

Abstract
This article examines the current use and future implications of stem cell therapy in treating Multiple Sclerosis (MS). MS is the most common neurological disease in young adults, affecting approximately two million people worldwide. Currently there is no cure for MS. The standard treatment of MS involves disease-modifying drugs, which work to alleviate the symptoms of MS. However, these drugs carry adverse side effects and are ineffective in preventing disease progression in many MS patients. Hematopoietic stem cell transplantation (HSCT) was first used in 1995 to treat patients with severe rapidly progressing MS. The HSCT treatment protocol has evolved into a less intense conditioning regimen that is currently demonstrating efficacy in treating patients with variable disease severity—with best results in early-stage rapidly progressing MS patients with active CNS inflammation. Mesenchymal stem cell therapy (MSCT) is an experimental stem cell therapy currently undergoing clinical trials. Animal models and early clinical trials have shown promise that MSCT might be a low risk treatment to precipitate neuroregeneration and immunomodulation in MS patients. Specifically, neuroprogenitor and placental-derived mesenchymal stem cells offer the best hope for a practical treatment for MS. Stem cell therapy, and perhaps a combinatorial therapeutic approach, holds promise for a better treatment for MS.
Keywords: Multiple sclerosis, stem cells, autologous hematopoietic stem transplantation, mesenchymal stem cell transplantation
Introduction
Multiple Sclerosis (MS) is a common autoimmune disease of the CNS which can lead to severe disability and neurological defects. In patients with MS, activated autoreactive T-cells enter the CNS, attacking myelin and producing inflammatory responses which cause multifocal demyelination, axonal loss, and scarring of white matter [1]. Following an acute demyelination episode, MS progression will then typically follow one of four courses: relapsing-remitting MS (RRMS), secondary progressive MS (SPMS), primary progressive MS (PPMS) and progressive-relapsing MS (PRMS).
Most patients will first have a single attack of symptoms, a neurological episode called a clinically isolated syndrome, suggestive of demyelination. Once a second attack occurs, the patient is considered to have RRMS. RRMS is the most common type of MS and is characterized by acute symptomatic attacks followed by periods of partial or complete recovery. These patients often transition to secondary progressive MS after 10-25 years: a state characterized by reduced remission periods and increased neurological deterioration without an acute attack. PPMS is characterized by neurological decline in the absence of acute attacks beginning from the onset of demyelination. There is also a less common subtype called PRMS where patients show a progressive functional decline between acute attacks [2]. Diagnosis is typically made through clinical neurological assessment and MRI [3].
MS is most common between ages 20-40, especially in women, and has a prevalence of 120 per 100,000 individuals. The etiological cause of MS remains unclear, and its complexity makes successful management difficult [4].The current hypothesis for the etiology of MS is the “fertile field hypothesis” in which a genetic predisposition combined with a number of harmful environmental factors prime the immune system for autoreactivity. Then a wide array or “fertile field” of other environmental factors trigger the acute or chronic autoimmune attack leading to demyelination and axonal loss [5] (Figure 1). While there are treatment options available, standard therapies are often short term and are used to treat the acute symptoms of the MS rather than the underlying cause. This has caused many patients to turn to alternative modes of therapy showing little to no scientific evidence, such as bee sting therapy and cranio-sacral therapy [6]. MS treatment has thus progressed into an increasingly active area of research as physicians, patients, and scientists continue to seek more ameliorative methods of therapy.
Figure 1.
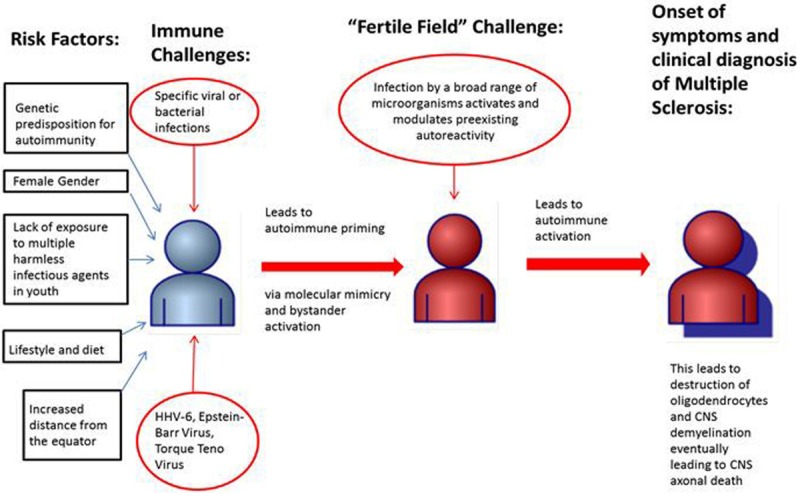
Proposed Etiology of Multiple Sclerosis. The current theory for the development of multiple sclerosis is that multiple risk factors combined with specific viral or bacterial infections activate the patient’s immune system and prime the patient for autoreactivity. Then infection with a fertile field of diverse microorganisms acutely and/or chronically triggers autoimmunity within a patient. The patient then suffers from acute or chronic CNS inflammatory attacks which destroy oligodendrocytes and lead to CNS neural demyelination. In the late stages the disease causes axonal loss [5].
The use of stem-cell therapy, specifically hematopoietic and mesenchymal stem cell transplantation, is a growing area of study that shows promise in MS treatment. Pre-clinical and early clinical studies involving mesenchymal stem cell therapy have demonstrated the potential of alleviating symptoms in MS, and combination therapies are beginning to be tested. While successful research in stem cell therapy warrants further exploration in this field, this potential treatment should serve as an additional option or contribution to current therapies that clinicians can pursue rather than an alternative. Although much more extensive research is necessary to achieve a full comprehension of MS and perhaps a cure, these stem cell transplant studies have produced effective results that are worth assessing, and may not only provide a viable treatment option with improved therapeutic benefit but also provide hope for further progress in MS research.
Current therapies
The current first-line therapies for RRMS are interferon beta or glatiramer acetate (GA). Interferon beta promotes anti-inflammatory cytokines and suppresses CD4+ activation, whereas GA, a synthetic molecule, mimics myelin basic protein, diverting the immune system’s attack away from its own myelin. GA also stimulates regulatory T-cell production, and produces anti-inflammatory and neuroprotective effects in the CNS [7].
A T-cell analysis of 84 RRMS patients undergoing treatment over the course of a year resulted in decreased CD4 T-cells and increased regulatory T-cell population for those on GA. Both interferon and GA treatments alter T-cell differentiation, decrease relapses by 30%, reduce the severity of the attacks, and slow the progress of MS [4]. Interferon exacerbates depression in patients, and thus may be a deciding factor when choosing between the two treatments. Though these standard therapies have proved somewhat effective and safe, there are many adverse effects. 75% of patients taking interferon have reported flu-like symptoms, and injection site skin reactions are common [8]. There is also risk for liver dysfunction, thyroid abnormalities, menstrual abnormalities, and a few rare disorders such as myasthenia gravis, hemolytic uremic syndrome, capillary leak syndrome, and hearing loss, among others. Additionally for pregnant women, there is a higher rate of miscarriages and stillbirths (39.1%) than in healthy controls (5%) [8]. GA differs in that it does not produce flu-like symptoms and instead is mostly associated with injection site reactions (73% of patients) involving pain, bruising, and inflammation. There are a few less common adverse effects such as chest pain, dyspnea, and lymphadenopathy [8].
In addition to interferon and GA, Several disease-modifying treatment options exist today for the management of MS. Recent studies have focused on the development of oral agents, such as fingolimod, the first oral treatment which has shown therapeutic benefit for MS patients in advanced clinical trials. Fingolimod acts directly on the CNS by crossing the blood brain barrier and drawing lymphocytes into lymph nodes, thereby preventing their migration to areas of the brain which would cause inflammation and damage [1]. Another more novel oral treatment recently approved by the FDA in September 2012, teriflunomide, is an inhibitor of pyrimidine synthesis, leading to a reduction of harmful T- and B-cell activation and proliferation. Teriflunomide has shown some promising effects with reduced relapse rates over a 372 week period, as well as reduced MRI activity (indicative of less demyelination) and decreased disability progression [9].
A variety of non-oral agents have proven beneficial in studies as well. Natalizumab, a monoclonal antibody, has shown a significant decrease in relapse rates by 60% over one year as well as an increased tissue volume indicative of remyelination. Natalizumab monotherapy comes with relatively higher risk than other drugs, such as the development of fatal progressive multifocal leukoencephalopathy, and thus is often used as a second-line of defense if initial treatments are failing [10]. Mitoxantrone is a unique therapy that targets patients with SPMS as well as PPMS, while the majority of MS treatment available is for RRMS. Mitoxantrone mechanisms are unclear, but are associated with blocked DNA synthesis. Though carrying high risk for cardiotoxicity, mitoxantrone shows reduced relapse rates as well as increased NK cell maturation which has been associated with successful treatment in studies of standard MS drugs [11].
Without a full understanding of MS, treatment options remain inevitably incomplete. Current treatment options do provide benefits; however, all have major risks and some are ineffective or even solely detrimental. Currently there are no effective therapies for PPMS and the only drugs shown to be effective in decreasing symptoms in SPMS are mitoxantrone and interferon beta [12]. Interferon Beta and GA are used primarily to reduce the amount of relapses in RRMS, but if the disease worsens to SPMS then these drugs lose their efficacy. Interferon Beta is the most widely prescribed treatment for RRMS, but there are large subsets of RRMS patients who do not response to the drug [13]. Additionally, in some RRMS patients interferon beta actually exacerbates their symptoms [14].
Stem cell therapy is a growing area of research that may contribute to additional treatment options, leading to more effective management of MS. Two Stem cell therapies currently being used to treat MS are Hematopoietic Stem Cell Transplantation (HSCT) and a newly emerging therapy of Mesenchymal Stem Cell Transplantation (MSCT).
Hematopoietic stem cell transplantation (HSCT)
HSCs are found within bone marrow in niches created by surrounding stromal cells. These stromal cells create a microenvironment where the HSCs are shielded from differentiation and apoptosis. HSCs have the potential to differentiate into the main hemato- and lymphopoietic precursors which then differentiate into mature cells. They are generated in large numbers throughout our lives and continually repopulate our blood and immune system.
The concept behind HSCT or autologous stem cell transplantation (ASCT) is that destroying the aberrant T-cells of an MS patient and then reconstituting the patient’s immune system with non-autoreactive T-cells would be a viable course of action in preventing future autoimmune damage. Replacing defective immune cell lineages with naïve cells harvested from the patient begins by administering drugs, typically granulocyte colony-stimulating factor (G-CSF) and cyclophosphamide (CY), to induce white blood cell proliferation in the peripheral blood. After proliferation, the patient’s hematopoietic stem cells are harvested by utilizing antibodies specific for the stem cell antigen CD34 that is present on hematopoietic progenitor cells [15]. After harvesting the graft, the patient undergoes a conditioning regime of variable intensity that uses pharmaceuticals and/or radiation to destroy part or all of the patient’s bone marrow. This is followed by the infusion of the previously harvested HSCs to then theoretically reconstitute a healthy immune system [16].
Due to the high variability of symptoms and disease progression associated with MS, a method of quantifying disease severity is necessary for objective evaluation of clinical success. The Extended Disability Status Scale (EDSS) is a system used to assess MS patient health in eight functional systems and then generate a score between 0-10. 0 is indicative of a normal neurological exam, 5 indicates the patient is ambulatory without aid for 200 meters but has a disruption of normal daily activities, and 10 indicates the patient has succumbed to death by MS. In the past this has been the clinical standard used to assess MS disease severity pre and post-treatment [17].
In 2001, Carreras conducted a phase II trial to evaluate the feasibility and toxicity of HSCT in MS patients. This trial, in which 15 advanced disability patients (9 with SPMS, 6 with RRMS) underwent HSCT, is demonstrative of the parameters of the initial HSCT trials [16]. The patient population for this study was chosen based upon lack of positive response to standard immunotherapy medications and disease severity indicated by a median EDSS of 6.0. Patient evaluations were conducted 1, 3, 6, 9, and 12 months following the HSCT. 1 patient did not hematologically reconstitute following 2 unsuccessful HSCT attempts and therefore was not included in the results. Complications included 1 patient who sustained a transient neurological deterioration, 3 patients who suffered from transient engraftment syndrome, and 2 who underwent a neurologic deterioration and high fever related to the initial myeloablative regimen. Other general side effects of the myeloablative process include loss of hair, premature menopause, increased risk of opportunistic infections, and adverse drug reactions. Post-treatment EDSS measurements, neurologic examinations, ambulatory index scores, and MRIs were used to assess both disease progression and treatment effectiveness. Out of the 14 patients who completed hematologic mobilization, 2 patients reported a worsening EDSS, 9 remained unchanged and 3 improved. Relapse occurred in 2 patients after the immunosuppressive therapy was removed. MRI identified new lesions following HSCT in 1 patient while 13 showed no new lesions. The study concluded that MS patients’ neurologic improvements were “unlikely” to be attributed to adaptive immune system re-education. Rather, the improvement was attributed to the immunosuppressive effects of the HSCT conditioning process. The conclusion drawn from this study was that the HSCT process was effective in reducing the progression of MS with an acceptable amount of toxicity and side effects.
Since 1997, when the first pilot study of MS treatment with HSCT began [18] the number of trials has increased and the treatment protocol has changed. Due to the side effects associated with HSCT, the initial trials were restricted to patients with severe rapidly progressing MS who were unresponsiveness to other MS medications. This was demonstrated by a retrospective survey done in 2006 that looked at all MS patients who underwent HSCT between the years of 1995-2000 registered in the database of the European Blood and Marrow Transplantation Group [19]. The survey found that before starting treatment the median EDSS score for the patient population was 6.5, indicating severe late stage MS.
More recently, researchers have conducted trials using less intense chemotherapeutic methods on a wider range of MS patients [20,21]. The rationale for this shift is a growing acceptance of the hypothesis that MS is caused by the interplay of genetics and specific sequential environmental triggers [22]. In this context, eradication of only self-reactive T cells (non-myeloablation) could itself halt MS progression. Full eradication of all of a patient’s hematopoietic cells (myeloablation) is therefore deemed unnecessary and inadvisable given the side effects [23]. If the fertile field hypothesis is correct then the patient’s remaining hematopoietic cells would not differentiate to self-reactive T cells since the environmental triggers inducing this state are unlikely to still be present. Additionally, HSCT is thought to have an immunomodulatory effect in addition to its immunosuppressive effect, evidenced by the fact that patients post-HSCT often have a more diverse T-cell clonal population [24,25]. This offers more evidence to support non-myeloablative HSCT because it demonstrates the potential of HSCT to modulate auto-immunity without the need to fully eradicate the patient’s bone marrow. Non-myeloablative HSCT carries less risk of lethal side effects and so the reduced risk-to-benefit ratio of these trials has allowed researchers to extend HSCT treatment to patients with less severe MS.
The “intensity” of the HSCT is based upon the conditioning regimen used. High intensity is a term used to describe conditioning regimens that use total body radiation (TBI) or busulphan-containing drugs, low intensity refers to regimens that consist of CY alone, melphalan alone and fludarabine-based regimens, and intermediate intensity includes most other combinations, such as BEAM (carmustine, etoposide, cytarabine, melphalan) and the combined use of Anti-T globulin (ATG) with high-dose CY or other chemotherapy [26]. Generally, the less intense the conditioning regimen, the less myeloablative it is, with low intensity regimens being considered truly non-myeloablative. The continued use of high intensity HSCT regimens is an area of contention. Some researchers argue against its use since clinical trials of high intensity HSCT have demonstrated equal efficacy as intermediate and low intensity regimens but have higher associated toxicity, prevalence of secondary autoimmune reactions, and mortality risk [27-29].
The results of recent non-myeloablative or low intensity HSCT trials show promise for an effective broader treatment for MS [20,21,30]. A study that compared the efficacy of low intensity vs. intermediate intensity HSCT in treating MS patients concluded that the low intensity HSCT is associated with similar clinical outcomes as the intermediate regimen but with lower toxicity and side effects [21]. In the study, clinicians performed HSCT on 41 patients with MS, 33 of which had SPMS. The median EDSS of the patients was 6.5. The study was initially designed to only test the efficacy of intermediate intensity HSCT in treating MS but was expanded to a comparative study involving less intense HSCT when three patients died in the early phases of the study from complications arising from the intermediate conditioning regimen. Systematic analysis reveals that transplant related mortality (TRM) in early clinical trials was 5-6% from 1995-2003, but dropped to 1-2% from 2003-2008 when low intensity trials first began. Patient outcomes are also extremely encouraging. In a 5 year prospective trial, 95 MS patients with differing disease severity underwent low intensity HSCT [30]. Progression-free survival after HSCT, measured by neurological improvement, decreased EDSS, and improved quality-of-life (QOL), was 82% at 5 years and at long term follow-up overall clinical response was 80%.
When interpreting results from HSCT trials, it is crucial to note that most of these trials are phase I/II safety and efficacy trials and therefore have limited clinical applicability. Additionally, the inclusion of healthier patients, as measured by a lower median EDSS (the median EDSS of patients in the Shevchenko trial was 3.5 pre-treatment), is most likely creating a selection bias that is distorting the true efficacy of the treatment. Looking forward, randomized clinical trials of low intensity HSCT with larger sample sizes, a broader diversity of MS patient types, and the inclusion of other parameters for judging treatment outcome, like the addition of a QOL assessment [31], will allow for a better portrayal of treatment efficacy and possibly lead to broader patient treatment. Given that there are a large number of patients who suffer from rapidly progressing MS unresponsive to conventional treatments and that the harmful side effects of low intensity HSCT are decreasing as researchers perfect the treatment protocol [30], implementation of these clinical trials is a realistic goal.
Looking forward, there are a few clinical HSCT trials currently ongoing, completed and compiling results, or planned that will offer more data on HSCT for MS that may lead to wider application (Table 1). The most noteworthy trial is being conducted by Richard Burt at Northwestern University. It is currently recruiting RRMS patients who have failed first-line therapies and projects to have 110 patients total. It is the first phase III clinical trial of HSCT for the treatment of MS and it will utilize a low intensity conditioning regimen. Also noteworthy is a planned trial sponsored by the North Bristol NHS Trust and run by Neil Scolding that will examine whether or not HSCT without any conditioning regimen may have efficacy in treating PPMS and SPMS patients. Depending on the results this trial may lead to a novel usage of HSCT in MS treatment.
Table 1.
Future HSCT Clinical Trials
| Principal Investigator(s) | Location | Number of Patients | Type of MS | Intensity of Conditioning | Status of Trial | ClinicalTrials.gov Reference # |
|---|---|---|---|---|---|---|
| Freedman and Atkins [49] | Ottawa Hospital Research Institute | 24 | Only RRMS and SPMS | High | Completed Awaiting Results | NCT01099930 |
| Burt | Northwestern University | 110 (projected) | Acute Inflammatory RRMS (failing conventional therapy) | Low | Recruiting Patients | NCT00273364 |
| Nash and Bown http://www.halt-ms.org | National Institute of Allergy and Infectious Disease (NIAID) | 25 | RRMS or PRMS | Intermediate | Ongoing | NCT00288626 |
| National Institute of Neurological Disorders and Stroke | Northwestern University and John Hopkins University | 34 | All types | Low | Completed | NCT00342134 |
| Lim | Texas Oncology Cancer Center | 50 (projected) | All types except PPMS (failing conventional therapy) | Low | Recruiting Patients | NCT01679041 |
| Scolding | Frenchay Hospital | 80 (projected) | PPMS or SPMS | None | Not yet Recruiting | NCT01815632 |
Relapsing-Remitting Multiple Sclerosis (RRMS); Secondary Progressive Multiple Sclerosis (SPMS); Primary Progressive Multiple Sclerosis (PPMS); Progressive-Relapsing Multiple Sclerosis (PRMS). Information complied from http://clinicaltrials.gov.
There are numerous other hurdles that still need to be overcome before determining whether or not HSCT is a feasible treatment for MS. Researchers need to further standardize other technical aspects of HSCT, specifically examining the efficacy of the mode of transplant delivery as well the timing [32]. The economics involved with a broad HSCT treatment plan also need to be examined since this type of approach is what many researchers perceive as the end goal of their research [33]. However, for this paper we chose to focus on the intensity of the conditioning regimen and the patient selection because those two variables are most directly linked with the safety and efficacy of HSCT clinical trials. Both safety and efficacy of HSCT need to be definitively resolved before any of these other hurdles can be addressed. A research focus on low intensity HSCT trials in a broader range of MS patients is the best immediate use of academic resources to approach this issue.
Nevertheless, HSCT does not represent a comprehensive treatment for MS. The current belief is that neurological deterioration in late-stage PPMS and SPMS is due primarily to axonal atrophy [34,35]. Thus reconstituting the patient’s immune system through HSCT prevents further damage but does not stop neurological deterioration in late-stage MS patients. This idea is supported by trials demonstrating the increased efficacy in stopping MS progression and promoting neurological improvement in patients with EDSS scores <6 and patients treated within 5 years of diagnosis [27,36]. These findings have led these researchers to conclude that HSCT is an effective treatment only for early-stage rapidly progressing MS that is unresponsive to conventional treatment. Specifically, the best candidates for HSCT are relatively young patients with recent disease onset, active inflammatory lesions of relatively short duration and rapidly progressive disease, but who still have low EDSS scores and are unresponsive to conventional therapy [24]. For late-stage MS, HSCT is not as effective and often not advisable given the inherent risks [30].
Mesenchymal stem cell transplantation (MSCT)
Mesenchymal stem cells (MSCs) are bone marrow- or placenta-derived stem cells that can differentiate into osteocytes, chondrocytes, adipocytes, astrocytes, etc. MSCs are immunomodulatory through the suppression of pathogenic lymphocytes in autoimmune disorders, and have been shown to induce localized remyelination in CNS lesions, postpone disease progression and reverse clinical disease symptoms in animal models [37]. The ability to use an autologous source of MSCs reduces the risk of immune reactions to prevent exacerbation of autoimmunity in MS patients [38].
Experimental Autoimmune Encephalomyelitis, or EAE, is the most commonly used animal analogue of MS, induced by the injection of the MOG peptide (self-antigen for the myelin oligodendrocyte glycine coprotein) into 6-8 week-old mice [39,40]. This injection induces host CD4+ T cell-mediated autoimmunity, leading to destruction of myelin and damage to axons within the CNS. Subsequent to the EAE induction, mice are evaluated for disease progression through scoring of neurological function [39]. On this 0-5 scale, 0 denotes asymptomatic EAE, 1-4 denotes varying deterioration of tail and limb control until paralysis and 5 denotes EAE-induced mortality [41].
Since EAE is characterized by CD4+ T lymphocyte-mediated autoimmunity, animal trials have been directed at attempting to hamper this aspect of EAE. A 2012 study proposed that the implantation of MSCs would affect the proliferation and pathogenesis of CD4+ T lymphocytes in the EAE model [40]. In this study, MSCs were isolated and cultured from test subject bone marrow. Mice received allogenic and syngenic MSC transplants following the induction of EAE, and cell samples taken 40 days post-transplant revealed several significant findings. First, the clinical disease scores of the MSCT recipients improved for a week following the transplant, regardless of MSC origin, then declined in parallel to the control group. Second, this improved neurological function following MSCT was associated with an upregulation of IL-10, TGF-beta, Foxp3 and the CD4+CD25+Foxp3+T cell in immune system tissues. The CD4+CD25+ regulatory lymphocytes suppress pathogenic, self-reactive T cells [40]. Thus, MSCT postpones the development of EAE in mice and might have a similar effect for MS.
Alternatively to bone marrow-derived MSCs, a 2012 animal study proposed the use of placental tissue as a more accessible and less invasive source for MSCs [39]. Female mice underwent EAE induction, were observed daily for disease progression, then underwent PL-MSC transplantation. PL-MSCT resulted in decreased disease progression, postponed onset of symptoms and increased survival rate compared to the control. Most importantly, PL-MSCs displayed similar immunomodulatory effects as Zhu et al. found with BM-MSCs by suppressing pathogenic T cells, protecting axons, and preventing demyelination. As with previous MSCT studies, transplanted PL-MSCs were found both at the transplant injection site, within the brain, as well as the test subjects’ spinal cords; indicating a capacity to migrate to damaged CNS tissue [37]. The combined suppression of neurodegeneration and immunomodulation in the CNS results in a local reduction in inflammation, which may have therapeutic potential for MS patients.
MSC-derived neural progenitors, or MSC-NPs, have also been shown to have a neuroprotective effect in EAE subjects via immunomodulatory mechanisms [42]. A recent study sought to determine if MSC-NPs could not only reduce the immune response in EAE models, but also induce remyelination to promote recovery [38]. MSCs were extracted from bone marrow, cultured, and differentiated into MSC-NPs. After EAE was induced and neural function was observed, mice were intrathecally injected with autologous MSC-NPs. Multiple transplants improved clinical symptoms, whereas a single transplant had no effect, and both mechanisms reduced immune cell infiltration. The findings suggest that the improved symptom score may not be contingent on immunomodulation, as was previously suggested, but rather a significant decline in areas of demyelination. The exact neural restoration mechanism is unknown, but repeated transplants might be needed to sustain the reversal of neural degradation [38].
Evidence from preclinical studies supports the immunomodulatory and neuroprotective nature of MSCTs, thus numerous clinical trials have emerged to test the safety and validity of this treatment for MS patients. First, a phase I clinical trial in 2010 isolated and cultured MSCs from healthy control individuals and MS patients in order to determine whether there were significant differences in MSCs that might have adverse implications for MSC therapy. Mallam et al. isolated and cultured MSCs from control and MS patient bone marrow, and found no significant differences in their histological or immunological phenotype, and in their in vitro differentiation and proliferation potential [43].
Additionally, a phase I/II open-safety clinical trial from 2010 examined the clinical potential of MSCT and any side effects in 15 patients with MS [37]. Patients received both intrathecal (via lumbar puncture) and intravenous autologous bone marrow-derived MSCs. Clinically, there was marked functional improvement for the first 6 months post-therapy without adverse reactions. Physiologically, the MSCT showed a rise in CD4+CD25+ regulatory T lymphocytes and decline in activated T lymphocytes, which is analogous to patterns in animal models [40]. Though the exact mechanism is difficult to ascertain, the immunomodulatory response of MSCT exceeds that of conventional disease-modifying therapies--authors hypothesized the migratory nature of MSCs may be responsible for the clinical improvement in these patients. Nonetheless, the trial validates results found in animal models and demonstrates the safety and potential of MSCT in human patients [37].
Additional studies focus on treating patients with secondary progressive MS, since there are limited treatment options for patients with progressive MS [44]. A recent open-label phase IIa proof-of-concept trial highlights the unmet demand for a successful treatment for secondary progressive MS and ascertains the neuroprotective benefit of MSCT therapy. Participants were specifically chosen for anterior optic lesions (retinal nerve demyelination) to more readily assess neuroprotective effects of intravenous autologous MSCs using optic nerve function. Patients showed both visual and general disability improvement, without major adverse effects. The mechanism by which MSCT improves clinical symptoms of progressive MS is unknown; however, the authors contend that remyelination might be responsible for the results [44].
Though several clinical trials support the safety and validity of MSCT in MS patients, there have yet to be clinical trials to assess the efficacy of MSC-derived neural progenitor transplants in MS patients, despite their therapeutic potential outlined in several animal trials [42,45]. In 2012, the first preclinical study to compare MSC-NPs from human donors with and without MS was completed [45]. Since autologous transplants are considered safest and most effective for patients with autoimmune disorders, it is vital to determine whether significant differences exist between MSC-NPs from MS or non-MS patients before autologous MSC-NP transplants can be used in clinical trials. MSCs were extracted from bone marrow, differentiated into MSC-NPs and studied for differentiation potential and cellular characteristics. This study determined that there are no differences between MSC-NPs, from MS or non-MS patients, and supports the potential safety of MSC-NP therapy in clinical trials [45].
The widespread research opportunities and rapid pace for both HSCT and MSCT have yielded promising results. There are several advantages to MSCT compared to HSCT that have shown promise in preclinical trials [41]. Like HSCT, the MSCT readily uses the patient as the donor and eliminates the risk of rejection and graft vs. host disease (GVHD). Animal studies with human MSCs (xenogeneic) as well as allogeneic and syngeneic transplants have shown that MSCs can exert their benefits before being phagocytized by immune cells. MSCs readily multiply in-vitro and maintain their multipotent properties until integrated into various tissues-even the central nervous system. This multipotency was further demonstrated when CD45-CD146+ cells established a hematopoietic environment similar to HSCs when introduced to blood and bone marrow [41].
In addition, the MSCs have shown immunomodulatory and immunosuppressive capabilities which decrease the inflammatory environment that degrades myelin. In mouse studies, this was observed when MSCs increased proliferation of CD4 cells while simultaneously reducing production of CD8 T-cells, plasma B-cells, NK cells, and the activity of antigen presenting cells [41]. Keeping immune cells in their quiescent, non-proliferative state prevents the secretion of pro-inflammatory cytokines such as interferon-γ, IL-2, and IL-17. Increased CD4 numbers correlate with increases in immune-regulatory cytokines IL-10, TGF-β1, and IL-4 [40].
In-vitro studies demonstrate that neurally-integrated MSCs have paracrine anti-apoptotic properties, promote cell body growth processes, and may induce neurogenesis and oligodendrogenesis through neurotropic factors. Our current inability to mark MSCs during in-vivo studies leads to challenges in deducing whether benefits are due to direct stem cell grafting or via a paracrine effect. Despite this ambiguity, quantifiable benefits have been observed in administrations of < 1 million MSCs [41]. Essentially, the presence of MSCs can create a less inflammatory and more anabolic microenvironment favorable to neural health and regeneration, without requiring the immunosuppressive procedures.
Looking forward, there are many upcoming clinical trials that test the safety and efficacy of MSCT in the treatment of MS (Table 2). These trials are all phase I clinical trials, which is demonstrative of the infancy of this treatment protocol; but due to the low toxicity associated with MSCT, these trials could spur rapid clinical and research developments.
Table 2.
Future MSCT Clinical Trials
| Principal Investigator(s) | Location | Number of Patients | Type of MS | Status of Trial | ClinicalTrials.gov Reference # |
|---|---|---|---|---|---|
| Connick and Chandran [50] | University of Cambridge | 10 | All types (with evidence of optic nerve damage) | Completed | NCT00395200 |
| Uccelli, Comi and Bonetti | University of Genova | 20 (projected) | RRMS, SPMS, PPMS | Recruiting Patients | NCT01854957 |
| Gourabi, Aghdami and Nabavi | Royan Institute | 30 (projected) | RRMS, SPMS, PPMS (failing conventional therapy) | Recruiting Patients | NCT01377870 |
| Shenzhen Beike Bio-Technology Co., Ltd. | Shenzhen Beike Bio-Technology Co., Ltd. | 20 (projected) | All types | Recruiting Patients | NCT01364246 |
| Fernandez and Ayuso | Fundación Pública Andaluza Progreso y Salud | 30 (projected) | SPMS | Ongoing | NCT01056471 |
| Cohen | University Hospital Case Medical Center | 24 | RRMS, SPMS, PRMS | Ongoing | NCT00813969 |
Relapsing-Remitting Multiple Sclerosis (RRMS); Secondary Progressive Multiple Sclerosis (SPMS); Primary Progressive Multiple Sclerosis (PPMS); Progressive-Relapsing Multiple Sclerosis (PRMS). Information complied from http://clinicaltrials.gov.
Combined therapies
Since the MS therapies discussed act through separate mechanisms, it is conceivable that the best treatment regimen for some MS patients might be a combination of multiple therapies. A pilot study [46] tested the benefits of intravenous infusion of MSCs in 5 patients receiving umbilical cord-derived HSCT. Compared to a control group undergoing only HSCT, patients receiving co-transplantation with MSCs had significantly faster hematopoietic recovery of neutrophils and platelets. The study concluded that MSC co-transplantation during HSCT could be a safe and effective way to improve HSCT treatment efficacy. In another trial, 5 female patients with RRMS who had failed to improve after non-myeloablative HSCT were treated with Natalizumab [47]. After starting Natalizumab, 4/5 patients showed a halt to MS progression as demonstrated by the lack of disease relapse and any new lesions on MRI, and 1 patient showed an initial relapse after Natalizumab infusion but then showed markedly improved neurological function evidenced by an EDSS score improvement from 6.5 pre-treatment to 4.0 post-treatment (40 months out). These two trials demonstrate that combination therapies involving stem cells are still in their infancy but offer hope of a safe and effective way to improve patient outcomes.
A recent trial examined the safety and efficacy of combining non-myeloablative HSCT with a consolidation therapy of Mitoxantrone [48]. 55 MS patients total were included in the trial; 32 RRMS, 13 SPMS, 9 PPMS and 1 PRMS patient. They had a median EDSS of 4.0. No transplant related mortality was observed. At mean follow-up of 26 months, the group with RRMS demonstrated improvement in 15 patients (58%) and stabilization in 11 (42%). In the group with progressive MS (PPMS, SPMS, RPMS), improvement was achieved in 15 patients (82%) and stabilization in 3 (18%). No relapses throughout the whole follow-up period were found. Researchers concluded that non-myeloablative HSCT with consolidation therapy by Mitoxantrone appears to be a safe and effective treatment for MS.
Conclusion
HSCT and MSCT offer hope for a new effective treatment for MS. Recent trials of low intensity HSCT indicate that as researchers improve the conditioning protocol, they can significantly reduce the adverse side effects without decreasing efficacy [30]. Similarly, a recent clinical trial evaluating MSCT concluded it was a feasible and safe treatment method for MS patients [37]. Given MSCT’s positive potential on neural restoration and immunomodulation demonstrated in animal models, MSCT offers a low risk/high reward treatment option for MS patients. Additionally, HSCT and MSCT could work synergistically to further decrease adverse side effects and increase patient outcomes.
Researchers are currently recommending that HSCT be used in patients with early-stage rapidly progressing MS unresponsive to first-line medications [30]. If current clinical trends continue and the risks continue to drop for HSCT and MSCT, these treatment options could be extended to a broader patient pool. Given the toxicity and ineffectiveness of many first-line MS medications it is conceivable that stem cell therapy could become the first-line treatment for MS patients. Larger randomized trials need to be conducted to determine the feasibility of such an approach and elucidate the different mechanisms of action of stem cell therapy. Recent safety trials have opened the door for future trials on MSC-NPs [38]. Placental derived MSCs appear to be more practically useful compared with bone marrow derived MSCs based upon accessibility and abundance [39]. Further clinical trials using placental derived MSCs and MSC-NPs will lead to insight on the feasibility of MSCT as a standard treatment for MS patient.
The large amount of MS patients unresponsive to current medications and in rapidly deteriorating health provides justification and impetus for these future trials in stem cell therapy. Presently, stem cell therapy is not a cure for MS but it offers great hope and promise.
References
- 1.Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33:91–101. doi: 10.1097/WNF.0b013e3181cbf825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Compston A, Coles A. Multiple sclerosis. Lancet. 2002;359:1221–1231. doi: 10.1016/S0140-6736(02)08220-X. [DOI] [PubMed] [Google Scholar]
- 3.Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, Fujihara K, Havrdova E, Hutchinson M, Kappos L, Lublin FD, Montalban X, O’Connor P, Sandberg-Wollheim M, Thompson AJ, Waubant E, Weinshenker B, Wolinsky JS. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292–302. doi: 10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Praksova P, Stourac P, Bednarik J, Vlckova E, Mikulkova Z, Michalek J. Immunoregulatory T cells in multiple sclerosis and the effect of interferon beta and glatiramer acetate treatment on T cell subpopulations. J Neurol Sci. 2012;319:18–23. doi: 10.1016/j.jns.2012.05.036. [DOI] [PubMed] [Google Scholar]
- 5.von Herrath MG, Fujinami RS, Whitton JL. Microorganisms and autoimmunity: making the barren field fertile? Nat Rev Microbiol. 2003;1:151–157. doi: 10.1038/nrmicro754. [DOI] [PubMed] [Google Scholar]
- 6.Berglund J. Alternative therapies: Desperate measures. Nature. 2012;484:S11. doi: 10.1038/nature11099. [DOI] [PubMed] [Google Scholar]
- 7.Arnold DL, Narayanan S, Antel S. Neuroprotection with glatiramer acetate: evidence from the PreCISe trial. J Neurol. 2013 doi: 10.1007/s00415-013-6903-5. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moses H Jr, Brandes DW. Managing adverse effects of disease-modifying agents used for treatment of multiple sclerosis. Curr Med Res Opin. 2008;24:2679–2690. doi: 10.1185/03007990802329959. [DOI] [PubMed] [Google Scholar]
- 9.Confavreux C, Li DK, Freedman MS, Truffinet P, Benzerdjeb H, Wang D, Bar-Or A, Traboulsee AL, Reiman LE, O’Connor PW Teriflunomide Multiple Sclerosis Trial Group. Long-term follow-up of a phase 2 study of oral teriflunomide in relapsing multiple sclerosis: safety and efficacy results up to 8.5 years. Mult Scler. 2012;18:1278–1289. doi: 10.1177/1352458512436594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zivadinov R, Dwyer MG, Hussein S, Carl E, Kennedy C, Andrews M, Hojnacki D, Heininen-Brown M, Willis L, Cherneva M, Bergsland N, Weinstock-Guttman B. Voxel-wise magnetization transfer imaging study of effects of natalizumab and IFNbeta-1a in multiple sclerosis. Mult Scler. 2012;18:1125–1134. doi: 10.1177/1352458511433304. [DOI] [PubMed] [Google Scholar]
- 11.Chanvillard C, Millward JM, Lozano M, Hamann I, Paul F, Zipp F, Dorr J, Infante-Duarte C. Mitoxantrone induces natural killer cell maturation in patients with secondary progressive multiple sclerosis. PLoS One. 2012;7:e39625. doi: 10.1371/journal.pone.0039625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castro-Borrero W, Graves D, Frohman TC, Flores AB, Hardeman P, Logan D, Orchard M, Greenberg B, Frohman EM. Current and emerging therapies in multiple sclerosis: a systematic review. Ther Adv Neurol Disord. 2012;5:205–220. doi: 10.1177/1756285612450936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, Naves R, Han M, Zhong F, Castellanos JG, Mair R, Christakos A, Kolkowitz I, Katz L, Killestein J, Polman CH, de Waal Malefyt R, Steinman L, Raman C. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med. 2010;16:406–412. doi: 10.1038/nm.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang AG, Lin YC, Wang SJ, Tsai CP, Yen MY. Early relapse in multiple sclerosis-associated optic neuritis following the use of interferon beta-1a in Chinese patients. Jpn J Ophthalmol. 2006;50:537–542. doi: 10.1007/s10384-006-0359-4. [DOI] [PubMed] [Google Scholar]
- 15.Rowley SD, Loken M, Radich J, Kunkle LA, Mills BJ, Gooley T, Holmberg L, McSweeney P, Beach K, MacLeod B, Appelbaum F, Bensinger WI. Isolation of CD34+ cells from blood stem cell components using the Baxter Isolex system. Bone Marrow Transplant. 1998;21:1253–1262. doi: 10.1038/sj.bmt.1701257. [DOI] [PubMed] [Google Scholar]
- 16.Carreras E, Saiz A, Marin P, Martinez C, Rovira M, Villamor N, Aymerich M, Lozano M, Fernandez-Aviles F, Urbano-Izpizua A, Montserrat E, Graus F. CD34+ selected autologous peripheral blood stem cell transplantation for multiple sclerosis: report of toxicity and treatment results at one year of follow-up in 15 patients. Haematologica. 2003;88:306–314. [PubMed] [Google Scholar]
- 17.Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS) Neurology. 1983;33:1444–1452. doi: 10.1212/wnl.33.11.1444. [DOI] [PubMed] [Google Scholar]
- 18.Fassas A, Anagnostopoulos A, Kazis A, Kapinas K, Sakellari I, Kimiskidis V, Tsompanakou A. Peripheral blood stem cell transplantation in the treatment of progressive multiple sclerosis: first results of a pilot study. Bone Marrow Transplant. 1997;20:631–638. doi: 10.1038/sj.bmt.1700944. [DOI] [PubMed] [Google Scholar]
- 19.Saccardi R, Kozak T, Bocelli-Tyndall C, Fassas A, Kazis A, Havrdova E, Carreras E, Saiz A, Lowenberg B, te Boekhorst PA, Gualandio F, Openshaw H, Longo G, Pagliai F, Massacesi L, Deconink E, Ouyang J, Nagore FJ, Besalduch J, Lisukov IA, Bonini A, Merelli E, Slavino S, Gratwohl A, Passweg J, Tyndall A, Steck AJ, Andolina M, Capobianco M, Martin JL, Lugaresi A, Meucci G, Saez RA, Clark RE, Fernandez MN, Fouillard L, Herstenstein B, Koza V, Cocco E, Baurmann H, Mancardi GL Autoimmune Diseases Working Party of EBMT. Autologous stem cell transplantation for progressive multiple sclerosis: update of the European Group for Blood and Marrow Transplantation autoimmune diseases working party database. Mult Scler. 2006;12:814–823. doi: 10.1177/1352458506071301. [DOI] [PubMed] [Google Scholar]
- 20.Burt RK, Loh Y, Cohen B, Stefoski D, Balabanov R, Katsamakis G, Oyama Y, Russell EJ, Stern J, Muraro P, Rose J, Testori A, Bucha J, Jovanovic B, Milanetti F, Storek J, Voltarelli JC, Burns WH. Autologous non-myeloablative haemopoietic stem cell transplantation in relapsing-remitting multiple sclerosis: a phase I/II study. Lancet Neurol. 2009;8:244–253. doi: 10.1016/S1474-4422(09)70017-1. [DOI] [PubMed] [Google Scholar]
- 21.Hamerschlak N, Rodrigues M, Moraes DA, Oliveira MC, Stracieri AB, Pieroni F, Barros GM, Madeira MI, Simoes BP, Barreira AA, Brum DG, Ribeiro AA, Kutner JM, Tylberi CP, Porto PP, Santana CL, Neto JZ, Barros JC, Paes AT, Burt RK, Oliveira EA, Mastropietro AP, Santos AC, Voltarelli JC. Brazilian experience with two conditioning regimens in patients with multiple sclerosis: BEAM/horse ATG and CY/rabbit ATG. Bone Marrow Transplant. 2010;45:239–248. doi: 10.1038/bmt.2009.127. [DOI] [PubMed] [Google Scholar]
- 22.Disanto G, Morahan JM, Ramagopalan SV. Multiple sclerosis: risk factors and their interactions. CNS Neurol Disord Drug Targets. 2012;11:545–555. doi: 10.2174/187152712801661266. [DOI] [PubMed] [Google Scholar]
- 23.Burt RK, Marmont A, Oyama Y, Slavin S, Arnold R, Hiepe F, Fassas A, Snowden J, Schuening F, Myint H, Patel DD, Collier D, Heslop H, Krance R, Statkute L, Verda L, Traynor A, Kozak T, Hintzen RQ, Rose JW, Voltarelli J, Loh Y, Territo M, Cohen BA, Craig RM, Varga J, Barr WG. Randomized controlled trials of autologous hematopoietic stem cell transplantation for autoimmune diseases: the evolution from myeloablative to lymphoablative transplant regimens. Arthritis Rheum. 2006;54:3750–3760. doi: 10.1002/art.22256. [DOI] [PubMed] [Google Scholar]
- 24.Rogojan C, Frederiksen JL. Hematopoietic stem cell transplantation in multiple sclerosis. Acta Neurol Scand. 2009;120:371–382. doi: 10.1111/j.1600-0404.2009.01168.x. [DOI] [PubMed] [Google Scholar]
- 25.Sun W, Popat U, Hutton G, Zang YC, Krance R, Carrum G, Land GA, Heslop H, Brenner M, Zhang JZ. Characteristics of T-cell receptor repertoire and myelin-reactive T cells reconstituted from autologous haematopoietic stem-cell grafts in multiple sclerosis. Brain. 2004;127:996–1008. doi: 10.1093/brain/awh117. [DOI] [PubMed] [Google Scholar]
- 26.Snowden JA, Saccardi R, Allez M, Ardizzone S, Arnold R, Cervera R, Denton C, Hawkey C, Labopin M, Mancardi G, Martin R, Moore JJ, Passweg J, Peters C, Rabusin M, Rovira M, van Laar JM, Farge D EBMT Autoimmune Disease Working Party (ADWP); Paediatric Diseases Working Party (PDWP) Haematopoietic SCT in severe autoimmune diseases: updated guidelines of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant. 2012;47:770–790. doi: 10.1038/bmt.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burt RK, Cohen BA, Russell E, Spero K, Joshi A, Oyama Y, Karpus WJ, Luo K, Jovanovic B, Traynor A, Karlin K, Stefoski D, Burns WH. Hematopoietic stem cell transplantation for progressive multiple sclerosis: failure of a total body irradiation-based conditioning regimen to prevent disease progression in patients with high disability scores. Blood. 2003;102:2373–2378. doi: 10.1182/blood-2003-03-0877. [DOI] [PubMed] [Google Scholar]
- 28.Nash RA, Bowen JD, McSweeney PA, Pavletic SZ, Maravilla KR, Park MS, Storek J, Sullivan KM, Al-Omaishi J, Corboy JR, DiPersio J, Georges GE, Gooley TA, Holmberg LA, LeMaistre CF, Ryan K, Openshaw H, Sunderhaus J, Storb R, Zunt J, Kraft GH. High-dose immunosuppressive therapy and autologous peripheral blood stem cell transplantation for severe multiple sclerosis. Blood. 2003;102:2364–2372. doi: 10.1182/blood-2002-12-3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loh Y, Oyama Y, Statkute L, Quigley K, Yaung K, Gonda E, Barr W, Jovanovic B, Craig R, Stefoski D, Cohen B, Burt RK. Development of a secondary autoimmune disorder after hematopoietic stem cell transplantation for autoimmune diseases: role of conditioning regimen used. Blood. 2007;109:2643–2548. doi: 10.1182/blood-2006-07-035766. [DOI] [PubMed] [Google Scholar]
- 30.Shevchenko JL, Kuznetsov AN, Ionova TI, Melnichenko VY, Fedorenko DA, Kartashov AV, Kurbatova KA, Gorodokin GI, Novik AA. Autologous Haematopoietic Stem Cell Transplantation with Reduced Intensity Conditioning in Multiple Sclerosis. Exp Hematol. 2012;40:892–8. doi: 10.1016/j.exphem.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 31.Noseworthy JH. Clinical scoring methods for multiple sclerosis. Ann Neurol. 1994;36(Suppl):S80–S85. doi: 10.1002/ana.410360718. [DOI] [PubMed] [Google Scholar]
- 32.Akiyama Y, Radtke C, Honmou O, Kocsis JD. Remyelination of the spinal cord following intravenous delivery of bone marrow cells. Glia. 2002;39:229–236. doi: 10.1002/glia.10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Atkins HL, Freedman MS. Hematopoietic stem cell therapy for multiple sclerosis: top 10 lessons learned. Neurotherapeutics. 2013;10:68–76. doi: 10.1007/s13311-012-0162-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating Oligodendrocytes in Chronic Lesions of Multiple Sclerosis. N Engl J Med. 2002;346:165–173. doi: 10.1056/NEJMoa010994. [DOI] [PubMed] [Google Scholar]
- 35.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal Transection in the Lesions of Multiple Sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 36.Bowen JD, Kraft GH, Wundes A, Guan Q, Maravilla KR, Gooley TA, McSweeney PA, Pavletic SZ, Openshaw H, Storb R, Wener M, McLaughlin BA, Henstorf GR, Nash RA. Autologous hematopoietic cell transplantation following high-dose immunosuppressive therapy for advanced multiple sclerosis: long-term results. Bone Marrow Transplant. 2012;47:946–951. doi: 10.1038/bmt.2011.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karussis D, Karageorgiou C, Vaknin-Dembinsky A, Gowda-Kurkalli B, Gomori JM, Kassis I, Bulte JW, Petrou P, Ben-Hur T, Abramsky O, Slavin S. Safety and Immunological Effects of Mesenchymal Stem Cell Transplantation in Patients With Multiple Sclerosis and Amyotrophic Lateral Sclerosis. Arch Neurol. 2010;67:1187–94. doi: 10.1001/archneurol.2010.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harris VK, Faroqui R, Vyshkina T, Sadiq SA. Characterization of autologous mesenchymal stem cell-derived neural progenitors as a feasible source of stem cells for central nervous system applications in multiple sclerosis. Stem Cells Transl Med. 2012;1:536–547. doi: 10.5966/sctm.2012-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fisher-Shoval Y, Barhum Y, Sadan O, Yust-Katz S, Ben-Zur T, Lev N, Benkler C, Hod M, Melamed E, Offen D. Transplantation of Placenta-Derived Mesenchymal Stem Cells in the EAE Mouse Model of MS. J Mol Neurosci. 2012;48:176–184. doi: 10.1007/s12031-012-9805-6. [DOI] [PubMed] [Google Scholar]
- 40.Zhu J, Zhang J, Li Q, Du Y, Qiao B, Hu X. Transplanting of mesenchymal stem cells may affect proliferation and function of CD4(+)T cells in experimental autoimmune encephalomyelitis. Exp Clin Transplant. 2012;10:492–500. doi: 10.6002/ect.2011.0197. [DOI] [PubMed] [Google Scholar]
- 41.Uccelli A, Prockop DJ. Why should mesenchymal stem cells (MSCs) cure autoimmune diseases? Curr Opin Immunol. 2010;22:768–774. doi: 10.1016/j.coi.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 42.Giannakopoulou A, Grigoriadis N, Polyzoidou E, Touloumi O, Michaloudi E, Papadopoulos GC. Inflammatory changes induced by transplanted neural precursor cells in a multiple sclerosis model. Neuroreport. 2011;22:68–72. doi: 10.1097/WNR.0b013e32834272eb. [DOI] [PubMed] [Google Scholar]
- 43.Mallam E, Kemp K, Wilkins A, Rice C, Scolding N. Characterization of in vitro expanded bone marrow-derived mesenchymal stem cells from patients with multiple sclerosis. Mult Scler. 2010;16:909–918. doi: 10.1177/1352458510371959. [DOI] [PubMed] [Google Scholar]
- 44.Connick P, Kolappan M, Crawley C, Webber DJ, Patani R, Michell AW, Du M, Luan S, Altmann DR, Thompson AJ, Compston A, Scott MA, Miller DH, Chandran S. Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: an open-label phase 2a proof-of-concept study. Lancet Neurol. 2012;11:150–156. doi: 10.1016/S1474-4422(11)70305-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harris VK, Yan QJ, Vyshkina T, Sahabi S, Liu X, Sadiq SA. Clinical and pathological effects of intrathecal injection of mesenchymal stem cell-derived neural progenitors in an experimental model of multiple sclerosis. J Neurol Sci. 2012;313:167–177. doi: 10.1016/j.jns.2011.08.036. [DOI] [PubMed] [Google Scholar]
- 46.Wu KH, Sheu JN, Wu HP, Tsai C, Sieber M, Peng CT, Chao YH. Cotransplantation of umbilical cord-derived mesenchymal stem cells promote hematopoietic engraftment in cord blood transplantation: a pilot study. Transplantation. 2013;95:773–7. doi: 10.1097/TP.0b013e31827a93dd. [DOI] [PubMed] [Google Scholar]
- 47.Capobianco M, Motuzova Y, Frau J, Cocco E, Mamusa E, Marrosu MG, Bertolotto A. Natalizumab in aggressive multiple sclerosis after haematopoietic stem cell transplantation. Neurol Sci. 2012;33:863–867. doi: 10.1007/s10072-011-0848-1. [DOI] [PubMed] [Google Scholar]
- 48.Novik AA, Kuznetsov AN, Melnichenko VY, Fedorenko DA, Ionova TI, Gorodokin GV. Non-myeloablative Autologous Haematopoietic Stem Cell Transplantation with Consolidation Therapy using Mitoxantrone as a Treatment Option in Multiple Sclerosis Patients. J Stem Cell Res Ther. 2011;1:102. [Google Scholar]
- 49.Saccardi R, Freedman MS, Sormani MP, Atkins H, Farge D, Griffith LM, Kraft G, Mancardi GL, Nash R, Pasquini M, Martin R, Muraro PA European Blood and Marrow Transplantation Group; Center for International Blood and Marrow Research; HSCT in MS International Study Group. A prospective, randomized, controlled trial of autologous haematopoietic stem cell transplantation for aggressive multiple sclerosis: a position paper. Mult Scler. 2012;18:825–34. doi: 10.1177/1352458512438454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Connick P, Kolappan M, Patani R, Scott MA, Crawley C, He XL, Richardson K, Barber K, Webber DJ, Wheeler-Kingshott CA, Tozer DJ, Samson RS, Thomas DL, Du MQ, Luan SL, Michell AW, Altmann DR, Thompson AJ, Miller DH, Compston A, Chandran S. The mesenchymal stem cells in multiple sclerosis (MSCIMS) trial protocol and baseline cohort characteristics: an open-label pre-test: post-test study with blinded outcome assessments. Trials. 2011;12:62. doi: 10.1186/1745-6215-12-62. [DOI] [PMC free article] [PubMed] [Google Scholar]