PA have mutations in PCCB rather than PCCA. An alternate explanation for the discrepancy could be due to the fact that PCC is a mitochondrial enzyme. Therefore, both PCCA and PCCB need to be coordinately targeted to the mitochondria for activity. It is therefore possible that imbalanced expression might disrupt mitochondrial targeting. Finally, our use of human PCCA protein may not optimally combine with endogenous mouse PCCB protein and limit full PCC enzymatic activity.
Previous testing in the Pcca−/− mice indicated that the liver-tropic AAV8 vector has utility for treating PA.29,30 Our study confirms this observation, and reinforces the promise of AAV8 or other liver-tropic vectors as possible treatments for PA. Currently, the most promising form of treatment is liver transplantation. Liver transplantation relieves most of the symptoms experienced by patients with PA and aids in normal development. However, liver transplantation itself poses significant risks to the patient and requires life-long administration of immunosuppressant drugs, which can be associated with an array of side effects themselves. Gene therapy with AAV or other gene therapy vectors may potentially be equally efficacious, but may have better safety margins, because they express one or a few genes rather than a whole organ of non-self antigens.
Materials and Methods
Generation of hPCCA hypomorph mice. Segments of human PCCA cDNA with mutations leading to A75P or A138T defects were synthesized by GenScript USA (Piscataway, NJ). These were used to replace wild-type Pcca in plasmid pShuttleCMV-FL-hPCCA-IRES-hrGFP. These mutant PCCA cDNAs were transferred to pCALL2-Δ-LoxP to generate plasmids pCALL2-Δ-LoxP-hPCCA-A75P and pCALL2-Δ-LoxP-hPCCA-A138T in which hPCCA is followed by an IRES-EGFP element to allow screening for transgenics (Figure 1). The pCALL2-Δ-LoxP plasmids were digested with BamHI and BsaWI and this transgene fragment was microinjected into the fertilized eggs of FVB mice by the Mayo Clinic Transgenic and Gene Targeted Mouse Shared Resource (TGTMSR). Founder mice were screened for GFP expression and by PCR using primers specific for the transgene cassette (F: CGGATTACGCGTAGCATGGTGAGCAA R: GCCTAAACGCGTTTACTTGTACAGCT). Positive mice were then crossed to Pcca+/− mice. All resulting progeny were screened using primers specific for the endogenous mPCCA gene, neomycin resistance gene (neo) and the transgene cassette described previously by Hofherr et al.29
Animals. All mice were housed at Mayo Clinic under the Association for Assessment and Accreditation of Laboratory Animal Care (AALAC) guidelines. The Mayo Clinic Animal Care and Use Committee approved all animal use protocols. All animal experiments were carried out according to the provisions of the PHS Animal Welfare Policy, Animal Welfare Act, the principles of the NIH Guide for the Care and Use of Laboratory Animals, and the policies and procedures of Mayo Clinic. Mice were fed standard PicoLab 5053 rodent chow (LabDiet, Brentwood, MO) with no substitutions throughout all experimental time points.
PCC enzyme activity assay. The mouse livers were each weighed and a volume of lysate buffer (50 mmol/l Tris pH 8.0, 1 mmol/l glutathione, 1 mmol/l EDTA, protease inhibitor cocktail) equal to three times the weight was added. The mouse tissues were homogenized using a handheld glass tissue homogenizer. The homogenized lysate was then spun at 15,000 rpm at 4 °C for 30 minutes. The supernatant was then used for the PCC radiometric activity assay performed as previously described.40 Protein concentration was determined by the Lowry method. The PCC assay used 75 µg of liver lysate in a total of 50 µl for 15 minutes at 37 °C.
Metabolite analysis in blood. Blood was obtained via submandibular puncture with a lancet and was spotted on Whatman 903 Protein Saver blood collection cards (GE Healthcare, Westborough, MA). Acylcarnitines and methylcitric acid (MeCit) were analyzed in the dried blood spots by tandem mass spectrometry following protocols established for newborn screening.11,14 Analysis of amino acid concentrations were also performed on plasma samples using tandem mass spectrometry.41
Plasma ammonia assay. Blood was obtained from anesthetized mice via cardiac puncture. Plasma was separated by spinning for 10 minutes at 1,400g and snap frozen in liquid nitrogen. Ammonia analysis was performed using the Sigma Ammonia Assay Kit (St Louis, MO).
Cardiac BNP assay. Assay for BNP was performed as described by Peche et al.42 SYBR Green Master Mix (Life Technologies, Grand Island, NY) was used with the previously published BNP primers and GAPDH reference primers (GAPDH Forward: AGCTGAACGGGAAGCTCACTGAPDH Reverse:GCTTCACCACCTTCTTGATGTC).
Adenovirus vector production. The human PCCA cDNA was codon optimized for mammalian expression and synthesized by GenScript USA. This hPCCAco cDNA was cloned into the pShuttle-CMV plasmid (Q-Biogene) and was recombined with pAd-Easy, then virus was rescued in 293A cells as described in Mok et al.43 All adenoviruses were purified using two sequential ultracentrifugation CsCl density gradients and quantitated by OD260.
AAV vector production. Codon-optimized human PCCA DNA (hPCCAco) was synthesized by GenScript USA and cloned into a CMV expression cassette (Figure 1) containing AAV2 inverted terminal repeats and packaged with an AAV2/8 packaging system. AAV particles were produced by a procedure similar to that of Lock et al. with some modifications.44 Briefly, polyethylenimine-based transfection of HEK 293 cells was performed in Corning (Corning, NY) 10 chamber CellSTACKs. Plasmids were transfected at a molar ratio of 1:1:1 (adenoviral helper plasmid: cis AAV Rep 2/Cap 8 plasmid: trans hPCCAco plasmid) and a polyethylenimine/DNA ratio of 3:1 (w/w) was utilized for transfection. The cell portion was subjected to three freeze-thaw cycles to form a lysate fraction. The media portion was clarified through 0.45 µm Nalgene (Thermo Fischer Scientific, Waltham, MA) bottle-top filters and then concentrated by tangential flow filtration through a disposable 0.01 m2 Sius-LS HyStream Hydrophilic membrane with a 100 kDa Molecular Weight Cutoff (TangenX Technology, Shrewsbury, MA) housed in a NovaSet-LS LHV holder (TangenX Technology) to a volume of ~50 ml. At this point, both the cell lysate and media fractions received DNAse and were incubated for 30 minutes at 37 °C. Discontinuous iodixanol gradients were formed as stated in Lock et al. and the tubes were sealed and centrifuged at 350,000g in a Beckman type 70.1 Ti rotor for 120 minutes. Cell and media fractions were pooled separately and concentrated by centrifugation using Amicon 100 kDa molecular weight cutoff filter units (EMD Millipore, Billerica, MA).
Vector titer. Real time PCR was performed using Sybr Green (Life Technologies) with primers specific for the CMV promoter region (Forward: CAAGTGTATCATATGCCAAGTACGCCCC, Reverse: CCCCGTGAGTCAAACCGCTATCCACGCC).
Vector administration. AAV vectors were diluted in phosphate-buffered saline to yield a final concentration injectable in a volume up to 100 µl. Viral genomes (vg) of 5 × 1011 per mouse of AAV8-hPCCAco were injected intravenously via the tail vein. Adenoviral vector was diluted in the same manner to yield a concentration injectable in a volume of 100 µl. Viral particles of 5 × 1010 were injected per mouse intravenously via the tail vein.
PCCA protein analysis. Protein blots were obtained by homogenizing sections of mouse liver in T-Per Reagent (Fischer Scientific, Rockford, IL). Total protein of 25 μg was loaded onto an SDS-PAGE gel from each sample. The subsequent blot was probed with Neutravidin-HRP, and imaged for luminescence. Total protein of 50 μg was loaded onto an SDS-PAGE gel from each sample for Western analysis using anti-PCCA antibody (ProteinTech, Chicago, IL). Quantitation of bands was performed using Kodak MI Software.
Data analysis. Graphing and statistical analysis was performed using GraphPad Prism software. Statistical significance was determined using the Student's two-tailed t-test for the bar graph in Figure 3, one-way ANOVA with Bonferroni post test for the line graphs and scatter plot in Figures 2–5, or log-rank tests for the Kaplan–Meier curve in Figure 2. Data are expressed as mean value ± SEM in bar, line, and scatter graphs in Figures 2–5.
Acknowledgments
This work was supported by grants to M.A.B. and J.P.K. from the Propionic Acidemia Foundation (PAF). It was also supported by a grant from the Department of Laboratory Medicine and Pathology (DLMP) at Mayo Clinic and the Liver Regeneration Program in the Center for Regeneration at Mayo Clinic. M.L.H. was supported by the Kidney Disease Research Training Program T32-DK007013. The authors declare no conflict of interest.
References
- Ravn K, Chloupkova M, Christensen E, Brandt NJ, Simonsen H, Kraus JP, et al. High incidence of propionic acidemia in greenland is due to a prevalent mutation, 1540insCCC, in the gene for the beta-subunit of propionyl CoA carboxylase. Am J Hum Genet. 2000;67:203–206. doi: 10.1086/302971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel RA, Lam KF, Mahuran D, Kronis A. Purification of human liver propionyl-CoA carboxylase by carbon tetrachloride extraction and monomeric avidin affinity chromatography. Arch Biochem Biophys. 1980;201:669–673. doi: 10.1016/0003-9861(80)90557-3. [DOI] [PubMed] [Google Scholar]
- Kalousek F, Darigo MD, Rosenberg LE. Isolation and characterization of propionyl-CoA carboxylase from normal human liver. Evidence for a protomeric tetramer of nonidentical subunits. J Biol Chem. 1980;255:60–65. [PubMed] [Google Scholar]
- Huang CS, Sadre-Bazzaz K, Shen Y, Deng B, Zhou ZH, Tong L. Crystal structure of the alpha(6)beta(6) holoenzyme of propionyl-coenzyme A carboxylase. Nature. 2010;466:1001–1005. doi: 10.1038/nature09302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez B, Desviat LR, Rodríguez-Pombo P, Clavero S, Navarrete R, Perez-Cerdá C, et al. Propionic acidemia: identification of twenty-four novel mutations in Europe and North America. Mol Genet Metab. 2003;78:59–67. doi: 10.1016/s1096-7192(02)00197-x. [DOI] [PubMed] [Google Scholar]
- Ugarte M, Pérez-Cerdá C, Rodríguez-Pombo P, Desviat LR, Pérez B, Richard E, et al. Overview of mutations in the PCCA and PCCB genes causing propionic acidemia. Hum Mutat. 1999;14:275–282. doi: 10.1002/(SICI)1098-1004(199910)14:4<275::AID-HUMU1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Desviat LR, Pérez B, Pérez-Cerdá C, Rodríguez-Pombo P, Clavero S, Ugarte M. Propionic acidemia: mutation update and functional and structural effects of the variant alleles. Mol Genet Metab. 2004;83:28–37. doi: 10.1016/j.ymgme.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Rincón A, Aguado C, Desviat LR, Sánchez-Alcudia R, Ugarte M, Pérez B. Propionic and methylmalonic acidemia: antisense therapeutics for intronic variations causing aberrantly spliced messenger RNA. Am J Hum Genet. 2007;81:1262–1270. doi: 10.1086/522376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavero S, Martínez MA, Pérez B, Pérez-Cerdá C, Ugarte M, Desviat LR. Functional characterization of PCCA mutations causing propionic acidemia. Biochim Biophys Acta. 2002;1588:119–125. doi: 10.1016/s0925-4439(02)00155-2. [DOI] [PubMed] [Google Scholar]
- Lehnert W, Sperl W, Suormala T, Baumgartner ER. Propionic acidaemia: clinical, biochemical and therapeutic aspects. Experience in 30 patients. Eur J Pediatr. 1994;153 7 Suppl 1:S68–S80. doi: 10.1007/BF02138781. [DOI] [PubMed] [Google Scholar]
- Turgeon CT, Magera MJ, Cuthbert CD, Loken PR, Gavrilov DK, Tortorelli S, et al. Determination of total homocysteine, methylmalonic acid, and 2-methylcitric acid in dried blood spots by tandem mass spectrometry. Clin Chem. 2010;56:1686–1695. doi: 10.1373/clinchem.2010.148957. [DOI] [PubMed] [Google Scholar]
- Deodato F, Boenzi S, Santorelli FM, Dionisi-Vici C. Methylmalonic and propionic aciduria. Am J Med Genet C Semin Med Genet. 2006;142C:104–112. doi: 10.1002/ajmg.c.30090. [DOI] [PubMed] [Google Scholar]
- Millington DS, Roe CR, Maltby DA. Application of high resolution fast atom bombardment and constant B/E ratio linked scanning to the identification and analysis of acylcarnitines in metabolic disease. Biomed Mass Spectrom. 1984;11:236–241. doi: 10.1002/bms.1200110508. [DOI] [PubMed] [Google Scholar]
- Turgeon C, Magera MJ, Allard P, Tortorelli S, Gavrilov D, Oglesbee D, et al. Combined newborn screening for succinylacetone, amino acids, and acylcarnitines in dried blood spots. Clin Chem. 2008;54:657–664. doi: 10.1373/clinchem.2007.101949. [DOI] [PubMed] [Google Scholar]
- Pena L, Franks J, Chapman KA, Gropman A, Ah Mew N, Chakrapani A, et al. Natural history of propionic acidemia. Mol Genet Metab. 2012;105:5–9. doi: 10.1016/j.ymgme.2011.09.022. [DOI] [PubMed] [Google Scholar]
- Henriquez H, el Din A, Ozand PT, Subramanyam SB, al Gain SI. Emergency presentations of patients with methylmalonic acidemia, propionic acidemia and branched chain amino acidemia (MSUD). Brain Dev. 1994;16 Suppl:86–93. doi: 10.1016/0387-7604(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Sutton VR, Chapman KA, Gropman AL, MacLeod E, Stagni K, Summar ML, et al. Chronic management and health supervision of individuals with propionic acidemia. Mol Genet Metab. 2012;105:26–33. doi: 10.1016/j.ymgme.2011.08.034. [DOI] [PubMed] [Google Scholar]
- Chapman KA, Gropman A, MacLeod E, Stagni K, Summar ML, Ueda K, et al. Acute management of propionic acidemia. Mol Genet Metab. 2012;105:16–25. doi: 10.1016/j.ymgme.2011.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barshes NR, Vanatta JM, Patel AJ, Carter BA, O'Mahony CA, Karpen SJ, et al. Evaluation and management of patients with propionic acidemia undergoing liver transplantation: a comprehensive review. Pediatr Transplant. 2006;10:773–781. doi: 10.1111/j.1399-3046.2006.00569.x. [DOI] [PubMed] [Google Scholar]
- Grünert SC, Müllerleile S, de Silva L, Barth M, Walter M, Walter K, et al. Propionic acidemia: neonatal versus selective metabolic screening. J Inherit Metab Dis. 2012;35:41–49. doi: 10.1007/s10545-011-9419-0. [DOI] [PubMed] [Google Scholar]
- Pena L, Burton BK. Survey of health status and complications among propionic acidemia patients. Am J Med Genet A. 2012;158A:1641–1646. doi: 10.1002/ajmg.a.35387. [DOI] [PubMed] [Google Scholar]
- Massoud AF, Leonard JV. Cardiomyopathy in propionic acidaemia. Eur J Pediatr. 1993;152:441–445. doi: 10.1007/BF01955907. [DOI] [PubMed] [Google Scholar]
- Bhan AK, Brody C. Propionic acidemia: a rare cause of cardiomyopathy. Congest Heart Fail. 2001;7:218–219. doi: 10.1111/j.1527-5299.2001.01011.x. [DOI] [PubMed] [Google Scholar]
- Baumgartner D, Scholl-Bürgi S, Sass JO, Sperl W, Schweigmann U, Stein JI, et al. Prolonged QTc intervals and decreased left ventricular contractility in patients with propionic acidemia. J Pediatr. 2007;150:192–7, 197.e1. doi: 10.1016/j.jpeds.2006.11.043. [DOI] [PubMed] [Google Scholar]
- Kakavand B, Schroeder VA, Di Sessa TG. Coincidence of long QT syndrome and propionic acidemia. Pediatr Cardiol. 2006;27:160–161. doi: 10.1007/s00246-005-1129-7. [DOI] [PubMed] [Google Scholar]
- Yorifuji T, Muroi J, Uematsu A, Nakahata T, Egawa H, Tanaka K. Living-related liver transplantation for neonatal-onset propionic acidemia. J Pediatr. 2000;137:572–574. doi: 10.1067/mpd.2000.108391. [DOI] [PubMed] [Google Scholar]
- Kayler LK, Merion RM, Lee S, Sung RS, Punch JD, Rudich SM, et al. Long-term survival after liver transplantation in children with metabolic disorders. Pediatr Transplant. 2002;6:295–300. doi: 10.1034/j.1399-3046.2002.02009.x. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Ohura T, Kobayashi M, Shigematsu Y, Yamaguchi S, Suzuki Y, et al. Fatal propionic acidemia in mice lacking propionyl-CoA carboxylase and its rescue by postnatal, liver-specific supplementation via a transgene. J Biol Chem. 2001;276:35995–35999. doi: 10.1074/jbc.M105467200. [DOI] [PubMed] [Google Scholar]
- Hofherr SE, Senac JS, Chen CY, Palmer DJ, Ng P, Barry MA. Short-term rescue of neonatal lethality in a mouse model of propionic acidemia by gene therapy. Hum Gene Ther. 2009;20:169–180. doi: 10.1089/hum.2008.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RJ, Chandrasekaran S, Carrillo-Carrasco N, Senac JS, Hofherr SE, Barry MA, et al. Adeno-associated virus serotype 8 gene transfer rescues a neonatal lethal murine model of propionic acidemia. Hum Gene Ther. 2011;22:477–481. doi: 10.1089/hum.2010.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Cerdá C, Merinero B, Rodríguez-Pombo P, Pérez B, Desviat LR, Muro S, et al. Potential relationship between genotype and clinical outcome in propionic acidaemia patients. Eur J Hum Genet. 2000;8:187–194. doi: 10.1038/sj.ejhg.5200442. [DOI] [PubMed] [Google Scholar]
- Filipowicz HR, Ernst SL, Ashurst CL, Pasquali M, Longo N. Metabolic changes associated with hyperammonemia in patients with propionic acidemia. Mol Genet Metab. 2006;88:123–130. doi: 10.1016/j.ymgme.2005.11.016. [DOI] [PubMed] [Google Scholar]
- Tuchman M, Yudkoff M. Blood levels of ammonia and nitrogen scavenging amino acids in patients with inherited hyperammonemia. Mol Genet Metab. 1999;66:10–15. doi: 10.1006/mgme.1998.2783. [DOI] [PubMed] [Google Scholar]
- Tsai SH, Lin YY, Chu SJ, Hsu CW, Cheng SM. Interpretation and use of natriuretic peptides in non-congestive heart failure settings. Yonsei Med J. 2010;51:151–163. doi: 10.3349/ymj.2010.51.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler CS, Ballard FJ. Regulation of the breakdown rates of biotin-containing proteins in Swiss 3T3-L1 cells. Biochem J. 1988;251:749–755. doi: 10.1042/bj2510749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs B, Nyhan WL, Borden M, Bard L, Cooke RE. Idiopathic hyperglycinemia and hyperglycinuria: a new disorder of amino acid metabolism. I. Pediatrics. 1961;27:522–538. [PubMed] [Google Scholar]
- Smeitink JA. Mitochondrial disorders: clinical presentation and diagnostic dilemmas. J Inherit Metab Dis. 2003;26:199–207. doi: 10.1023/a:1024489218004. [DOI] [PubMed] [Google Scholar]
- Gompertz D, Storrs CN, Bau DC, Peters TJ, Hughes EA. Localisation of enzymic defect in propionicacidaemia. Lancet. 1970;1:1140–1143. doi: 10.1016/s0140-6736(70)91216-x. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ertl HC, Wilson JM. MHC class I-restricted cytotoxic T lymphocytes to viral antigens destroy hepatocytes in mice infected with E1-deleted recombinant adenoviruses. Immunity. 1994;1:433–442. doi: 10.1016/1074-7613(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Jiang H, Rao KS, Yee VC, Kraus JP. Characterization of four variant forms of human propionyl-CoA carboxylase expressed in Escherichia coli. J Biol Chem. 2005;280:27719–27727. doi: 10.1074/jbc.M413281200. [DOI] [PubMed] [Google Scholar]
- Lacey JM, Magera M, Casetta B, Daniels S, Eckerman JS, Rinaldo P, Matern D. Evaluation of an iTRAQ amino acid analysis kit for the rapid quantitation of amino acids in biological fluids. Mol Genet Metab. 2009;98 [Google Scholar]
- Peche VS, Holak TA, Burgute BD, Kosmas K, Kale SP, Wunderlich FT, et al. Ablation of cyclase-associated protein 2 (CAP2) leads to cardiomyopathy. Cell Mol Life Sci. 2013;70:527–543. doi: 10.1007/s00018-012-1142-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok H, Palmer DJ, Ng P, Barry MA. Evaluation of polyethylene glycol modification of first-generation and helper-dependent adenoviral vectors to reduce innate immune responses. Mol Ther. 2005;11:66–79. doi: 10.1016/j.ymthe.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Lock M, Alvira M, Vandenberghe LH, Samanta A, Toelen J, Debyser Z, et al. Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum Gene Ther. 2010;21:1259–1271. doi: 10.1089/hum.2010.055. [DOI] [PMC free article] [PubMed] [Google Scholar]