

Abstract
We showed earlier that nutritional stress like starvation or high-fat diet resulted in phenotypic changes in the lipidomes of hepatocyte lipid droplets (LDs), representative for the pathophysiological status of the mouse model. Here we extend our former study by adding genetic stress due to knockout (KO) of adipocyte triglyceride lipase (ATGL), the rate limiting enzyme in LD lipolysis. An intervention trial for 6 weeks with male wild-type (WT) and ATGL-KO mice was carried out; both genotypes were fed lab chow or were exposed to short-time starvation. Isolated LDs were analyzed by LC-MS/MS. Triacylglycerol, diacylglycerol, and phosphatidylcholine lipidomes, in that order, provided the best phenotypic signatures characteristic for respective stresses applied to the animals. This was evidenced at lipid species level by principal component analysis, calculation of average values for chain-lengths and numbers of double bonds, and by visualization in heat maps. Structural backgrounds for analyses and metabolic relationships were elaborated at lipid molecular species level. Relating our lipidomic data to nonalcoholic fatty liver diseases of nutritional and genetic etiologies with or without accompanying insulin resistance, phenotypic distinction in hepatocyte LDs dependent on insulin status emerged. Taken together, lipidomes of hepatocyte LDs are sensitive responders to nutritional and genetic stress.
Keywords: adipocyte triglyceride lipase, fasting, nutritional stress, super stress, steatosis, mass spectrometry, Lipid Data Analyzer, principal component analysis, lipid species
In a preceding study, we carried out intervention trials with wild-type (WT) mice fed normal chow, exposed to short-time starvation, or treated with high-fat diet (HFD). The aim was to assess the effect of these forms of nutritional stress on the lipidome of lipid droplets (LDs) isolated from hepatocytes. Lipidomic analysis by LC-MS enabled profiling of all species from glycerolipid and glycerophospholipid classes and revealed particularly the triacylglycerol (TG) lipidome to be best suited for phenotyping nutritional stresses applied to the animals. In addition, structural analysis by MS/MS at lipid molecular species level provided metabolic relationships characteristic for hepatocyte lipid metabolism (1). To further probe the phenotypic information potential inherent in the lipidome of hepatocyte LDs, we have now extended the question to genetic stress; we have characterized a mouse model lacking adipocyte triglyceride lipase (ATGL), which shows a marked defect in lipid metabolism in many tissues including the liver.
ATGL was discovered in 2004 to be the rate-limiting enzyme for degradation of TGs to diacylglycerols (DGs) stored in adipocyte LDs (2). It is an enzyme located on the LD surface and starts liberating fatty acids by hydrolyzing TGs upon activation via protein-protein interaction with comparative gene identification 58 (CGI-58). Adrenergic stimulation of adipocytes initiates this first step in lypolysis via generation of cAMP, which in turn activates PKA for phosphorylation of perilipin 1 (and of hormone sensitive lipase for translocation to the LD surface enabling the second step of lipolysis). Phosphorylated perilipin 1 at the LD surface then releases CGI-58 for interaction with ATGL (3). As LDs are quite ubiquitously present in mammalian cells, ATGL is broadly expressed as well. Yet in contrast to adipocytes, expression of ATGL in hepatocytes is low. By the same token, a body of information is known about regulation of ATGL activity in adipocytes, but its regulation in hepatocytes is unexplored. Nonetheless, data on functional properties of ATGL in liver available from systemic and liver-specific knockout (KO) mouse models, or from humans with ATGL deficiency suggest an important role of this enzyme in hepatocytes (4).
It was shown in many animal models that surplus accumulation of liver TGs, generally termed nonalcoholic fatty liver disease, is caused by excess food intake, inflammation, or ER stress and is always accompanied by insulin resistance (5–7). ATGL acting on these surplus TG stores produces intracellular DGs that have been considered to inhibit insulin signaling by activation of PKC isoforms δ, ϵ, and θ, thus blocking in turn phosphorylation of insulin receptor kinase of insulin receptor substrates 1 and 2 (8, 9). Accordingly, studies with obese humans revealed a positive correlation of hepatic insulin resistance with intrahepatic DG levels (10) and a strong correlation of hepatic PKCϵ activation with DG amounts in hepatocyte LDs, but not with total amounts of hepatocyte or liver DGs (11). Of note, PKC signaling is attained by interaction of the kinase with sn-1,2-DGs only, whereas direct products of ATGL-catalyzed degradation of TGs are sn-2,3- and sn-1,3-DGs (12).
The first murine model lacking ATGL was reported by our group in 2006 (13) and featured the prominent though surprising finding of TG accumulation in multiple tissues including liver without systemic insulin resistance. These mice, in addition, developed a torpor-like state with lowered body temperature and oxygen consumption in response to fasting. Furthermore, it was found that ATGL-KO animals displayed enhanced glucose tolerance and insulin sensitivity, although insulin signaling in liver was somewhat impaired by reduced Akt phosphorylation (14). The latter finding, however, was not supported by a subsequent study (15). Also, hepatic ATGL-KO uncoupled glucose intolerance from liver TG accumulation (16, 17).
We found by MS profiling in our preceding study that fasting WT mice resulted in a TG lipidome in hepatocyte LDs enriched in species having very long-chain (VLC)-polyunsaturated fatty acids (PUFAs). HFD administration, however, produced a TG lipidome with less unsaturated TG species in addition to characteristic TG marker species. At lipid molecular species level we recognized that DG species with VLC-PUFAs were precursor candidates for phosphatidylcholine (PC) species, the remainder for TG species (1). Here, we extend these studies to genetic stress-induced lipidomes in hepatocyte LDs that emerge from aberrant lipid metabolism in ATGL-deficient mice. Furthermore, we explore whether lipidomic data can be related to liver steatoses of different etiologies.
The experimental conduct to test our working hypotheses consisted of an intervention study with male WT and ATGL-KO mice that received control chow (FED) or were fasted before sacrifice (FAS). From excised livers we prepared hepatocytes from which LDs were isolated, lipids extracted and subjected to lipidomic LC-MS/MS analysis. Lipid class-specific analyses were carried out at levels of lipid species (profiling) and of lipid molecular species. Samples analyzed were from mouse sample groups WT-FED (control), WT-FAS, KO-FED, and KO-FAS.
MATERIALS AND METHODS
Materials
Materials used were the same as described in our preceding paper (1).
Animals, diets, and intervention study
All animal experiments were performed in compliance with the Austrian animal protection law. The mice were housed and handled in accordance with good animal practice as defined by FELASA (1). The animal welfare committees of the University of Graz and the national authorities approved all animal experiments. Male WT ATGL-KO C57BL/6 mice (13) aged 6 weeks were used in this study. Mice were maintained on a regular light (14 h)-dark (10 h) cycle and fed standard laboratory chow diet (sniff® Spezialdiäten, Soest, Germany) for 6 weeks (FED groups), or were fasted 14 h prior to sacrifice (FAS groups). Three animals per group were kept in one cage with ad libitum access to food and water. During the intervention period, mouse weights were monitored regularly. At the end of the 6 week trial period, animals were euthanized at 8:00 AM under anesthesia.
Isolation procedures, microscopy, and enzyme tests
Isolation of hepatocytes and subsequently hepatic LDs, confocal laser scanning microscopy of isolated hepatocytes, and measurement of total acylglycerols and of proteins present in isolated LDs were carried out essentially as described earlier (1).
Determination of LD lipid profiles and molecular species by LC-MS/MS
LDs isolated from hepatocytes were measured individually (three each for WT-FED, WT-FAS, KO-FED, and KO-FAS sample groups) according to the hyphenated approach described by us earlier (18). In brief, samples were spiked with 40 nmol TG 17:0/17:0/17:0, 1.2 nmol PC 12:0/12:0, 2.4 nmol phosphatidylethanolamine (PE) 12:0/12:0, and 4 nmol phosphatidylserine (PS) 12:0/12:0 as internal standards and lipids were extracted with methyl tert-butyl ether (19). This ether extract was taken to dryness and the remainder solubilized in around 2 ml CHCl3/methanol 1:1 (v/v). For TG class determination 1:100, for all other class determinations 1:4 dilutions were prepared using the same solvent. For LC-MS/MS measurements, 200 μl aliquots were always taken from the dilutions and a solution mix of 45 LIPID MAPS internal standards (supplementary Table I) was added, taken to dryness and resuspended in 200 μl solvent as above.
LC-MS/MS and data analysis
Ultra high-performance liquid chromatography, lipid mass spectrometry, and analysis of lipid molecular species with Lipid Data Analyzer (LDA) (20) were carried out with the same instruments and methods as described before (1).
Statistical treatment of data
Relative values for mass spectrometric data of each lipid species (in percent or per thousand) per lipid class pertain to the intervention study and are means ± SD of three animals per sample group. For mass spectrometry data, total class amounts were calculated as sum from individual lipid species, standardized by sets of internal standards for each lipid class (supplementary Table I) as described previously (18). The standard deviation of the lipid class is a result of the individual standard deviations of each species determined by the law of error propagation (21). An independent two-sample t-test for equal sample sizes and equal variance was used to assess statistical significance of the observed changes at levels of P < 0.05, 0.01, and 0.001, respectively. Quantitative mass spectrometric data for each lipid species in nmol (or μmol) per LD sample are presented as means ± SD (n = 3). These additional values were calculated by LDA based on the same sets of internal standards mentioned above. For better comparability, data were normalized to the amount of acylglycerols measured by the TG kit (InfinityTM Triglycerides Reagent). Principal component analysis (PCA) was performed in R (R Development Core Team, R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria) using the function prcomp with default parameters. Principal component 1 (PC1) and principal component 2 (PC2) loadings are recorded in supplementary Table II.
Supplementary information
Detailed lipidomic data elaborated together with their statistical treatment are listed in supplementary Tables III–X. Data represent quantitative values for lipid species in both their relative contribution (in percent or per thousand) to respective lipid classes and their absolute contribution in nmol or μmol/g total acylglycerols. These data are complemented with those on lipid molecular species resulting from MS/MS analysis of individual lipid species when sufficient amounts were available.
RESULTS
Lipid deposition in hepatocyte LDs in response to genetic and nutritional stresses
Freshly isolated hepatocytes were treated with “green” BODIPY to stain hydrophobic acylglycerols of LDs for inspection by confocal laser scanning microscopy. Figure 1A demonstrates that ATGL deficiency results in accumulation of LDs with enhanced diameters (KO-FED). Diameters were even more enhanced due to nutritional stress exerted by fasting WT animals (WT-FAS). Interestingly, combined genetic and nutritional stresses (KO-FAS, “super stress”) reversed these pathogenic phenotypes almost to a phenocopy of the control (WT-FED), which is likely due to defective adipose tissue lipolysis causing a reduced flux of fatty acids to the liver.
Fig. 1.
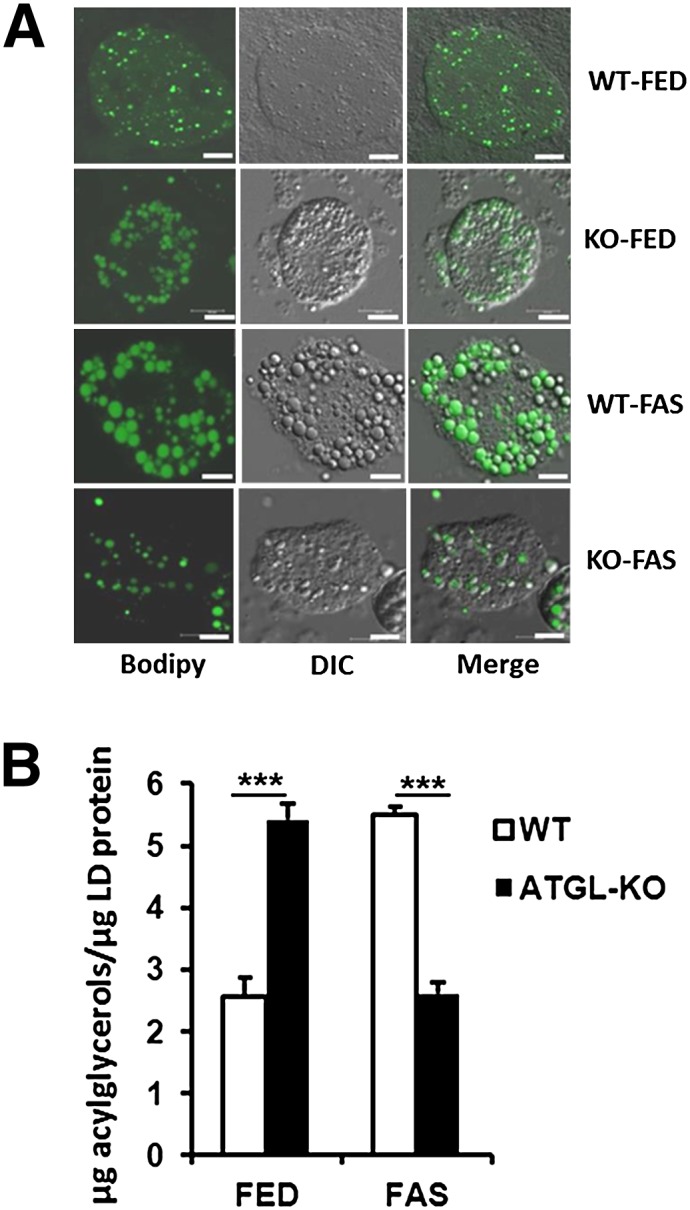
Gross phenotyping of animal model for genetic, nutritional, and super stresses. A: Characterization of stained LDs in isolated primary hepatocytes (representative examples) by confocal laser scanning microscopy. Cells were treated with neutral lipid-specific fluorescence dye BODIPY® 493/503 for 10 min. Z-stacks through entire cells were acquired using a 40× oil immersion objective. BODIPY® 493/503 was excited at 488 nm and detected between 500 and 550 nm. Transmission images were recorded with differential interference contrast (DIC) optics. Fluorescence images represent maximum-intensity projections of generated z-stacks. B: Amount of LD acylglycerols relative to LD protein found in isolated LDs, determined by enzyme tests (mean ± SD, n = 3). ***P < 0.001.
Next, we isolated LDs from freshly prepared hepatocytes by nitrogen cavitation and gradient centrifugation. Absolute amounts of acylglycerols (mostly TGs) in relation to LD protein revealed a 2-fold increase for WT-FAS and KO-FED samples, whereas combined effects in KO-FAS brought back TG amounts close to WT-FED samples. These quantitative data are shown in Fig. 1B; they also support the interpretation of the micrographs.
In Table 1 we present quantitative analyses of hepatocyte LD lipid classes as determined by LC-MS. For better comparison, data are expressed relative to total lipids for each sample group. As expected, the TG class with over 97 mol% is most prominent; statistically different amounts are not seen in this mode of presentation. Yet, this mode revealed that all other classes except phosphatidylinositol (PI) respond statistically differently to genetic, nutritional, and super stresses. In comparison to the control WT-FED group, DG class increased relatively in all other sample groups, whereas all phospholipid classes examined decreased significantly.
TABLE 1.
Effect of fasting and ATGL-KO on lipid class composition in isolated hepatocyte LDs
| Lipid Class | Lipid Class Relative to Total Lipids (mol%) |
|||
| WT-FED | KO-FED | WT-FAS | KO-FAS | |
| TG | 97.9 ± 15.5 | 98.8 ± 14.1 | 97.4 ± 13.8 | 98.6 ± 21.5 |
| DG | 0.43 ± 0.06 | 0.61 ± 0.1a | 1.60 ± 0.25a,bb | 0.62 ± 0.14a,cc |
| PC | 0.85 ± 0.12 | 0.28 ± 0.03a | 0.51 ± 0.05a,bb | 0.33 ± 0.05aa,c |
| PE | 0.50 ± 0.06 | 0.18 ± 0.02a | 0.31 ± 0.04a,b | 0.30 ± 0.05a |
| PI | 0.20 ± 0.06 | 0.07 ± 0.01 | 0.12 ± 0.012b | 0.10 ± 0.02 |
| PS | 0.06 ± 0.007 | 0.02 ± 0.003aa | 0.03 ± 0.003aa,b | 0.03 ± 0.005aa,b |
| SM | 0.024 ± 0.003 | 0.006 ± 0.001a | 0.014 ± 0.002a,bb | 0.010 ± 0.002a |
Data are presented in mol% from total lipids in each group and are calculated from absolute amounts of individual lipid species obtained by LC-MS and then normalized to acylglycerols (supplementary Tables III–X). Values are expressed as means ± SD (n = 3). P values: a refers to WT-FED group, b to KO-FED group, c to WT-FAS group (a,b,cP < 0.5, aa,bb,ccP < 0.01).
This first step in phenotyping allowed evaluation of gross differences with respect to the pathophysiological state of the animals. In the following, we highlight our lipidomic data obtained by LC-MS/MS analysis at levels of lipid species profiles and of lipid molecular species in hepatocyte LDs. Profiles of a lipid class are based on lipid species annotated by number of carbon atoms:number of double bonds (e.g., TG 52:3, PC 36:3). In lipid molecular species, by definition, constituent acyl groups are known (e.g., TG 16:0_18:1_18:2, PC 18:1_18:2). The “_” instead of a “/” indicates that sn-positions of acyl groups on glycerol are not identified (22).
PCA of LD lipidomes and its structural basis
Lipid species of each class in the four sample groups as shown in Table 1 were subjected to PCA. In a nutshell, PCA takes the abundance matrix of observed variables (species of a lipid class) and calculates new variables called principal components that account for most of the variance in the data. By this, data patterns are identified and visualized by scatter plots. In addition, lipid species contributing most to differences between sample groups can be identified (supplementary Table II). We found common denominators; lipid species mainly responsible for shifts in PC1 and PC2 were calculated and are presented always in descending order.
All lipid species could be separated into the four sample groups and thus phenotyped except those from PI and PS classes. Focusing on main TG, DG, and PC classes, Fig. 2A illustrates for TG species that nutritional stress led to positive shifts in PC1 (empty red squares) seen from WT sample groups (full red squares), ATGL KO provoked negative shifts (full blue squares). Super stress almost “neutralized” individual genetic and nutritional stresses as KO-FAS (empty blue squares) became almost identical to WT-FED in PC1. In PC2, negative shifts clearly characterized this super stress by separation from all other sample groups. PCA of DG species shown in Fig. 2B separated in PC1 genetic (blue squares) from nutritional stress (red squares), whereas PC2 separated WT-FED (control, full red squares) by a huge positive shift from all other sample groups. Analysis of the PC lipidome (Fig. 2C) resulted in a separation of FED (full squares) from FAS (empty squares) sample groups in PC1 and of KO (blue squares) from WT (red squares) sample groups in PC2.
Fig. 2.

Phenotyping by PCA of TG, DG, and PC species from WT-FED (full red), WT-FAS (empty red), KO-FED (full blue), and KO-FAS (empty blue) sample groups. Data for each species is based on per thousand relative to total amount of TG, DG, and PC class, taken from supplementary Tables III–V, respectively (n = 3). Corresponding PCA loadings are shown in supplementary Table II. Panels (A), (B), and (C) demonstrate that sample groups can be clearly separated by PC1 and PC2. The cumulative variance explained by the two principal components is over 88% indicating that they already contain most of the information provided by TG, DG, and PC profiles. In comparison to FED sample groups, the KO effect always generated a clear shift in PC1.
Our understanding of long-chain and VLC fatty acid biosynthesis indicates a correlation between chain extension and increased desaturation, i.e., with more and longer VLC acyl residues in the lipid molecule, higher numbers of double bonds are found. Based on the quantitative values (in percent or per thousand) for each species relative to total amounts of TG, DG, and PC, respectively (reported in supplementary Tables III, IV, and V), the bars in the histograms shown in Fig. 3 are the result of calculations of average chain-lengths and average numbers of double bonds in TG, DG and PC species and reflect such correlation. Most important, in comparison to WT-FED all other sample groups of TG and DG glycerolipid classes are characterized by a significant average chain-length and number of double bonds. Of note, the drastic reduction, in particular on the average number of double bonds due to the KO effect, is reversed by additional fasting (KO-FAS sample group) as the average double bond number is now closer to that of the WT-FED sample group. This is in accordance with the neutralizing effect alluded to above.
Fig. 3.

Phenotyping by calculation of average chain-lengths and numbers of double bonds of TG, DG, and PC species. Whereas DG patterns follow somewhat TG patterns, PC patterns are opposite to both of them, but all with high significances. Data are based on the sum of species values per thousand relative to total amount of TG, DG, and PC class taken from supplementary Tables III–V, respectively. *P < 0.05; **P < 0.01; ***P < 0.001 in comparison to WT-FED; +P < 0.05; +++P < 0.001 in comparison to WT-FAS (n = 3).
Figure 3 also demonstrates that the situation is totally different for PC species. ATGL-KO led to highly significant increases of average values for chain-length and number of double bonds. Fasting has a rather moderate impact in comparison to WT-FED, but in combination with the KO effect led to a drastic reduction of average chain-lengths and numbers of double bonds in comparison to all other sample groups (super stress).
TG lipidomes in hepatocyte LDs are markers for the mode of stress applied to the animals
Lipidomic analysis of TG species from hepatocyte LDs from control animals (WT-FED) and from those kept under gene and/or nutritional stress (KO-FED, WT-FAS, KO-FAS) resulted in up to 100 species ranging in either sample group from TG 32:0 to 60:14. C50 to C58 species predominated the TG species profile for the WT-FED group as expected from our previous study (1). Yet, individual features for each sample group over the whole carbon number range were found upon construction of respective heat maps. They are based on the quantitative TG species data deposited in supplementary Table III, i.e., calculated relative to 100% (sum of all species) that proved to be superior to the use of absolute amounts.
Thus, the heat map shown in Fig. 4 highlights changes relating to total TG content for a given TG species. First, ATGL KO and/or exposure of the animals to fasting nearly eliminated TG species from C32 to C48 (red cells in Fig. 4 only in WT-FED column); lipidomic analyses at TG molecular species level (supplementary Table III) demonstrated that these are all species containing a medium-chain fatty acid residue. Second, the KO effect became visible by the dark to bright green colors for species of the KO-FED sample group, particularly for the losses in the C32–C46 and C58–C62 ranges. Third, WT-FAS data reveal an enrichment of highly unsaturated TG species from C48 to C60 at the high end of double bond numbers. Finally, fasting of ATGL-KO animals resulted in a resurgence of VLC-PUFA. This was again in accordance with the neutralizing effect observed in PCA, placing TG species from KO-FAS near to the WT-FED sample group (Fig. 2A). Taken together, these data indicate that nutritional stress, as well as genetic stress, results in a phenotypic signature of TG species in hepatocyte LDs.
Fig. 4.

Heat map visualizing phenotypes of TG lipidomes detected in WT-FED, KO-FED, WT-FAS, and KO-FAS sample groups (n = 3 each). Calculations are based on per thousand of a given TG species relative to total TG content (supplementary Table III). Colors encoding the rectangular cells are the ratio of the species value relative to the mean of the four groups of one lipid species. If in a row all of the four cells are black, there is no or little difference in the relative contribution of this lipid species. Red and green correspond to higher and lower ratios, respectively. Gray cells represent species not detected.
A specific point is shown in Fig. 5, where values of marker TG species for steatosis after HFD administration are compared with those of respective values of WT-control, short-time fasting, and the ATGL-KO effect. The first two data sets are taken from the preceding study (1), the latter two from the present study. Apart from WT control, liver steatosis was a parameter for the animals’ pathophysiological status. But only HFD administration led to insulin resistance in contrast to steatosis without insulin resistance observed after fasting and ATGL ablation.
Fig. 5.

Marker TG species identified after HFD administration are markers for insulin resistance as well. Data are taken from TG lipidomes of hepatocyte LDs from mice fed a HFD (WT-HFD) in relation to respective TG species from mice fed chow (WT-FED, controls), from ATGL-KO mice (KO-FED), and from mice exposed to short-time fasting (WT-FAS). Bars represent mean values (n = 3). Data for WT-HFD and WT-FED sample groups are taken from our preceding study (1), those for KO-FED and WT-FAS sample groups from the current study. For simplicity error bars are omitted, but can be retrieved from supplementary tables in either publication.
DG and PC lipidomes are stress sensitive as well
The heat maps for both lipid classes are shown in Fig. 6 in absolute amounts (nmol or μmol) based on internal standards per LD sample and normalized to acylglycerols (supplementary Tables IV and V). Differences in sample groups can be more easily recognized in this mode. The DG heat map revealed WT-FAS as the most abundant species and WT-FED as the least abundant species. Moreover, super stress ameliorated to some extent the KO stress moving the abundance of DG species toward those of the WT-FED sample group.
Fig. 6.
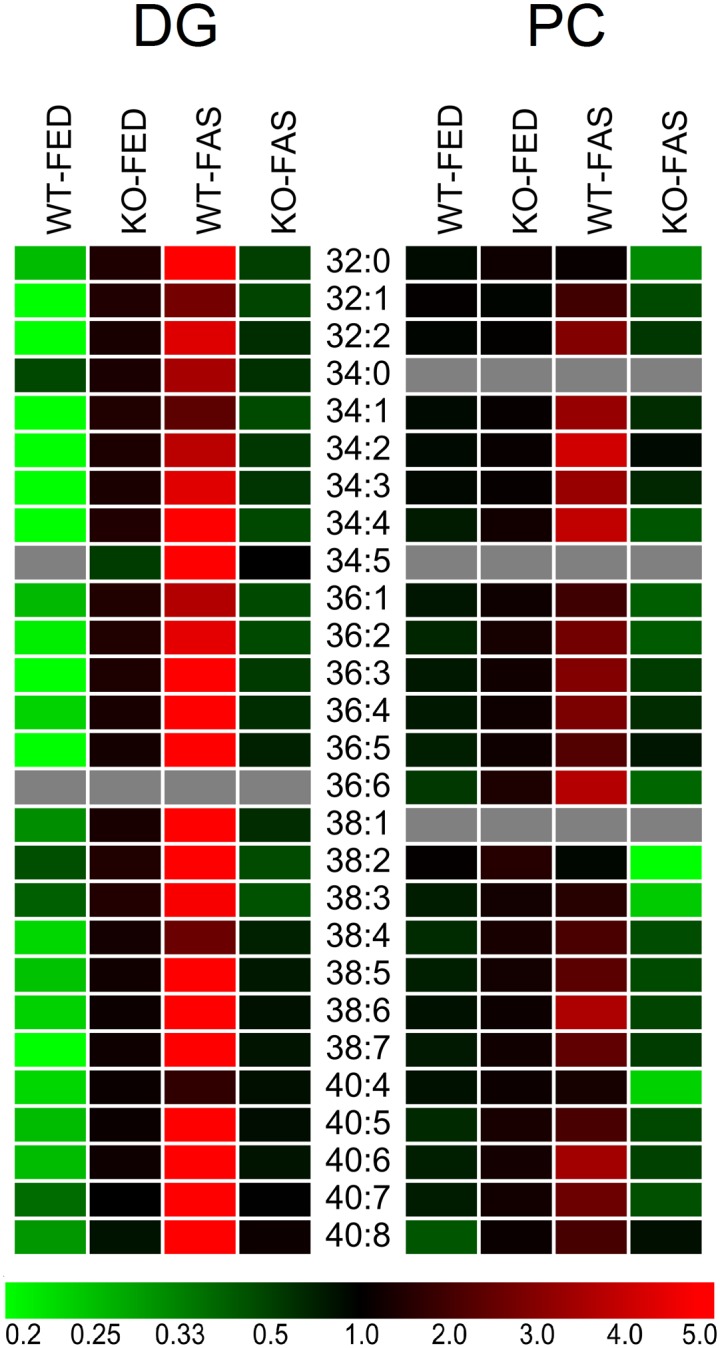
Heat maps of DG and PC species detected in WT-FED, KO-FED, WT-FAS, and KO-FAS sample groups (n = 3 each). Calculations are based on absolute amounts determined by LC-MS and then normalized to acylglycerols in nmol or μmol for DG and PC species (supplementary Tables IV and V, respectively). Colors encoding the rectangular cells are the ratio of the species value relative to the mean of the four groups of one lipid species. Clear phenotypes can be recognized for DG lipidomes, to a lesser extent for PC lipidomes.
Metabolic relationships between molecular species of DGs, PCs, and TGs
In the preceding study we demonstrated that nutritional stress strongly affected profiles of lipid species of some lipid classes found in hepatocyte LDs. In addition, structures of lipid molecular species derived from respective lipid species by MS/MS analysis allowed recognition of metabolic relationships between molecular species of these lipid classes (1). Similarly, the effects of genetic stress studied here led to lipid species and lipid molecular species, particularly for DG and PC classes (data in supplementary Tables IV and V, respectively), whose structures provided insights on metabolic relationships as presented in Table 2.
TABLE 2.
DG and PC molecular species identified in hepatocyte LDs from all sample groups
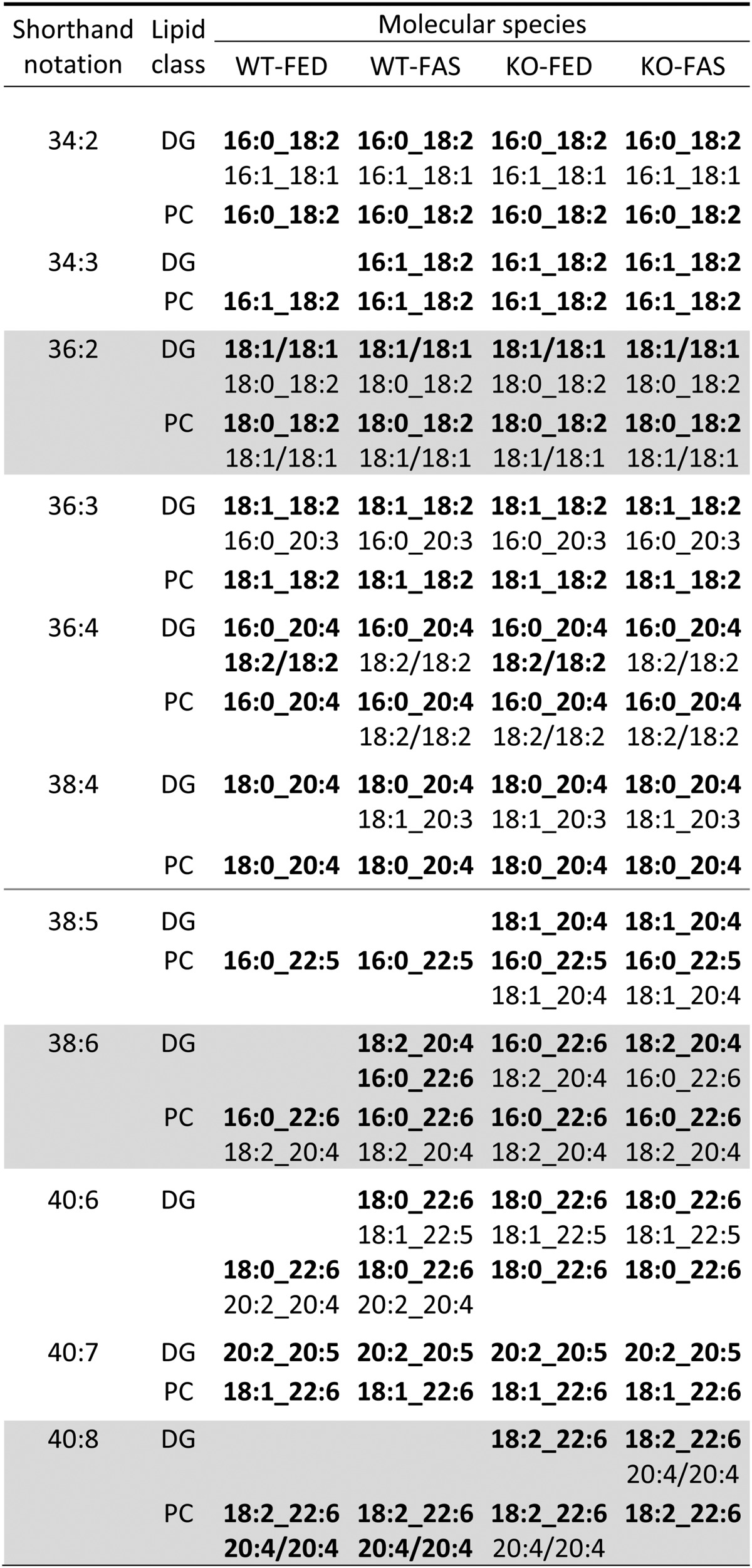
From the metabolic point of view, the data in Table 2 can be interpreted for all four sample groups that DG molecular species up to 38:4 can serve as precursors for PC molecular species. Vice versa, PC molecular species can be degraded by phospholipase C to DG molecular species. Interestingly, from 38:5 to 40:8 (below gray line in Table 2) respective DG molecular species in WT-FED sample groups were not detected, but still under fasting and KO conditions (WT-FAS, KO-FED, and KO-FAS). Consequently, DG and PC metabolic relationships can be possible here only under genetic, nutritional, or super stress. Further inspection of Table 2 reveals differences at the molecular species level between sample groups (gray shaded) for a given species: A case in point were species 40:8, where DG molecular species were not detected in WT samples, whereas DG and PC 18:2_22:6 molecular species were identified in KO samples; the latter, however, differed in the presence of minor 20:4/20:4 molecular species. The example of species 38:6 indicated that the abundance of constituent acyl groups can also characterize sample groups: A DG molecular species was not identified in WT-FED, in WT-FAS molecular species 16:0_22:6 and 18:2_20:4 were equally present, whereas in KO-FED 16:0_22:6 and in KO-FAS 18:2_20:4 molecular species predominated.
In the previous paper we reported impressive product-precursor relationships between DG and TG molecular species after HFD administration [Table 3 in (1)]. This was not so prominently observed in this study. Considering the data for TG molecular species identified here (supplementary Table III), we conclude for 36:2 species (gray shaded in Table 2) that predominant DG 18:1/18:1 is mainly converted to TG molecular species. In contrast, minor DG 18:0_18:2 is converted to respective and predominant PC molecular species. In WT samples, DG molecular species could also be the result of the degradation of TG molecular species by ATGL and, consequently, in ATGL-KO samples DG molecular structures could be different. This was not the case; DG molecular structures shown in Table 2 were not different as determined by MS/MS.
DISCUSSION
By the criteria of genetic background, age, maintenance, and diet regimes for the male mouse models, the current intervention trial with three animals per sample group corresponded with those of the two preceding trials (1). In fact, WT-FED and WT-FAS sample groups in the current trial corresponded to the WT-FED and WT-FAS sample groups in the nutritional stress study. Comparisons of respective data at lipid species levels revealed similar percent or per thousand contributions to total lipid class within biological tolerances. Thus, at lipid species level maximum differences were recognized to be 5% for very few TG and PC species only. At lipid molecular species level structural determination by MS/MS of constituent fatty acids agreed in all details for major species, yet some differences were observed for minor species regarding constituent PUFA and VLC-PUFA residues. Overall, the comparisons confirmed our conclusions drawn in the former study. This offered a solid basis for interpretation of data obtained in the genetic stress study here.
TG lipidomes certainly were the phenotypic highlights in this study. Gross investigation of absolute TG amounts by enzymatic test and microscopy (Fig. 1) revealed huge effects in response to both genetic and nutritional stresses. Calculation of quantitative MS data relative to total amount of lipid species per sample group did not indicate the differences for TG amounts between sample groups. It did reveal a clear preponderance of the TG class (sum of all TG species amounts) over all other lipid classes in the four sample groups (Table 1).
Inspection at TG and molecular species levels allowed gauging the information for separation of and distinction between the four sample groups. Interestingly, in-depth analysis using PCA did single out main TG species (see supplementary Table II) responsible for the separation of the stress conditions (Fig. 2A). Predominant increases of TG 54:5, 54:6, and 54:4 were characteristic for the WT-FAS state (empty red squares), which was in accordance with our finding that PUFA-containing TG species become enriched under fasting conditions. Conversely, enrichments in TG 52:2 and 52:3 were the major determinants for the negative shift in PC1 (full blue squares), indicating still the presence of prominent TG species having shorter chain-lengths and lower numbers of double bonds as a result of ATGL KO. Finally, separation of the super stress (torpor-like pathophysiological status) from all other sample groups became possible in PC2 (empty blue squares), affected predominantly by TG 56:7 and 56:8. The main TG species listed here were characteristic for both genetic and nutritional stresses and implicated that double bonds were the main determinants for separation by PCA.
Figure 3 shows for the TG lipidome of the KO-FED sample that the values for average chain-lengths and numbers of double bonds are 52.7 and 3.65, respectively. This is in support for the selection of TG 52:2 and 52:3 by PCA as main separation determinants for the defect in TG catabolism present in ATGL-KO mice. The HFD effect on WT animals reported in the preceding paper lowered the average double bond number further to 3.4 (1). All this indicated that evaluation of average numbers of quantitative lipidomic species data enables phenotyping a pathophysiological status of the animal model. In addition, the same quantitative MS data allowed exploring information at the levels of individual TG species in the four sample groups; this is illustrated by the heat map shown in Fig. 4. Of note, the values shown do not represent the direct values determined by MS (supplementary Table III), rather the MS values for each TG species from the four sample groups are averaged and displayed as ratios representing gains or losses against the average value. This is visualized by the tint and intensity of the colors. Clearly, TG lipidomes emerging from this approach are specific for genetic, nutritional, and super stresses. When we included the HFD data from the previous study (1) as the fifth lipidome and recalculated the values, again each TG lipidome had a distinguishable pattern (data not shown).
Ample evidence exists in the literature that TG accumulation in the liver is a result of obesity and diabetes. Furthermore, nonalcoholic fatty liver diseases of different etiologies are associated with insulin resistance. An exception is the ATGL-KO animal model exhibiting high insulin sensitivity despite liver TG storage. Figure 5 shows that HFD application to WT animals produced marker TG species (1). These species were not predominant in fasted WT animals and ATGL-KO animals despite steatosis. Consequently, species identified are a marker for HFD-induced steatosis and insulin resistance. As mentioned before, in 2006 our group found that ATGL-KO led to fatty liver but not to systemic insulin resistance (13). This finding was supported by a very recent study, where HFD feeding to liver-specific ATGL-KO mice enhanced liver TG contents and primary hepatocytes derived from these livers exhibited improved glucose tolerance despite unaltered insulin signaling (16). Importantly, diseases are known to be caused by inactive ATGL protein due to a genetic defect (neutral lipid storage disease with myopathy) (23), or to be caused by functional ATGL yet inactive by lack of functional coactivator CGI-58 (neutral lipid storage disease with ichtyosis, Chanarin-Dorfman syndrome) (24). In either case, enhanced liver TG levels were observed, but without insulin resistance and glucose intolerance (25). We daresay that these diseases will have a clearly distinguishable phenotypic imprint on the hepatocyte lipidome.
Turning to the DG lipidome, we report in Table 1 that fasting produced a distinct effect on DG amounts present in hepatocyte LDs. An almost 4-fold increase relative to total lipid content was measured upon short-time fasting, an 0.5-fold increase upon KO-effect, whereas super stress returned the fasting DG level to levels measured in ATGL-KO mice. We conclude that substantial enhancement of DG amounts in the liver (6, 26), particularly in hepatocyte LDs (11), can be considered a further indicator of insulin resistance; this does not apply to the smaller DG increases caused by ATGL deficiency, however. In line with this, a long-term study with liver-specific ATGL-KO mice revealed progressive hepatosteatosis, but unchanged insulin tolerance; a compensatory mechanism was suggested by reduced DG acyltransferase 2 mRNA (responsible for TG synthesis) and increased lipolysomal activity for TG degradation (17). The opposite approach, liver-specific overexpression of ATGL, resulted in amelioration of steatosis combined with increased β-oxidation and improved insulin signaling, but did not affect insulinemia (17, 27). Also, we demonstrated here that various types of fatty livers resulting from genetic and nutritional stresses can be characterized at DG lipidome level. This was evidenced by PCA (Fig. 2), average values for chain-length and number of double bonds (Fig. 3), and heat map (Fig. 6) of DG species. Moreover, similar distinction for the DG lipidome from HFD-caused fatty liver (1) with insulin resistance can be recognized in a respective DG heat map as well (data not shown).
Including the PC lipidome into the discussion, it is interesting to note that PCA of DG and PC species, respectively, separated sample groups class specifically, i.e., totally different in PC1 as well as in PC2 (Fig. 2B, C). Figure 3, however, indicates with respect to average calculations that the fasting effect did matter to some extent in the DG class, but not in the PC class. In contrast, the KO effect led to drastic changes in both average values, but in opposite directions for DG and PC classes. As deduced from Table 2, DG molecular species with higher chain-lengths and numbers of double bonds were preferably channeled into PC class as a result of ATGL-KO, and more so under super stress condition. These shifts of constituent VLC-PUFA could be the determinants for the different PCA separation of sample groups in DG and PC classes.
Taken together, our study provides evidence that the pathophysiological status of the animal model exposed to genetic or nutritional stress, or both together, can be assessed by lipid class lipidomes of hepatocyte LDs, in particular those of TG, DG, and PC species. Moreover, TG and DG lipidomes reflect the insulin status of the animal.
Supplementary Material
Footnotes
Abbreviations:
- ATGL
- adipocyte triglyceride lipase
- CGI-58
- comparative gene identification 58
- DG
- diacylglycerol
- FAS
- fasted before sacrifice
- FED
- received control chow
- HFD
- high-fat diet
- KO
- knockout
- LD
- lipid droplet
- LDA
- Lipid Data Analyzer
- PC
- phosphatidylcholine
- PCA
- principal component analysis
- PC1
- principal component 1
- PC2
- principal component 2
- PE
- phosphatidylethanolamine
- PI
- phosphatidylinositol
- PS
- phosphatidylserine
- TG
- triacylglycerol
- VLC
- very long-chain
- WT
- wild-type
- WT-FED, WT-FAS, KO-FED, KO-FAS
- hepatocyte LD sample groups from wild-type or knockout mice fed chow or kept in fasted condition, respectively
The research leading to these results received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement number 202272, the Austrian Ministry of Science and Research GEN-AU project BIN (FFG Grant 820962), and FWF Project Grant Z136, Wittgenstein. The Austrian Centre of Industrial Biotechnology (ACIB) contribution was supported by FFG, BMWFJ, BMVIT, ZIT, Zukunftsstiftung Tirol, and Land Steiermark within the Austrian COMET programme (FFG Grant 824186).
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of 10 tables.
REFERENCES
- 1.Chitraju C., Trötzmüller M., Hartler J., Wolinski H., Thallinger G. G., Lass A., Zechner R., Zimmermann R., Köfeler H. C., Spener F. 2012. Lipidomic analysis of lipid droplets from murine hepatocytes reveals distinct signatures for nutritional stress. J. Lipid Res. 53: 2141–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zimmermann R., Strauss J. G., Haemmerle G., Schoiswohl G., Birner-Gruenberger R., Riederer M., Lass A., Neuberger G., Eisenhaber F., Hermetter A., et al. 2004. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 306: 1383–1386 [DOI] [PubMed] [Google Scholar]
- 3.Granneman J. G., Moore H. P., Krishnamoorthy R., Rathod M. 2009. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J. Biol. Chem. 284: 34538–34544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schweiger M., Lass A., Zimmermann R., Eichmann T. O., Zechner R. 2009. Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am. J. Physiol. Endocrinol. Metab. 297: E289–E296 [DOI] [PubMed] [Google Scholar]
- 5.Shulman G. I. 2000. Cellular mechanisms of insulin resistance. J. Clin. Invest. 106: 171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samuel V. T., Petersen K. F., Shulman G. I. 2010. Lipid-induced insulin resistance: unravelling the mechanism. Lancet. 375: 2267–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Savage D. B., Semple R. K. 2010. Recent insights into fatty liver, metabolic dyslipidaemia and their links to insulin resistance. Curr. Opin. Lipidol. 21: 329–336 [DOI] [PubMed] [Google Scholar]
- 8.Schmitz-Peiffer C., Biden T. J. 2008. Protein kinase C function in muscle, liver, and beta-cells and its therapeutic implications for type 2 diabetes. Diabetes. 57: 1774–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samuel V. T., Liu Z. X., Qu X., Elder B. D., Bilz S., Befroy D., Romanelli A. J., Shulman G. I. 2004. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 279: 32345–32353 [DOI] [PubMed] [Google Scholar]
- 10.Magkos F., Su X., Bradley D., Fabbrini E., Conte C., Eagon J. C., Varela J. E., Brunt E. M., Patterson B. W., Klein S. 2012. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology. 142: 1444.e2–1446.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumashiro N., Erion D. M., Zhang D., Kahn M., Beddow S. A., Chu X., Still C. D., Gerhard G. S., Han X., Dziura J., et al. 2011. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA. 108: 16381–16385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eichmann T. O., Kumari M., Haas J. T., Farese R. V., Jr, Zimmermann R., Lass A., Zechner R. 2012. Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone-sensitive lipase, and diacylglycerol-O-acyltransferases. J. Biol. Chem. 287: 41446–41457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haemmerle G., Lass A., Zimmermann R., Gorkiewicz G., Meyer C., Rozman J., Heldmaier G., Maier R., Theussl C., Eder S., et al. 2006. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 312: 734–737 [DOI] [PubMed] [Google Scholar]
- 14.Kienesberger P. C., Lee D., Pulinilkunnil T., Brenner D. S., Cai L., Magnes C., Koefeler H. C., Streith I. E., Rechberger G. N., Haemmerle G., et al. 2009. Adipose triglyceride lipase deficiency causes tissue-specific changes in insulin signaling. J. Biol. Chem. 284: 30218–30229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turpin S. M., Hoy A. J., Brown R. D., Rudaz C. G., Honeyman J., Matzaris M., Watt M. J. 2011. Adipose triacylglycerol lipase is a major regulator of hepatic lipid metabolism but not insulin sensitivity in mice. Diabetologia. 54: 146–156 [DOI] [PubMed] [Google Scholar]
- 16.Ong K. T., Mashek M. T., Bu S. Y., Mashek D. G. 2013. Hepatic ATGL knockdown uncouples glucose intolerance from liver TAG accumulation. FASEB J. 27: 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J. W., Wang S. P., Alvarez F., Casavant S., Gauthier N., Abed L., Soni K. G., Yang G., Mitchell G. A. 2011. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology. 54: 122–132 [DOI] [PubMed] [Google Scholar]
- 18.Fauland A., Köfeler H. C., Trötzmüller M., Knopf A., Hartler J., Eberl A., Chitraju C., Lankmayr E., Spener F. 2011. A comprehensive method for lipid profiling by liquid chromatography-ion cyclotron resonance mass spectrometry. J. Lipid Res. 52: 2314–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matyash V., Liebisch G., Kurzchalia T. V., Shevchenko A., Schwudke D. 2008. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 49: 1137–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartler J., Trötzmüller M., Chitraju C., Spener F., Köfeler H. C., Thallinger G. G. 2011. Lipid Data Analyzer: unattended identification and quantitation of lipids in LC-MS data. Bioinformatics. 27: 572–577 [DOI] [PubMed] [Google Scholar]
- 21.Larionov A., Krause A., Miller W. 2005. A standard curve based method for relative real time PCR data processing. BMC Bioinformatics. 6: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liebisch G., Vizcaino J. A., Köfeler H., Trötzmüller M., Griffiths W. J., Schmitz G., Spener F., Wakelam M. J. O. 2013. Shorthand notation for lipid structures derived from mass spectrometry. J. Lipid Res. 54: 1523–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer J., Lefevre C., Morava E., Mussini J. M., Laforet P., Negre-Salvayre A., Lathrop M., Salvayre R. 2007. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat. Genet. 39: 28–30 [DOI] [PubMed] [Google Scholar]
- 24.Lass A., Zimmermann R., Haemmerle G., Riederer M., Schoiswohl G., Schweiger M., Kienesberger P., Strauss J. G., Gorkiewicz G., Zechner R. 2006. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman syndrome. Cell Metab. 3: 309–319 [DOI] [PubMed] [Google Scholar]
- 25.Brown J. M., Betters J. L., Lord C., Ma Y., Han X., Yang K., Alger H. M., Melchior J., Sawyer J., Shah R., et al. 2010. CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. J. Lipid Res. 51: 3306–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorden D. L., Ivanova P. T., Myers D. S., McIntyre J. O., VanSaun M. N., Wright J. K., Matrisian L. M., Brown H. A. 2011. Increased diacylglycerols characterize hepatic lipid changes in progression of human nonalcoholic fatty liver disease; comparison to a murine model. PLoS ONE. 6: e22775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reid B. N., Ables G. P., Otlivanchik O. A., Schoiswohl G., Zechner R., Blaner W. S., Goldberg I. J., Schwabe R. F., Chua S. C., Jr, Huang L. S. 2008. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J. Biol. Chem. 283: 13087–13099 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.