

Abstract
Acyl-CoA thioesterase 12 (ACOT12) is the major enzyme known to hydrolyze the thioester bond of acetyl-CoA in the cytosol in the liver. ACOT12 contains a catalytic thioesterase domain at the N terminus and a steroidogenic acute regulatory protein-related lipid transfer (START) domain at the C terminus. We investigated the effects of lipids (phospholipids, sphingolipids, fatty acids, and sterols) on ACOT12 thioesterase activity and found that the activity was inhibited by phosphatidic acid (PA) in a noncompetitive manner. In contrast, the enzymatic activity of a mutant form of ACOT12 lacking the START domain was not inhibited by the lipids. These results suggest that the START domain is important for regulation of ACOT12 activity by PA. We also found that PA could bind to thioesterase domain, but not to the START domain, and had no effect on ACOT12 dissociation. ACOT12 is detectable in the liver but not in hepatic cell lines such as HepG2, Hepa-1, and Fa2N-4. ACOT12 mRNA and protein levels in rat primary hepatocytes decreased following treatment with insulin. These results suggest that cytosolic acetyl-CoA levels in the liver are controlled by lipid metabolites and hormones, which result in allosteric enzymatic and transcriptional regulation of ACOT12.
Keywords: acyl-CoA thioesterase 12, phosphatidic acid, steroidogenic acute regulatory protein-related lipid transfer domain protein, lipid biosynthesis
Acetyl-CoA plays a key role in many aspects of metabolism. In mammalian cells, there are two distinct subcellular pools of acetyl-CoA, the cytosolic and mitochondrial pools. ATP-citrate lyase and cytosolic acetyl-CoA synthetase in the cytosol are known to be important for synthesis of cytosolic acetyl-CoA, which in turn is valuable for biosynthesis of fatty acids and cholesterol in the cytosol (1–3). Cytosolic acetyl-CoA is also a precursor for acetylcholine (4), platelet-activating factor (5), and protein acetylation of both histone and nonhistone proteins (6). In contrast, maintenance of acetyl-CoA levels in mitochondria is mainly achieved via oxidation of pyruvate by pyruvate dehydrogenase and β-oxidation of fatty acids.
The amount of acetyl-CoA in the cytosol is also regulated by the rate of acetyl-CoA degradation. Acyl-CoA thioesterase 12 (ACOT12), known as StarD15 or cytoplasmic acetyl-CoA hydrolase, was originally identified via purification, cDNA cloning, and identification of a protein with acetyl-CoA thioesterase activity. ACOT12 appears to be the major cytoplasmic enzyme in the liver that preferentially hydrolyzes the thioester bond of acetyl-CoA, producing acetate and CoA, and does not hydrolyze long chain acyl-CoA (7–10). It was previously reported that ACOT12 is an allosteric enzyme regulated by ATP (as an activator) and ADP (as an inhibitor), and its enzymatic activity is cold labile (11, 12). ACOT12 activity in the liver can also be stimulated by various treatments including thyroid hormones, CS-514 (3-hydroxy-3-methylglutaryl-CoA reductase inhibitor), and clofibrate (an anti-obesity drug that acts via downregulation of fatty acid synthase, ATP-citrate lyase, and acetyl-CoA carboxylase, and upregulation of β-oxidation) (13, 14). However, the physiological functions of the enzyme have yet to be fully understood.
The ACOT12 enzyme consists of a catalytic domain, a pair of thioesterase domains, referred to as hotdog-fold domains at the N terminus (15), and a steroidogenic acute regulatory protein-related lipid transfer (START) domain at the C terminus (Fig. 1A). The N-terminal thioesterase domains are required for enzymatic activity. The C-terminal START domain is a protein module of ∼210 amino acid residues thought to act as a lipid binding, transferring, and sensing domain for phospholipids, sterols, and sphingolipids (16–19). There are 15 distinct human proteins containing START domains, termed StarD1–StarD15. The lipids or sterols bound by nine of the START domain-containing proteins (StarD1–7, -10, and -11) have been identified (16, 18). In contrast, neither lipid ligands nor the biological significance of the START domains of six other START proteins (StarD8, -9, and -12–15), including ACOT12 (StarD15), are known.
Fig. 1.

Expression of ACOT12 and mutant proteins. A: The structures of cDNA constructs used in this study are shown. Each construct includes a 6×His-Tag at the N terminus. B: Purified proteins were separated by SDS-PAGE. The gel was stained with Coomassie Brilliant Blue.
Because ACOT12 appears to be the major enzyme responsible for determining the rate of degradation of cytosolic acetyl-CoA in the liver, the enzymatic regulation of ACOT12 is important for the control of lipid biosynthesis. In this study, we found that acetyl-CoA hydrolase activity of ACOT12 can be inhibited by phosphatidic acid (PA) and lysophosphatidic acid (LPA). Furthermore, the START domain in ACOT12 plays an important role in inhibition. We also report that in rat primary hepatocytes, ACOT12 mRNA and protein levels are downregulated following treatment with insulin. These results suggest that acetyl-CoA levels for lipid biosynthesis are regulated by lipid metabolites and hormones via control of the enzymatic activity and transcription of ACOT12.
MATERIALS AND METHODS
Biochemicals
Egg yolk PA and soybean phosphatidylinositol were obtained from Nacalai Tesque (Kyoto, Japan). Insulin, 1-oleoyl LPA, glycerol 3-phosphate (G3P), oleoyl-CoA, acetoacetyl-CoA, and 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) were purchased from Sigma-Aldrich (St. Louis, MO). Dioleoyl phosphatidic acid (DOPA); dioleoyl phosphatidylcholine (DOPC); 1-stearoyl, 2-oleoyl-phosphatidylcholine (PC); 1-stearoyl, 2-oleoyl-phosphatidylserine; 1-stearoyl, 2-oleoyl-phosphatidylethanolamine (PE); and dimyristoyl phosphatidylglycerol (DMPG) were obtained from Avanti Polar Lipids (Alabaster, AL). Acetyl-CoA, clofibrate, and 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) were obtained from Wako Pure Chemicals (Tokyo, Japan). Butyryl (C4)-CoA and octanoyl (C8)-CoA were from DOOSAN Serdary Research Laboratories (Seoul, Korea). All reagents used were of the highest purity available.
Expression and purification of ACOT12
To obtain the V5-tagged constructs for expression in mammalian cells, a DNA fragment encoding mouse wild-type (WT) ACOT12 (GenBank accession number NM_028790) was prepared by PCR using a forward primer (5′-ATGGAGTCGATGGTGGCGCCGGGTGAGGTG-3′) and a reverse primer (5′-TAACACACTTTTAAGTCCATCAGGAGTAGC-3′) using mouse cDNA library as template. A DNA fragment encoding the thioesterase domain (THIO) was amplified using the same forward primer and a reverse primer (5′-CCACTGTGCACTGAGTGGAACTTCTTTCTT-3′). These fragments were cloned into pcDNA3.1/V5-His (Invitrogen, Carlsbad, CA). To obtain the 3xFlag-tagged constructs, a DNA fragment encoding WT was amplified by PCR using a forward primer containing an NotI site (5′-ATTTGCGGCCGCGATGGAGTCGATGGTGGCGCC-3′) and a reverse primer containing an XbaI site and a stop codon (5′-GCTCTAGATTATAACACACTTTTAAGTCCA-3′). A DNA fragment encoding the THIO was amplified using the same forward primer and a reverse primer containing an XbaI site and a stop codon (5′-GCTCTAGATTACCACTGTGCACTGAGTGGA-3′). These fragments were cloned into the NotI-XbaI sites of p3xFLAG-CMV-10 (Sigma-Aldrich).
For expression in Escherichia coli, a DNA fragment encoding mouse WT ACOT12 was prepared by PCR using a forward primer containing an XhoI site (5′-CCGCTCGAGATGGAGTCGATGGTGGCGCCG-3′) and a reverse primer containing an XbaI site (5′-GCTCTAGATTATAACACACTTTTAAGTCCA-3′) using ACOT12 cDNA cloned into pcDNA3.1/V5-His. A DNA fragment encoding the THIO (Met1-Trp344) was amplified using the same forward primer and a reverse primer containing an XbaI site (5′-GCTCTAGATTACCACTGTGCACTGAGTGGA-3′). A DNA fragment encoding the START domain (Asp345-Leu556) was amplified using a forward primer containing an XhoI site (5′-CCGCTCGAGGATATAAGCAAAAAGGGATCC-3′) and the same reverse primer used for WT amplification. The DNA fragments were cloned into the XhoI-XbaI sites of a bacterial expression vector, pColdDNAI (Novagen, Madison, WI). The vectors were constructed with insertions of 6×His-Tag at an N terminus of expressed proteins. Nucleotide sequences were confirmed using the BigDye Terminator Cycle Sequencing kit (Applied Biosystems) and a DNA sequencer (ABI PRISM 377-XL, Applied Biosystems).
The insert-containing vectors were then transformed into E. coli strain JM109 and grown at 37°C to an O.D. A600 of 0.5 in 200 ml of Luria-Bertani broth supplemented with 50 μg/ml ampicillin. Next, protein expression was induced by the addition of 0.1 mM isopropyl β-D-thiogalactopyranoside followed by incubation for 20 h at 15°C. Subsequently, cells were collected, resuspended, and sonicated in 10 ml of 50 mM Tris-HCl buffer, pH 8.0, containing 0.5 mM phenylmethylsulfonyl fluoride (PMSF). The protein purification was performed at room temperature to avoid cold inactivation (20). After removing cell debris by centrifugation at 10,000 g for 10 min, the supernatant was applied to a NTF Sepharose column (GE Healthcare Bio-Sciences Corp., Uppsala, Sweden). The expressed His-tagged proteins were adsorbed and eluted with 20 mM Tris-HCl buffer, pH 8.0, containing 500 mM imidazole. Imidazole was then removed by applying the purified proteins to a PD-10 column (BioRad, Hercules, CA) and the proteins were eluted with 20 mM Tris-HCl buffer, pH 8.0, containing 150 mM NaCl. Purified protein was stored in 5 μl aliquots at −80°C and thawed immediately prior to use.
Acetyl-CoA hydrolase activity
Acetyl-CoA thioesterase activity was measured on a Model 680 microplate reader (BioRad) using DTNB. Briefly, acetyl-, acetoacetyl-, butyryl-, octanoyl-, or HMG-CoA at 1 mM or another concentration (as indicated) was incubated at 37°C for the indicated time with an appropriate amount of tag-purified enzyme (25–120 ng) in 50 μl of 20 mM Tris-HCl buffer, pH 8.0, containing 150 mM NaCl, 1 mM ATP, and 1 mM DTNB. Immediately following, the absorbance of the reaction mixture at 415 nm was analyzed using a spectrophotometer. To analyze the effects of phospholipids, fatty acids, or sphingolipids on enzyme activity, the lipids were dissolved in chloroform, dried under nitrogen, dissolved in 20 mM Tris-HCl buffer, pH 8.0, containing 150 mM NaCl, and sonicated using a probe-type sonicator. Sterols were dissolved in the same buffer containing 3 mM hydroxypropyl-β-cyclodextrin (final concentration).
Binding assay for phospholipid-conjugated beads
Phospholipid-conjugated butyl-Sepharose beads were prepared as described previously (21) with some modifications. Briefly, a clear suspension of DOPC or DOPA (0.5 mM) was formed by dissolving each in Tris-buffered saline (TBS) using a probe-type sonicator. Next, butyl-Sepharose equilibrated with TBS (200 μl) was combined with the phospholipid solutions (800 μl, 400 nmol). After incubation for 1 h with shaking, the slurry was extensively washed with TBS, and then with 5 vol of TBS containing 0.05% Triton X-100. The amount of phospholipids conjugated to the gel was quantified by measuring inorganic phosphorous following perchloric acid digestion (22). In this procedure, about 300 nmol of phospholipids (75% of phospholipids added) were conjugated with 200 μl of the gel. When G3P was used instead of phospholipids, about 30 nmol of G3P (10% of G3P added) were conjugated to the beads, indicating that fatty acid chains in phospholipids are important for binding to the gel. Purified WT, THIO, or START proteins were mixed with PC- or PA-conjugated beads and incubated for 30 min at room temperature to allow for binding. Proteins bound to the beads or remaining in the supernatants were separated by centrifugation at 3,000 g for 5 min. The proteins bound to the lipid-conjugated beads were then extracted by boiling in SDS-PAGE sample buffer. Proteins from both fractions were analyzed by immunoblotting using an anti-ACOT12 antibody. To assess the effect of acetyl-CoA on ACOT12-DOPA interaction, ACOT12-bound DOPA gel was incubated with the indicated concentrations of acetyl-CoA in TBS containing 0.05% Triton X-100 for 30 min. After centrifugation, proteins in the supernatant and precipitated fractions were analyzed as described above.
Antibody and immunoblot analysis
Anti-ACOT12 serum was prepared by injection of 0.5 mg of the purified ACOT12 protein four times into a New Zealand White rabbit. Preimmune serum was obtained before immunization and used as a negative control. To perform immunoblot analysis, proteins were first separated by SDS-PAGE and then transferred to nitrocellulose membranes using a semi-dry electroblotter (Nihon Eido Co., Ltd., Tokyo, Japan). Next, the membranes were incubated with 1% (w/v) skim milk in TBS for 1 h and washed three times with TBS containing 0.02% Tween 20 (T-TBS). The membranes were then incubated with antiserum for 12 h at 4°C, washed three times with T-TBS, and incubated with horseradish peroxidase-conjugated anti-rabbit IgG for 1 h at room temperature. Finally, the membranes were washed three times with T-TBS and signals were observed using a SuperSignal West Pico Substrate peroxidase staining kit (Pierce, Rockford, IL), according to the manufacturer's instructions.
Protein oligomerization and dissociation
Oligomerization of recombinant enzymes produced in E. coli was assessed by gel filtration using a HiLoad 16/600 Superdex 200 pg column (GE Healthcare). The column was equilibrated with 50 mM phosphate buffer, pH 6.8, containing 150 mM NaCl. Proteins were eluted from the column at a flow rate of 1.0 ml/min at room temperature using a Waters 650 system (Waters Corp., Milford, MA). To induce oligomerization, proteins were preincubated with 2 mM ATP for 30 min at 37°C. The column was calibrated with gel filtration molecular weight markers (BioRad). Fractions were precipitated with 10% trichloroacetic acid and analyzed by Western blotting using anti-ACOT12 antibody. Band density was quantified using the ChemiDoc XRS system (Bio-Rad).
Oligomerization and dissociation of recombinant proteins expressed in mammalian cells were assessed as follows. Hepa-1 cells, a murine-derived hepatoma cell line, were maintained in 5% CO2 at 37°C in DMEM (Wako Chemicals) with 10% fetal bovine serum (FBS). Cells in 60 mm plates were cotransfected with V5- and 3xFlag-tagged constructs using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. After 18 h, cells were lysed with lysis buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.1% Triton X-100) containing 1 mM PMSF and 10 μg/ml of leupeptin, pepstatin, and chymostatin at room temperature. After centrifugation at 25,000 g for 15 min, 3xFlag-tagged protein was immunoprecipitated using anti-Flag M2 affinity gel (Sigma-Aldrich) at room temperature. The immunoprecipitated protein complex was washed five times with TBS to remove any remaining detergent completely, and then incubated with or without 100 μM of DOPA in TBS at room temperature, or with TBS on ice for 1 h. The proteins bound to the gel or released into the supernatants were separated by centrifugation at 3,000 g for 5 min and analyzed by immunoblotting using anti-V5 (Invitrogen) or anti-Flag (Sigma-Aldrich) antibody.
Primary culture of rat hepatocytes and quantitative PCR
Hepatocytes were isolated from Wistar rats using a modified two-step collagenase perfusion in situ (23). To do this, cells were plated at 2 × 104 cells/0.25 ml/cm2 in collagen (type I)-coated 12-well plates (Asahi Glass Co., Japan) and cultured in Williams’ medium E (Sigma-Aldrich) supplemented with 10% FBS (Invitrogen), 100 units/ml penicillin, 100 μg/ml streptomycin, 1 μM dexamethasone, and 1 μM insulin under 5% CO2 at 37°C. Four hours after plating the cells, the medium was replaced with Williams’ medium E without dexamethasone and insulin, and cells were cultured in the presence or absence of 10% FBS for the number of days indicated. The culture medium was changed daily. To analyze the effects of insulin and clofibrate, cells plated as described above were cultured in the absence of FBS, dexamethasone, and insulin for 20 h, then cells at day 1 were cultured with or without insulin (100 nM) or clofibrate (100 μM) for 2–10 h. Total RNA was collected using RNA extraction kits (Qiagen, Valencia, CA) according to the manufacturer's instructions. To quantify the mRNA levels for ACOT12 and β-actin, total RNA was reverse-transcribed at 37°C for 15 min using PrimeScript™ reverse transcriptase (Takara Bio Inc., Shiga, Japan) and then subjected to 40 cycles of amplification (7300 Real Time PCR System, Applied Biosystems, Indianapolis, IN) using a FastStart Universal SYBR Green Master (Roche). β-actin was used as an internal control for the verification of ACOT12. The following primers were used for quantitative PCR: 5′-CATGGCGTGGATGGAGACA-3′/5′-TTGACGATGGCATTGAAGACAAG-3′ for rat ACOT12; 5′-GACTCATCGTACTCCTGCTTGCTG-3′/5′-GGAGATTACTGCCCTGGCTCCTA-3′ for rat β-actin.
HepG2 cells were cultured in DMEM with 10% FBS. Fa2N-4 cells were purchased from XenoTech LLC (Lenexa, KS) and cultured in MFE support medium according to the manufacturer's instructions.
RESULTS
Expression and oligomerization of recombinant proteins
Protein constructs used in this study are shown in Fig. 1A. cDNA fragments encoding mouse WT ACOT12 (556 amino residues), the THIO (Met1-Trp344), or the START domain (Asp345-Leu556) were expressed in E. coli, then purified using a Ni-Sepharose column (Fig. 1B).
As shown in Fig. 2A, the thioesterase activities of WT and THIO increased in a nearly linear manner, dependent upon the amount of purified protein. Figure 2 panels B and C show a time course of the hydrolysis of acetyl-CoA at various concentrations by WT (25 ng) and THIO (120 ng). The thioesterase reactions proceeded linearly up to 15 min and then enzyme activities were saturated via a 60 min incubation with 1 mM acetyl-CoA. To determine the initial velocity rate for hydrolysis of CoA compounds by the purified proteins, enzyme activities were assayed at 5 min or less in subsequent experiments using a substrate concentration of 1 mM. As shown in Fig. 2D, the specific activity of the WT protein (74.4 ± 2.5 μmol/mg/min) was about five times higher than that of THIO (15.7 ± 0.4 μmol/mg/min). START did not have enzymatic activity. Figure 2E shows a Lineweaver-Burk plot of WT and THIO at various concentrations of acetyl-CoA (0.125–2.0 mM). The apparent Vmax and Km values were as follows. WT (Vmax = 125 ± 7.5 μmol/min/mg, Km = 0.36 ± 0.03 mM), THIO (Vmax = 23.4 ± 0.6 μmol/min/mg, Km = 0.36 ± 0.01 mM). As mentioned in below (Fig. 3), 45.3% of WT forms active tetramers, and 55.7% of THIO forms a mixture of active tetramer (31.8%) and dimer (23.9%) in the reaction. Considering this, kcat values of WT and THIO are calculated as 7.26 ± 0.44 × 104 S−1 and 1.99 ± 0.05 × 103 S−1, respectively. Thus, Vmax and kcat values of THIO were reduced compared with those of WT. Therefore, the START domain is important for typical catalytic activity of the enzyme. In contrast, Km values of WT and THIO were the same. Thus, the START domain has little effect on the affinity between ACOT12 and substrates.
Fig. 2.

Characterization of ACOT12 and mutant proteins. A: Thioesterase activity was measured with various amounts of WT (12.5–100 ng) and THIO (30–240 ng) proteins using 1 mM acetyl-CoA for 5 min. Open circles, WT; closed circles, THIO. B, C: Time course of acetyl-CoA thioesterase activity. Acetyl-CoA at the indicated concentration was incubated with WT (25 ng) (B) or THIO (120 ng) (C) for the indicated time (5–60 min or 5–100 min). Closed circles, 2 mM (acetyl-CoA); open squares, 1 mM; closed triangles, 0.5 mM; open diamonds, 0.25 mM; closed squares, 0.125 mM. D: Specific activities of WT, THIO, and START proteins. E: Lineweaver-Burk plot. Open circles, WT; closed circles, THIO. F: Substrate specificity of WT against various short chain CoA compounds (1 mM). Acetyl-CoA, open squares; acetoacetyl-CoA, closed circles; butyryl (C4)-CoA, open triangles; octanoyl (C8)-CoA, closed triangles; HMG-CoA, open circles. Values are shown as the mean ± SD (n = 3).
Fig. 3.

Oligomerization of ACOT12. Elution profile of (A) WT or (B) THIO by gel filtration. To induce oligomerization, proteins were preincubated with 2 mM ATP for 30 min, and then applied to the column. Fractions were analyzed by immunoblotting using an anti-ACOT12 antibody. The chemiluminescence signal for each protein band was calculated. Open squares, without ATP treatment; closed squares, with ATP treatment. Arrowheads indicate the molecular weight of standard proteins. Arrows indicate the apparent molecular weight of each protein.
We next analyzed the substrate specificity of WT using various short carbon hydrate chains as provided by CoA compounds (1 mM). As shown in Fig. 2F, acetyl-CoA was the most favorable substrate, followed by butyryl-CoA and acetoacetyl-CoA. No hydrolysis was observed for HMG-CoA or octanoyl-CoA. Long chain acyl-CoA substrates such as palmitoyl-CoA and oleoyl-CoA, as well as malonyl-CoA, were also not hydrolyzed (data not shown). The apparent Vmax and Km values were as follows: acetoacetyl-CoA (Vmax = 30.5 ± 3.9 μmol/min/mg, Km = 0.33 ± 0.1 mM), and butyryl-CoA (Vmax = 44.7 ± 4.0 μmol/min/mg, Km = 0.41 ± 0.1 mM).
It was reported that ACOT12 usually forms oligomers (dimers or tetramers) at room temperature and is only active when it forms these oligomers (24). In addition, ATP has been shown to induce tetramerization of ACOT12 (24). To assess protein oligomerization, gel filtration was performed using a HiLoad 16/600 Superdex 200 pg column. As shown in Fig. 3A, when WT was applied to the column without preincubation with ATP, it was eluted from the column at an apparent molecular weight of 77 kDa, the expected molecular weight of a monomer. When WT was preincubated with ATP, a protein peak corresponding to an apparent molecular weight at 322 kDa was observed. This molecular weight is consistent with a tetramer. The peak areas of tetramer and monomer formation after ATP treatment were estimated to be 45.3% and 49.6%, respectively. As shown in Fig. 3B, THIO was eluted from the column at an apparent molecular weight of 44 kDa, consistent with a monomer. After preincubation with ATP, protein peaks corresponding to apparent molecular weights of 182 and 88 kDa were observed, consistent with a tetramer and a dimer formation, respectively. The peak areas of tetramer, dimer, and monomer formation after ATP treatment were 31.8%, 23.9%, and 43.1%, respectively. Therefore, about 56% of THIO forms active tetramer or dimer following treatment with ATP. These results indicate that almost half of WT or THIO proteins can form active oligomers in the presence of ATP. It should also be noted that THIO formed both dimers and tetramers, whereas WT formed only tetramers after treatment with ATP. These results also suggest that the START domain in ACOT12 is important for proper tetramer formation after dimer formation.
Effect of lipids on ACOT12 activity
The START domain is thought to act as a lipid binding, transferring, and sensing domain for phospholipids, sterols, and sphingolipids. With this in mind, we next tested the acetyl-CoA thioesterase activity of WT and THIO enzymes in the presence of various types of lipids (phospholipids, fatty acids, sphingolipids, and sterols) or G3P. Phospholipids were sonicated with a probe-type sonicator to form small unilamellar phospholipid vesicles, which were then added to the reaction mixture. Monomer or micellar forms of 1-oleoyl LPA, palmitic acid, or oleic acid were added to the reaction mixture. The critical micelle concentrations (CMCs) of 1-oleoyl LPA, palmitic acid, and oleic acid in the reaction were around 60, 4, and 6 μM, respectively (25, 26). Sterols were dissolved as cyclodextrin complexes and added to the reaction mixtures. As shown in Fig. 4A, the enzymatic activity of WT was dramatically inhibited by DOPA and egg yolk PA vesicles. These lipids showed inhibitory effects at a concentration of 1 μM. 1-Oleoyl LPA at a concentration of 25, 100, or 500 μM also had inhibitory effects. Because the CMC of 1-oleoyl LPA was around 60 μM, it is thought that both monomer and micelle forms of 1-oleoyl LPA have inhibitory effects on WT. We found that 500 μM of DMPG and oleic acid (micellar form) also inhibited WT activity. Other phospholipids, sphingolipids, and sterols had no significant effects. In contrast to what was observed for WT, the THIO fragment, which lacks a START domain, exhibited an increase in thioesterase activity following treatment with DOPA, egg PA, 1-oleoyl LPA, or DMPG (Fig. 4B). Oleic acid at 500 μM inhibited THIO similar to WT. Egg yolk PA, which has heterogeneous fatty acid residues in a diacylglycerol moiety, was as effective as DOPA. In contrast, DOPC, which has the same diacylglycerol moiety as DOPA, did not show significant effects on either enzyme. G3P also did not show any significant effects. These results indicate that the head moiety, possibly the phosphate group of PA or LPA, and hydrophobicity of at least one acyl chain are important for enzymatic regulation of WT and THIO.
Fig. 4.

Effect of lipids on thioesterase activity of ACOT12. The thioesterase activities of WT (25 ng) (A) or THIO (120 ng) (B) were measured in the presence of control vehicle (Cont), phospholipids, sphingolipids, fatty acids, sterols, or G3P at 1, 5, 25, 100, and 500 μM. Sterols were used at concentrations of 100 μM or below due to insolubility. Enzyme activities were measured using 1 mM acetyl-CoA for 5 min. Values are shown as the mean ± SD (n = 3). *P < 0.001 as compared with control. PG, phosphatidylglycerol; PI, phosphatidylinositol; PS, phosphatidylserine.
To assess the contribution of the START domain on ACOT12 activity, we next investigated the enzymatic activity of THIO in the presence or absence of isolated START protein. If the isolated START protein is efficient, THIO activity should be activated (Fig. 2E), or inhibited by DOPA similar to WT. However, no significant change was observed (supplementary Fig. I).
We next explored the enzymatic properties of the inhibition of WT activity by DOPA and 1-oleoyl LPA. As shown in Fig. 5A, WT activity could be inhibited robustly (20% of control levels) with addition of only 3 μM DOPA. WT activity was also inhibited by 1-oleoyl LPA. Inhibition of WT with 1-oleoyl LPA was less robust than inhibition by DOPA. WT activity was strongly inhibited by the addition of the monomer form of 1-oleoyl LPA and less inhibition was observed for a micellar form of the lipid; i.e., maximum inhibition by LPA was obtained at a concentration lower than its CMC (60 μM). The half-maximal inhibitory concentration (IC50) was 1.2 μM for DOPA and 16.1 μM for 1-oleoyl LPA (Fig. 5A). In contrast, acetyl-CoA thioesterase activity of THIO activity was significantly enhanced (almost 200% of control values) following the addition of DOPA up to about 5 μM (Fig. 5B). 1-Oleoyl LPA also enhanced THIO activity under CMC. Although the LPA concentration increased more than the CMC, the enhancement of THIO activity by the lipid did not increase. These results suggest that monomer forms of LPA actually activate THIO activity than its micellar forms.
Fig. 5.

Effects of acetyl-CoA and PA/LPA concentrations on the thioesterase activity of ACOT12. Thioesterase activity of WT (25 ng) (A) and THIO (120 ng) (B) was measured in the presence of DOPA (closed circles) or 1-oleoyl LPA (open triangles) at the indicated concentrations (0.78–100 μM). Enzyme activities were analyzed using 1 mM acetyl-CoA for 5 min. Saturation curve (C) and Lineweaver-Burk plot (D) in the presence of DOPA. WT was incubated with various concentrations of acetyl-CoA (0.0625, 0.125, 0.25, 0.5, 1.0, or 2.0 mM) without (open circles) or with DOPA (1 μM, closed circles; 2 μM, closed squares; or 4 μM, closed triangles) for 5 min. Values are shown as the mean ± SD (n = 3).
We next looked at the mechanism of inhibition of WT activity by DOPA. To address this, we performed kinetic experiments based on parameters obtained for thioesterase activity at various concentrations of acetyl-CoA in the absence or presence of DOPA (1, 2, and 4 μM). As shown in Fig. 5C, a typical Michaelis-Menten curve was obtained when the initial velocity was plotted against the acetyl-CoA concentration. Lineweaver-Burk plots for the inhibition of DOPA are shown in Fig. 5D. Vmax, Km, and Vmax/ Km are also shown in Table 1. Both Vmax and Vmax/ Km values decreased in a DOPA concentration-dependent manner, whereas Km values were nearly constant. These results clearly indicate that DOPA is a noncompetitive inhibitor of WT thioesterase activity. The slope (Km /Vmax) was plotted against inhibitor concentration (Fig. 5D, inset). The apparent Ki value for DOPA was estimated to be 0.50 ± 0.11 μM.
TABLE 1.
Kinetics of ACOT12 inhibition by DOPA
| Inhibitor | Vmax (μmol/min/min) | Km (mM) | Vmax/ Km |
| Control | 125.0 ± 7.5 | 0.36 ± 0.03 | 34.9 × 10 |
| 1 μM DOPA | 44.6 ± 3.0 | 0.32 ± 0.03 | 13.9 × 10 |
| 2 μM DOPA | 26.5 ± 1.9 | 0.40 ± 0.06 | 6.6 × 10 |
| 4 μM DOPA | 20.3 ± 3.0 | 0.43 ± 0.17 | 4.7 × 10 |
Thioesterase activity of ACOT12 (WT) was measured in the presence of DOPA at the indicated concentrations. Values are shown as the mean ± SD (n = 3).
Interaction of ACOT12 with PA
Because PA is a noncompetitive inhibitor, it can interact with an allosteric site of ACOT12 that is distinct from the substrate binding site. We next assessed lipid-protein interactions between PA and ACOT12 using butyl-Sepharose beads conjugated to DOPC or DOPA. Purified WT, THIO, or START proteins were mixed with DOPC- or DOPA-conjugated beads and incubated for 30 min at room temperature to allow for binding. Proteins bound to the beads were then pelleted (beads), and both these and proteins remaining in the supernatant were analyzed by immunoblotting using an anti-ACOT12 antibody. The results clearly demonstrated that both the WT and THIO proteins could specifically bind to DOPA-conjugated beads, although both showed nonspecific weak binding to the control and DOPC-conjugated beads (Fig. 6A). START could not interact with PA-conjugated beads. These results are in good agreement with the level of enzyme activity in the supernatant (Fig. 6B). These results suggest that the N-terminal thioesterase domain is essential for a direct interaction with PA. We subsequently asked if acetyl-CoA interferes with the interaction between ACOT12 and PA. ACOT12-bound DOPA beads were incubated with the indicated concentrations of acetyl-CoA. As shown in Fig. 6C, the ACOT12-DOPA interaction was not affected by acetyl-CoA, suggesting that the binding site for PA is different from that for acetyl-CoA. This is consistent with the finding that the enzymatic activity of ACOT12 is inhibited by PA allosterically and in a noncompetitive manner.
Fig. 6.
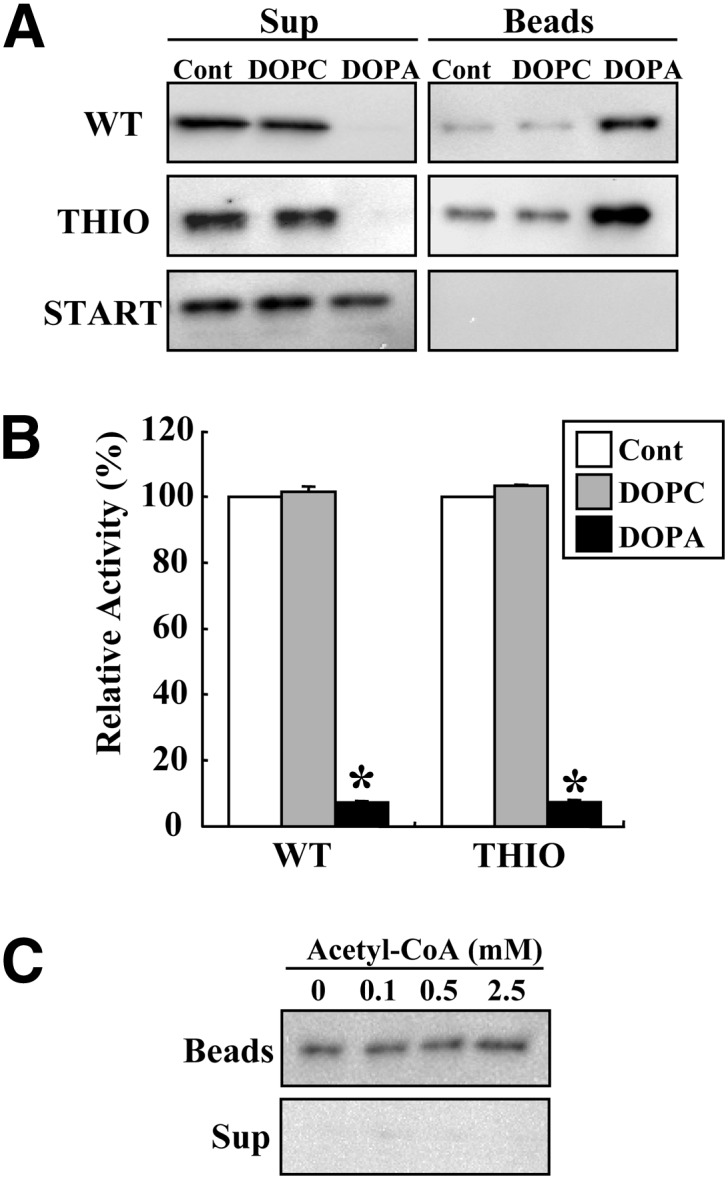
Interaction of ACOT12 with PA. A: Tag-purified WT, THIO, and START were incubated with butyl-Sepharose control (Cont) or butyl-Sepharose conjugated with DOPC or DOPA (20 μl) for 30 min. After centrifugation at 3,000 g for 5 min, proteins bound to the lipid-conjugated beads (Beads) or remaining in the supernatant (Sup) were analyzed by immunoblotting using an anti-ACOT12 antibody. B: Enzymatic activity in the supernatant was assayed using acetyl-CoA (1 mM) as a substrate for 5 min. Values are shown as the mean ± SD (n = 3). *P < 0.001 as compared with control. C: ACOT12-bound DOPA beads were incubated with the indicated concentration of acetyl-CoA, and proteins bound to the beads (Beads) or in the supernatant (Sup) after centrifugation were analyzed by immunoblotting using an anti-ACOT12 antibody.
ACOT12 oligomerization and dissociation
Next, we investigated the effect of PA on ACOT12 dissociation. We prepared a protein complex of 3xFlag-ACOT12 and V5-ACOT12, and analyzed the effects of PA on its interaction with ACOT12. Briefly, both 3xFlag-tagged protein and V5-tagged protein were cotransfected into Hepa-1 cells, and 3xFlag-protein was immunoprecipitated with anti-Flag antibody-conjugated agarose beads at room temperature. Then, V5-tagged proteins that coimmunoprecipitated with Flag-tagged proteins were detected by immunoblotting with anti-V5 antibody. As shown in Fig. 7A, V5-WT coprecipitated with 3xFlag-WT. Addition of DOPA to the V5- and 3xFlag-tagged protein complexes did not show any significant effects on dissociation. It has been reported that ACOT12 dissociates into an inactive monomer when incubated at low temperature (24). Consistent with this, we found that if the beads were incubated on ice, V5-WT dissociated from the beads and eluted with the supernatant. These results indicate that the inhibition of ACOT12 by PA is not due to disruption of protein oligomers.
Fig. 7.

Oligomerization and dissociation of ACOT12. Hepa-1 cells were cotransfected with 3xFlag-WT and V5-WT (A) or with 3xFlag-THIO and V5-THIO or 3xFlag-WT and V5-THIO (B). Protein complexes were immunoprecipitated using anti-Flag antibody-conjugated beads at room temperature. After beads were washed and incubated with TBS containing 100 μM DOPA at room temperature or with TBS on ice for 60 min, proteins bound to the beads (Ppt) or dissociated into the supernatant (Sup) were analyzed using anti-Flag or anti-V5 antibodies. IP, immunoprecipitation; WB, Western blot.
We next analyzed protein interactions between 3xFlag-WT and V5-THIO or 3xFlag-THIO and V5-THIO. As shown in Fig. 7B, V5-THIO coimmunoprecipitated with 3xFlag-WT and 3xFlag-THIO. This indicates that the THIO domain, not the START domain, is necessary for ACOT12 oligomerization. The addition of DOPA did not disrupt WT-THIO or THIO-THIO protein interactions.
Detection of ACOT12 protein in rat tissues or cultured hepatic cell lines
To determine if ACOT12 protein is present in various tissues, we generated anti-ACOT12 antisera and performed immunoblot analysis with a variety of rat tissues. As shown in Fig. 8A, ACOT12 is detected at high levels in the liver, with the next highest intensity of staining detected in the kidney and the intestine. The presence of ACOT12 was also assayed in the following hepatic cell lines: Fa2N-4 (an SV40 immortalized human hepatocyte cell line), Hepa-1 (a murine-derived hepatoma cell line), and HepG2 (a human liver carcinoma cell line) (Fig. 8B). Despite the fact that ACOT12 protein was detectable in liver tissue, ACOT12 was not detected by immunoblotting in hepatic cell lines.
Fig. 8.

Distribution of ACOT12. The presence of ACOT12 in (A) rat tissues or (B) hepatic cell lines was analyzed by immunoblot analysis using an anti-ACOT12 antibody. β-actin was used as a loading control.
Transcriptional regulation of hepatic ACOT12 by insulin
Because ACOT12 is the major enzyme known to hydrolyze cytosolic acetyl-CoA in the liver, which is essential for lipid biosynthesis, we reasoned that transcription of ACOT12 might be regulated by hormones involved in energy homeostasis. To investigate this, we next analyzed the effects of insulin on ACOT12 mRNA levels. We used rat primary hepatocytes, as based on the results of our immunoblot analysis; hepatic cell lines do not appear to express ACOT12 protein. The mRNA and protein levels of ACOT12 of freshly isolated primary hepatocytes before plating, indicated as day 0, and the cells cultured for the day indicated (day 1, 2, 4, or 6) in the presence or absence of FBS, were analyzed by quantitative PCR and immunoblotting, respectively. As shown in Fig. 9A, ACOT12 mRNA of day 0 was drastically decreased after plating. The ACOT12 mRNA at day 1 was about 2% of day 0. ACOT12 mRNA levels were continuously decreased over time. As shown in Fig. 9B, the protein levels of ACOT12 were also decreased in good accordance with mRNA expression, and ACOT12 expression was not preserved by FBS treatment. Thus, we decided to use primary hepatocytes at day 1 to analyze hormonal control. As shown in Fig. 9C, D, a significant decrease in ACOT12 mRNA and protein levels was observed following treatment with insulin (100 nM). It was previously reported that subcutaneous injection of clofibrate increased ACOT12 activity in the rat liver (14). We confirmed that treatment of rat hepatocytes at day 1 with this drug in vitro also increased both ACOT12 mRNA and protein levels (Fig. 9E, F). Next, we investigated whether the drug can also enhance ACOT12 mRNA expression of cells at day 6. As shown in supplementary Fig. II, the level of ACOT12 mRNA was increased about 1.6-fold by treatment of day 6 cells with clofibrate, although no visible increase of ACOT12 protein was observed.
Fig. 9.

Transcriptional regulation of ACOT12 in rat hepatocytes. Rat primary hepatocytes were first cultured with 10% FBS and 1 μM of dexamethasone and insulin. Then, 4 h after plating, cells were cultured for the indicated number of days in Williams’ medium E without insulin and dexamethasone in the presence (+) or absence (−) of 10% FBS. Finally, the levels of ACOT12 mRNA (A) and protein (B) were determined by quantitative PCR and immunoblot analysis, respectively. Day 0 means freshly isolated hepatocytes before plating. β-Actin was used as a loading control (Cont). *P < 0.001 as compared with day 0 cells. Rat primary hepatocytes (day 1) cultured without FBS were incubated with or without 100 nM insulin (C, D) or 100 μM clofibrate (E, F) for the time indicated. Finally, the levels of ACOT12 mRNA (C, E) and protein (D, F) were determined. Values are shown as the mean ± SD from three independent culture dishes. *P < 0.05 and **P < 0.001 as compared with control cells incubated with vehicle.
DISCUSSION
Cytosolic acetyl-CoA is the starting compound for biosynthesis of both fatty acids and cholesterol. Because ACOT12 appears to be the major enzyme responsible for determining the rate of degradation of cytosolic acetyl-CoA in the liver, the enzymatic and transcriptional regulation of ACOT12 is important for the control of lipid biosynthesis. After screening with several types of lipids here, we determined that the enzymatic activity of ACOT12 is inhibited by PA in vitro in a noncompetitive manner. The results clearly show that the START domain of ACOT12 plays an important role in the inhibitory effects of PA, as enzyme activity of THIO, which does not contain the START domain, was enhanced rather than inhibited by PA (Figs. 4, 5). Although START domains have been thought to be involved in binding, transferring, and sensing lipids, the START domain of ACOT12 does not appear bind to PA, whereas our data show that THIO, a catalytic domain of ACOT12, is essential for PA binding (Fig. 6). As shown in Fig. 2E, Km values of WT and THIO were the same. Thus, the START domain has little effect on the affinity between ACOT12 and substrates. On the other hand, Vmax and kcat values of THIO were reduced compared with those of WT. Therefore, the START domain is important for typical catalytic activity of the enzyme.
Oligomerization is important for enzymatic activity of ACOT12 (9, 20). In this study, we showed that the THIO domain is necessary for proper oligomerization and that ACOT12 oligomers are not disrupted following the addition of PA (Fig. 7). The results of our gel filtration study clearly show that WT forms tetramers after ATP treatment whereas THIO forms both dimers and tetramers (Fig. 3). This suggests that the START domain of ACOT12 is important to facilitate the formation of tetramer. The detailed mechanisms underlying how the START domain affects enzyme activity or oligomerization have yet to be fully characterized. Nevertheless, it seems reasonable to propose that the START domain might modulate the whole enzyme and/or the active site, for example by interacting with PA at the N-terminal catalytic domain.
There are 12 ACOT protein family members in the mouse. Based on structure predictions, five of these can be placed in the hotdog-fold enzyme superfamily (7, 15, 27). Among these, ACOT13, also known as thioesterase superfamily member 2, contains a 140 amino acid hotdog-fold thioesterase domain, and interestingly, its enzymatic activity can be inhibited by PC vesicles (28). Moreover, addition of PA to PC vesicles reportedly led to a more significant inhibitory effect (molar ratio of PA/PC, 0.2 or 0.3) on enzymatic activity than addition of vesicles containing only PC. Additionally, binding of ACOT13 to PC vesicles increased with increasing PA content in PC vesicles. Thus, PA shows similar inhibitory effects on the enzymatic activity of both ACOT12 and ACOT13, although PC was not effective with ACOT12. It will be interesting to determine if the enzymatic activity of other members of the hotdog-fold thioesterase superfamily can also be inhibited by PA and whether or not they bind to PA.
The thioesterase activity of ACOT11, known as thioesterase superfamily member 1 or StarD14, another hotdog-fold ACOT member with a C-terminal START domain, was recently characterized (29). This enzyme preferentially hydrolyzes long chain acyl-CoA compounds such as palmitoyl-CoA and myristoyl-CoA. An ACOT11 mutant lacking the START domain showed a higher Km value and lower Vmax value with long chain acyl-CoA than intact ACOT11. Interestingly, mixing the mutant with the deleted START domain led to reduced Km and increased Vmax values, close to what was observed for intact ACOT11. It was also reported that the enzymatic activity of ACOT13 was increased by PC-transfer protein, which consists entirely of a START domain (30). We do not yet have a detailed understanding of how START domain-containing proteins facilitate enzymatic activity of ACOT11 or ACOT13. However, we have shown that the enzymatic activity of THIO, a mutant form of ACOT12 lacking the START domain, did not change in the presence of deleted START domain (supplementary Fig. I). Thus, we suggest that although the START domains in ACOT11 and ACOT12 are each important for optimal thioesterase activity, their specific roles or mechanisms of action might be different.
In this study, we found that the addition of insulin led to a decrease in the expression of ACOT12 in rat hepatocytes. We speculate on the relationship between downregulation of ACOT12 by insulin and inhibition of ACOT12 activity by PA and LPA as follows. When lipogenesis is elevated by insulin, the biosynthesis of LPA and then PA (precursors of diacylglycerol and triacylglycerol) is upregulated via activation of the de novo synthesis pathway (31–35). Insulin can also lead to an increase in PA production by PC degradation via the activation of phospholipase D, or of phospholipase C and diacylglycerol kinase (35–37). Higher levels of PA or LPA might in turn act as negative regulators of ACOT12 (i.e., create a negative feedback loop), such that the amount of cytosolic acetyl-CoA available for lipogenesis would then increase. At the same time, insulin treatment also leads to downregulation of ACOT12 transcription, leading to a subsequent increase in cytosolic acetyl-CoA. Taken as a whole, these results suggest that ACOT12 contributes to the regulation of lipid biosynthesis by controlling the rate of degradation of cytosolic acetyl-CoA, especially in the liver.
Much has been learned about the synthetic regulation of cytosolic acetyl-CoA such as by ATP-citrate lyase or acetyl-CoA synthetase. In contrast, degradative regulation has not been well characterized. The results of the present study may facilitate research regarding the biosynthesis of lipids or other compounds synthesized from acetyl-CoA.
Supplementary Material
Acknowledgments
The authors thank Drs. Hisashi Ueta and Kenjiro Matsuno of the Department of Anatomy (Macro) for technical support with primary culture of rat hepatocytes, and Dr. Takashi Namatame of the Clinical Research Center for DNA sequencing.
Footnotes
Abbreviations:
- ACOT12
- acyl-CoA thioesterase 12
- CMC
- critical micelle concentration
- DMPG
- dimyristoyl phosphatidylglycerol
- DOPA
- dioleoyl phosphatidic acid
- DOPC
- dioleoyl phosphatidylcholine
- G3P
- glycerol 3-phosphate
- LPA
- lysophosphatidic acid
- PA
- phosphatidic acid
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- PMSF
- phenylmethylsulfonyl fluoride
- START
- steroidogenic acute regulatory protein-related lipid transfer
- THIO
- thioesterase domain
- WT
- wild type
This study was supported by the Naito Foundation Subsidy for the Promotion of Specific Research Projects (Y.H.), an Investigator-Initiated Research Grant from Dokkyo Medical University (No. 2012-01-2), and a Grant-in-Aid for Young Scientists (B) from the Ministry of Education, Culture, Sports, Science and Technology, Japanese Government.
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of two figures.
REFERENCES
- 1.Beigneux A. P., Kosinski C., Gavino B., Horton J. D., Skarnes W. C., Young S. G. 2004. ATP-citrate lyase deficiency in the mouse. J. Biol. Chem. 279: 9557–9564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luong A., Hannah V. C., Brown M. S., Goldstein J. L. 2000. Molecular characterization of human acetyl-CoA synthetase, an enzyme regulated by sterol regulatory element-binding proteins. J. Biol. Chem. 275: 26458–26466 [DOI] [PubMed] [Google Scholar]
- 3.Ikeda Y., Yamamoto J., Okamura M., Fujino T., Takahashi S., Takeuchi K., Osborne T. F., Yamamoto T. T., Ito S., Sakai J. 2001. Transcriptional regulation of the murine acetyl-CoA synthetase 1 gene through multiple clustered binding sites for sterol regulatory element-binding proteins and a single neighboring site for Sp1. J. Biol. Chem. 276: 34259–34269 [DOI] [PubMed] [Google Scholar]
- 4.Berrard S., Brice A., Lottspeich F., Braun A., Barde Y. A., Mallet J. 1987. cDNA cloning and complete sequence of porcine choline acetyltransferase: in vitro translation of the corresponding RNA yields an active protein. Proc. Natl. Acad. Sci. USA. 84: 9280–9284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shindou H., Hishikawa D., Nakanishi H., Harayama T., Ishii S., Taguchi R., Shimizu T. 2007. A single enzyme catalyzes both platelet-activating factor production and membrane biogenesis of inflammatory cells. Cloning and characterization of acetyl-CoA:LYSO-PAF acetyltransferase. J. Biol. Chem. 282: 6532–6539 [DOI] [PubMed] [Google Scholar]
- 6.Van Damme P., Hole K., Pimenta-Marques A., Helsens K., Vandekerckhove J., Martinho R. G., Gevaert K., Arnesen T. 2011. NatF contributes to an evolutionary shift in protein N-terminal acetylation and is important for normal chromosome segregation. PLoS Genet. 7: e1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hunt M. C., Yamada J., Maltais L. J., Wright M. W., Podesta E. J., Alexson S. E. 2005. A revised nomenclature for mammalian acyl-CoA thioesterases/hydrolases. J. Lipid Res. 46: 2029–2032 [DOI] [PubMed] [Google Scholar]
- 8.Suematsu N., Isohashi F. 2006. Molecular cloning and functional expression of human cytosolic acetyl-CoA hydrolase. Acta Biochim. Pol. 53: 553–561 [PubMed] [Google Scholar]
- 9.Prass R. L., Isohashi F., Utter M. F. 1980. Purification and characterization of an extramitochondrial acetyl coenzyme A hydrolase from rat liver. J. Biol. Chem. 255: 5215–5223 [PubMed] [Google Scholar]
- 10.Suematsu N., Okamoto K., Shibata K., Nakanishi Y., Isohashi F. 2001. Molecular cloning and functional expression of rat liver cytosolic acetyl-CoA hydrolase. Eur. J. Biochem. 268: 2700–2709 [DOI] [PubMed] [Google Scholar]
- 11.Isohashi F., Nakanishi Y., Sakamoto Y. 1983. Factors affecting the cold inactivation of an acetyl-coenzyme-A hydrolase purified from the supernatant fraction of rat liver. Eur. J. Biochem. 134: 447–452 [DOI] [PubMed] [Google Scholar]
- 12.Söling H. D., Rescher C. 1985. On the regulation of cold-labile cytosolic and of mitochondrial acetyl-CoA hydrolase in rat liver. Eur. J. Biochem. 147: 111–117 [DOI] [PubMed] [Google Scholar]
- 13.Matsunaga T., Isohashi F., Nakanishi Y., Sakamoto Y. 1985. Physiological changes in the activities of extramitochondrial acetyl-CoA hydrolase in the liver of rats under various metabolic conditions. Eur. J. Biochem. 152: 331–336 [DOI] [PubMed] [Google Scholar]
- 14.Ebisuno S., Isohashi F., Nakanishi Y., Sakamoto Y. 1988. Acetyl-CoA hydrolase: relation between activity and cholesterol metabolism in rat. Am. J. Physiol. 255: R724–R730 [DOI] [PubMed] [Google Scholar]
- 15.Dillon S. C., Bateman A. 2004. The Hotdog fold: wrapping up a superfamily of thioesterases and dehydratases. BMC Bioinformatics. 5: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alpy F., Tomasetto C. 2005. Give lipids a START: the StAR-related lipid transfer (START) domain in mammals. J. Cell Sci. 118: 2791–2801 [DOI] [PubMed] [Google Scholar]
- 17.Lavigne P., Najmanivich R., Lehoux J. G. 2010. Mammalian StAR-related lipid transfer (START) domains with specificity for cholesterol: structural conservation and mechanism of reversible binding. Subcell. Biochem. 51: 425–437 [DOI] [PubMed] [Google Scholar]
- 18.Clark B. J. 2012. The mammalian START domain protein family in lipid transport in health and disease. J. Endocrinol. 212: 257–275 [DOI] [PubMed] [Google Scholar]
- 19.Horibata Y., Sugimoto H. 2010. StarD7 mediates the intracellular trafficking of phosphatidylcholine to mitochondria. J. Biol. Chem. 285: 7358–7365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suematsu N., Okamoto K., Isohashi F. 2003. Simple and unique purification by size-exclusion chromatography for an oligomeric enzyme, rat liver cytosolic acetyl-coenzyme A hydrolase. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 790: 239–244 [DOI] [PubMed] [Google Scholar]
- 21.Edwards H. C., Crumpton M. J. 1991. Ca(2+)-dependent phospholipid and arachidonic acid binding by the placental annexins VI and IV. Eur. J. Biochem. 198: 121–129 [DOI] [PubMed] [Google Scholar]
- 22.Bartlett G. R. 1959. Phosphorus assay in column chromatography. J. Biol. Chem. 234: 466–468 [PubMed] [Google Scholar]
- 23.Seglen P. O. 1976. Preparation of isolated rat liver cells. Methods Cell Biol. 13: 29–83 [DOI] [PubMed] [Google Scholar]
- 24.Isohashi F., Nakanishi Y., Matsunaga T., Sakamoto Y. 1984. A cold-labile acetyl-coenzyme-A hydrolase from the supernatant fraction of rat liver. Reactivation and reconstitution of the active species from the inactive monomer. Eur. J. Biochem. 142: 177–181 [DOI] [PubMed] [Google Scholar]
- 25.Li Z., Mintzer E., Bittman R. 2004. The critical micelle concentrations of lysophosphatidic acid and sphingosylphosphorylcholine. Chem. Phys. Lipids. 130: 197–201 [DOI] [PubMed] [Google Scholar]
- 26.Richieri G. V., Ogata R. T., Kleinfeld A. M. 1992. A fluorescently labeled intestinal fatty acid binding protein. Interactions with fatty acids and its use in monitoring free fatty acids. J. Biol. Chem. 267: 23495–23501 [PubMed] [Google Scholar]
- 27.Kirkby B., Roman N., Kobe B., Kellie S., Forwood J. K. 2010. Functional and structural properties of mammalian acyl-coenzyme A thioesterases. Prog. Lipid Res. 49: 366–377 [DOI] [PubMed] [Google Scholar]
- 28.Wei J., Kang H. W., Cohen D. E. 2009. Thioesterase superfamily member 2 (Them2)/acyl-CoA thioesterase 13 (Acot13): a homotetrameric hotdog fold thioesterase with selectivity for long-chain fatty acyl-CoAs. Biochem. J. 421: 311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han S., Cohen D. E. 2012. Functional characterization of thioesterase superfamily member 1/Acyl-CoA thioesterase 11: implications for metabolic regulation. J. Lipid Res. 53: 2620–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanno K., Wu M. K., Agate D. S., Fanelli B. J., Wagle N., Scapa E. F., Ukomadu C., Cohen D. E. 2007. Interacting proteins dictate function of the minimal START domain phosphatidylcholine transfer protein/StarD2. J. Biol. Chem. 282: 30728–30736 [DOI] [PubMed] [Google Scholar]
- 31.Farese R. V., Konda T. S., Davis J. S., Standaert M. L., Pollet R. J., Cooper D. R. 1987. Insulin rapidly increases diacylglycerol by activating de novo phosphatidic acid synthesis. Science. 236: 586–589 [DOI] [PubMed] [Google Scholar]
- 32.Farese R. V., Cooper D. R., Konda T. S., Nair G., Standaert M. L., Davis J. S., Pollet R. J. 1988. Mechanisms whereby insulin increases diacylglycerol in BC3H-1 myocytes. Biochem. J. 256: 175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baldini P. M., Zannetti A., Donchenko V., Dini L., Luly P. 1992. Insulin effect on isolated rat hepatocytes: diacylglycerol-phosphatidic acid interrelationship. Biochim. Biophys. Acta. 1137: 208–214 [DOI] [PubMed] [Google Scholar]
- 34.Nagle C. A., Klett E. L., Coleman R. A. 2009. Hepatic triacylglycerol accumulation and insulin resistance. J. Lipid Res. 50(Suppl.): S74–S79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farese R. V. 2001. Insulin-sensitive phospholipid signaling systems and glucose transport. Update II. Exp. Biol. Med. (Maywood). 226: 283–295 [DOI] [PubMed] [Google Scholar]
- 36.Donchenko V., Zannetti A., Baldini P. M. 1994. Insulin-stimulated hydrolysis of phosphatidylcholine by phospholipase C and phospholipase D in cultured rat hepatocytes. Biochim. Biophys. Acta. 1222: 492–500 [DOI] [PubMed] [Google Scholar]
- 37.Novotná R., De Vito P., Currado L., Luly P., Baldini P. M. 2003. Involvement of phospholipids in the mechanism of insulin action in HEPG2 cells. Physiol. Res. 52: 447–454 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.