

Abstract
Cholesteryl ester transfer protein (CETP) activity results in a proatherogenic lipoprotein profile. In cholestatic conditions, farnesoid X receptor (FXR) signaling by bile acids (BA) is activated and plasma HDL cholesterol (HDL-C) levels are low. This study tested the hypothesis that FXR-mediated induction of CETP contributes to this phenotype. Patients with cholestasis and high plasma BA had lower HDL-C levels and higher plasma CETP activity and mass compared with matched controls with low plasma BA (each P < 0.01). BA feeding in APOE3*Leiden transgenic mice expressing the human CETP transgene controlled by its endogenous promoter increased cholesterol within apoB-containing lipoproteins and decreased HDL-C (each P < 0.01), while hepatic CETP mRNA expression and plasma CETP activity and mass increased (each P < 0.01). In vitro studies confirmed that FXR agonists substantially augmented CETP mRNA expression in hepatocytes and macrophages dependent on functional FXR expression (each P < 0.001). These transcriptional effects are likely mediated by an ER8 FXR response element (FXRE) in the first intron. In conclusion, using a translational approach, this study identifies CETP as novel FXR target gene. By increasing CETP expression, FXR activation leads to a proatherogenic lipoprotein profile. These results have clinical relevance, especially when considering FXR agonists as emerging treatment strategy for metabolic disease and atherosclerosis.
Keywords: nuclear receptor, lipoproteins, bile acids, hepatocyte, macrophage
The cholesteryl ester transfer protein (CETP) promotes the heteroexchange of cholesteryl esters and triglycerides (TG) between plasma high density lipoproteins (HDL) and apoB-containing lipoproteins, resulting in a proatherogenic plasma lipid profile with increased VLDL and LDL cholesterol levels and decreased HDL cholesterol (HDL-C) (1, 2). Given the key importance of dyslipidemia for the development of atherosclerosis (3), most (1, 2, 4–6) but not all (7, 8) human studies consequently reported a positive association between plasma CETP activity and atherosclerotic cardiovascular disease (CVD) morbidity and mortality. Especially in the setting of the metabolic syndrome, including type 2 diabetes mellitus and hypertriglyceridemia, the predictive value of increased CETP mass and activity for CVD events is particularly strong (9). For this target group, activators of the farnesoid X receptor (FXR), for which bile acids (BA) are the natural ligands, have been recently proposed as a novel therapeutic strategy (10).
However, patients with cholestasis have high plasma BA levels and decreased plasma HDL-C (11, 12). In addition, BA administration (e.g., chenodeoxycholic acid, CDCA) to patients with gallstones or cerebrotendinous xanthomatosis (CTX) decreases HDL-C (13, 14). These metabolic effects have thus far been attributed to the suppression of apoA-I expression by BA-mediated FXR activation (11), while a potential contribution of CETP to the shift in plasma lipoprotein profiles in patients with elevated plasma BA has not been explored.
Therefore, the present study tested the hypothesis that CETP represents a novel FXR target gene. Our results demonstrate in humans and transgenic mice in vivo as well as in hepatocytes and macrophages in vitro that BAs upregulate CETP gene expression and activity dependent on the functional expression of FXR, likely via an ER8 FXR response element (FXRE) in the first intron.
MATERIALS AND METHODS
Human subjects
Twenty patients with histologically proven liver cirrhosis were studied, divided into two groups according to their plasma BA levels, either below (noncholestatic group) or above (cholestatic group) 30 μmol/l (each n = 10; for details, see Table 1). The two groups of patients were exactly matched for gender, age, and body mass index (BMI) as well as for the clinical stage of liver disease assessed by the Child-Pugh score and the hepatic protein synthesis function to exclude the degree of liver function decline as a potential confounding factor. As parameters for hepatic protein synthesis, capacity plasma albumin (none of the patients received albumin infusions) and prothrombin time were used. The underlying etiologies were in the noncholestatic group: viral hepatitis (n = 6), alcohol (n = 3), and cryptogenic (n = 1), and in the cholestatic group, biliary (primary biliary cirrhosis / primary sclerosing cholangitis, n = 10). All cirrhotic patients were studied while hospitalized for evaluation before liver transplantation. All subjects included were in a stable clinical condition before entering the study and had been following a weight-maintaining diet containing 80 g of protein daily for at least one week. Subjects with proteinuria, suspected infections, clinically overt diabetes mellitus, thyroid dysfunction, or any other endocrine disorder were excluded from the study. No hormone, antidiabetic, or thyroid regulatory medication was administered. Patency of portal vein and hepatic artery was documented by Doppler ultrasound before entering the study. Blood was obtained in the morning from fasted subjects. Plasma samples were stored at −80°C prior to determination of BA, CETP mass levels, CETP activity, and HDL-C levels (see below). All patients gave written, informed consent before entering the study. The study protocol has been approved by the Ethics Committee of the Hannover Medical School.
TABLE 1.
Clinical characterization of the patients with liver disease
| Characteristic | Noncholestatic (Low BA) | Cholestatic (High BA) |
| n = 10 | n = 10 | |
| Male/female | 3/7 | 3/7 |
| Age (years) | 49 ± 1 | 50 ± 1 |
| BMI (kg/m2) | 22.8 ± 2.7 | 22.4 ± 2.6 |
| Child stage (A/B/C)a | 0/8/2 | 0/8/2 |
| Bilirubin (µmol/l) | 50 ± 36 | 96 ± 110* |
| BA (µmol/l) | 26 ± 1 | 88 ± 14† |
| Albumin (g/l) | 32 ± 3 | 32 ± 2 |
| Prothrombin time (%) | 72 ± 4 | 76 ± 4 |
| ALT (U/l) | 36 ± 20 | 47 ± 22 |
| γ-GT (U/l) | 61 ± 42 | 162 ± 68† |
Values are means ± SD. Significantly different from the low BA group: *P < 0.01, †P < 0.001. ALT, alanine aminotransferase; γ-GT, γ glutamyltransferase.
Grading system for the clinical severity of liver disease, with increasing severity from A to C.
Animals and diets
The human CETP transgenic mice expressing the transgene under the control of natural flanking regions (15) and APOE*3-Leiden.CETP (E3L.CETP) mice derived from crossing these mice with APOE*3-Leiden (E3L) mice (16) were all on a C57BL/6J background. E3L.CETP mice represent a well-established mouse model for human-like lipoprotein metabolism and have been shown to respond in a human-like manner to the HDL-modulating effect of several drugs (17). Three-month-old male mice were used. The mice were kept in animal rooms with alternating 12 h periods of light (from 7:00 AM to 7:00 PM) and dark (from 7:00 PM to 7:00 AM), and they had ad libitum access to food and water throughout the experiments. Food intake and body weight were monitored daily. Animals first followed a run-in period of one week during which they received standard chow (A03 containing 5.1% of total fat and 0.01% of cholesterol, Scientific Animal Food and Engineering, Villemoisson-sur-Orge, France). Subsequently, animals were divided into two groups, with the control group receiving standard A03 chow, the other group chow supplemented with 0.5% w/w taurocholic acid (TCA, Sigma). After six days of diet, mice were anesthetized with isoflurane after a 4 h fast. Blood samples were drawn by cardiac puncture and collected into heparin-containing tubes. Plasma was separated by centrifugation and stored at –80°C before analysis. Livers were excised and weighed. A part of the liver was immediately homogenized in 500 µl Trizol (Invitrogen, Paisley, UK), and the homogenate was stored at –80°C before RNA isolation. All procedures were approved by the local ethics committees for animal experimentation of the Leiden University Medical Center and the University Medical Center Groningen.
Measurement of CETP activity
Plasma CETP activity under the dependence of endogenous plasma lipoproteins (plasma cholesteryl ester transfer activity, CETA) in mouse and human plasma was measured with a fluorescent assay that was performed in microplates by using donor liposomes enriched with nitrobenzoxadiazole-labeled cholesteryl esters (NBD-CE) (Roar Biomedical, New York, NY) as described previously (18). In brief, incubation media contained 4 µl of donor liposomes and 10 µl of total plasma, as a source of CETP and endogenous lipoprotein substrates, in a final volume of 200 µl TBS. Incubations were performed in triplicate for 1 h at 37°C in a FL600 Microplate Fluorescence Reader (Bio-Tek, Winooski, VT). The CETP-mediated transfer of NBD-CEs from self-quenched donors to acceptor endogenous plasma lipoproteins was monitored by the increase in fluorescence intensity (excitation, 465 nm; emission, 535 nm). Results were expressed as the initial transfer rate of NBD-CEs after deduction of blank values (wild-type mouse plasma devoid of CETP activity). In addition, CETP activity measurements with diluted plasma in the presence of an excess of exogenous lipoprotein substrates (diluted CETP activity) were performed. Under these conditions, CETP activity is an almost direct reflection of CETP protein levels. These measurements were carried out as described above, except that incubation media contained 0.5 µl of plasma (CETP source), 5 µl of donor liposomes, and 5 µl of acceptor VLDL provided by the manufacturer in a final volume of 200 µl PBS.
Determination of plasma CETP mass levels
CETP mass levels in patient plasmas were measured by a specific enzyme-linked immunosorbent assay with TP1 monoclonal anti-CETP primary antibodies as previously described (19).
CETP mass levels in mouse plasmas were determined by a specific immunoassay with TP2 anti-CETP monoclonal antibodies (Heart Institute, Ottawa, Canada). In brief, plasma samples were diluted (1:9, v/v) in TBS (100 mM; pH 6.8) containing SDS (25 g/l), and then incubated for 15 min at 80°C. Samples were subsequently electrophoresed using 8–12% discontinuous polyacrylamide gels in a Mini Protean device (Bio-Rad Laboratories) and then transferred to nitrocellulose membranes (Trans Blot, Bio-Rad Laboratories). The resulting blots were blocked for 1 h in 5% low-fat dry milk in PBS (100 mM; pH 7.4) containing 0.1% Tween, and then washed with PBS/Tween. Human CETP was revealed by successive incubations with TP2 anti-CETP antibodies and horseradish peroxidase-coupled secondary antibodies (DakoCytomation, Glostrup, Denmark). Blots were finally developed using the SuperSignal Chemiluminescent kit (Pierce, Rockford, IL). The CETP mass level in each plasma sample was estimated by comparison with a calibration curve that was obtained with serial dilutions of purified CETP (kind gift of Dr. J. P. Pais de Barros, INSERM U866, Dijon, France) submitted to electrophoresis together with the samples.
Plasma analyses
Commercially available kits were used for the determination of total cholesterol, TG (Roche Molecular Biochemicals, Mannheim, Germany), and phospholipids (Wako, Neuss, Germany) in plasma. To assess the distribution of cholesterol and TG over the different lipoprotein subclasses, pooled plasma samples of all animals from one group were used for lipoprotein separation by fast-protein liquid chromatography with a Superose 6 HR 10/30 column (Amersham Biosciences) as described previously (20). Fractions 1–8, 9–16, and 17–30 contained VLDL, IDL/LDL, and HDL, respectively. Cholesterol content of HDL from individual plasma samples was determined after selective precipitation of apoB-containing lipoproteins as previously described (21). Briefly, 10 µl of a 4% phosphotungstic acid solution in 0.16M NaOH was mixed with 100 µl of plasma before adding 2.5 µl of a 2M MgCl2 solution and incubating for 30 min at 4°C. Precipitated apoB-containing lipoproteins were then pelleted by a 30 min spin at 1,500 g and the HDL-containing supernatant was assayed for cholesterol. HDL-C concentrations in patients were determined by lipid electrophoresis (REP-HDL-plus cholesterol electrophoresis, Helena Diagnostika, Hartheim, Germany). Total plasma BA concentrations were measured using an enzymatic fluorimetric assay essentially as described (22).
Cell culture and treatments
Human monocytic THP-1 cells (ATCC, Rockville, Maryland) were maintained in RPMI 1640 medium containing 10% of fetal calf serum and penicillin/streptomycin (100 µg/ml, Lonza, Verviers, Belgium). THP-1 cells were seeded in 6-well plates (1 × 106 cells/well), and differentiation into macrophages was achieved by 48 h incubation with 100 nM PMA. Human hepatoma HepG2 cells (ATCC, Rockville, MD) as well as HepG2 cells stably transfected with Na+-taurocholate cotransporting polypeptide (HepG2-NTCP cells, a kind gift of Dr. Gerd Kullak-Ublick, University Hospital Zurich, Switzerland) (23) were maintained in DMEM with 10% of fetal calf serum and penicillin/streptomycin (100 µg/ml) and treated when they reached 80% of confluence. For the isolation of primary hepatocytes, mouse livers were perfused with a collagenase solution essentially as described before (21). Viability of the cells was greater than 85% (Trypan Blue exclusion). Hepatocytes were plated on 24-well plates (120,000 cells/well) precoated with collagen (Serva Feinbiochemika, Heidelberg, Germany) in William's E medium (Invitrogen, Breda, The Netherlands) supplemented with penicillin/streptomycin. During the attachment period (4 h) 50 nmol/l dexamethasone (Novo Nordisk Pharma BV, Amsterdam, The Netherlands) and 10% fetal calf serum (Invitrogen) were added to the medium. All cell models were maintained at 37°C in 5% CO2 / 95% air in a water-saturated atmosphere.
For treatments, cells were switched to serum-free medium following extensive washing with PBS. The bile acids TCA and CDCA were dissolved in ethanol, while the FXR agonist GW4064 (Sigma), the RXR agonist 9-cis-retinoic acid (Sigma), and the inhibitor of protein synthesis CHX were dissolved in DMSO prior to addition to the cells at the indicated concentrations for 24 h. Vehicle control cells received ethanol or DMSO as appropriate. For the FXR knockdown experiments, SMART pool FXR siRNA and control siRNA (Dharmacon, Lafayette; CO) were transfected into HepG2 cells according to the manufacturer's instructions. After an 8 h transfection, treatments were performed as detailed above. Following the indicated incubation times, cells were washed with PBS before adding Trizol for RNA extraction.
RNA isolation and quantitative real-time PCR
Total RNA was isolated with Trizol (Invitrogen) and quantified using a NanoDrop system (Thermo Fisher Scientific, Walthman, MA). One microgram of RNA was reverse-transcribed into cDNA using M-MLV reverse transcriptase, random primers, and RNaseOUT inhibitor (Invitrogen). Real-time quantitative PCR was performed using an ABI PRISM 7700 sequence detection system (Applied Biosystems, Darmstadt, Germany) with the default settings (20). Primers were obtained from Invitrogen (Breda, The Netherlands). Fluorogenic probes were synthesized by Eurogentec (Seraing, Belgium). All expression data were subsequently standardized for 18S rRNA analyzed in separate runs and further normalized for the respective expression values of the control groups.
Reporter assays
For whole-promoter assays, the 3.5 kb genomic promoter of the human CETP gene (kindly provided by Dr. A. Tall, Columbia University, NY) (24) was cloned into the BglII/NheI restriction sites of a pGL3 plasmid (Promega) to obtain a hCETP-Luc plasmid. HepG2 cells were maintained in DMEM supplemented with 10% FBS. One day before transfection, cells were seeded at a density of 1 × 105 cells/cm2, and transfection was performed 24 h later with Lipofectamine (Invitrogen). For each well of 24-well plates, 50 ng pSG5-hFXR plasmid, 200 ng hCETP-Luc plasmid, and 25 ng pRL Renilla Luciferase Vector as control (Promega) were cotransfected. Twenty-four hours after transfection, cells were supplied with DMEM with 10% charcoal-stripped serum containing GW4064 (2.5 µM). After 24 h incubation, luciferase activities were measured with the Dual-Luciferase Reporter Assay System (Promega) on a Victor3 Multilabel Counter (Perkin Elmer) and normalized with Renilla luciferase activities according to the manufacturer's instructions.
Putative FXREs in the promoter and in the first intron of the CETP gene were detected by using the NHR-Scan computational analysis program (25). For reporter assays with these sequences, human embryonic kidney 293T (HEK293T) cells were transiently transfected with expression vector pSG5-FXR (50 ng) and pCMV-β-galactosidase (50 ng) and reporter vectors TkpGL3 containing a single copy of each respective putative response element present in the promoter or in the first intron of the CETP gene (100 ng for each construct) using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. Medium was changed 24 h after transfection, and cells were treated with or without 2.5 µM GW4064 for 24 h. Luciferase activity was measured using the ONE-Glo Luciferase Assay System (Promega) according to the manufacturer's instructions. Values obtained on a luminometer (Wallac 1420 Multilabel Counter; PerkinElmer) were normalized with β-galactosidase activity determined by measuring hydrolyzation of chlorophenolred-β-D-galactopyranoside (CPRG; Roche Diagnostics, Germany) at 560 nm using a spectrophotometer (Wallac 1420 Multilabel Counter; PerkinElmer).
Statistical analyses
All data are reported as means ± SD. Statistical analyses were performed using SPSS 12.0 for Windows (SPSS, Inc., Chicago, IL). Differences between groups were analyzed by the Mann-Whitney U-test. Differences with a P value below 0.05 were considered statistically significant.
RESULTS
Plasma CETP activity and mass are increased in patients with high plasma BA levels
To assess the impact of plasma BA on CETP expression, cholestatic patients with liver cirrhosis (high BA group) were investigated. To exclude a potential confounding effect of reduced liver synthesis function in these patients, controls (low BA group) were matched not only for age, sex, and BMI but also for liver function and clinical stage of the liver disease (Table 1). On average, plasma BA levels were 3.7-fold higher in the high BA compared with the low BA group (Table 1). As expected, patients with high BA had 43% lower HDL-C levels than controls (20 ± 10 versus 35 ± 8 mg/dl, P = 0.001, Fig. 1A), while LDL cholesterol levels were increased (177 ± 53 versus 109 ± 41 mg/dl, P < 0.01, Fig. 1B). Next, CETA was determined in plasma of these subjects. Patients with high BA displayed 69% higher plasma CETA compared with the low BA group (initial transfer rates, 60.7 ± 10.6 versus 35.9 ± 8.3 pmol/h, P < 0.001, Fig. 1C), and plasma CETA correlated with plasma BA levels (r = 0.72, P < 0.001, Fig. 1E). To determine whether the elevated CETA in the high BA group was caused by an increase in the amount of circulating CETP, plasma CETP mass was measured by competitive ELISA. As shown in Fig. 1D, plasma CETP mass was 36% higher in the high BA group compared with the low BA group (2.66 ± 0.37 versus 1.96 ± 0.56 mg/l, P < 0.01, Fig. 1D) and was also correlated with plasma BA levels (r = 0.52, P = 0.019, Fig. 1F). These data suggest that BA might increase CETP expression.
Fig. 1.

Patients with high plasma BAs have increased CETA and CETP mass and decreased HDL-C levels. Plasma HDL-C (A), plasma LDL-cholesterol (B), plasma CETA (C), and CETP mass (D) in patients with liver cirrhosis. A group of patients with low plasma BA (≤30 µmol/l) was compared with a group with high plasma BA (>30 µmol/l). Groups were matched for age, sex, BMI, hepatic protein synthesis function, and the clinical stage of liver disease. CETP mass was assayed with a specific ELISA, and plasma CETA was measured by a fluorescent method as detailed in Materials and Methods. Data are presented as means ± SD, n = 10 patients/group. Significantly different from the low BA group, **P < 0.01, ***P < 0.001. Correlation between plasma bile acids and (E) plasma CETA and (F) CETP mass in the patients investigated.
BA feeding increases CETP expression and activity in E3L.CETP mice in vivo
To determine whether BAs indeed stimulate CETP expression, thereby increasing plasma CETP mass and activity in vivo, E3L mice expressing the human CETP transgene under the control of its natural (i.e., human) promoter were fed chow supplemented with 0.5% (w/w) TCA or control chow for six days. The treatment induced no change in body weight (Table 2) or food intake (data not shown), but plasma BAs were significantly increased (P < 0.01, Table 2). Plasma total cholesterol levels were significantly higher in the TCA-treated group (+46%, P < 0.01, Table 2), while plasma TG and phospholipids were lower compared with controls (−53%, P < 0.001 and −16%, P < 0.01, respectively). In addition, HDL-C levels were also decreased in the TCA-receiving group compared with controls (−27%, P < 0.01).
TABLE 2.
Body weight and plasma parameters in E3L.CETP mice fed either control chow or chow supplemented with 0.5% (w/w) TCA for six days
| Parameter | Control | TCA |
| n = 9 | n = 8 | |
| Body weight (g) | 28.9 ± 1.9 | 28.4 ± 2.3 |
| BA (µmol/l) | 42 ± 3 | 245 ± 116* |
| Total cholesterol (mmol/l) | 2.45 ± 0.20 | 3.58 ± 0.85* |
| Triglycerides (mmol/l) | 2.24 ± 0.28 | 1.06 ± 0.36† |
| Phospholipids (mmol/l) | 2.19 ± 0.22 | 1.84 ± 0.24* |
| HDL-C (mmol/l) | 0.59 ± 0.13 | 0.43 ± 0.14* |
Values are means ± SD. Significantly different from the respective chow-treated control groups: *P < 0.01, †P < 0.001.
FPLC analysis showed for both groups the overall lipid profile of E3L.CETP mice with accumulation of cholesterol within apoB-containing lipoproteins (Fig. 2A). However, TCA treatment resulted in an increase in VLDL and LDL cholesterol compared with controls (+32%), while HDL-C was decreased (−24%, Fig. 2A). TCA administration also reduced the TG content of apoB-containing lipoproteins (−52%, Fig. 2A). Although the effect of TCA treatment on TG levels might reflect changes in TG lipase activities (26), the changes in plasma cholesterol distribution observed in response to TCA treatment are consistent with the metabolic effects of increased CETP activity.
Fig. 2.

BA feeding increases CETP expression and CETA in CETP-transgenic APOE*3-Leiden mice. APOE*3-Leiden.CETP mice received either chow or chow supplemented with 0.5% (w/w) TCA for six days as indicated. (A) Cholesterol and triglyceride distribution over the different lipoprotein subfractions was analyzed by fast protein liquid chromatography using pooled plasma samples from mice receiving control diet (open circles) or TCA-containing diet (filled circles). (B) Plasma CETA and (C) diluted CETP activity measured with an excess of exogenous lipoprotein substrates in control (open circles) and TCA-treated (filled circles) mice measured by a fluorescent assay. (D) Plasma CETP mass determined with a specific immunoassay in control (open bars) and TCA-administered mice (filled bars). The lower panel shows representative Western blots for CETP in plasma from three control and three TCA-treated mice. (E) Hepatic mRNA expression of CETP, Shp, Cyp7a1, and Abcb11 (Bsep) in control (open bars) and TCA-receiving mice (filled bars) determined by quantitative real-time PCR. Experimental procedures are detailed in Materials and Methods. Values are given as means ± SD, n = 8–9 animals/group. *Significantly different from controls, at least P < 0.05.
Indeed, plasma CETA was significantly increased in TCA-receiving mice compared with controls (initial transfer rates, 137 ± 34 versus 43 ± 20 pmol/h, P < 0.01, Fig. 2B). In addition, diluted CETP activity measured with diluted plasma samples in the presence of an excess of exogenous lipoprotein substrates was also significantly higher in the TCA-fed group than in controls (83 ± 20 pmol/h versus 32 ± 15 pmol/h, P < 0.01, Fig. 2C), suggesting that the amount of active CETP in plasma is increased. Subsequent measurements of plasma CETP mass by immunoassay confirmed significantly higher CETP levels in TCA-treated mice compared with controls (6.2 ± 2.5 versus 2.3 ± 0.8 mg/l, P < 0.01, Fig. 2D).
Because most of plasma CETP originates from liver (15, 24), hepatic RNA was extracted from TCA-treated and control mice to determine whether the increase in plasma CETP levels was caused by increased CETP mRNA expression. As shown in Fig. 2E, CETP mRNA levels were markedly increased in TCA-treated mice compared with controls (+140%, P < 0.01). The observed increases in Shp and Abcb11 (Bsep) gene expression (+213% and +40%, respectively), together with the marked repression of the Cyp7a1 gene (−94%) in the TCA group (Fig. 2E) confirmed successful FXR activation by the BA treatment. TNFα expression was identical in both groups (data not shown), suggesting that changes in the inflammatory state did not affect the results. In addition, hepatic mRNA expression of apoA-I remained unchanged (1.00 ± 0.12 in controls versus 1.12 ± 0.23 in TCA-treated mice, nonsignificant) consistent with a previous notion that in contrast to human apoA-I mouse apoA-I is not an FXR target gene (11, 27). Taken together, these data suggest that the increase in plasma CETP activity and mass by TCA treatment in vivo in transgenic mice is transcriptionally mediated, likely via activation of the nuclear receptor FXR.
FXR agonists increase CETP gene expression in hepatocytes and macrophages in vitro
To further explore the mechanisms of the regulation of CETP expression by BA, first the FXR-dependent regulation of CETP was investigated in HepG2 cells stably transfected with the BA transporter Na+/taurocholate cotransporting polypeptide (NTCP/SLC10A1) (HepG2-NTCP cells) in order to allow efficient transport of TCA into the cells. Consistent with the results obtained in E3L.CETP mice, CETP gene expression was upregulated in a dose-dependent manner in the presence of increasing amounts of TCA, with a maximal 2.5-fold increase at TCA concentrations of 100 µM (P < 0.05, Fig. 3A). In addition, treatment of HepG2-NTCP cells with 2.5 µM of the synthetic nonsteroidal FXR agonist GW4064 resulted in a significant increase in CETP gene expression compared with vehicle-treated controls (+67%, Fig. 3B). Finally, also in primary hepatocytes from CETP-transgenic mice, FXR activation with 2.5 µM of GW4064 resulted in a significant 40% increase in CETP mRNA levels (+40%) and a similar increase in Abcb11 (Bsep) (+36%, P < 0.05), representing a “model” FXR target gene (Fig. 3C). In addition, we investigated whether the FXR-mediated activation of the CETP gene requires de novo protein synthesis. Treatment of HepG2 cells with GW4064 or the natural FXR ligand CDCA caused a similar increase in CETP mRNA expression in the absence (+60% for GW4064 and +61% for CDCA versus vehicle-treated cells, P < 0.05 in both cases) or presence (+49% for GW4064 and +60% for CDCA versus CHX-treated cells, P < 0.05 in both cases) of the protein synthesis inhibitor CHX (10 µg/ml). These results demonstrate that the increase in CETP gene expression by FXR agonists occurs independent of de novo protein synthesis, likely representing a direct transcriptional activation of FXR (Fig. 3D). To further confirm the dependency of the induction of CETP expression by FXR agonists on functional FXR expression, knockdown experiments were performed. Compared with control siRNA-transfected HepG2 cells, CETP mRNA expression in cells transfected with siRNA directed against FXR could no longer be induced by either taurocholate or GW4064 (Fig. 3E). For the known FXR target gene SHP, identical results were observed, while the negative control gene cyclophilin did not show any significant changes in expression under these assay conditions (supplementary Fig. I).
Fig. 3.

FXR ligands increase CETP mRNA expression in hepatocytes in vitro. (A and B) CETP mRNA levels in the HepG2-NTCP hepatoma cell line after treatment with vehicle (open bars), 10–100 µM TCA (panel A, filled bars), or 2.5 µM of the nonsteroidal FXR agonist GW4064 (panel B, filled bar). (C) CETP and Abcb11 (Bsep) mRNA levels in primary hepatocytes isolated from human CETP-transgenic mice treated with vehicle (open bars) or 2.5 µM GW4064 (filled bars). (D) CETP mRNA levels in the HepG2 hepatoma cell line after treatment with vehicle, 2.5 µM GW4064, or 100 µM CDCA in the absence or presence of 10 µg/ml CHX. (E) CETP mRNA levels in the HepG2-NTCP hepatoma cell line transfected with control siRNA (control) or FXR-specific siRNA (FXR) (see Materials and Methods) and treated with vehicle (open bars), 25 µM TCA, or 2.5 µM GW 4064 (GW, filled bars). Expression levels were related to the vehicle-treated control siRNA group (control). All experiments were performed at least in triplicate. Data are given as means ± SD. *Significantly different from controls, at least P < 0.05.
Since Kupffer cells, the resident liver macrophages, also contribute substantially to hepatic CETP production (28), the effect of FXR activation on CETP gene expression was investigated in vitro using human THP-1 monocytes differentiated into macrophages. In our hands, FXR expression was readily detectable in differentiated THP-1 macrophages, although we failed to detect expression of SHP (data not shown). As shown in Fig. 4A, the FXR-specific agonist GW4064 induced a marked dose-dependent upregulation of CETP expression, with a maximum 20-fold increase in CETP mRNA levels at concentrations of 5 µM. In addition, this stimulating effect of GW4064 on CETP mRNA levels was further potentiated in the presence of the retinoid X receptor (RXR) agonist 9-cis retinoic acid (Fig. 4B). The effect of GW4064 on the expression of CETP thus seems to follow a classical FXR activation pattern involving the heterodimerization of FXR with RXR. However, 9-cis retinoic acid could also induce CETP expression by acting on the RXR/liver X receptor (LXR) heterodimer (29).
Fig. 4.
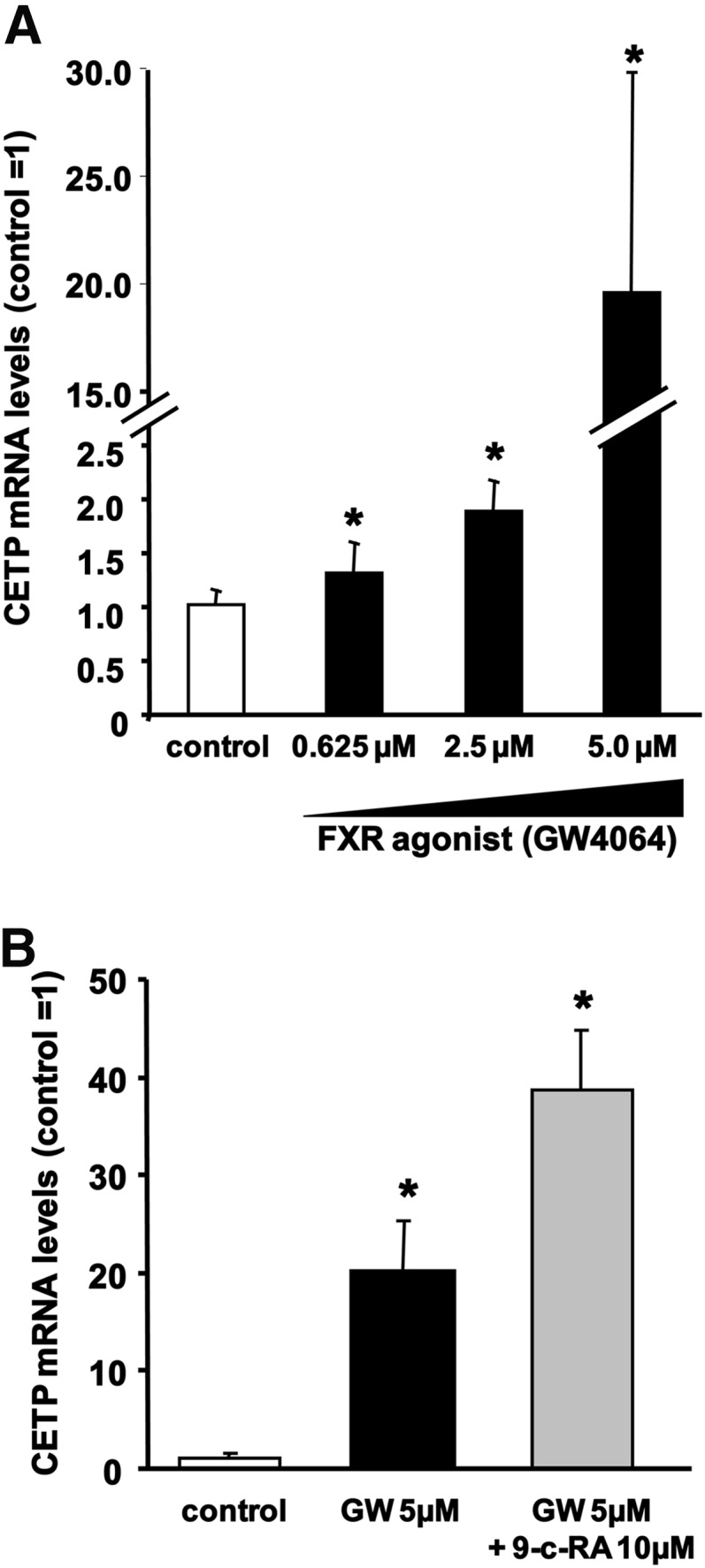
FXR ligands increase CETP mRNA expression in macrophages in vitro. (A) CETP mRNA levels in human THP-1 monocyte-derived macrophages treated with vehicle (open bars) or 0.625 to 5 µM GW4064 (filled bars). (B) CETP mRNA levels in human THP-1 monocyte-derived macrophages treated with vehicle (white bar), 5 µM GW4064 alone (GW, black bar), or in combination with 10 µM 9-cis-retinoic acid (9-c-RA, gray bar). All experiments were performed at least in triplicate. Data are given as means ± SD. *Significantly different from vehicle-treated controls, at least P < 0.05.
Taken together, these data demonstrate that FXR activation increases CETP mRNA expression in vitro in two relevant cell types.
FXR activation involves an ER8 element in the first intron of the CETP gene
In a first attempt to get insights into the molecular mechanisms and the sequences involved in the upregulation by FXR, we analyzed the sequence of the proximal promoter and the first intron of the CETP gene by using the NHR-scan analysis program. As shown in Fig. 5A, the software detected, in addition to the previously described LXR response element, which is a DR4 sequence at position −384 to −399 (29), two putative FXREs [i.e., one IR1 sequence in the promoter (position −2415 to −2427) and one ER8 in the first intron (position +317 to +336)] (30). The functionality of the proximal promoter of CETP containing the IR1 sequence was tested in HepG2 cells transfected with the first 3.5 kb upstream of the first exon and treated with GW4064. However, this sequence showed only limited responsiveness to FXR stimulation (+21% in luciferase activity compared with untreated controls, Fig. 5B). The isolated IR1 sequence of the CETP promoter was then tested in HEK293T (Fig. 5C) and appeared to be unresponsive to FXR stimulation, with similar reporter luciferase activities with native, mutated, and deleted CETP IR1 as well as with empty vector, whereas the ileal bile acid binding protein (IBABP) IR1, which was used as positive control (31), showed a good responsiveness to GW4064 treatment (3-fold increase compared with empty vector, P < 0.05). These results suggest that the proximal region (3.5 kb) of the CETP promoter, including the IR1 sequence at position −2415 to −2427, is not involved in the response to FXR. Conversely, the isolated ER8 sequence of the first intron of the CETP gene appeared to be functional in terms of FXR response, with a 2.6-fold increase in reporter luciferase activity after GW4064 treatment compared with empty vector (P < 0.05, Fig. 5D). Taken together, these data demonstrate that the upregulation of CETP in response to FXR stimulation occurs via an ER8 element that is present in the first intron at position +317 to + 336.
Fig. 5.

FXR regulates CETP gene expression through an ER8 sequence located in the first intron of the CETP gene. (A) Schematic representation of the proximal promoter and of the first intron and exon of the CETP gene. Promoter and introns are shown in gray and exons in black. The previously described LXR response element (DR4) and the TATA box are shown in addition to two putative FXR response elements, which were detected with the NHR-Scan software: one IR1 sequence in the promoter (position −2415/−2427) and one ER8 in the first intron (position +317/+336). (B) Reporter assay in HepG2 cells transfected with the proximal 3.5 kb of the human CETP promoter. Cells were treated with 2.5 µM of the FXR agonist GW4064 or vehicle for 24 h before performing dual luciferase assays. Results are shown as fold induction by GW4064 compared with cells treated with vehicle only. (C and D) Reporter assays with putative FXR response elements of the CETP gene in HEK293T cells after 24 h of treatment with vehicle or 2.5 µM GW 4064. Luciferase activities were corrected with β-Gal activities, and results are expressed as fold-induction compared with vehicle-treated cells after correction with cells transfected with empty vector. (C) Reporter assay with wild-type (IR1 CETP), mutated (IR1mut CETP), and deleted (IR1del CETP) IR1 sequences of the proximal promoter of the CETP gene. The functional IR1 sequence of IBABP was used as positive control. (D) Reporter assay with the wild-type ER8 sequence of the first intron of the CETP gene. The dysfunctional IR1 sequence of the CETP gene and the functional IR1 sequence of IBABP were used as negative and positive controls, respectively. Bars and values in panels B, C, and D are means of four determinations ± S.D. *Significantly different from empty vector, P < 0.05.
DISCUSSION
The results of this study demonstrate in vivo in patients and transgenic mice as well as in vitro in hepatocytes and macrophages that human CETP is positively regulated by FXR activation. Thus, based on these data using FXR agonists in humans, e.g. to target atherosclerosis or metabolic diseases, would be expected to result in a proatherogenic lipoprotein profile.
Endogenous BAs (11, 12) and exogenously supplied BAs (13, 14) have been recognized as negative regulators of plasma HDL-C levels in humans. These observations have thus far been largely attributed to FXR-mediated repression of apoA-I, the major structural apolipoprotein component of HDL, by BA (11, 32). Since CETP activity also lowers plasma HDL-C, we hypothesized that CETP might contribute to decreased HDL levels in conditions when plasma BAs are increased. Thus far, the impact of FXR on CETP has not been systematically explored, likely because studies addressing FXR signaling were predominantly conducted in mice (11, 33), which, in contrast to humans, represent a CETP-deficient species (1).
We first demonstrated that in cholestatic patients, plasma CETP levels as well as activity are significantly increased compared with control patients exactly matched for sex, age, BMI, and liver protein synthesis capacity. Thus far, only a few studies addressed alterations of CETP in patients with increased plasma BA compared with controls. One study assessed CETP activity in patients with cholestasis and found, in contrast to our study, a decrease in CETP activity compared with control subjects (34). However, in that study, controls were not carefully matched and decreased hepatic protein synthesis capacity in the patient group might conceivably represent a confounding factor, explaining the difference between these previous results and our study. In line with our data are anecdotal case reports presenting circumstantial evidence for a potential positive association between plasma BA and CETP. One report indicated that plasma CETP mass is positively correlated with plasma BA levels in three patients with Alagille syndrome (35), a liver disease associated with elevated plasma BA. Another case report on a single patient with cerebrotendinous xanthomatosis showed that plasma CETP activity increased after the initiation of CDCA treatment (36).
As a second step, we confirmed in vivo in mice expressing the human CETP transgene under the control of its endogenous promoter and in vitro in cell culture studies that FXR agonism increases CETP gene expression. Thus far, CETP gene expression has been described to be positively regulated mainly by cholesterol-driven mechanisms, either via SREBP (37, 38) or LXR (29), the latter potentiated by LRH-1 expression (39). On the other hand, CETP expression is negatively regulated by inflammation (40, 41). However, we ensured that TCA feeding did not cause a proinflammatory response in our experimental model, as opposed to the effects of CA feeding (33); therefore, we do not consider inflammation a confounding factor potentially affecting our experimental results.
Several studies have addressed the involvement of FXR in the development of atherosclerosis in mice. Results obtained with the use of FXR-deficient mice not expressing CETP are controversial (42–44). However, FXR agonists were demonstrated in mouse models without (44) or with CETP expression on cholesterol-enriched Western-type diets (45) to exert antiatherogenic effects, in a large part via decreasing levels of proatherogenic apoB-containing lipoproteins. These metabolic effects are likely mediated by decreased intestinal cholesterol absorption in response to FXR activation (46), which represents a significant contributing factor to total cholesterol levels in mice on a Western-type diet. In contrast, the mice in our study were fed chow and subsequently showed an increase in VLDL/LDL cholesterol as a reflection of increased CETP activity. However, in Ldlr-deficient mice with transgenic CETP expression, as well as in cynomolgous monkeys, FXR activation consistently resulted in a strong decrease in plasma HDL-C levels (45), similar to our results in the E3L.CETP model. Because CETP promotes the catabolism of antiatherogenic HDL and because our results show that FXR upregulates CETP expression, a reconsideration of the previous conclusion that FXR is antiatherogenic might be required, at least before extrapolating these results to the human situation.
Overall, our results provide strong evidence that the BA-mediated transcriptional upregulation of CETP expression observed in vivo is FXR dependent. Our in vitro results show that CETP expression in hepatocytes from CETP-transgenic mice and humans as well as in human macrophages is upregulated not only by BA, which might also have certain FXR-independent effects, but also with the specific nonsteroidal FXR agonist GW4064. In addition, we demonstrate that the upregulation of CETP by FXR agonists is direct and does not depend on de novo protein synthesis. Furthermore, knockdown experiments proved that the transcriptional regulation of CETP by BAs and GW4064 is dependent on functional FXR expression. Further support of a direct role of FRX/RXR heterodimers in the upregulation of CETP gene expression is provided by our observation that the GW4064-induced upregulation of CETP gene expression was further potentiated by the RXR agonist 9-cis retinoic acid. However, 9-cis retinoic acid can also induce CETP via the RXR/LXR heterodimer (29).
Regarding the molecular mechanism whereby FXR upregulates CETP gene expression, we were able to identify an ER8 element in the first intron that is responsive to FXR agonists. In contrast, a predicted inverted repeat (IR-1) sequence at nucleotides −2415 to −2427 in the proximal promoter closely resembling the consensus FXR/RXR binding sequence (A/G)GGTCA(A/G)(T/A)G(A/G)CCT (47) was unresponsive to FXR activation.
In summary, our study demonstrates in vivo in humans and transgenic mice as well as in vitro that FXR agonists upregulate human CETP expression, likely via an ER8 FXRE in the first intron. The FXR-mediated increase in CETP mass and activity results in the development of a proatherogenic lipoprotein profile. Thus, these data are not only of relevance for disease states linked to alterations in BA metabolism and excretion but also especially when considering FXR activation as a novel therapeutic strategy against metabolic diseases and atherosclerotic CVD.
Supplementary Material
Footnotes
Abbreviations:
- BA
- bile acid
- BMI
- body mass index
- CDCA
- chenodeoxycholic acid
- CETA
- cholesteryl ester transfer activity
- CETP
- cholesteryl ester transfer protein
- CHX
- cycloheximide
- CVD
- cardiovascular disease
- FXR
- farnesoid X receptor
- FXRE
- FXR response element
- HDL-C
- HDL cholesterol
- IBABP
- ileal bile acid binding protein
- LXR
- liver X receptor
- NBD-CE
- nitrobenzoxadiazole-labeled cholesteryl ester
- RXR
- retinoid X receptor
- TCA
- taurocholic acid
- TG
- triglyceride
This work was supported by grants from the Netherlands Organization for Scientific Research (VIDI Grant 917-56-358 to U.J.F.T., VIDI Grant 917-36-351 to P.C.N.R., VIDI Grant 917-66-301 to M.V.E.); the Fondation Recherche Médicale (FRM SPE20040300452 to T.G.); the Institut National de la Santé et de la Recherche Médicale and the French National Research Agency under the program “Investissements d'Avenir” (ANR-11-LABX-0021 to T.G., J.G., and L.L.); the Netherlands Heart Foundation (NHS 2003B136 to P.C.N.R.); and TopInstitute Pharma (Project T2-110 to F.K. and T.J.C.V.B.). P.C.N.R. (Grant 2009T038) and M.V.E. (Grant 2007T56) are established investigators of the Netherlands Heart Foundation.
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of one figure.
REFERENCES
- 1.Masson D., Jiang X. C., Lagrost L., Tall A. R. 2009. The role of plasma lipid transfer proteins in lipoprotein metabolism and atherogenesis. J. Lipid Res. 50(Suppl.): S201–S206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barter P. J., Brewer H. B., Jr, Chapman M. J., Hennekens C. H., Rader D. J., Tall A. R. 2003. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 23: 160–167 [DOI] [PubMed] [Google Scholar]
- 3.Rader D. J., Daugherty A. 2008. Translating molecular discoveries into new therapies for atherosclerosis. Nature. 451: 904–913 [DOI] [PubMed] [Google Scholar]
- 4.Boekholdt S. M., Kuivenhoven J. A., Wareham N. J., Peters R. J., Jukema J. W., Luben R., Bingham S. A., Day N. E., Kastelein J. J., Khaw K. T. 2004. Plasma levels of cholesteryl ester transfer protein and the risk of future coronary artery disease in apparently healthy men and women: the prospective EPIC (European Prospective Investigation into Cancer and nutrition)-Norfolk population study. Circulation. 110: 1418–1423 [DOI] [PubMed] [Google Scholar]
- 5.Zeller M., Masson D., Farnier M., Lorgis L., Deckert V., Pais de Barros J. P., Desrumaux C., Sicard P., Grober J., Blache D., et al. 2007. High serum cholesteryl ester transfer rates and small high-density lipoproteins are associated with young age in patients with acute myocardial infarction. J. Am. Coll. Cardiol. 50: 1948–1955 [DOI] [PubMed] [Google Scholar]
- 6.Ridker P. M., Paré G., Parker A. N., Zee R. Y., Miletich J. P., Chasman D. I. 2009. Polymorphism in the CETP gene region, HDL cholesterol, and risk of future myocardial infarction: genomewide analysis among 18,245 initially healthy women from the Women's Genome Health Study. Circ Cardiovasc Genet. 2: 26–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vasan R. S., Pencina M. J., Robins S. J., Zachariah J. P., Kaur G., D'Agostino R. B., Ordovas J. M. 2009. Association of circulating cholesteryl ester transfer protein activity with incidence of cardiovascular disease in the community. Circulation. 120: 2414–2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ritsch A., Scharnagl H., Eller P., Tancevski I., Duwensee K., Demetz E., Sandhofer A., Boehm B. O., Winkelmann B. R., Patsch J. R., et al. 2010. Cholesteryl ester transfer protein and mortality in patients undergoing coronary angiography: the Ludwigshafen Risk and Cardiovascular Health study. Circulation. 121: 366–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Vries R., Perton F. G., Dallinga-Thie G. M., van Roon A. M., Wolffenbuttel B. H., van Tol A., Dullaart R. P. 2005. Plasma cholesteryl ester transfer is a determinant of intima-media thickness in type 2 diabetic and nondiabetic subjects: role of CETP and triglycerides. Diabetes. 54: 3554–3559 [DOI] [PubMed] [Google Scholar]
- 10.Porez G., Prawitt J., Gross B., Staels B. 2012. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J. Lipid Res. 53: 1723–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Claudel T., Sturm E., Duez H., Torra I. P., Sirvent A., Kosykh V., Fruchart J. C., Dallongeville J., Hum D. W., Kuipers F., et al. 2002. Bile acid-activated nuclear receptor FXR suppresses apolipoprotein A-I transcription via a negative FXR response element. J. Clin. Invest. 109: 961–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sorokin A., Brown J. L., Thompson P. D. 2007. Primary biliary cirrhosis, hyperlipidemia, and atherosclerotic risk: a systematic review. Atherosclerosis. 194: 293–299 [DOI] [PubMed] [Google Scholar]
- 13.Kuriyama M., Tokimura Y., Fujiyama J., Utatsu Y., Osame M. 1994. Treatment of cerebrotendinous xanthomatosis: effects of chenodeoxycholic acid, pravastatin, and combined use. J. Neurol. Sci. 125: 22–28 [DOI] [PubMed] [Google Scholar]
- 14.Leiss O., von Bergmann K. 1982. Different effects of chenodeoxycholic acid and ursodeoxycholic acid on serum lipoprotein concentrations in patients with radiolucent gallstones. Scand. J. Gastroenterol. 17: 587–592 [DOI] [PubMed] [Google Scholar]
- 15.Jiang X. C., Agellon L. B., Walsh A., Breslow J. L., Tall A. 1992. Dietary cholesterol increases transcription of the human cholesteryl ester transfer protein gene in transgenic mice. Dependence on natural flanking sequences. J. Clin. Invest. 90: 1290–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Westerterp M., van der Hoogt C. C., de Haan W., Offerman E. H., Dallinga-Thie G. M., Jukema J. W., Havekes L. M., Rensen P. C. 2006. Cholesteryl ester transfer protein decreases high-density lipoprotein and severely aggravates atherosclerosis in APOE*3-Leiden mice. Arterioscler. Thromb. Vasc. Biol. 26: 2552–2559 [DOI] [PubMed] [Google Scholar]
- 17.Quintao E. C. R., Cazita P. M. 2010. Lipid transfer proteins: past, present and perspectives. Atherosclerosis. 209: 1–9 [DOI] [PubMed] [Google Scholar]
- 18.Gautier T., Tietge U. J. F., Boverhof R., Perton F. G., Le Guern N., Masson D., Rensen P. C., Havekes L. M., Lagrost L., Kuipers F. 2007. Hepatic lipid accumulation in apolipoprotein C–I-deficient mice is potentiated by cholesteryl ester transfer protein. J. Lipid Res. 48: 30–40 [DOI] [PubMed] [Google Scholar]
- 19.Guyard-Dangremont V., Lagrost L., Gambert P., Lallemant C. 1994. Competitive enzyme-linked immunosorbent assay of the human cholesteryl ester transfer protein (CETP). Clin. Chim. Acta. 231: 147–160 [DOI] [PubMed] [Google Scholar]
- 20.Wiersma H., Gatti A., Nijstad N., Oude Elferink R. P., Kuipers F., Tietge U. J. F. 2009. Scavenger receptor class B type I mediates biliary cholesterol secretion independent of ATP-binding cassette transporter G5/G8 in mice. Hepatology. 50: 1263–1272 [DOI] [PubMed] [Google Scholar]
- 21.Wiersma H., Nijstad N., de Boer J. F., Out R., Hogewerf W., Van Berkel T. J., Kuipers F., Tietge U. J. 2009. Lack of ABCG1 results in decreased plasma HDL cholesterol levels and increased biliary cholesterol secretion in mice fed a high cholesterol diet. Atherosclerosis. 206: 141–147 [DOI] [PubMed] [Google Scholar]
- 22.Hulzebos C. V., Renfurm L., Bandsma R. H., Verkade H. J., Boer T., Boverhof R., Tanaka H., Mierau I., Sauer P. J., Kuipers F., et al. 2001. Measurement of parameters of cholic acid kinetics in plasma using a microscale stable isotope dilution technique: application to rodents and humans. J. Lipid Res. 42: 1923–1929 [PubMed] [Google Scholar]
- 23.Glasova H., Berghaus T. M., Kullak-Ublick G. A., Paumgartner G., Beuers U. 2002. Tauroursodeoxycholic acid mobilizes alpha-PKC after uptake in human HepG2 hepatoma cells. Eur. J. Clin. Invest. 32: 437–442 [DOI] [PubMed] [Google Scholar]
- 24.Drayna D., Jarnagin A. S., McLean J., Henzel W., Kohr W., Fielding C., Lawn R. 1987. Cloning and sequencing of human cholesteryl ester transfer protein cDNA. Nature. 327: 632–634 [DOI] [PubMed] [Google Scholar]
- 25.Sandelin A., Wasserman W. W. 2005. Prediction of nuclear hormone receptor response elements. Mol. Endocrinol. 19: 595–606 [DOI] [PubMed] [Google Scholar]
- 26.Claudel T., Inoue Y., Barbier O., Duran-Sandoval D., Kosykh V., Fruchart J., Fruchart J. C., Gonzalez F. J., Staels B. 2003. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology. 125: 544–555 [DOI] [PubMed] [Google Scholar]
- 27.Gardès C., Blum D., Bleicher K., Chaput E., Ebeling M., Hartman P., Handschin C., Richter H., Benson G. M. 2011. Studies in mice, hamsters, and rats demonstrate that repression of hepatic apoA-I expression by taurocholic acid in mice is not mediated by the farnesoid-X-receptor. J. Lipid Res. 52: 1188–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Eck M., Ye D., Hildebrand R. B., K. Kruijt J., de Haan W., Hoekstra M., Rensen P. C., Ehnholm C., Jauhiainen M., Van Berkel T. J. 2007. Important role for bone marrow-derived cholesteryl ester transfer protein in lipoprotein cholesterol redistribution and atherosclerotic lesion development in LDL receptor knockout mice. Circ. Res. 100: 678–685 [DOI] [PubMed] [Google Scholar]
- 29.Luo Y., Tall A. R. 2000. Sterol upregulation of human CETP expression in vitro and in transgenic mice by an LXR element. J. Clin. Invest. 105: 513–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edwards P. A., Kast H. R., Anisfeld A. M. 2002. BAREing it all: the adoption of LXR and FXR and their roles in lipid homeostasis. J. Lipid Res. 43: 2–12 [PubMed] [Google Scholar]
- 31.Grober J., Zaghini I., Fujii H., Jones S. A., Kliewer S. A., Willson T. M., Ono T., Besnard P. 1999. Identification of a bile acid-responsive element in the human ileal bile acid-binding protein gene. Involvement of the farnesoid X receptor/9-cis-retinoic acid receptor heterodimer. J. Biol. Chem. 274: 29749–29754 [DOI] [PubMed] [Google Scholar]
- 32.Srivastava R. A., Srivastava N., Averna M. 2000. Dietary cholic acid lowers plasma levels of mouse and human apolipoprotein A-I primarily via a transcriptional mechanism. Eur. J. Biochem. 267: 4272–4280 [DOI] [PubMed] [Google Scholar]
- 33.Lefebvre P., Cariou B., Lien F., Kuipers F., Staels B. 2009. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 89: 147–191 [DOI] [PubMed] [Google Scholar]
- 34.Iglesias A., Arranz M., Alvarez J. J., Perales J., Villar J., Herrera E., Lasunción M. A. 1996. Cholesteryl ester transfer activity in liver disease and cholestasis, and its relation with fatty acid composition of lipoprotein lipids. Clin. Chim. Acta. 248: 157–174 [DOI] [PubMed] [Google Scholar]
- 35.Nagasaka H., Miida T., Hirano K., Ota A., Murayama K., Yorifuji T., Kobayashi K., Takatani T., Tsukahara H., Hui S. P., et al. 2008. Fluctuation of lipoprotein metabolism linked with bile acid-activated liver nuclear receptors in Alagille syndrome. Atherosclerosis. 198: 434–440 [DOI] [PubMed] [Google Scholar]
- 36.Kinoshita M., Kawamura M., Fujita M., Hirota D., Suda T., Taki M., Kusano J., Takao K., Takenaka H., Kubota S., et al. 2004. Enhanced susceptibility of LDL to oxidative modification in a CTX patient: role of chenodeoxycholic acid in xanthoma formation. J. Atheroscler. Thromb. 11: 167–172 [DOI] [PubMed] [Google Scholar]
- 37.Gauthier B., Robb M., Gaudet F., Ginsburg G. S., McPherson R. 1999. Characterization of a cholesterol response element (CRE) in the promoter of the cholesteryl ester transfer protein gene: functional role of the transcription factors SREBP-1a, -2, and YY1. J. Lipid Res. 40: 1284–1293 [PubMed] [Google Scholar]
- 38.Chouinard R. A., Jr, Luo Y., Osborne T. F., Walsh A., Tall A. R. 1998. Sterol regulatory element binding protein-1 activates the cholesteryl ester transfer protein gene in vivo but is not required for sterol up-regulation of gene expression. J. Biol. Chem. 273: 22409–22414 [DOI] [PubMed] [Google Scholar]
- 39.Luo Y., Liang C. P., Tall A. R. 2001. The orphan nuclear receptor LRH-1 potentiates the sterol-mediated induction of the human CETP gene by liver X receptor. J. Biol. Chem. 276: 24767–24773 [DOI] [PubMed] [Google Scholar]
- 40.Masucci-Magoulas L., Moulin P., Jiang X. C., Richardson H., Walsh A., Breslow J. L., Tall A. 1995. Decreased cholesteryl ester transfer protein (CETP) mRNA and protein and increased high density lipoprotein following lipopolysaccharide administration in human CETP transgenic mice. J. Clin. Invest. 95: 1587–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lakomy D., Rébé C., Sberna A. L., Masson D., Gautier T., Chevriaux A., Raveneau M., Ogier N., Nguyen A. T., Gambert P., et al. 2009. Liver X receptor-mediated induction of cholesteryl ester transfer protein expression is selectively impaired in inflammatory macrophages. Arterioscler. Thromb. Vasc. Biol. 29: 1923–1929 [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y., Wang X., Vales C., Lee F. Y., Lee H., Lusis A. J., Edwards P. A. 2006. FXR deficiency causes reduced atherosclerosis in Ldlr-/- mice. Arterioscler. Thromb. Vasc. Biol. 26: 2316–2321 [DOI] [PubMed] [Google Scholar]
- 43.Hanniman E. A., Lambert G., McCarthy T. C., Sinal C. J. 2005. Loss of functional farnesoid X receptor increases atherosclerotic lesions in apolipoprotein E-deficient mice. J. Lipid Res. 46: 2595–2604 [DOI] [PubMed] [Google Scholar]
- 44.Hartman H. B., Gardell S. J., Petucci C. J., Wang S., Krueger J. A., Evans M. J. 2009. Activation of farnesoid X receptor prevents atherosclerotic lesion formation in LDLR-/- and apoE-/- mice. J. Lipid Res. 50: 1090–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hambruch E., Miyazaki-Anzai S., Hahn U., Matysik S., Boettcher A., Perovic´-Ottstadt S., Schlüter T., Kinzel O., Krol H. D., Deuschle U., et al. 2012. Synthetic farnesoid X receptor agonists induce high-density lipoprotein-mediated transhepatic cholesterol efflux in mice and monkeys and prevent atherosclerosis in cholesteryl ester transfer protein transgenic low-density lipoprotein receptor (-/-) mice. J. Pharmacol. Exp. Ther. 343: 556–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gardes C., Chaput E., Staempfli A., Blum D., Richter H., Benson G. M. 2013. Differential regulation of bile acid and cholesterol metabolism by the farnesoid-X-receptor in Ldlr-/- mice versus hamsters. J. Lipid Res. 54: 1283–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Forman B. M., Goode E., Chen J., Oro A. E., Bradley D. J., Perlmann T., Noonan D. J., Burka L. T., McMorris T., Lamph W. W., et al. 1995. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 81: 687–693 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.