

Abstract
Rheumatic fever (RF) is an autoimmune disease triggered by Streptococcus pyogenes infection frequently observed in infants from developing countries. Rheumatic heart disease (RHD), the major sequel of RF, leads to chronic inflammation of the myocardium and valvular tissue. T cells are the main population infiltrating cardiac lesions; however, the chemokines that orchestrate their recruitment are not clearly defined. Here, we investigated the expression of chemokines and chemokine receptors in cardiac tissue biopsies obtained from chronic RHD patients. Our results showed that CCL3/MIP1α gene expression was upregulated in myocardium while CCL1/I-309 and CXCL9/Mig were highly expressed in valvular tissue. Auto-reactive T cells that infiltrate valvular lesions presented a memory phenotype (CD4+CD45RO+) and migrate mainly toward CXCL9/Mig gradient. Collectively, our results show that a diverse milieu of chemokines is expressed in myocardium and valvular tissue lesions and emphasize the role of CXCL9/Mig in mediating T cell recruitment to the site of inflammation in the heart.
KEY WORDS: rheumatic fever, rheumatic heart disease, chemokines, CXCL9 (Mig), valvular lesions
INTRODUCTION
Rheumatic fever (RF) is a post-infectious autoimmune disease triggered by the Gram-positive bacteria Streptococcus pyogenes. The disease manifests initially as polyarthritis, carditis/valvulitis, chorea, erythema marginatum, and/or subcutaneous nodules [1]. Arthritis, the earliest feature of the disease, is present in 60–80 % of patients [2]. Rheumatic carditis occurs a few weeks after the streptococcal pharyngeal infection in 30 to 45 % of RF patients and can lead to permanent disability. Recurrent acute cardiac lesions frequently evolve into chronic rheumatic heart disease (RHD), of which valvular deformities are the most important chronic sequel, and lead to mitral and aortic regurgitation and/or stenosis. Valve replacement surgery is usually the only treatment remaining for chronic RHD patients.
Previous reports have described rheumatic valvulitis as a mononuclear cellular infiltrate made up mainly of CD4+ and CD8+ T cells [3–6]. The expression of major histocompatibility complex class II molecules has been observed on vessel endothelium, valvular fibroblasts, and macrophage-like cells within the rheumatic lesions [5–7]. Aschoff nodules/bodies, observed in the cardiac tissue of some RF patients, are considered hallmarks of active rheumatic carditis [8]. Aschoff bodies are granulomatous structures consisting of fibrinoid changes, lymphocytic infiltrate, occasional plasma cells, and macrophages surrounding necrotic centers. Aschoff body development is composed of three distinct phases. The earliest phase is represented by degenerate and fibrinoid changes in the cardiac collagenous tissue. In the intermediate proliferative or granulomatous phase, the main histological features are inflammatory cell proliferation and the presence of large macrophages termed Anitschkow or caterpillar cells. The late or cicatricial phase is represented by fibrosis or healing of the lesions [9].
Although substantial progress has been made in the understanding of RF as an autoimmune disease, the precise pathogenic mechanisms of RF have not been completely defined. Nevertheless, the current knowledge in the field emphasizes T and B cell activation by streptococcal antigens as the main event triggered by the infection.
Lymphocytic infiltration through the valve surface endothelium appears to be the initiating step for tissue damage and disease pathogenesis. A number of evidences show that B cells, through the production of antibodies that cross-recognize streptococcal antigens and cardiac tissue proteins, such as laminin, vimentin, and myosin [10, 11], are responsible for the valvular endothelium upregulation of the adhesion molecule vascular cell adhesion molecule-1. The binding of this molecule to very late antigen-4 on activated T cells is apparently crucial for the extravasation of CD4+ and CD8+ T cells through the valve-activated endothelium [12].
The perpetuation of valvular injury however is carried out by T cells that simultaneously recognize streptococcal antigens and valvular tissue proteins through molecular mimicry [6, 13]. Heart-infiltrating T cells that contribute to the tissue inflammation mainly secrete inflammatory cytokines such as IFN gamma and TNF alpha [14].
T lymphocytes seem to be the main players in the maintenance and perpetuation of valve damage; however, the molecules that orchestrate the recruitment of inflammatory mononuclear cells and specific T cell subsets to the myocardium and subsequently to the valve lesion are not clearly defined.
Chemokines are a group of small (8–14 kDa), structurally related molecules that regulate cell trafficking through interactions with a subset of seven-transmembrane G protein-coupled receptors. They are important mediators of cellular migration, contributing not only to homeostatic migration but also to cellular entry into sites of acute and chronic inflammation. The milieu of chemokines and chemokine receptors expressed by endothelial cells and leukocyte subsets determines leukocyte recruitment for participation in specific inflammatory pathologies.
In the present study, we investigated the expression of chemokines and their respective receptors in myocardium and valvular tissue from chronic RHD patients and evaluated their role in the recruitment of T cells to cardiac lesions.
MATERIALS AND METHODS
Patients
Severe chronic RHD patients who underwent cardiac surgery for valve replacement were included in the study (n = 23). Patients were followed at the Heart Institute, University of São Paulo (SP) and at the Márcio Cunha Hospital, Ipatinga (MG), Brazil. This study was approved by both the Heart Institute Ethics Committee (CAPPesq) from Clinical Hospital, School of Medicine, University of São Paulo and Márcio Cunha Hospital Ethics Committee. Informed consent was assigned from all patients participating in the study or from the parents or legal responsible of the patients under 18 years old.
The studied cohort was composed of 10 males and 13 females with a mean age of 24.31 ± 16.06 years. All patients had morphologic cardiac valve lesions (Table 1) and most of them presented severe mitral and/or aortic valve regurgitation (56.5 %: patients 1 to 13) and severe mitral and aortic valve regurgitation and stenosis (26.1 %; patients 14 to 19). Isolated valve stenosis was found in 17.4 % of the patients (patients 20 to 23). Only few patients were operated during acute RF episode (patients 2, 4, 6, and 22) (Table 1). None of the patients present medical history of other autoimmune disease and were also not under any immunosuppressive drug treatment.
Table 1.
Identification, Clinical, and Histopathological Date of RHD Patients
| Patients (#) | Gender | Age | Clinical date | Histopathology |
|---|---|---|---|---|
| 1 | M | 50 | Severe mitral and aortic valve regurgitation | Chronic valvulitis and fibrosis |
| 2 | F | 13 | Severe mitral and aortic valves regurgitation | Acute valvulitis, Aschoff bodies in aortic valve, and fibrosis |
| 3 | F | 14 | Severe mitral valve regurgitation | Chronic valvulitis and fibrosis |
| 4 | F | 13 | Severe mitral valve regurgitation | Acute valvulitis, Aschoff bodies mitral valve, and fibrosis |
| 5 | F | 8 | Severe mitral valve regurgitation | Not done |
| 6 | M | 10 | Severe mitral, aortic, and tricuspid valves regurgitation | Acute valvulitis, Aschoff bodies in mitral valve, and fibrosis |
| 7 | M | 11 | Severe mitral and aortic valves regurgitation | Acute valvulitis and Aschoff bodies in LA |
| 8 | F | 5 | Severe mitral and moderate aortic valves regurgitation | Chronic valvulitis and Aschoff bodies in LA |
| 9 | F | 18 | Severe mitral valve regurgitation | Chronic valvulitis and fibrosis |
| 10 | M | 13 | Severe mitral and aortic valves regurgitation | Chronic valvulitis and fibrosis |
| 11 | F | 13 | Severe mitral and aortic valves regurgitation | Chronic valvulitis and fibrosis |
| 12 | M | 16 | Severe aortic valve regurgitation | Fibrosis |
| 13 | M | 19 | Severe aortic valve regurgitation | Chronic valvulitis and fibrosis |
| 14 | M | 55 | Severe mitral valve regurgitation and stenosis | Chronic valvulitis, fibrosis, and calcification |
| 15 | F | 25 | Severe mitral valve regurgitation and stenosis | Chronic valvulitis and fibrosis |
| Severe mitral valve stenosis | ||||
| 16 | M | 34 | Severe mitral valve regurgitation and stenosis | Fibrosis |
| 17 | F | 16 | Severe mitral valve regurgitation/moderate stenosis | Chronic valvulitis and fibrosis |
| 18 | M | 30 | Severe aortic valve regurgitation/stenosis; moderate mitral valve regurgitation | Chronic valvulitis and fibrosis |
| 19 | F | 49 | Aortic valve regurgitation/mitral valve stenosis | Chronic valvulitis, fibrosis, and calcification |
| 20 | F | 33 | Severe mitral valve stenosis | Chronic valvulitis and fibrosis |
| 21 | F | 55 | Severe mitral valve stenosis | Fibrosis |
| 22 | M | 15 | Mitral valve stenosis | Not done |
| 23 | F | 44 | Mitral valve stenosis | Fibrosis |
Heart Tissue Samples
Heart tissue samples were collected during cardiac surgery procedures for valve corrections in accordance with the Hospital Ethics Committee.
Histopathology
Histological sections from cardiac tissue samples were stained with hematoxylin–eosin and were reviewed by two independent pathologists to evaluate histological features of acute and chronic RHD, such as inflammatory infiltrates, rheumatic activity, neovascularization, fibrosis, and calcification.
Real-time qPCR
Total RNA was extracted from heart tissue fragments using Trizol® (Gibco, Life Technologies, Invitrogen Corporation, Carlsbad, CA) according to the manufacturer’s protocol. Tissue disruption and homogenization was performed using a rotor-stator homogenizer device (Power Gen1000, Fischer Scientific, Hampton, NH). After determining the optical density at 260 nm, RNA was treated with DNase I (Invitrogen, Carlsbad, CA). Five micrograms of RNA was reverse-transcribed into cDNA using Superscript™ II Reverse Transcriptase (Invitrogen Corporation, Carlsbad, CA). Gene expression levels of chemokines and chemokine receptors were determined by real-time qPCR using the ABI 7500 Sequence Detection System (Applied Biosystems, Foster City, CA). Amplification was carried out for 40 cycles of 15 s at 95 °C and 1 min at 60 °C, and the product was detected using SYBR Green dye (Molecular Probes Inc., Eugene, OR). Glyceraldehyde-3-phosphate dehydrogenase was used as an endogenous control gene. Gene expression levels of heart tissue fragments from patients who underwent cardiac surgery due to non-inflammatory cardiac diseases were used as reference controls. A list of the genes studied and the primer sequences used is presented in Table 2. Samples were run in triplicate and a dissociation curve analysis was performed in each run to ensure specificity of the primers. The relative gene quantification of each gene was calculated using the 2−ΔΔCt method [15].
Table 2.
List of Genes Studied and Primers Sequences Used for Real-Time PCR Amplification
| Genes | Accession number | Sequence | Amplicon (bp) | Concentration (nM) |
|---|---|---|---|---|
| CCL1 | M57502 | (F): GCTCCAATGAGGGCTTAATATTCA | 91 | 200 |
| (R): ATTTTTCTGTGCCTCTGAACCCAT | ||||
| CCL3 | AF043339 | (F): ACCAGTTCTCTGCATCACTTGCT | 110 | 200 |
| (R): GCTGCTCGTCTCAAAGTAGTCAGC | ||||
| CCL4 | J04130 | (F): GCTTCCTCGCAACTTTGTGGT | 110 | 300 |
| (R): CACTGGGATCAGCACAGACTTG | ||||
| CCL5 | M21121 | (F): CGTGCCCACATCAAGGAGTATT | 91 | 400 |
| (R): CACACACTTGGCGGTTCTTTC | ||||
| CXCL9 | X72755 | (F): TCTGATTGGAGTGCAAGGAACC | 98 | 100 |
| (R): GGTCTTTCAAGGATTGTAGGTGGA | ||||
| CXCL10 | X02530 | (F): TCCACGTGTTGAGATCATTGCTA | 93 | 300 |
| (R): GCTTTCAGTAAATTCTTGATGGCC | ||||
| CCL17 | D43767 | (F): CACATCCACGCAGCTCGA | 98 | 200 |
| (R): TGGTACCACGTCTTCAGCTTTCTA | ||||
| CCL22 | U83171 | (F): CTGCGCGTGGTGAAACACTT | 91 | 200 |
| (R): CACAGATCTCCTTATCCCTGAAGGT | ||||
| CCR4 | X85740 | (F): CCCTTCCTGGCTTTCTGTTCA | 91 | 200 |
| (R): TTCCACGTCGTGGAGTTGAGA | ||||
| CCR5 | U57840 | (F): TCCGCTCTACTCACTGGTGTTCA | 91 | 100 |
| (R): CATGCTCTTCAGCCTTTTGCAG | ||||
| CCR8 | NM_005201 | (F): ATGCCCTAAAGGTGAGGACGAT | 91 | 200 |
| (R): ACTAGCAATGGGATGGTAGCCA | ||||
| CXCR3 | NM_001504 | (F): GTCCTTGAGGTGAGTGACCACC | 106 | 200 |
| (R): ACGAGTCACTCTCGTTTTCTCCA | ||||
| GAPDH | NM_002046 | (R): TGGTCTCCTCTGACTTCA | 117 | 200 |
| (F): AGCCAAATTCGTTGTCAT |
(F) forward sequence, (R) reverse sequence, bp base pair, GAPDH glyceraldehyde-3-phosphate dehydrogenase
Immunofluorescence
Frozen heart tissue sections of 5 μm were fixed with acetone and rinsed with PBS containing 0.1 % Triton X-100. Subsequently, the sections were incubated with PBS containing 2 % BSA followed by an overnight incubation at 4 °C with the primary antibodies. The following primary antibodies were used: CCL1 (clone 35305), CCL3 (clone 93321), CXCL9 (clone 49106), CCR5 (clone 45523), CXCR3 (clone 49801), FITC-conjugated CD14 (clone TuK4), Alexa Fluor 488-conjugated CD4 (clone S3.5), and Alexa Fluor 488-conjugated CD8 (clone 3B5; all unconjugated primary antibodies were purchased from R&D Systems Inc., Minneapolis, MN, and all conjugated primary antibodies were purchased from Invitrogen Corporation Carlsbad, CA). After washing, the slides were incubated with Alexa Fluor 633-conjugated anti-mouse or Alexa Fluor 633-conjugated anti-rat secondary antibodies (both from Invitrogen) and DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride, Sigma-Aldrich Corp., St. Louis, MO) was used for nuclear staining. Fluorescent images were acquired using UV/Laser excitation on an LSM/Meta 510 Zeiss microscope and analysis was performed using LSM Image Examiner software (Carl Zeiss, Standort Göttingen, Germany). Unspecific staining was excluded using sections stained only with primary antibody as well as sections only with secondary antibody.
Chemotaxis Assays
Chemotaxis assays were performed using intralesional T cell lines established from mitral or aortic valve surgical fragments as previously described [6]; 2 × 106 T cells were plated in a 5-μm transwell collagen-coated device (Corning Costar Corp., Acton, MA). The migration capabilities of T cells were evaluated in the presence of CCL1/I-309, CCL3/MIP1α CCL17/TARC, and CXCL9/Mig (all recombinant proteins were purchased from R&D), which were added to the lower chamber diluted in DMEM (Invitrogen, Life Technologies) at 50, 100, and 250 nM. After 3 h incubation, the absolute number of cells that transmigrated to the lower chamber was counted using a Neubauer chamber. To determine which subsets of cells transmigrated, a fraction of the cells that transmigrated to the lower chamber was stained and analyzed by flow cytometry using a FACS Calibur cytometer for acquisition and Cell Quest software for analysis (Becton & Dickinson). The cells were stained with antibodies against CD3, CD4, CD8, and CD45RO (all purchased from Becton & Dickinson).
Statistics
Nonparametric tests specified in the figures legends were used for comparisons of the gene expression of chemokines and their receptors in myocardial and valvular tissue as well as for migration assay using T cell lines. P values of <0.05 were considered statistically significant.
RESULTS
Fibrosis, Inflammation, and Neovascularization are the Main Features of Rheumatic Heart Lesions
Cardiac tissue sections from 23 RHD patients who underwent valve replacement surgery were analyzed for the presence of rheumatic activity, inflammation, neovascularization, fibrosis, and calcification. Histological analysis showed the presence of inflammation in 18 out of 26 fragments analyzed. Fibrosis was observed in 16 out of 26 fragments and neovascularization was also frequently observed (12 out of 26 fragments). Additionally, Aschoff bodies, the hallmarks of rheumatic activity, were observed in five tissue fragments of patients 2, 4, 6, 7, and 8 with acute RF episodes (Table 3).
Table 3.
Histopathological Data of Cardiac Tissue Fragments from RHD Patients
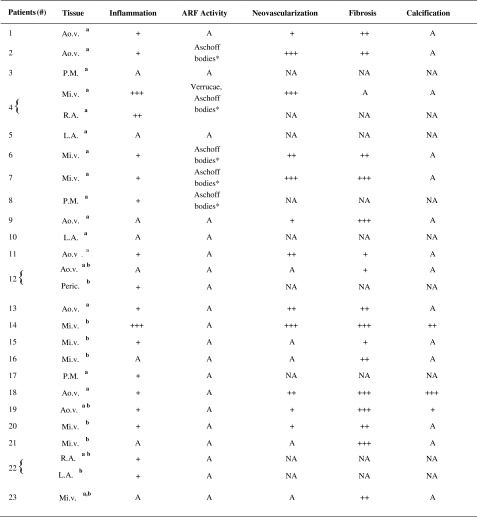
Mi.v. mitral valve, Ao.v. aortic valve, R.A. right atrium, L.A. left atrium, P.M papillary muscle, Peric. Pericardium, A absent, NA not analyzed, “+” mild, “++” moderate, “+++” severe, ARF acute rheumatic fever
aChemokines and chemokine receptors gene expression determinates by real-time qPCR
bChemokines and chemokine receptors expression determinates by confocal microscopy
cAschoff bodies in proliferative phase
CCL3/MIP1α, CCL1/I-309, and CXCL9/Mig are Differentially Expressed in Myocardium and Valvular Tissue Lesions
In order to identify whether distinct chemokines and their respective receptors are involved in cell recruitment to different sites of rheumatic lesions, we compare gene expression of samples obtained from myocardium and valvular tissue lesions from RHD patients. Samples obtained from patients who underwent cardiac surgery due to non-inflammatory disorders were used as reference controls. The list of chemokines and receptors analyzed is presented in Table 2.
Gene expression analysis showed that CCL1/I-309 and CXCL9/Mig were up-regulated in valvular tissue compared with myocardium (P = 0.034 and 0.005, respectively) (Fig. 1). In contrast, CCL3/MIP1α gene expression was highly upregulated in myocardium samples when compared with valvular tissue (P = 0.001) (Fig. 1). Of the chemokine receptors examined, CCR5 and CCR8 gene expression was upregulated in valvular tissue when compared with myocardium (P = 0.021 and 0.013, respectively) (Fig. 1). Although the CXCR3 receptor gene expression did not reach a statistical significance we observed that 75 % (9 out of 12) valve fragments showed an increased expression of this receptor when compared with myocardium tissue.
Fig. 1.

Chemokines and receptors gene expression at different sites of rheumatic heart disease lesions. Chemokine and chemokine receptor gene expression was analyzed by real-time qPCR. Myo myocardium biopsies, Valve mitral and/or aortic valve biopsies. Statistical analysis was performed using nonparametric Mann–Whitney U test, and P values of <0.05 were considered significant and are depicted in the figure.
Aiming to validate gene expression results, in situ expression of CCL1/I-309, CCL3/MIP1α and CXCL9/Mig, as well as CCR5 and CXCR3, was investigated by immunofluorescence and confocal microscopy. For this purpose, myocardium and valvular tissue fragments were stained with specific antibodies and subsequently analyzed by microscopy. Although high gene expression of CCL1/I-309 was observed in valvular tissue samples as mentioned above, we observed CCL1-positive cells only in heart tissue samples of patients 2, 4, and 22 when analyzed by immunofluorescence (Table 4). In contrast, most of cardiac tissue samples analyzed presented CXCL9-positive cells (Table 4). CCL3-positive cells were observed in myocardium sections (patient 22) (Table 4). Additionally, CCR5- and CXCR3-positive cells were observed in most of the tissue sections analyzed (Table 4). CCR8 expression was not determined due to technical problems. Figure 2 depicts some examples of chemokines and receptors expression in heart tissue sections.
Table 4.
In Situ Expression of Chemokines and Chemokine Receptors
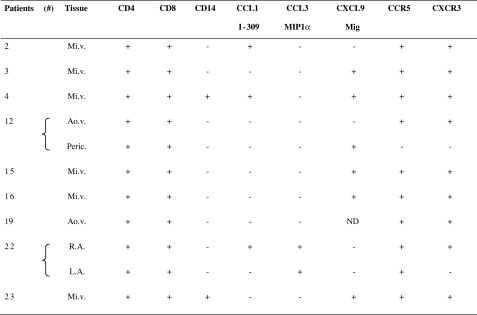
“+” occurrence of positive cells; “−” absence of positive cells, Mi.v. mitral valve, Ao.v. aortic valve, R.A. right atrium, L.A. left atrium, Peric. pericardium
Fig. 2.

In situ expression of chemokine and chemokine receptors. Cardiac tissue sections from RHD patients were stained with primary antibodies against CD4-Alexa Fluor 488, CD8-Alexa Fluor 488, CCL3, CXCL9, CCR5, and CXCR3 followed by incubation with Alexa Fluor 633-conjugated secondary antibodies. DAPI was used for nuclear staining. Fluorescent images were obtained using an LSM/Meta 510 Zeiss microscope and analysis was performed using LSM Image Examiner software (Zeiss). The figure depicts some examples of positive staining from different patients. a, b CD4+ and CD8+ staining (green) in the mitral valve of patient 15; c, d CCR5 and CCL3/MIP1α staining (pink and red, respectively) in the myocardium of patient 22; e, f CXCL9/Mig and CXCR3 staining (red) in the mitral valve of patient 16.
In Situ Identification of Cell Subsets Represented in the Cardiac Lesions
Cell subsets represented in the cardiac lesions were stained by immunofluorescence using monoclonal antibodies against T cells (anti-CD4 and anti-CD8) and macrophages (anti-CD14) (Table 4; Fig. 2). CD4 and CD8-positive cells were observed in all tissue sections analyzed; nevertheless, CD14-positive cells were rarely observed (Table 4). CD4 and CD8 T cells staining are illustrated in Fig. 2.
Heart-Infiltrating T cells from Valvular Tissue Migrate Toward CXCL9/Mig Gradient In Vitro
To investigate whether CCL1/I-309 and CXCL9/Mig, chemokines (upregulated in the valvular tissue), CCL3/MIP1α (upregulated in myocardium), and CCL17/TARC (equally expressed in valvular and myocardium tissue, data not shown) would affect the recruitment of T cells to the lesion sites, we performed a migration assay using pre-established valvular tissue-infiltrating T cell lines in the presence of different concentrations of chemokines. Valvular tissue-derived T cell lines were established from tissue fragments from chronic RHD patients 1, 14, 18, and 19 as previously described [6].
Phenotypic characterization of the valvular tissue-infiltrating T cell lines prior to chemokine-induced migration showed that T cell lines from patients 1, 14, and 19 were essentially CD4+CD45RO+ T cells, a phenotype representative of memory T cells. T cell line from patient 18 presented similar percentages of CD4+ and CD8+ T cells; however, only a few memory CD4+ T cells were identified (data summarized on the legend of Table 5).
Table 5.
Phenotypic Characterization of Valvular Tissue-Derived T Cells that Migrate upon Specific Chemokine Gradient
| Valvular tissue-derived cell lines from RHD patients | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | Patient 14 | Patient 18 | Patient 19 | Patient 14 | Patient 18 | Patient 19 | Patient 18 | Patient 1 | Patient 18 | Patient 14 | |
| CXCL9/Mig | CCL1/I-309 | CCL3/MIP1α | CCL17/TARC | ||||||||
| 100 nM | 250 nM | 100 nM | 250 nM | 50 nM | 100 nM | 100 nM | 250 nM | ||||
| T cells post-migration (%) | 2.9 | 3.7 | 4.8 | 6.7 | 4.4 | 4.5 | 5.0 | 4.8 | 5.4 | 7.6 | 4.2 |
| CD4+ (%) | 89.6 | 66.7 | 38.6 | 98.2 | 50.2 | 38.9 | 59.0 | 48.0 | 98.3 | 95.0 | 98.2 |
| CD4+CD45RO+ (%) | 89.6 | 57.5 | 0 | 98.3 | 85.4 | 92.7 | 68.1 | 0 | 93.2 | 92.0 | 97.6 |
| CD8+ (%) | 7.9 | 27.5 | 61.3 | 1.3 | 45.6 | 45.0 | 33.1 | 47.4 | 1.2 | 0 | 0 |
| CD8+CD45RO+ (%) | 9.1 | 30 | 0 | 0 | 96.4 | 97.4 | 99.5 | 0 | 1.0 | 0 | 0 |
Valvular tissue-derived T cell lines obtained from heart lesions of RHD patients were assessed for their capability to migrate toward chemokine gradient and their phenotype are indicated. The phenotype of heart-tissue infiltrating T cells before migration was determined by flow cytometry and the frequencies (in percent) were the following: patient 1, CD4+CD45RO+ (98.31 %) and CD8+CD45RO+ (0.93 %); patient 14, CD4+CD45RO+ (90.30 %) and CD8+CD45RO+ (9.44 %); patient 19 CD4+CD45RO+ (99.01 %) and CD8+CD45RO+ (0.89 %); patient 18, CD4+ (58.90 %) and CD8+ (42.0 %) and low numbers (3.10 %) of memory T cells
Notably, CXCL9/Mig gradient induced a significant increase in the migration of valvular-tissue infiltrating T cells (Fig. 3a). CCL1/I-309 and CCL17/TARC gradients induced migration of T cells in a dose-dependent manner however the results did not reach statistical significance (Fig. 3a). Different concentrations of CCL3 (MIP1α) seems to not affect valvular-tissue-derived T cells migration. Of note, CCL3 (MIP1α gene expression was mostly upregulated in myocardium tissue samples (Fig. 1).
Fig. 3.

Heart-infiltrating T cells migrate towards specific chemokines gradient. a Absolute numbers of heart infiltrating T cells that migrated toward CCL1/I-309, CCL3/MIP1α, CXCL9/Mig, and CCL17/TARC gradient compared with untreated cells (controls). Data represent mean ± SD of three to four valvular tissue-derived T cell lines from different RHD patients that transmigrated toward chemokine gradient. Statistical analysis was performed using nonparametric unpaired t test. P values are depicted in the figure. b The phenotype of heart tissue-infiltrating T cells that migrated toward CCL1/I-309 gradient (250 nM). The percentage of cells was determined by flow cytometric analysis (FACS) and the results of patient 19 are depicted. Pre- and post-migration CD4+CD45RO+ and CD8+CD45RO+ subpopulations are shown.
The percentage of transmigrating cells varied from 2.9 to 7.6 % and were characterized mainly as CD4+CD45RO+ (Table 5), except cells from patient 18 who presented few CD4+CD45RO+ memory T cells (3.10 %) before migration (legend Table 5). Figure 3b shows a representative dot plot of the T cell line derived from patient 19 pre- and postmigration toward CCL1/I-309 gradient.
DISCUSSION
Lymphocytic infiltration through the valve surface endothelium is thought to be the initial step for tissue damage and pathogenesis of RHD. Yet, chemokines and receptors that mediate cell recruitment to the inflammatory sites are unclear. In the present article, we investigated the expression of several chemokines and their receptors in clinical tissue samples obtained from chronic RHD patients. Our results showed a differential chemokine expression in distinct sites of lesions. CCL3/MIP1α expression was upregulated in samples from myocardium; in contrast, CCL1/I-309 and CXCL9/Mig were upregulated in valvular tissue lesions and can be considered as important inflammatory chemokines that enhance the severity of valve damage. In addition, CD4+CD45RO+ valvular tissue-infiltrating T cells migrated toward CXCL9/Mig gradient in vitro.
CCL3/MIP1α is a chemokine produced by several cell types, such as macrophages, dendritic cells (DCs), lymphocytes, and endothelial cells. This chemokine induces recruitment of CCR5+ cells, including activated and memory T cells, monocytes, macrophages and immature DCs [16, 17]. Our observation that CCL3/MIP1α is highly expressed in the myocardium of RHD patients may indicate that the recruitment of T cells to the myocardium is the initial step in the development of the disease, with migration to the valvular tissue occurring later in disease progression. CCR5 was also highly expressed in tissue samples analyzed from rheumatic valvular lesions. This receptor is upregulated by IFN-γ and TNF-α in tissue-specific inflammation and Th1 cells [17, 18].
CCL1/I-309 is a chemokine that specifically binds to the chemokine receptor CCR8. CCL1/I-309 is mainly secreted by monocytes, activated lymphocytes and endothelial cells [19–21], and it is described as a potent chemoattractant for the same cells [20–23]. Moreover, CCL1/I-309 has been implicated in the induction of angiogenesis and the activation of endothelial cells as a mechanism of valve repair [23]. In our study, CCL1/I-309 and its receptor CCR8 were upregulated in the valvular tissue samples; however, CCL1/I-309 gradient was not effective in inducing significant migration of valvular tissue infiltrating T cells. Despite of these findings, histological analyses performed in the present study showed neovascularization as a frequent observation in most of the valvular tissue samples analyzed. Hence it is tempting to speculate that the expression of CCL1/I-309 and CCR8 may be a marker of angiogenesis as mechanism of valve repair. In line with that, increased expression of factors that induce angiogenesis, such as VEGF, and decreased expression of anti-angiogenesis factors, such as condromodulin-I, have already been described in RHD [24].
CXCL9/Mig is an IFN-γ-inducible chemokine produced mainly by DCs, B lymphocytes, and macrophages [25]. CXCL9/Mig binds to the receptor CXCR3, which is expressed on multiple cell types but predominantly on memory phenotype cells and primed effector T cells producing IFN-γ [26]. Our results showed upregulation of CXCL9/Mig in the valvular tissue and importantly, CXCL9/Mig gradient induced significant migration of T cells isolated from valvular tissue lesions. It is important to note that in this study, in situ expression of CXCR3 was observed in most of the samples analyzed from valvular tissue. The heart-tissue infiltrating cells resulted from S. pyogenes oligoclonal primed expansions that are able of recognize valve-derived proteins as previously described [6, 27] and are maintained in the valvular tissue upon inflammatory cytokines in line with our previous work in which we showed that IFNγ-positive mononuclear cells are one of the major cell types present in cardiac rheumatic lesions [14]. The in situ production of IFNγ by mononuclear cells that infiltrated both myocardium and valvular tissue of rheumatic lesions would be the driving factor for the secretion of CXCL9/Mig by valvular tissue-resident antigen presenting cells that subsequently recruits valvular-tissue autoreactive T cells expressing CXCR3. Altogether, these cells favor a milieu that lead an inflammatory reaction perpetuating the valvular lesions, consequently leading to the loss of function and heart failure.
Interestingly, it was demonstrated that combined blockade of CXCR3 and CCR5 was effective in preventing acute and chronic allograft rejection in murine model [28, 29]. In the case of RF, blocking of CXCR3 could be an effective strategy to control inflammatory activity within the valves. However, additional studies will be necessary to achieve specific blockade of IFN-γ producing T cells recruitment to the valvular lesions without impairment of effector T cell responses.
Collectively, this study provides novel evidences that defined chemokines and receptors that are expressed in different sites of rheumatic lesions, i.e., myocardium and valvular tissue. In addition, CXCL9/Mig is the main chemokine driving T cell recruitment to valvular tissue, site in which the cardiac deformities are extremely severe causing permanent damage.
Acknowledgment
The authors thank Simone R. Santos and Karen Köhler for technical assistance.
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
Funding
This work was supported by “Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP)” and “Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq)”, Brazil. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Dajani AS, Ayoub E, Bierman FZ, Bisno AL, Denny FW, Durack DT, Ferrieri P, Freed M, Gerber MD, Kaplan E, Karchmer AW, Markowitz M, Rahimtoola SH, Shurman ST, Stollerman G, Takahashi M, Taranta A, Taubert KA, Wilson W, Durack DT. Guidelines for the diagnosis of rheumatic fever. Jones criteria 1992 Update. Special writing group of the Committee on Rheumatic fever, Endocarditis and Kawasaki disease of the Council of Cardiovascular disease in the young of the American Heart Association. Journal of the American Medical Association. 1992;268:2069–2073. doi: 10.1001/jama.1992.03490150121036. [DOI] [PubMed] [Google Scholar]
- 2.Lee JL, Naguwa SM, Cheema GS, Gershwin ME. Acute rheumatic fever and its consequences: a persistent threat to developing nations in the 21st century. Autoimmunity Reviews. 2009;9:117–123. doi: 10.1016/j.autrev.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Raizada V, Williams RC, Jr, Chopra P, Gopinath N, Prakash K, Sharma KB, Cherian KM, Panday S, Arora R, Nigam M, Zabriskie JB, Husby G. Tissue distribution of lymphocytes in rheumatic heart valves as defined by monoclonal anti-T cell antibodies. American Journal of Medicine. 1983;74:90–96. doi: 10.1016/0002-9343(83)91124-5. [DOI] [PubMed] [Google Scholar]
- 4.Marboe CC, Knowles DM, Weiss MB, Ursell PC, Fenoglio J., Jr Monoclonal antibody identification of mononuclear cells in endomyocardial biopsy specimens from a patient with rheumatic carditis. Human Pathology. 1985;16:332–338. doi: 10.1016/S0046-8177(85)80227-6. [DOI] [PubMed] [Google Scholar]
- 5.Kemeny E, Grieve T, Marcus R, Sareli P, Zabriskie JB. Identification of mononuclear cells and T cell subsets in rheumatic valvulitis. Clinical Immunology and Immunopathology. 1989;52:225–237. doi: 10.1016/0090-1229(89)90174-8. [DOI] [PubMed] [Google Scholar]
- 6.Guilherme L, Cunha-Neto E, Coelho V, Snitcowsky R, Pommerantzeff PMA, Assis RV, Pedra FF, Neumann J, Goldberg AC, Patarroyo ME, Pillegi F, Kalil J. Human heart-filtrating T cell clones from rheumatic heart disease patients recognize both streptococcal and cardiac proteins. Circulation. 1995;92:415–420. doi: 10.1161/01.CIR.92.3.415. [DOI] [PubMed] [Google Scholar]
- 7.Amoils B, Morrison RC, Wadee AA, Marcus R, Ninin D, King P, Sareli P, Levin S, Rabson AR. Aberrant expression of HLA-DR antigen on valvular fibroblasts from patients with active rheumatic carditis. Clinical and Experimental Immunology. 1986;66:88–94. [PMC free article] [PubMed] [Google Scholar]
- 8.Narula J, Chopra P, Talwar KK, Reddy KS, Vasan RS, Tandon R, Bhatia ML, Southern JF. Does endomyocardial biopsy aid in the diagnosis of active rheumatic carditis? Circulation. 1993;88:2198–2205. doi: 10.1161/01.CIR.88.5.2198. [DOI] [PubMed] [Google Scholar]
- 9.Gross I, Ehrlich JC. Studies on the myocardial Aschoff body. American Journal of Pathology. 1934;10:467–488. [PMC free article] [PubMed] [Google Scholar]
- 10.Cunningham MW. Pathogenesis of group A streptococcal infections. Clinical Microbiology Reviews. 2000;13:470–511. doi: 10.1128/CMR.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cunningham MW. Pathogenesis of group A streptococcal infections and their sequelae. Advances in Experimental Medicine and Biology. 2008;609:29–42. doi: 10.1007/978-0-387-73960-1_3. [DOI] [PubMed] [Google Scholar]
- 12.Roberts S, Kosanke S, Terrence Dunn S, Jankelow D, Duran CM, Cunningham MW. Pathogenic mechanisms in rheumatic carditis: focus on valvular endothelium. Journal of Infectious Diseases. 2001;183:507–511. doi: 10.1086/318076. [DOI] [PubMed] [Google Scholar]
- 13.Faé KC, Silva DD, Oshiro SE, Tanaka AC, Pomerantzeff PM, Douay C, Charron D, Toubert A, Cunningham MW, Kalil J, Guilherme L. Mimicry in recognition of cardiac myosin peptides by heart-intralesional T cell clones from rheumatic heart disease. Journal of Immunology. 2006;176:5662–5670. doi: 10.4049/jimmunol.176.9.5662. [DOI] [PubMed] [Google Scholar]
- 14.Guilherme L, Cury P, Demarchi LM, Coelho V, Abel L, Lopez AP, Oshiro SE, Aliotti S, Neto EC, Pomerantzeff PM, Tanaka AC, Kalil J. Rheumatic heart disease: proinflammatory cytokines play a role in the progression and maintenance of valvular lesions. Americal Journal of Pathology. 2004;165:1583–1591. doi: 10.1016/S0002-9440(10)63415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C (T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 16.Sallusto F, Mackay CR, Lanzavecchia A. The role of chemokine receptors in primary, effector, and memory immune responses. Annual Review of Immunology. 2000;18:593–620. doi: 10.1146/annurev.immunol.18.1.593. [DOI] [PubMed] [Google Scholar]
- 17.Baggiolini M, Dewald B, Moser B. Interleukin-8 and related chemotactic cytokines—CXC and CC chemokines. Advances in Immunology. 1994;55:97–109. doi: 10.1016/S0065-2776(08)60509-X. [DOI] [PubMed] [Google Scholar]
- 18.Bonecchi R, Bianchi G, Bordignon PP, D’Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, Sinigaglia F. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. The Journal of Experimental Medicine. 1998;187:129–134. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/S1074-7613(00)80165-X. [DOI] [PubMed] [Google Scholar]
- 20.Haque NS, Zhang X, French DL, Li J, Poon M, Fallon JT, Gabel BR, Taubman MB, Koschinsky M, Harpel PC. CC chemokine I-309 is the principal monocyte chemoattractant induced by apolipoprotein (a) in human vascular endothelial cells. Circulation. 2000;102:786–792. doi: 10.1161/01.CIR.102.7.786. [DOI] [PubMed] [Google Scholar]
- 21.Miller MD, Hata S, De Waal Malefyt R, Krangel MS. A novel polypeptide secreted by activated human T lymphocytes. Journal of Immunology. 1989;143:2907–2916. [PubMed] [Google Scholar]
- 22.Haque NS, Fallon JT, Taubman MB, Harpel PC. The chemokine receptor CCR8 mediates human endothelial cell chemotaxis induced by I-309 and Kaposi sarcoma herpesvirus-encoded vMIP-I and by lipoprotein (a)-stimulated endothelial cell conditioned medium. Blood. 2001;97:39–45. doi: 10.1182/blood.V97.1.39. [DOI] [PubMed] [Google Scholar]
- 23.Bernardini G, Spinetti G, Ribatti D, Camarda G, Morbidelli L, Ziche M, Santoni A, Capogrossi MC, Napolitano M. I-309 binds to and activates endothelial cell functions and acts as an angiogenic molecule in vivo. Blood. 2000;96:4039–4045. [PubMed] [Google Scholar]
- 24.Yoshioka M, Yuasa S, Matsumura K, Kimura K, Shiomi T, Kimura N, Shukunami C, Okada Y, Mukai M, Shin H, Yozu R, Sata M, Ogawa S, Hiraki Y, Fukuda K. Chondromodulin-I maintains cardiac valvular function by preventing angiogenesis. Nature Medicine. 2006;12:1151–1159. doi: 10.1038/nm1476. [DOI] [PubMed] [Google Scholar]
- 25.Park MK, Amichay D, Love P, Wick E, Liao F, Grinberg A, Rabin RL, Zhang HH, Gebeyehu S, Wright TM. The CXC chemokine murine monokine induced by IFN-gamma (CXC chemokine ligand 9) is made by APCs, targets lymphocytes including activated B cells, and supports antibody responses to a bacterial pathogen in vivo. Journal of Immunology. 2002;169:1433–1443. doi: 10.4049/jimmunol.169.3.1433. [DOI] [PubMed] [Google Scholar]
- 26.Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, Baggiolini M, Moser B. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. The Journal of Experimental Medicine. 1996;184:963–969. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guilherme L, Dulphy N, Douay C, Coelho V, Cunha-Neto E, Oshiro SE, Assis RV, Tanaka AC, Pomerantzeff PM, Charron D, Toubert A, Kalil J. Molecular evidence for antigen-driven immune responses in cardiac lesions of rheumatic heart disease patients. International Immunology. 2000;12:1063–1074. doi: 10.1093/intimm/12.7.1063. [DOI] [PubMed] [Google Scholar]
- 28.Akashi S, Sho M, Kashizuka H, Hamada K, Ikeda N, Kuzumoto Y, Tsurui Y, Nomi T, Mizuno T, Kanehiro H, Hisanaga M, Ko S, Nakajima Y. A novel small-molecule compound targeting CCR5 and CXCR3 prevents acute and chronic allograft rejection. Transplantation. 2005;80:378–384. doi: 10.1097/01.tp.0000166338.99933.e1. [DOI] [PubMed] [Google Scholar]
- 29.Schnickel GT, Bastani S, Hsieh GR, Shefizadeh A, Bhatia R, Fishbein MC, Belperio J, Ardehali A. Combined CXCR3/CCR5 blockade attenuates acute and chronic rejection. Journal of Immunology. 2008;180:4714–4721. doi: 10.4049/jimmunol.180.7.4714. [DOI] [PubMed] [Google Scholar]