

Abstract
Vif forms a complex with Elongin B/C, Cullin-5 and Rbx-1 to induce the polyubiquitination and proteasome-mediated degradation of human APOBEC3G (hA3G). These interactions serve as potential targets for anti-HIV-1 drug development. We have developed a cell culture-based assay to measure Vif-induced A3G degradation. The assay is based on α-complementation, the ability of β-galactosidase fragments to complement in trans. A3G expressed with a fused α-peptide was enzymatically active, complemented a coexpressed ω fragment and could be targeted for degradation by Vif. Vif reduced β-galactosidase activity in the cell by 10-30-fold. The assay was validated by testing various A3G and Vif point mutants. The assay accurately detected the effects of D128 in A3G, and the BC box, Cul5 box, and HCCH motifs of Vif. The results showed a strict association of Vif biological function with hA3G degradation. These findings support hA3G degradation as a requirement for Vif function. The Vif α-complementation assay may be a useful tool for the identification of Vif inhibitors.
Keywords: HIV-1, Vif, APOBEC3G, α-complementation, cell-based assay, protein degradation, high throughput screening
Introduction
APOBEC3 cytidine deaminases constitute an arm of the innate immune system that protects against HIV-1 and the transposition of endogenous retroelements (Cullen, 2006; Harris and Liddament, 2004; Navarro and Landau, 2004). In HIV-1 that is deleted for Vif (Δvif), human APOBEC3G (hA3G) as well as other APOBEC3 family members can be packaged into virions in an infected cell. In the next round of replication, the packaged hA3G deaminates the viral DNA as it is synthesized. C→U deamination of the viral DNA minus-strand is manifested as G→A hypermutation of the plus-strand. Wild-type HIV-1 encodes the accessory protein Vif, which binds APOBEC3G and APOBEC3F to induce their rapid proteasomal degradation prior to encapsidation.
To cause hA3G degradation, Vif associates with a specific ECS-type E3 ligase which consists of Cullin-5 (Cul5), Elongin B, Elongin C and Rbx1 subunits (Mehle et al., 2004a; Yu et al., 2003). The Vif/Elongin BC/Cul5 complex catalyzes polyubiquitination of A3G which is followed by proteasomal degradation. Association of the E3 ligase with Vif is mediated by a sequence motif homologous to that of SOCS-box proteins, a family of proteins that mediates the binding of substrate to the E3 ligase. The SLQ sequence in Vif is highly conserved and has homology to the SOCS box motif (Marin et al., 2003; Mehle et al., 2004a; Yu et al., 2004). An HCCH motif in primate lentiviral Vif is required for Vif interaction with Cul5, but not with Elongin B or Elongin C (Luo et al., 2005).
We report here on the development of a cell-based assay to measure Vif-induced hA3G degradation. The assay is based on α-complementation of β-galactosidase in which an 84 amino acid, amino-terminal α–fragment of β-galactosidase and a carboxy-terminal ω-fragment, associate to form the catalytically active tetrameric enzyme. The method was validated by testing A3G and Vif mutations that are involved in Vif-induced A3G degradation. This assay can be easily adapted to a high-throughput format and used to perform large-scale screening for small chemical molecules that interrupt Vif-induced hA3G degradation. Various Vif and hA3G molecules that contained missense amino acid changes in conserved amino acid motifs were tested for α-complementation, biological function and protein stability by immunoblotting. The results show a strict association of hA3G degradation with Vif biological function, strongly supporting degradation of hA3G as a requirement for Vif function.
Materials and Methods
Plasmids
pA3G-α was generated by amplification of a human hA3G cDNA with primers containing Xba-I and Sma-I restriction sites. The amplicon was digested with Xba-I and Sma-I and ligated to similarly cleaved pcDNA3.1-α. To generate pcDNA3.1-α, the DNA encoding the α-fragment was PCR amplified with primers 5’-GAGTCTAGAGTCGACCTGCAGCCCAAGCTTGGGCTG-3’ and 5’-GAGGGATCCCTCAGGAAGATCGCACTCCAGCCAGCT-3’. The fragment was cleaved with Xba-I and BamH-1 and ligated to cleaved pcDNA3.1(-). phVif, pVif(co) and pVif(co)-HA were described previously (Nguyen et al., 2004; Schrofelbauer et al., 2006). phVifLF was constructed by ligating a Vif fragment amplified from phVif with pcDNA3.1(-). A3G and Vif mutants were generated using the QuickChange Site-Directed Mutagenesis Kit (Stratagene).
Cell lines
293T, 293T-ω (Holland et al., 2004), and HOS cells were maintained in DMEM/10% fetal bovine serum supplemented with penicillin and streptomycin.
α-complementation assay
293T or 293T-ω cells (5×105) were seeded in 6-well plates. The next day, the cells were transfected using Lipofectamine 2000 (Invitrogen) with Vif and A3G expression vectors. After 48 hours, the cells were rinsed twice with 2 ml cold phosphate buffered saline (PBS), then scraped off the plates in 250 μl 1% NP-40 lysis buffer. The lysates were incubated on ice for 30 min and centrifuged at 14,000 rpm for 15 min at 4°C. The protein concentration of the supernatant was measured using coomassie blue protein assay solution (Bio-Rad). Cell lysate containing 3 μg of protein was mixed with 100 μl of GalactoStar reagent (Applied Biosystems) in a 96-well plate and incubated at room temperature for 1 hr. Luminescence was quantitated in triplicate with a TopCount luminometer (Packard Instrument Co.). For colorimetric measurement, 12 μg of cell lysate was mixed with 100 μl reaction buffer (0.32 mg/ml 2-Nitrophenyl β-D-galactopyranoside (ONPG), 0.1 M phosphate buffer (pH 7.5)) and incubated at room temperature. The plate was read at A405 using a VersaMax microplate reader (Molecular Devices).
Luciferase reporter virus assay
293T cells were cotransfected with 1 μg of wild-type or Δvif pNL-LucE-R- luciferase reporter virus plasmid (Connor et al., 1995), 0.5 μg of pVSV-G, and with or without 1μg of A3G expression vector. For Vif mutant experiments, 1 μg of Vif expression vector was also cotransfected. Reporter viruses were harvested and the luciferase activity was measured as described previously (Schrofelbauer, Chen, and Landau, 2004).
Western blots
Cell lysate (10 μg) and solubilized virions (100 ng p24) were separated by SDS-PAGE and transferred to PVDF membranes. Proteins were detected with anti-human APOBEC3G mAb (1:3,000 dilution, Immunodiagnostic, Inc), anti-HIV Vif mAb (1:3000 dilution, AIDS Research and Reference Program), anti-p24 mAb (1:500 dilution, AIDS Research and Reference Program), anti-tubulin mAb (1:5000, Sigma), as previously described (Mariani et al., 2003; Schrofelbauer, Chen, and Landau, 2004).
Results
We previously reported on an α-complementation assay to measure viral envelope glycoprotein-mediated fusion (Holland et al., 2004). In that assay, the α-fragment and envelope glycoprotein were expressed in one cell and the ω-fragment and CD4/coreceptor in another. Upon co-culture of the cells, envelope glycoprotein-mediated fusion caused mixing of the cells’ cytoplasm, resulting in α-complementation. β-galactosidase activity could then be measured rapidly and accurately using luminescent substrate.
To establish an assay to measure Vif induced degradation of A3G, we adapted the α-complementation fusion assay (Holland et al., 2004). An hA3G-α fusion protein was expressed in cells that stably express the ω-fragment. α-complementation of the hA3G-α and the ω-fragment was predicted to generate high levels of β-galactosidase. In addition, the activity would be reduced by Vif as a result of the proteasomal degradation of the hA3G-α. The amount of β-galactosidase activity in the cells with and without Vif would be a measure of Vif-induced A3G degradation.
For the assay, the vector pA3G-α was constructed that expressed hA3G with the α-peptide fused to the C-terminus. To test whether hA3G-α was competent to mediate α-complementation, 293T and 293T-ω cells were transfected with pA3G-α plasmid alone or with pCMV-ω. As negative controls, the cells were transfected separately with pCMV-α and pCMV-ω. As positive controls, the cells were either cotransfected with pCMV-α and pCMV-ω or transfected with pCMV-LacZ which encodes full-length β-galactosidase. pCMV-α, pA3G-α and hA3G without the fused α-peptide (data not shown) yielded background levels of β-galactosidase activity (Fig. 1). Cotransfection of pA3G-α and pCMV-ω resulted in a high level of β-galactosidase activity, yielding a signal that was more than 100-fold above background and nearly as high as the native β-galactosidase. As the amount of transfected pA3G-α was increased, the enzyme activity levels increased further. The amount of β-galactosidase activity generated was similar in 293T-ω cells, suggesting that ω-fragment expression was not limiting (Fig. 1). These results showed that hA3G-α efficiently complemented the ω-fragment in the transfected cells.
FIG. 1. A3G-α complements ω-fragment in transfected cells.
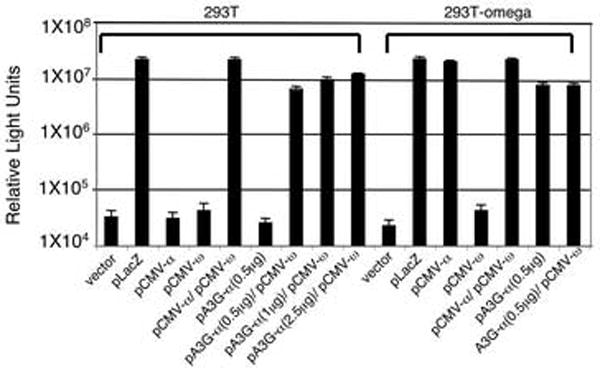
293T or 293T-ω cells were transfected with pA3G-α in triplicate. After two days, lysates were prepared and normalized for protein and β-galactosidase activity was measured by luminescence. The results are shown with the standard deviation.
Next, we tested whether hA3G-α maintained antiviral function. This was tested using single-cycle wild-type and Δvif luciferase reporter viruses. To produce the reporter viruses, 293T cells were cotransfected with wild-type or Δvif pNL-LucE-R- and either pA3G-α or control HA-tagged hA3G expression vector. pA3G-α was found to reduce virus infectivity to an extent comparable to hA3G and was found to be sensitive to Vif (Fig. 2A). These results suggested hA3G-α to be fully competent to be packaged into virions and to bind to Vif. Immunoblot analysis of the cell lysates showed that the hA3G-α was present at reduced quantity in cells that expressed Vif (Fig. 2B). The reaction was comparable to that of HA-tagged hA3G. Thus, the hA3G-α maintained the properties of the native protein.
FIG. 2. A3G-α maintains antiviral function.
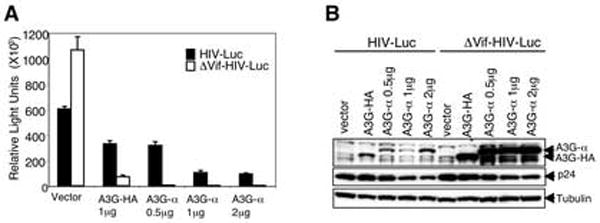
(A) Antiviral activity was measured with single-cycle HIV luciferase reporter virus. Reporter viruses were prepared in 293T cells cotransfected with wild-type or Δvif NL-Luc and A3G expression vector. Infectivity of the viruses was tested by infection of HOS cells and measurement of intracellular luciferase activity three days postinfection. (B) A3G-α in the transfected cells was detected on an immunoblot of cell lysates probed with anti-A3G mAb.
To determine how effectively Vif would reduce α-complementation, 293T-ω cells were transfected with pA3G-α, with or without HIV-1 Vif expression vector. Two days later, β-galactosidase activity was measured. In the absence of Vif, increasing amounts of hA3G-α expression vector caused corresponding increases in β-galactosidase activity levels (Fig. 3A). Vif caused a 10-fold decrease in β-galactosidase activity and this reduction was maintained over a range of hA3G-α expression levels. Immunoblot analysis of the cell lysates showed that Vif had caused a reduction of hA3G-α to undetectable levels (Fig. 3B). This was the case even in the transfection with the least amount of hA3G-α. As hA3G was increased, Vif decreased. This result confirmed the earlier findings of Mehle et al. that Vif is degraded with hA3G-α (Mehle et al., 2004b). Thus, the α-complementation assay accurately reflected hA3G levels and was more sensitive and quantitative than immunoblot analysis.
FIG. 3. Measurement Vif function by α-complementation.
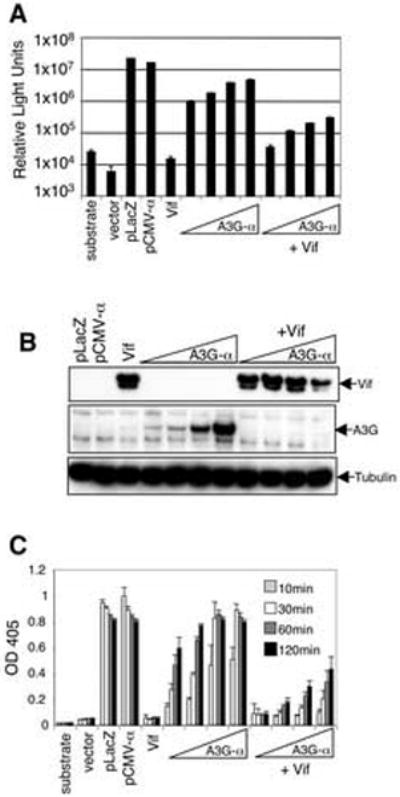
(A) 293T-ω cells were transfected with pcA3G-α alone (0.16, 0.4, 0.7, or 1.0 μg) or cotransfected with Vif expression vector (4 μg). β-galactosidase activity was measured two days later by luminescence. (B) Quantitation of Vif-induced A3G-α degradation by immunoblot analysis. (C) β-galactosidase activity in the α-complementation assay was determined by colorimetric analysis with ONPG substrate. Cell lysates were prepared in triplicate and normalized for protein concentration. ONPG substrate was added and A405 was measured over time.
β-galactosidase activity can also be measured colorimetrically using ONPG substrate, which is less expensive and produces a stable signal. To determine whether this method was suitable for the α-complementation assay, cell lysates from the previous experiment were incubated with ONPG and read at time points from 10-120 minutes. The signal increased over 60 minutes and then reached a plateau (Fig. 3C). The measurements agreed with the luminescent substrate results, also showing the 10-fold decrease in signal caused by Vif.
Several Vif expression vectors were tested for efficiency of hA3G-α degradation. phVif (Nguyen et al., 2004) and phVifLF express a partially codon-optimized HIV-1 Vif in pcDNA3.1-Myc-His and pcDNA3.1, respectively. pcVif(co)-HA and pcVif(co) express a fully codon optimized HIV-1 Vif with or without a C-terminal HA tag (Schrofelbauer et al., 2006). The Vif expression vectors were tested in the α-complementation assay. The fully codon-optimized Vifs were more efficient, reducing β-galactosidase activity 20-30-fold (Fig. 4).
FIG. 4. Comparison of Vif expression vectors in the α-complementation assay.
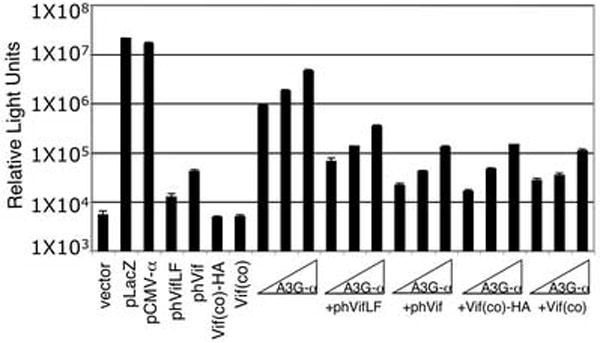
293T-ω cells were cotransfected with 4 μg of Vif expression vectors phVifLF, phVif, pcVif(co)-HA, or pcVif(co) and with or without pA3G-α (0.1, 0.2, or 0.5 μg).
Amino acid 128 of hA3G controls the species-specificity of the interaction with Vif (Bogerd et al., 2004; Mangeat et al., 2004; Schrofelbauer, Chen, and Landau, 2004; Xu et al., 2004). To test whether the D128K mutant, which contains the African green monkey-specific amino acid residue, was associated with resistance to Vif induced degradation, the mutation was introduced into hA3G-α and tested in the α-complementation assay. Over a range of hA3G-α concentrations, hA3G D128K-α was largely resistant to Vif (Fig. 5A). β-galactosidase was slightly reduced by the D128K mutant, indicating that it is still slightly sensitive to HIV-1 Vif. Luciferase reporter virus assay confirmed that the hA3G D128K-α was functional (Fig. 5B). The effect of Vif on the steady state level of D128K hA3G-α was tested by immunoblot analysis. This showed what appeared to be a more substantial degradation of hA3G D128K-α by Vif than what was detected by α-complementation (Fig. 5C). Thus, the D128K hA3G molecules that escape Vif degradation are sufficient to block viral infectivity. The α-complementation assay more accurately represents the antiviral function of Vif than does the immunoblot. These results further support an association of A3G degradation with Vif function.
FIG. 5. α-complementation accurately detects the effect of the D128K mutation on hA3G degradation.
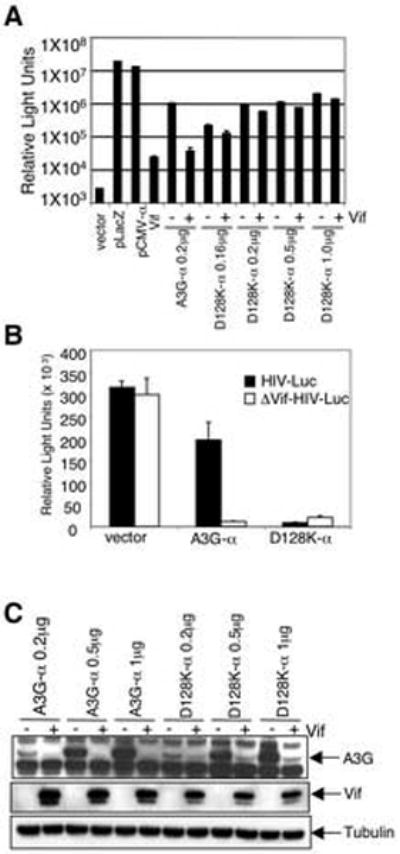
(A) 293T-ω cells were transfected with wild type A3G-α or A3G-α D128K with or without pcVif(co) and β-galactosidase was measured after two days. (B) Analysis of D128K hA3G-α antiviral activity by luciferase reporter virus assay. 293T cells were cotransfected with wild-type or Δvif NL-Luc with wild type A3G-α or A3G-α D128K. Virus was harvested, normalized for p24 and measured for infectivity in HOS cells. (C) Immunoblot analysis of the effect of Vif on steady-state levels of D128K A3G-α. Transfected cell lysates were probed with anti-A3G mAb.
Amino acid motifs of Vif that are required for Vif function include a BC-box that has similarity to the SOCS6 motif (SLQ144-146), a Cul5-box (L163P164L169) (Mehle et al., 2004a; Shirakawa et al., 2006; Yu et al., 2004) and an HCCH motif (Luo et al., 2005). To determine whether the role of these motifs could be detected in the α-complementation assay, the mutations were introduced into codon-optimized Vif and the mutants were tested in the assay. As shown in Fig. 6A, the BC-box triple SLQ→AAA mutation abolished Vif function in the assay although single amino acid changes in the motif did not affect Vif function. Single amino acid mutations in the HCCH motif abolished Vif function. These effects were confirmed by immunoblot analysis, which showed that the mutations that blocked Vif function in the α-complementation assay failed to induce hA3G-α degraded. Analysis of the mutants in the luciferase reporter virus assay showed that the α-complementation assay accurately determined Vif function in the virus. The SLQ triple mutant and the HCCH single mutants were nonfunctional in both assays (Fig. 6A, top and bottom). Analysis of mutants in the Cul5-box showed that the triple mutation (L163AP164AL169A) was inactive. A single mutation in the Cul5-box at amino acid 163 was still active while the single mutations at P164A and L169A inactivated the protein. Immunoblot analysis confirmed that these mutations were active at the level of hA3G degradation. Luciferase reporter virus analysis of the mutants showed that these motifs are required for Vif function in the virus and that function was closely associated with hA3G degradation.
FIG. 6. The role of the BC-box, HCCH and Cul5-box in Vif-induced hA3G-α degradation.
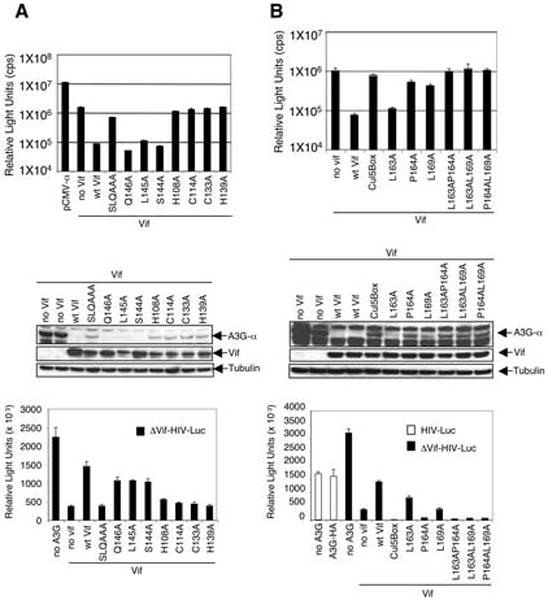
(A, top) 293T-ω cells were cotransfected with pA3G-α (0.3 μg) with Vif BC box (S144, Q145, and L146) and HCCH (H108, C114, C133, and H139) mutant expression vectors (3 μg) and β-galactosidase activity was measured two days posttransfection. (A, middle) The effect of the Vif mutants on hA3G-α degradation was detected by immunoblot analysis of the transfected cell lysates. (A, bottom) The effect of the Vif mutants on viral infectivity of Δvif virus was tested with luciferase reporter virus. (B, top, middle, and bottom). Vif with mutations in the Cul5-box (L163, P164, and L169) were similarly tested.
Finally, we tested a panel of polymorphic patient-derived Vifs identified by Simon et al. (Simon et al., 2005). The point mutations were introduced into the codon-optimized Vif and tested in the luciferase reporter virus assay (Fig. 7A, top). Of the six tested, four were inactive while two (W11R and G143R) maintained wild-type function, in agreement with the previous report (Simon et al., 2005). Immunoblot analysis of the virions showed that that the biologically inactive Vif mutants failed to prevent the packaging of hA3G while W11R and G143R mutants did not (Fig. 7A, bottom). In the cell lysates, there was no significant difference in hA3G degradation between the active (W11R and G413R) and inactive Vifs. In the α-complementation assay W11R and G143R Vifs were active, while the others were inactive (Fig. 7B, top), in agreement with the luciferase report virus assay. Immunoblot analysis of α-complementation samples confirmed the hA3G-α degradation by active W11R and G143R Vifs, but not other mutant Vifs (Fig. 7B, bottom). The discrepancy between the α-complementation and the reporter virus results was likely due to the difference in Vif:hA3G ratio in the two assays. In the α-complementation assay the ratio was at 10:1 while in the luciferase reporter virus assay a 1:1 ratio was used. These results showed a close correlation of Vif function in the viral infectivity with hA3G degradation, supporting the importance of degradation for Vif function.
FIG. 7. Association of Vif function with hA3G degradation as determined using natural Vif point mutants from primary viruses.
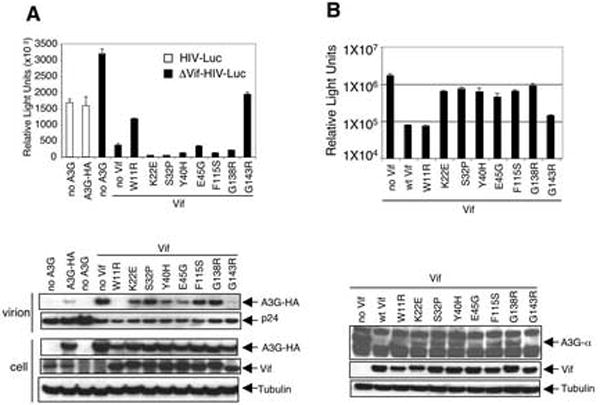
(A) Luciferase reporter viruses were prepared in 293T cells cotransfected with 1 μg of wild-type or Δvif NL-LucR-E- and 1 μg of A3G-HA expression vector. Vif point mutant expression vectors (1 μg) were included in the transfections and tested with luciferase reporter virus (top) and by immunoblot analysis (bottom). (B) 293T-ω cells were cotransfected with pA3G-α (0.3 μg) and Vif point mutant expression vectors (3 μg) and were tested by α-complementation (top) and immunoblot analysis (bottom).
Discussion
We have developed a cell-based assay for Vif-induced degradation of hA3G that is more rapid and accurate than immunoblot analysis. The assay is dependent upon the functional amino acid motifs of Vif including the Cul5-box, BC-box and HCCH motif, and can be adapted to high-throughput screening in small well formats. In principal, hA3G degradation could be measured by the more straight-forward use of an hA3G-luciferase or hA3G-EGFP fusion protein; however, initial tests of an hA3G-EGFP fusion protein were unsuccessful (not shown). The fusion protein maintained its antiviral activity but was insensitive to Vif, presumably as a result of steric hindrance to Vif binding. The much smaller α-peptide did not have this problem. In addition, our previous findings demonstrated the high sensitivity of the α-complementation system (Holland et al., 2004).
In our current analysis of the D128K hA3G, a surprisingly large proportion of the mutant protein was degraded by HIV-1 Vif. The unexpected amount of D128K hA3G degradation was probably the result of over-expression of Vif from the codon-optimized expression vector. This result suggests that amino acid 128 is not the sole determinant for the interaction with Vif. The finding is consistent with our earlier report which showed that the D128K hA3G could form a weak complex with Vif (Schrofelbauer, Chen, and Landau, 2004). The weak interaction is not sufficient to fully induce hA3G degradation and as a result, D128K hA3G scores as Vif-resistant in antiviral activity assays (Bogerd et al., 2004; Mangeat et al., 2004; Schrofelbauer, Chen, and Landau, 2004; Xu et al., 2004).
Most of the Vif mutants with alterations in the known amino acid motifs were nonfunctional in the α-complementation assay, although one difference from published results was noted. In contrast to previous reports, we found that L145A Vif, which is altered at the highly conserved SLQ motif, was active (Mehle et al., 2004a; Yu et al., 2004). In our study, L145A rescued infectivity and induced hA3G degradation. Alteration of the three amino acids of the motif was needed to block Vif function. This difference could be caused by the expression of high levels of Vif in our system.
Our data demonstrate a close association of the ability of Vif to rescue viral infectivity with hA3G degradation. Vif mutants that were inactive failed to induce hA3G degradation while functional mutants efficiently induced degradation. One exception to this finding was the analysis of Vif mutants W11R and G143R. At a 1:1 ratio of Vif to hA3G, W11R and G143R Vif did not reduce hA3G levels in the cell. However, the mutant Vifs restored viral infectivity and prevented hA3G encapsidation, in agreement with a previous report (Tian et al., 2006). At a 10:1 ratio, both mutants induced efficient hA3G degradation. This could be interpreted as a dissociation of hA3G degradation from biological function; however, an alternative explanation is more likely. At reduced levels, Vif may preferentially degrade hA3G molecules that are near assembling virions in the cell. Vif is localized in the cell to regions that contain Gag (Simon et al., 1999; Simon et al., 1997) and this localization could be a means of catching hA3G molecules that are in the proximity of an assembling virion. This would allow Vif to block hA3G packaging without depleting hA3G from the cell. With higher levels of Vif, hA3G might be depleted from the cell. Our results are consistent with hA3G degradation as the key mechanism for Vif function.
In a small molecule screen, the α-complementation assay would detect cell permeable compounds that act either on Vif or hA3G to block their interaction or compounds that inhibit the E3 ligase or proteasome function. Small molecules that target the protein degradation pathway could be cytotoxic by interfering with the role of these proteins in cellular metabolism. However, many of these proteins are redundant (Ciechanover, 2005; Schuberth et al., 2004) and their partial inhibition might be sufficient to rescue the antiviral function of hA3G. Thus, small molecules specific for Cul5/ElonginBC/Rbx1 complexes might block Vif function without having serious cytotoxic effects.
Current antiretrovirals target reverse transcriptase, protease or envelope glycoprotein. The interaction of Vif with hA3G is an additional promising target for drug development. The α-complementation assay is rapid and accurate making it useful for the discovery of molecules that target this interaction.
Acknowledgments
We thank Qin Yu, Bärbel Schrofelbauer, Hui Chen, and Yoshiyuki Hakata for helpful discussions, Klaus Strebel for Vif expression vector phVif. We thank Jody Chou for technical support and Erica Dhuey for critical reading of the manuscript. This work was funded by N.I.H grants AI51686 and DA14494, the UCSD Center for AIDS Research and a post-doctoral fellowship to L. F. from the American Foundation for AIDS Research. N.R.L is an Elizabeth Glaser Fellow of the Pediatric AIDS Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bogerd HP, Doehle BP, Wiegand HL, Cullen BR. A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc Natl Acad Sci U S A. 2004;101(11):3770–4. doi: 10.1073/pnas.0307713101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6(1):79–87. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206(2):935–44. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- Cullen BR. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J Virol. 2006;80(3):1067–76. doi: 10.1128/JVI.80.3.1067-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol. 2004;4(11):868–77. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- Holland AU, Munk C, Lucero GR, Nguyen LD, Landau NR. Alpha-complementation assay for HIV envelope glycoprotein-mediated fusion. Virology. 2004;319(2):343–52. doi: 10.1016/j.virol.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Luo K, Xiao Z, Ehrlich E, Yu Y, Liu B, Zheng S, Yu XF. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc Natl Acad Sci U S A. 2005;102(32):11444–9. doi: 10.1073/pnas.0502440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Liao S, Trono D. A single amino acid determinant governs the species-specific sensitivity of APOBEC3G to Vif action. J Biol Chem. 2004;279(15):14481–3. doi: 10.1074/jbc.C400060200. [DOI] [PubMed] [Google Scholar]
- Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114(1):21–31. doi: 10.1016/s0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9(11):1398–403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- Mehle A, Goncalves J, Santa-Marta M, McPike M, Gabuzda D. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 2004a;18(23):2861–6. doi: 10.1101/gad.1249904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 2004b;279(9):7792–8. doi: 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- Navarro F, Landau NR. Recent insights into HIV-1 Vif. Curr Opin Immunol. 2004;16(4):477–82. doi: 10.1016/j.coi.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Nguyen KL, llano M, Akari H, Miyagi E, Poeschla EM, Strebel K, Bour S. Codon optimization of the HIV-1 vpu and vif genes stabilizes their mRNA and allows for highly efficient Rev-independent expression. Virology. 2004;319(2):163–75. doi: 10.1016/j.virol.2003.11.021. [DOI] [PubMed] [Google Scholar]
- Schrofelbauer B, Chen D, Landau NR. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif) Proc Natl Acad Sci U S A. 2004;101(11):3927–32. doi: 10.1073/pnas.0307132101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrofelbauer B, Senger T, Manning G, Landau NR. Mutational alteration of human immunodeficiency virus type 1 Vif allows for functional interaction with nonhuman primate APOBEC3G. J Virol. 2006;80(12):5984–91. doi: 10.1128/JVI.00388-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuberth C, Richly H, Rumpf S, Buchberger A. Shp1 and Ubx2 are adaptors of Cdc48 involved in ubiquitin-dependent protein degradation. EMBO Rep. 2004;5(8):818–24. doi: 10.1038/sj.embor.7400203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa K, Takaori-Kondo A, Kobayashi M, Tomonaga M, Izumi T, Fukunaga K, Sasada A, Abudu A, Miyauchi Y, Akari H, Iwai K, Uchiyama T. Ubiquitination of APOBEC3 proteins by the Vif-Cullin5-ElonginB-ElonginC complex. Virology. 2006;344(2):263–6. doi: 10.1016/j.virol.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Simon JH, Carpenter EA, Fouchier RA, Malim MH. Vif and the p55(Gag) polyprotein of human immunodeficiency virus type 1 are present in colocalizing membrane-free cytoplasmic complexes. J Virol. 1999;73(4):2667–74. doi: 10.1128/jvi.73.4.2667-2674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JH, Fouchier RA, Southerling TE, Guerra CB, Grant CK, Malim MH. The Vif and Gag proteins of human immunodeficiency virus type 1 colocalize in infected human T cells. J Virol. 1997;71(7):5259–67. doi: 10.1128/jvi.71.7.5259-5267.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon V, Zennou V, Murray D, Huang Y, Ho DD, Bieniasz PD. Natural Variation in Vif: Differential Impact on APOBEC3G/3F and a Potential Role in HIV-1 Diversification. PLoS Pathog. 2005;1(1):e6. doi: 10.1371/journal.ppat.0010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C, Yu X, Zhang W, Wang T, Xu R, Yu XF. Differential requirement for conserved tryptophans in human immunodeficiency virus type 1 Vif for the selective suppression of APOBEC3G and APOBEC3F. J Virol. 2006;80(6):3112–5. doi: 10.1128/JVI.80.6.3112-3115.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Svarovskaia ES, Barr R, Zhang Y, Khan MA, Strebel K, Pathak VK. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc Natl Acad Sci U S A. 2004;101(15):5652–7. doi: 10.1073/pnas.0400830101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302(5647):1056–60. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu XF. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 2004;18(23):2867–72. doi: 10.1101/gad.1250204. [DOI] [PMC free article] [PubMed] [Google Scholar]