

Abstract
Individual saposin A (A−/−) and saposin B (B−/−)-deficient mice show unique phenotypes caused by insufficient degradation of myelin-related glycosphingolipids (GSLs): galactosylceramide and galactosylsphingosine and sulfatide, respectively. To gain insight into the interrelated functions of saposins A and B, combined saposin AB-deficient mice (AB−/−) were created by knock-in point mutations into the saposins A and B domains on the prosaposin locus. Saposin A and B proteins were undetectable in AB−/− mice, whereas prosaposin, saposin C and saposin D were expressed near wild-type (WT) levels. AB−/− mice developed neuromotor deterioration at >61 days and exhibited abnormal locomotor activity and enhanced tremor. AB−/− mice (∼96 days) lived longer than A−/− mice (∼85 days), but shorter than B−/− mice (∼644 days). Storage materials were observed in Schwann cells and neuronal processes by electron microscopy. Accumulation of p62 and increased levels of LC3-II were detected in the brainstem suggesting altered autophagy. GSL analyses by (liquid chromatography) LC/MS identified substantial increases in lactosylceramide in AB−/− mouse livers. Sulfatide accumulated, but galactosylceramide remained at WT levels, in the AB−/− mouse brains and kidneys. Brain galactosylsphingosine in AB−/− mice was ∼68% of that in A−/− mice. These findings indicate that combined saposins A and B deficiencies attenuated GalCer-β-galactosylceramidase and GM1-β-galactosidase functions in the degradation of lactosylceramide preferentially in the liver. Blocking sulfatide degradation from the saposin B deficiency diminished galactosylceramide accumulation in the brain and kidney and galctosylsphingosine in the brain. These analyses of AB−/− mice continue to delineate the tissue differential interactions of saposins in GSL metabolism.
INTRODUCTION
Four saposins (A, B, C and D) are derived from their precursor, prosaposin (1–3), by proteolytic processing in the late endosomes and lysosomes (4,5). Each saposin is an 80 amino acids heat stable protein with specific roles in the glycosphingolipid (GSL) degradation pathway (6,7). Saposin A enhances the activity of β-galactosylceramidase that degrades galatosylceramide (GalCer) and galactosylsphingosine (8). Saposin B presents substrates to multiple lysosomal GSL hydrolases, including, sulfatide (arylsulfatase A), lactosylceramide [LacCer; (GM1-β-galactosylceramidase)] and globotriaosylceramide [Gb3; (α-galactosidase A)] (9,10). Saposin C optimizes acid β-glucosidase activity and protects the enzyme from proteolytic degradation (11,12). Saposin D enhances acid ceramidase function (13). In humans, mutations of the prosaposin gene leading to a complete deficiency of prosaposin and saposins result in severe neurological deficits with complex GSLs storage in neuronal and visceral organs (14–17). Saposin B-deficient patients present with metachoromatic leukodystrophy disease-like phenotypes (16,18) that are caused by a variety of mutations in the saposin B region at the prosaposin locus (16,19). Saposin C-deficient patients present as neuronal variants of Gaucher's disease-like phenotypes (20–22). Saposin A-deficient patients develop abnormal myelination resembling Krabbe's disease (23). Combined heteroallelic mutations in both saposin D and saposin C have also been identified in patients (24). Due to the rarity of these diseases, a limited number of human samples are available for physiological analyses of saposin functions.
Prosaposin- and individual saposin-deficient mouse models have been generated (25–29). The phenotypes of complete prosaposin- and saposin-deficient mice resemble those in human patients (25). Severe leukodystrophy with multiple GSLs storage occurs in the brain and visceral organs. Individual saposin (A, B, C and D)-deficient mice were created by knock-in mutations targeting a conserved cysteine residue located in the specific saposin domain (26–29). This strategy results in disruption of one of the three disulfide bridges and the deficiency of the mutated saposin leaving prosaposin and other saposins intact. Saposin A-deficient mice develop a chronic form of globoid cell leukodystrophy around 2.5 months and survive by about 5 months (26). Saposin B-deficient mice present neurological defects that mimic the biochemistry and phenotype of the human disease with onset around 1 year of age and life span up to 23 months (29). These mice exhibit unique myelin defects caused by insufficient degradation of the glycolipids: GalCer, galactosylsphingosine or sulfatide (26,29). Saposin C-deficient mice show slow progressive neurological deterioration and cerebellar atrophy with moderate GSLs storage in spinal cord and cerebellum at ∼3 months and survive up to 2 years of age (28). Saposin D deficiency in mice results in accumulation of ceramide and hydroxy fatty acid ceramide and leads to progressive polyuria and Purkinje cell degeneration (27). These mice develop ataxia at 4 months and live to ∼15 months. Simple summation of individual saposin deficiencies does not predict the phenotype observed in complete deficiency of prosaposin and saposins. Mice with combined saposin deficiencies were created to gain insight into the in vivo interaction of saposins. Mice with combined saposin C and D deficiencies alter prosaposin trafficking and secretion and potentiate the accumulation of ceramide and α-hydroxyl ceramide (30).
Here, combined saposin A and B-deficient mice (AB−/−) were created by knock-in point mutations into the saposin A and saposin B domains of prosaposin. Characterization of GSLs, histology and behavioral phenotypes in AB−/− mice provide insight into the tissue differential interactions of saposin A and saposin B in the GSL metabolism.
RESULTS
Combined saposin A and B targeting in mice
Each saposin contains six cysteines that form three disulfide bridges that provide protein stability (31). Knock-in mutations of a cysteine (Cys) break one of the three bonds in the selected saposin and lead to deficiency of such saposins (26,29,30). Saposin A and saposin B-deficient mice (AB−/−) were created using the same strategy. The codons for Cys 4 of saposins A and B located in exon 4 (saposin A domain) and exon 7 (saposin B domain) of the prosaposin locus were point mutated to encode phenylalanine (Supplementary Material, Fig. S1A). The homologous recombinant embryo stem (ES) cells were verified by Southern blot (Supplementary Material, Fig. S1B). The mutations (Cys→Phe) of saposins A and B were confirmed by PCR and direct DNA sequencing (Supplementary Material, Fig. S1C). The neo gene in the targeting vector was removed subsequently by cross-breeding of heterozygous F1 mice with ZP3-Cre transgenic mice (29). The resultant mice were then back-crossed with wild-type (WT) C57BL6 mice to eliminate the ZP3 transgene that was confirmed by PCR (Supplementary Material, Fig. S1D).
Expression of prosaposin and saposin proteins in AB−/− mice
The effects of the mutations of saposins A and B on the other two saposins (C and D) and prosaposin expression were evaluated by immunoblotting using antibodies against individual mouse saposins. Saposin A and B proteins were not detectable in any tissue of AB−/− mice (Fig. 1). In AB−/− cerebrum and liver, saposin C proteins were above WT levels, whereas saposin D was at WT levels (Fig. 1A). Prosaposin in AB−/− mouse cerebrum was at or above WT levels and increased in AB−/− mouse fibroblasts and livers (Fig. 1A and B). The intermediate processing forms of prosaposin to saposins were identified in the cerebrum using anti-saposin A, B, C and D antibodies (Fig. 1A, arrows). The levels of the intermediate forms in AB−/− mice cerebrum were elevated when compared with WT mice (Fig. 1A). Anti-saposin A antibody detected a non-specific protein (arrow head) above the prosaposin. Additional non-specific proteins were reacted with anti-saposin A antibody (Fig. 1A). These results suggested that deficiency of saposin A and B had minor effects on prosaposin processing. Importantly, prosaposin, saposin C and D proteins in AB−/− mice were expressed at or above WT mice levels.
Figure 1.
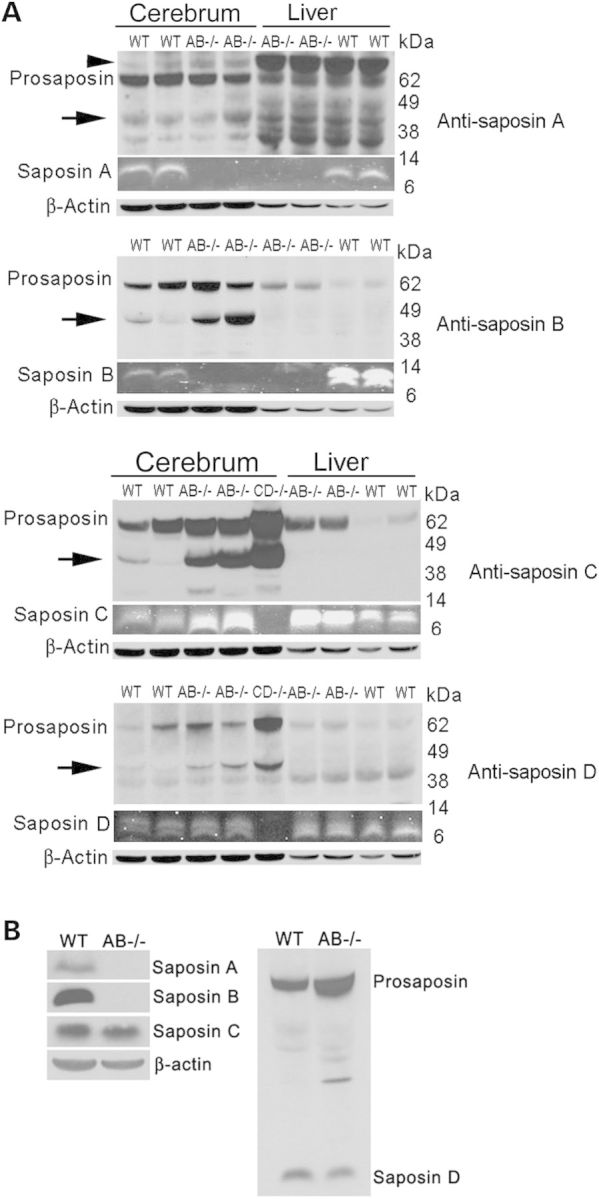
Immunoblot analysis of saposin proteins in AB−/− mice. Saposins and prosaposin proteins in the cerebrum, liver and fibroblast cells from AB−/− and WT mice were determined using rabbit anti-mouse saposin A, B, C and D antibodies (labeled on the right of each panel in A). (A) Cerebrum and liver. (Top two panels) Saposin A and B were not present in AB−/− mice, whereas prosaposin was expressed. (Bottom two panels) Saposin C levels in AB−/− mice cerebrum and livers were higher than WT mice. Saposin D proteins in AB−/− mice were expressed at WT mice levels. Saposin CD−/− mouse cerebrum was included as control and showed no detection of saposin C and D proteins. Prosaposin level was expressed at or above WT levels in AB−/− liver and cerebrum. Increased intermediate forms (arrows) of prosaposin processing to saposin were detected by anti-saposin A, B, C or D antibodies in AB−/− mouse cerebrum. Anti-saposin A antibody detected a non-specific protein above prosaposin (arrow head) and additional non-specific proteins. Saposin proteins were imaged by Odyssey Infrared Imaging System using 800 nm fluorescence channel and converted to gray color. Signals for prosaposin, intermediated forms and β-actin were developed using ECL detection reagent. Protein molecular weights were labeled for top (prosaposin) and middle (saposin) gels. Each lane represents individual mouse. AB−/− and WT mice were at 14 days and CD−/− mouse was at 28 days. (B) Fibroblasts. Saposin A and saposin B proteins were not detected, whereas prosaposin, saposin C and saposin D were present in AB−/− mice fibroblasts. Signals were developed using ECL detection reagent. β-actin was a loading control.
Phenotypes of AB−/− mice
AB−/− mice showed the onset of motor defects between 61 and 88 days. The first apparent abnormality was hind limb clasping during tail suspension (Fig. 2A). The average life span of AB−/− mice was 96 days that was slightly longer than A−/− mice (∼85 days), but substantially shorter than B−/− mice (∼644 days) (Fig. 2B). A decline in body weight of AB−/− mice started simultaneously with onset of the phenotype and progressed to 86% of age-matched WT mice levels by the terminal stage (Fig. 2C). No phenotypic difference was observed between male and female AB−/− mice. AB+/− mice showed no observable phenotype, and their biochemical characteristics were comparable to WT mice. The AB+/− mice breeding resulted in offspring having the expected Mendelian ratio. Both male and female AB−/− mice were fertile and produced one or two litters.
Figure 2.
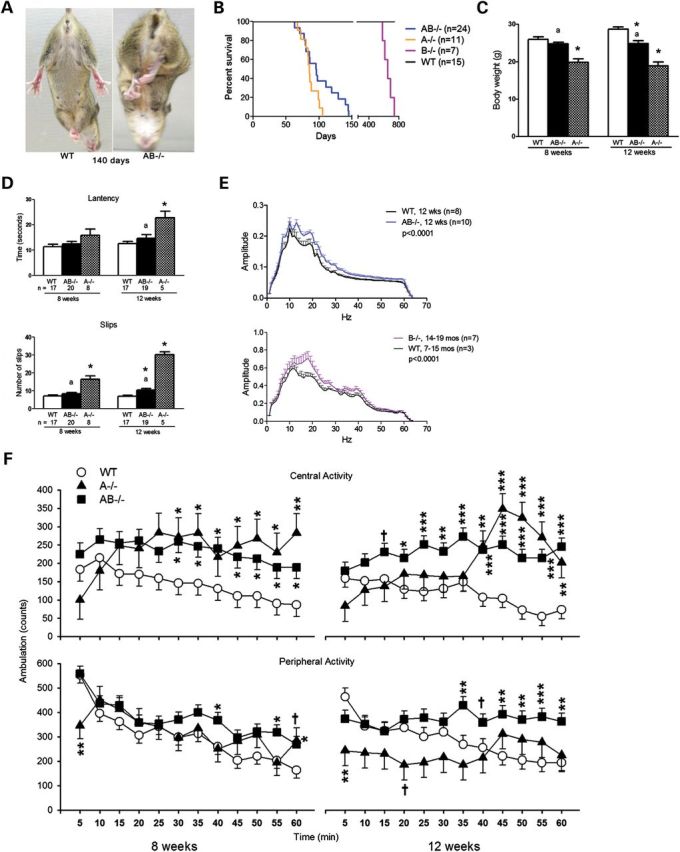
Phenotypes and behavioral performance of AB−/− mice. (A) AB−/− mice exhibited shaking and hind-limb clasping during tail suspension. (B) Life span of AB−/− mice. Compared with saposin A (A−/−)-deficient mice (n = 11), AB−/− mice (n = 24) had longer life spans on average, but much shorter than B−/− mice (n = 7). (C) Body weight. Male mice used for behavior tests were recorded for their body weight at 8 and 12 weeks. AB−/− mice (n = 19) had significantly reduced weight at 12 weeks when compared with WT mice (n = 17). A−/− mice (n = 5) showed body weight decreases at 8 and 12 weeks. (D) Narrow bridges test. Data are shown for AB−/− and A−/− mice tested on the 5 mm2 square beam. The latency (top panel) and foot slips (bottom panel) in A−/− mice were significantly different from WT and AB−/− at 12 weeks. AB−/− mice had a significant increase in number of foot slips, but no difference in latency at 12 weeks. The numbers of mice are indicated under the columns. ANOVA test (P < 0.05, *, differ significantly from WT; a, differ significantly between A−/− and AB−/− mice). (E) Tremor. Tremor activity in the frequency range of 10–30 Hz was recorded as amplitude for 34 min. AB−/− mice at 12 weeks (top panel) and B−/− mice at 14–19 months (bottom panel) showed significantly increased tremor activity when compared with age-matched WT mice. (AB−/− mice compared with WT: genotype × interval interaction F(1,1024) = 1.94, P < 0.0001; B−/− mice compared with WT: genotype × interaction F(1,496) = 1.42, P < 0.0001). (F) Locomotor activity. AB−/−, A−/− and WT mice were tested for 60 min at 8 and 12 weeks, and the data analyzed in 5 min intervals. Both A−/− and B−/− mice had significant hyperactivity in central regions at 8 and 12 weeks (8 weeks: genotype × interval interaction F(22,357) = 2.13, P < 0.01; 12 weeks: genotype main effect F(2,40.4) = 10.29, P < 0.001 and genotype × interval interaction F(22,385) = 3.06, P < 0.0001). By comparison, A−/− (n = 6) and AB−/− mice (n = 19) at 12 weeks were less active in peripheral regions relative to the WT mice (n = 16) [genotype main effect F(2,39.5) = 4.47, P < 0.02 and genotype × interval interaction F(22,382) = 2.61, P < 0.0001], whereas the AB−/− mice had increased locomotor activity in the periphery at specific intervals at both the 8 and 12 week time points. A−/− (n = 6) and AB−/− mice (n = 19) at 8 weeks had nearly WT mice (n = 17) levels of peripheral activity except that A−/− mice were hypoactive at the beginning of the test [genotype × interval interaction F(22,364) = 2.17, P < 0.01] and AB−/− mice showed hyperactivity at the later intervals of the test.
Neurobehavioral assessment
AB−/− mice were assessed for their motor function using narrow bridge and locomotor activity tests and resting tremor. In the narrow bridge tests, round and square wood beams (1 m in length) were used. Round beams had diameters of 11 and 17 mm, and square beams had cross sections of 5 mm2 and 12 mm2. Foot slips and latency (seconds) to cross each beam were recorded. A−/− mice showed age-dependent increases of foot slips on the 12 mm2 square and 17 mm round beams when compared with WT mice (data not shown). On the 5 mm2 square beam, AB−/− mice at 12 weeks and A−/− mice at 8 and 12 weeks had a significant increase in foot slips (Fig. 2D). On the 11 mm round beam, only 12-week-old A−/− mice showed a significant increase in foot slips (data not shown). On all beams tested, A−/− mice had an increased number of slips when compared with AB−/− mice. AB−/− mice showed no significant changes in latency to cross the beams relative to WT mice. A−/− mice had significantly increased latencies at 12 weeks on the 5 mm2 square beam (Fig. 2D) and the other three beams when compared with WT mice. By comparison, the number of slips for 12-week-old AB−/− mice were about the same for B−/− mice at 12 months (29).
Tremor analyses were conducted in AB−/− and WT male mice at 8 and 12 weeks. Data were recorded for 34 min. Tremor-associated motion power is presented as amplitude with bandpass filters ranging from 4 to 64 Hz. AB−/− mice at 8 weeks showed no difference in amplitude from WT mice (data not shown). By 12 weeks, AB−/− mice showed significantly increased amplitude at 10–30 Hz (Fig. 2E, top), indicating increased tremor. Head shaking was evident in B−/− mice by 15 months (29). Tremor was also detected at 10–30 Hz in B−/− mice at 14–19 months of age (Fig. 2E, bottom). The comparisons between AB−/− and B−/− mice were not attempted because the two groups of mice were recorded with different instruments, but clearly the B−/− mice had much later onset of tremor.
Locomotor activity was tested in the same mice for general exploration. Compared with age-matched WT mice, both A−/− and AB−/− mice at 8 and 12 weeks were hyperactive in central regions (Fig. 2F). A−/− mice were hypoactive in peripheral regions at 12 weeks and did not show normal habituation when compared with WT mice (Fig. 2F). The AB−/− mice showed peripheral hyperactivity especially at 12 weeks when compared with WT mice, and they also showed less habituation. B−/− mice did not show any significant differences from WT mice on locomotor activity at 15 months (29).
These results demonstrate that deficiency of both saposin A and B, or only saposin A or B, in mice affected neuromotor function and caused tremor. The progression of neuromotor deterioration in AB−/− mice was slower than in A−/− mice, but more rapid than in B−/− mice.
Neuronal pathology in AB−/− mice
AB−/− mice exhibited abnormal morphology in the central and peripheral nervous systems (Fig. 3A). Dorsal root ganglion in AB−/− spinal cords contained inclusion material (Fig. 3A–D). As assessed by anti-CD68 antibody staining, activated macrophages were present in the dorsal root ganglion, sciatic nerve and spinal cord (Fig. 3A–C, A–E and A–F), and CD68-positive microglia were in various regions of the brain (Fig. 3C). Astrogliosis in AB−/− mice was assessed using anti-glial fibrillary acidic protein (GFAP) antibody staining (Fig. 3B). Increased GFAP signals were in the thalamus (Fig. 3B-B), corpus callosum (Fig. 3B–D), brain stem (Fig. 3B–F) and spinal cord (data not shown). Enhanced expression of CD68 in macrophage/microglia and astrogliosis reflect proinflammatory responses in AB−/− mice.
Figure 3.
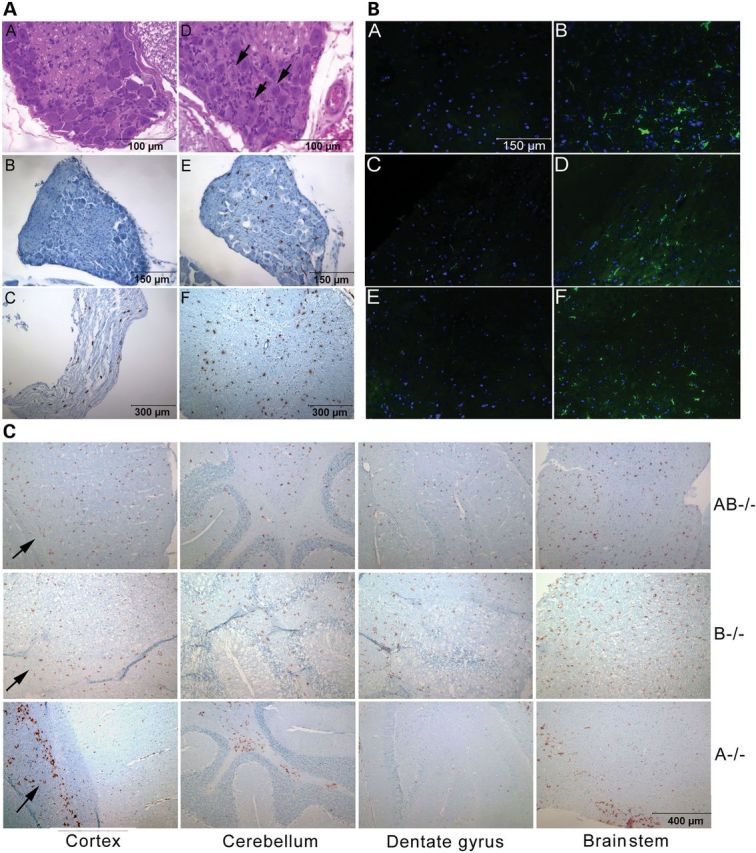
CNS pathology. (A) H&E stained dorsal root ganglion in 12 weeks WT (A–A) and AB−/− mice (A–D). The ganglion neurons of AB−/− mice contained storage inclusions (arrows). Sections (B, C, E and F) were stained by anti-CD68 antibody (brown) and counter stained with hematoxylin (blue). No CD68 positivity in WT mouse dorsal root ganglion (A–B). CD68-positive macrophage/microglial cells in dorsal root ganglion (A-E), sciatic nerve (A-C) and spinal cord (A–F) of AB−/− mice. (B) Astrogliosis in AB−/− mice brains. The brain sections of 12 weeks WT mouse thalamus (B–A), WT mouse corpus callosum (B–C), WT mouse brainstem (B-E), AB−/− mouse thalamus (B–B), AB−/− mouse corpus callosum (B–D) and AB−/− mouse brainstem (B-F) were stained with anti-glial fibrillary acidic protein (GFAP) (green) antibody. Enhanced GFAP signals were seen in AB−/− mice. The nucleus was labeled by 4′,6-diamidino-2-phenylindole (DAPI) (blue). The scale 150 μm is for all six images. (C) Differential distribution of activated macrophage/microglial cells in AB−/−, B−/− and A−/− mice brains stained by anti-CD68 antibody (brown). (Top panels) CD68-positive cells in AB−/− mice (12 weeks) were uniformly distributed in cortex, cerebellum, dentate gyrus and brainstem. (Middle panels) Uniform distribution of CD68-positive cells in B−/− mice brains (12 months) was in similar regions as shown in AB−/− mice. (Bottom panels) CD68-positive cells in A−/− mouse brain (12 weeks) were found in corpus callosum (arrow) below cortex region, white matter of cerebellum and selected regions of brainstem. The sections were counter stained with hematoxylin (blue). The scale 400 μm is for all 12 images.
Distinct brain regions showed CD68-positive microglia in AB−/− mice and A−/− mice. In sagittal sections, CD68-positive microglial cells were uniformly distributed throughout AB−/− mice brains, similar to those in B−/− mice (Fig. 3C). In contrast, CD68-positive microglial cells in A−/− mice were restricted to selected myelin enriched regions, including the corpus callosum below the cortex, white matter of the cerebellum and the brain stem (Fig. 3C). Such differential regional involvement of enhanced expression of CD68 in microglia may reflect the degree of toxicity from substrate accumulation in B−/− (Sulfatide), AB−/− (sulfatide and galactosylsphingosine) and A−/− (GalCer and large amounts of galactosylsphingosine) mice.
Autophagy, protein degradation and apoptosis were studied in AB−/− mice brains at terminal stages. Autophagy markers, p62 and LC3, were increased in AB−/− mouse brains. A polyubiquitin-binding protein, p62, participates in autophagic clearance of ubiquitinated protein. In AB−/− mice, p62-positive aggregates were found in brain stem, thalamus and gray matter of spinal cord and were mostly colocalized in the same cell with Lamp 2, a late endosome/lysosomal marker (Fig. 4A). LC3, a microtubule-associated protein 1A/1B-light chain 3, is an autophagosome membrane marker. In comparison with WT mice, AB−/− mice had LC3 aggregates along the nerve fiber tracks in the brainstem (Fig. 4A) and thalamus (data not shown). Western blotting using anti-LC3 antibody confirmed the increased level of LC3 II in the AB−/− mice brainstems (Fig. 4C), indicating an increase in overall autophagy. Increased p62 was detected in A−/− thalamus (Fig. 4B) and brainstem (data not shown). LC3 II was also detected in A−/− brainstem at terminal stages (Fig. 4C). The change of p62 and LC3 II level in B−/− mice was not apparent (Fig. 4B and C). Because B+/− brain had normal morphology and phenotype, low level of LC3 II in both 18 month B−/− and B+/− brainstem suggests an age-related autophagic activity in aging mice and not caused by deficiency of saposin B. Enhanced ubiquitin signals were found in AB−/− mice brainstems (Fig. 4A). AB−/− mice brains showed increased amyloid precursor protein (APP) deposition in thalamus (Fig. 4A), pons, corpus callosum and cerebellar white matter (data not shown). TUNEL assays for apoptosis were negative in AB−/− mice brains and spinal cords. These results suggest that the deficiencies of saposins A and B, primarily saposin A, can affect protein degradation and autophagy in the central nervous system (CNS).
Figure 4.
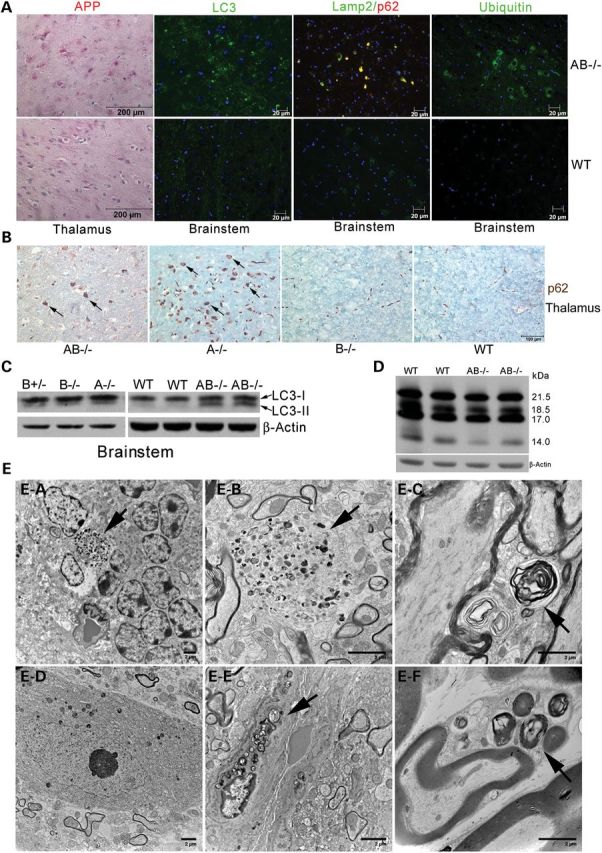
Neuronal pathology and ultrastructure studies in AB−/− mice. (A) Altered autophagy-lysosome degradation in AB−/− brain (12 weeks). Immunohistostaining showed the enhanced signals of APP (red) in AB−/− mouse thalamus. Immunofluorescence signals of LC3, p62, Lamp2 and ubiquitin were enhanced in AB−/− mouse brainstems when compared with WT. Some of p62 (red) were colocalized with Lamp2 (green) in the cells showing yellow signals. (B) p62 signals (brown) in the cells (arrow) of thalamus are shown in AB−/− (14 weeks) and A−/− (10 weeks) mice and not detected in B−/− (10 months) and WT (10 months) mice. (C) Immunoblot of LC3. LC3-I protein (19 kDa) presented in WT, A−/−, B−/−, B+/− and AB−/− mice brainstems. Increased LC3-II (17 kDa) was detected in AB−/− and A−/− mice brainstems. Low level LC3-II was detected in B−/− and B+/− brainstems. β-actin was used as loading control. The samples were from 12 weeks old AB−/− and WT mice, 10 weeks old A−/− mice and 18 months old B−/− and B+/− mice. (D) Immunoblot of MBP in AB−/− mice brains (12 weeks). MBP was resolved as four protein bands (14, 17, 18.5 and 21.5 kDa). All MBP isoforms in AB−/− mice brains were 85% of WT level after normalization to β-actin. Most reduced MBP isoform in AB−/− mouse brain was 14 kDa. (E) Ultrastructural studies of AB−/− mice nervous systems. (E–A) Degenerating cells in granular cell layer of AB−/− mouse cerebellum accumulated undigested materials containing dense inclusion bodies (arrow). (E–B) Inclusion bodies (arrow) were in AB−/− mouse brainstem. (E–C) Myelinated and unmyelinated processes in AB−/− mouse brainstem accumulated membrane whirl-like inclusion bodies (arrow). (E–D) Neuron in AB−/− mouse spinal cord had normal morphology. (E–E) Endothelial cell in AB−/− mouse spinal cord contained heterogeneous undigested membrane inclusions (arrow). (E–F) Schwann cells in AB−/− mouse sciatic nerve contained membrane whirl-like inclusions (arrow). The myelin sheath was normal. The tissues were from 14 weeks old mice. The images are representative of 10–26 images for each tissue sections from 2 AB−/− mice. No inclusion bodies were observed in WT mice.
Myelin basic protein (MBP) is essential for myelination by forming a protein diffusion barrier to regulate biosynthesis of the myelin membrane (32). AB−/− mice cerebrum at terminal age showed mildly reduced MBP levels (85% of WT) (Fig. 4D) as determined using immunoblots. The 14 kDa isoform was the most reduced. Such reductions in MBP are likely caused by disrupted GSLs composition in AB−/− mice nervous systems.
Ultrastructurally, inclusions in degenerating cells were found in the cerebellum (Fig. 4E-A and E-B). Such undigested membranous inclusions were present in the brainstem, spinal cord and thalamus. Myelinated and unmyelinated neuronal processes in brainstem (Fig. 4E-C) and Schwann cells in sciatic nerve (Fig. 4E-F) contained membranous whorls. The myelin sheaths had normal structure in AB−/− mice. No inclusion bodies were found in neurons of AB−/− spinal cord (Fig. 4E-D) and brains. Vascular endothelial cells in AB−/− spinal cords did contain membrane inclusion bodies (Fig. 4E-E). There was an average of two inclusions per EM image in AB−/− tissues. No inclusion bodies were observed in WT mice.
Cells stained positive with alcian blue showed the presence of acidic sulfated substance in several regions of the AB−/− miceCNS, including the cerebral cortex, hippocampus, corpus callosum, brainstem, midbrain, cerebellum, spinal cord and sciatic nerves (Supplementary Material, Fig. S2).
Visceral pathology in AB−/− mice
In the liver of AB−/− mice, large engorged storage cells appeared at ≥4 weeks. These were identified as macrophages by anti-CD68 antibody staining (Fig. 5A). Such storage macrophages were present in clusters with multiple nuclei stained with hematoxcylin (Fig. 5A). Alcian blue-positive staining was in AB−/− mice kidney tubules indicating sulfatide accumulation (Supplementary Material, Fig. S2). Ultrastructurally, the storage materials showed tubular structures with a twisted filamentous appearance (Fig. 5BC) that were similar to those seen in globoid cells of Krabbe's disease (33). Engorged macrophages were also present in A−/− mice livers, whereas the liver of B−/− mice had normal morphology (Fig. 5A). B−/− mice kidneys had storage materials in both distal and proximal tubules (29), whereas AB−/− mice kidneys showed storage bodies only in distal tubules (Fig. 5B-E). The storage materials had mixed forms of membrane whorl lamination and multivesicular bodies (Fig. 5B-F and B-G) that were similar to those reported in kidney tubular cells of Fabry mice (34). The podocytes and mesangial cells in AB−/− mice kidneys appeared normal. The histology of AB−/− mice lungs, spleen and pancreas were indistinguishable from WT mice (data not shown).
Figure 5.

Visceral tissue pathology. (A) H&E-stained liver sections showed the storage cells (arrows) in AB−/− (12 weeks) and A−/− (12 weeks) mice. Those storage cells were Kupffer cells stained positive for CD68 (brown), a macrophage marker. Age-matched WT and B−/− mice livers had normal morphology. Cell nuclei stained with hematoxylin (blue). (B) Ultrastructural studies of AB−/− mice liver and kidney. (B-A) WT mouse liver showed normal sinusoidal lining cells and hepatocytes. (B-B) Multiple sinusoidal lining cells contained tubular inclusions (arrow) in AB−/− mouse liver. The undigested materials were present in the vacuoles (arrow head). (B-C) The storage inclusions shown in (B-B) had twister filamentous appearance (arrow). (B-D) WT mouse kidney distal tubule. (B-E) AB−/− mouse kidney distal tubule contained membrane inclusions (arrows). (B-F and B-G) Enlarge view of the inclusions in B-E showed laminated bodies (B–F) and multivesicular inclusions bodies (B-G). The tissues were from 14 weeks old mice.
GSLs analyses
Saposin B binds sulfatide and presents it to arylsulfatase A for degradation to GalCer (35). Deficiency of arylsulfatase A or saposin B leads to sulfatide accumulation and metachromatic leukodystrophy phenotypes (29,36). Sulfatide levels in AB−/− mice were evaluated at three ages, 4 weeks, 8 weeks and terminal stages and compared with age-matched A−/−, B−/− and WT mice (Fig. 6). Terminal age groups included, A−/− mice at 10–13 weeks, AB−/− mice at 12–14 weeks and WT and B−/− mice at 14 weeks. Total non-hydroxyl fatty acid sulfatide (NFA sulfatide) and hydroxyl fatty acid sulfatide (HFA sulfatide) were increased in AB−/− mice brains, kidneys and livers with age similar to that seen in B−/− mice (Fig. 6). The accumulation of NFA sulfatide and HFA sulfatide in AB−/− mice were found at 4 weeks and progressively increased with age. By terminal age (10–14 weeks), NFA sulfatide and HFA sulfatide levels in AB−/− were ∼1.5 -fold of WT mice levels in the brain. AB−/− kidney at 10–14 weeks had higher level of sufatides accumulation than brain and liver with 11- and 19-fold increases in NFA sulfatide and HFA sulfatide, respectively. Increases in NFA sulfatide and HFA sulfatide in AB−/− mice liver were significantly above WT mice levels that had nearly detectable sulfatides. By terminal stage, AB−/− mice had more sulfatide accumulation in the kidney (2-fold) and liver (2- to 5-fold) than B−/− mice. Slight increases in NFA sulfatide were detected in A−/− mice kidneys and livers by terminal stage at 10–13 weeks (Fig. 6).
Figure 6.

Liquid chromatography (LC)/MS analyses of sulfatides accumulation in AB−/− mice. NFA sulfatide and HFA sulfatide levels were increased in the brain, kidney and liver of B−/− and AB−/− mice from 4 to 14 weeks. (A) Brain sulfatide level in B−/− and AB−/− mice increased by 1.4- and 1.5-fold relative to age-matched WT, respectively. (B) AB−/− mice kidneys had more sulfatide accumulation than B−/− mice kidneys by terminal stage. The level of HFA sulfatide was higher than NFA sulfatide in AB−/− mice kidneys. A−/− mice kidneys had consistent increases in NFA sulfatide with age. (C) The sulfatides were undetectable in WT mice livers. Significantly increased sulfatides were detected in B−/− and AB−/− mice livers. Fold changes were not assessed for the liver. Fold increases in sulfatide in A−/− and AB−/− mice samples relative to WT were indicated above the column for brain and kidney. A−/− and AB−/− mice in 10–14 weeks group were at terminal stages. The ages of mice in 10–14 weeks group are A−/− mice, 10–13 weeks; B−/− mice, 14 weeks, AB−/− mice, 12–14 weeks; and WT mice, 14 weeks. The amount of sulfatide was normalized by milligram of tissue weight (n = 3 mice). The data were analyzed by one-way ANOVA with Tukey's post hoc test (P < 0.05): *, differ significantly from WT; a, differ significantly between A−/− and AB−/− mice; b, differ significantly between B−/− and AB−/− mice. Student's t-test was compared between WT and mutant (#, P < 0.05).
Tissue-specific accumulation of sulfatide species was evident in AB−/− and B−/− mice. The content of NFA sulfatide was about 2-fold higher than HFA sulfatide in the AB−/− mice brains, whereas brain NFA and HFA sulfatides in A−/− mice were at WT mice levels (Fig. 6). The sulfatide species that accumulated in AB−/− mice brain contained NFAs with C16:0, C18:1, C18:0 and C20:0 fatty acid acyl chains and HFAs with C18:0, C20:0 and C22:0 chains (Supplementary Material, Fig. S3). In comparison to brain, the levels of HFA sulfatides in the kidney were higher than NFA sulfatide by ∼2-fold (Fig. 6). Long acyl chain species of HFA sulfatide, C20:0, C22:0 and C24:0, were the major accumulated species in AB−/− mice kidneys (Supplementary Material, Fig. S3). Sulfatide species were below the level of detection in WT mouse livers. Detectable sulfatide levels were in B−/− and AB−/− mice livers, but were only ∼1% of those in the kidney (Fig. 6). Sulfatide species were below the level of detection in spleen of WT or mutant (AB−/−, A−/−, B−/−) mice.
Saposin A enhances the activity of β-galactosylceramidase for the degradation of GalCer and galactosylsphingosine (26). A−/− mice showed accumulation of GalCer in the brain (1.9-fold) and kidney (4-fold) of WT mice level by terminal stage at 10–13 weeks (Fig. 7). The accumulated GalCer species in A−/− mice brains contained C18:0 and C24:1 fatty acid acyl chains (Supplementary Material, Fig. S4). Most GalCer species were elevated in A−/− mice kidneys and included chain lengths from C16:0 to C24:0. Total GalCer contents were about 200 times higher in the AB−/− mice brains when compared with those in kidneys. In comparison with A−/− mice, GalCer contents in AB−/− mice brains or kidneys were reduced by 40 or 80%, respectively. No detectable changes of GalCer levels were found in A−/− and AB−/− mice livers (data not shown). Glucosylsphingosine and galactosylsphingosine are toxic metabolites and elevated in patients with neuronopathic Gaucher's disease and Krabbe's disease, respectively (37,38). After separation of these two glycosphingosines, significant increases in galactosylsphingosine were found in A−/− and AB−/− mice brains (Fig. 7B), but in AB−/− mice, brains levels were reduced to 68% of those in A−/− mice brains. Glucosylsphingosine was below the levels of detection in the brains of these mice (data not shown). These results show that deficiency of saposin B blocked the conversion of sulfatide to GalCer and diminished the accumulation of GalCer, as well as galactosylsphingosine, caused by saposin A deficiency.
Figure 7.

Galactosylceramide (GalCer) and galactosylsphingosine levels in 10–14 weeks group by LC/MS analysis. (A) Increased total GalCer was in A−/− mice brains (1.9-fold) and (B) kidneys (4-fold). In comparison to A−/− mice, AB−/− mice had decreased GalCer level in the brain and kidney. (C) Significantly increased galactosylsphingosine levels were in A−/− and AB−/− mice brains at terminal stage (10–14 weeks). Galactosylsphingosine level in AB−/− mouse brain was reduced to 68% of A−/− mice level. Brain and kidney GalCer was separated for GluCer, and galactosylsphingosine was separated from glucosylsphingosine by LC/MS. Tissue samples and data analysis are as described in Figure 6.
LacCer is hydrolyzed by GM1-β-galactosidase and β-galactosylceramidase. Saposins B and C are involved in LacCer hydrolysis (35). Saposin A, as the activator for β-galactosylceramidase, can also participate in LacCer degradation (39). LacCer levels were analyzed in AB−/− mice brains, livers, kidneys and spleens. The LacCer species, C18:0, showed major increases in AB−/− mice brains (Supplementary Material, Fig. S5). Small, but significant increases, in LacCer were detected in the kidneys of A−/−, B−/− and AB−/− mice with C16:0, C18:0 and C24:0 being the most abundant species (Fig. 8 and Supplementary Material, Fig. S5). Liver LacCer accumulations were age dependent (Supplementary Material, Fig. S6) and were substantially increased in AB−/− (102-fold) and A−/− (90-fold) mice above WT mice levels in the terminal stage group (10–14 weeks) (Fig. 8). Long acyl chain LacCer species (C22:0, C24:1 and C24:0) accumulated in A−/− and AB−/− mice livers (Supplementary Material, Fig. S5). LacCer in B−/− mice livers at 14 weeks was at nearly WT mice levels ,and minor increases were detected by 15 months (29). These results indicate preferential tissue LacCer degradation by selected saposins. Deficiency of either saposin A or B disrupted LacCer hydrolysis in the kidney, whereas combined deficiencies of saposins A and B affected LacCer degradation in the brain. Saposin A appeared to be a major saposin involved in liver LacCer degradation.
Figure 8.

LacCer levels in 10–14 weeks group by LC/MS analysis. Total LacCer was slightly increased in AB−/− mice brains (A), A−/−, B−/− and AB−/− mice kidneys (C), highly accumulated in A−/− and AB−/− mice livers (B) and elevated in A−/− and AB−/− mice spleens at (D) 10–14 weeks of age. Tissues samples and data analysis are as described in Figure 6.
GluCer is the product of LacCer hydrolysis, and GluCer levels were increased by ∼ 2-fold in AB−/− mice brains at terminal stage, but not in A−/− and B−/− mice brains (Supplementary Material, Fig. S7). Total kidney and spleen GluCer levels of AB−/− mice were at WT mice levels, whereas GluCer levels were increased in A−/− and AB−/− mice livers. The major GluCer species in brain was C18:0, and longer acyl chain (C20:0–C24:0) contents were in the livers and spleens (Supplementary Material, Fig. S7).
In the GSL degradation pathway, ceramide is generated from the hydrolysis of GluCer, GalCer and sphingomyelin. A−/− and AB−/− mice brains showed decreased levels of ceramide relative to WT and B−/− mice brains (Supplementary Material, Fig. S8) which suggest that GalCer is the major source of ceramide in the brain. In comparison with the brain, ceramide levels were significantly increased in A−/− mice livers and spleens, but not in AB−/− mice livers and spleens (Supplementary Material, Fig. S8). No differences of ceramide levels were found between saposin mutants (A−/−, B−/− and AB−/−) and WT mice kidneys. Ceramide levels in B−/− mice were at WT mice levels in these four tissues (Supplementary Material, Fig. S8). No hydroxyl ceramide was detected in these tissues (data not shown). The results indicate that ceramide levels were affected indirectly by deficiency of saposin A and B in a tissue-specific manner.
Saposin B plays a role in promoting α-galactosidase A activity for hydrolysis of globotriaosylceramide (Gb3) and participates in GM3 degradation (35). By thin-layer chromatography (TLC) analyses, elevated Gb3 was detected in AB−/− mice livers and kidneys (Supplementary Material, Fig. S9), supporting the in vivo role of saposin B in Gb3 degradation in those organs. Ganglioside (GM1, GM2 and GM3) levels in AB−/− mice brain samples were not changed relative to WT mice (Supplementary Material, Fig. S9). These data are consistent with the finding of no gangliosides accumulation in saposin B-deficient mice brain by TLC analyses (29).
Lipid analyses of AB−/− mice tissues revealed that these saposins had tissue-specific roles in GSL metabolism. Disruption of sulfatide degradation, due to saposin B deficiency, diminished GalCer and galactosylsphingosine accumulation caused by saposin A deficiency. Blockage of GalCer degradation in the brain led to reduced ceramide levels. Multiple saposins (A, B and C) play a role in LacCer hydrolysis. Saposin A was the dominant saposin for liver LacCer degradation, whereas all three saposins had compensating roles for brain LacCer hydrolysis.
DISCUSSION
In the GSL degradation pathway, sulfatide is converted to GalCer by arylsulfatase A and saposin B. GalCer is further hydrolyzed to ceramide by GalCer-β-galactosylceramidase and saposin A (35). Lacking saposin A or saposin B leads to GalCer or sulfatide accumulation, respectively, indicating the importance of these saposins for maintaining homeostasis of GalCer and sulfatide, the major lipids in myelin (26,29). Combined deficiencies of saposins A and B in mice revealed that their interrelated actions in GSL degradation alter the effects of either saposin alone on the development of CNS disease. Disruption of sulfatide degradation in AB−/− mice due to saposin B deficiency diminished the GalCer accumulation resulting from saposin A deficiency. Decreased degradation of CNS GalCer also resulted in diminished ceramide levels. Deficiency of saposin A potentiated sulfatide accumulation resulting from saposin B deficiency in the liver and kidney, but not in the brain. Although saposins A, B and C modulated LacCer hydrolysis in the CNS, saposin A had the dominant role in liver LacCer degradation. Disrupted GSL degradation in AB−/− mice led to inclusion materials in neuronal processes and Schwann cells, neurological deterioration and shortened life spans. The deficiency of saposins A and B had minor effects on prosaposin processing in having increased levels of prosaposin and saposins C and D. The increased level of saposins C in AB−/− liver had no detectable effect on GluCer levels. In addition to these effects, analyses of the AB−/− mice showed the tissue-specific functions of the saposins in GSLs metabolism (Fig. 9), including their essential role in maintaining normal neuronal function. Interestingly, the combined deficiencies of saposins A and B did not affect brain formation at gross level. In comparison to the A−/− and B−/− mice, the AB−/− mice demonstrated the tissue differential interactions of dual saposins in GSL metabolism, and saposin A deficiency dominated the phenotype at a gross and biochemical level (Table 1).
Figure 9.
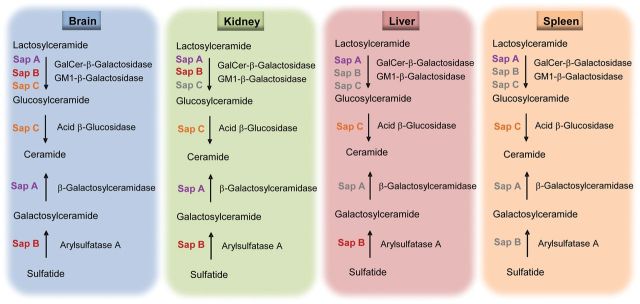
Tissue differential functions of saposins A and B in GSL degradation. The findings from the saposin A and B-deficient mouse models provide in vivo data that both saposin A and B participate in lactosylceramide degradation in the brain and kidney, whereas saposin A is the major saposin for liver and spleen lactosylceramde hydrolysis. Based on tissue environmental GSL synthetic rate, saposin B is essential for sulfatide degradation in the brain, kidney and liver, but not in the spleen. Saposin A participates in the degradation of galacosylceramide in the brain and kidney and not in the liver and spleen. Saposins labeled in color had in vivo roles in GSL degradation in that tissue. Saposins in gray color indicate no major roles in vivo.
Table 1.
Major characteristics of AB−/−, A−/− and B−/− mice
| AB−/− | A−/− | B−/− | |
|---|---|---|---|
| Life span (days) | 96 | 85 | 644 |
| Body weight (% WT) | 86 | 64 | 100 |
| Neuromotor deterioration | Rapid | More rapid | Slow |
| Brain pathology | Severe | Severe | Severe |
| Liver pathology | Severe | Severe | Normal |
| GSL accumulations | |||
| Sulfatide (brain and kidney) | High | Normal | High |
| GalCer (brain and kidney) | Normal | High | Normal |
| Galactosylsphingosine (brain) | Low | High | Normal |
| LacCer (liver) | High | High | Normal |
Relative to the A−/− mice, the AB−/− mice had a milder phenotype with a somewhat longer life span, slower weight loss rate and better preserved behavioral functions. This might be explained by the decreased rate of GalCer and galactosylsphingosine accumulation in the CNS of the AB−/− mice. However, brain sulfatides (NFA and HFA sulfatides) in AB−/− mice accumulated at same age (14 weeks) slightly more than those in the saposin B-deficient mice. A similar histopathological distribution of proinflammation was present in B−/− and AB−/− mice, but at a much earlier age in the AB−/− mice as shown by the regional presence of enhanced CD68 expression in microglia. The concordance of the B−/− and AB−/− mouse pathologies lends credence to sulfatide being an injurious lipid in AB−/− mice brains. However, the saposin A−/− effects in the AB−/− mice were dominant over the B−/− effects. This was evidenced by the more rapid degenerative course in the AB−/− mice and the accumulation of galactosylsphingosine, a toxic lipid. Galactosy- and glucosyl-sphingosines have been postulated as neurodegenerative agents in Krabbe's and Gaucher's diseases, respectively (37,38,40). Galactosylsphingosine was increased in saposin A-deficient mice (26) and not detected in B−/− mice. Thus, the dual presence of galactosylsphingosine and sulfatide in the AB−/− mice brains suggests a dominant role of the former lysolipid in increasing the pathological progression. The concordant decrease in GalCer and galactosylsphingosine (∼68% relative to A−/−) supports a diminished flux of sulfatide and lysosulfatide to GalCer and galactosylsphingosine, respectively, through β-galactosylceramidase. As a result, the deficiency of saposin B somewhat attenuated the CNS phenotype and altered regional involvement of the A−/− mice to produce the AB−/− mice manifestations. These results indicate the coordinated roles of these two saposins in the galactosyl-based sphingolipid pathways and their influence on the flux of these lipids through these pathways.
Two enzymes, GM1-β-galactosidase and GalCer-β-galactosylceramidase, have overlapping functions in the degradation of LacCer (35). Ablating either enzyme alone has no effect on LacCer levels, whereas knocking-out both enzymes leads to LacCer accumulation in liver and brain (41). Genetic mutations of β-galactosylceramidase lead to Krabbe's disease in humans and the twitcher phenotype in mice (42,43). The Matsuda A−/− mice had a shorter life span in our laboratory than previously reported (26). Similar to these A−/− mice, the AB−/− mice developed large clusters of storage cells in the liver with increases in LacCer. LacCer levels in AB−/− mice kidney and brain showed significant increases, but at overall low levels. In vitro studies showed effects of saposins B and C on GM1-β-galactosidase and GalCer-β-galactosylceramidase for hydrolysis of LacCer (10). In vivo, LacCer levels had minor elevations in B−/− mouse livers, whereas C−/− mice livers did not, and only small increases of LacCer were present in the cerebella of C−/− mice (28). Overall in mouse models, saposins A, B and C participate in LacCer hydrolysis in a tissue-specific manner. Liver LacCer hydrolysis is dominated by saposin A with saposins B or C. In brain, saposins A, B and C appear to substitute for each other in LacCer degradation, and both saposins A and B are important to LacCer hydrolysis in the kidney (Fig. 9).
In the GSL degradation pathway, ceramide can be produced from GalCer, GluCer or sphingomyelin hydrolysis. Because sphingomyelin levels were not changed in AB−/− mice (unpublished observation), altered ceramide levels in AB−/− mice are likely to arise from GalCer or GluCer hydrolysis. Analysis of ceramide levels in AB−/− mice revealed a tissue-specific effect. Reduced ceramide level in AB−/− and A−/− mice brains could be due to the blockage of GalCer and sulfatide degradation, suggesting that ceramide is mainly generated from GalCer in the brain. In contrast to the brain, ceramide levels were increased in the liver and spleen of A−/− and AB−/− mice. Large amounts of accumulated LacCer, as a substrate to enhance Vmax, might account for an increase in the products such as GluCer and ceramide.
Evidence that GSL accumulations affect autophagy has recently emerged (44–48). Aggregation of two autophagy markers, p62 and LC3, along with enhanced Lamp 2, a lysosomal membrane protein, in AB−/− mice brains implicates altered autophagy and lysosomal degradation. The enhanced p62 and LC3-II in A−/−, but not in B−/− mice, suggest that saposin A deficiency is the primary factor for autophagy alteration. Most importantly, the majority of such aggregates were distributed in the brainstem and thalamus, where inclusion bodies were found. The accumulation of APP and ubiquitin in these regions could result from altered autophagic flux (49). The results indicate that neuropathologic mechanisms in AB−/− mice brains were complex with involvement of the CLEAR system (50), protein ubiquitination and proinflammation. These neuropathologic changes correlate well with the sequential deterioration of neurological function and poor behavioral performance, impairment of motor coordination, tremor, increased central and peripheral locomotor activity and failure to habituate.
The AB−/− model did not exactly resemble either A−/− or B−/− mice by GSLs, neuromotor behavior and histopathological analyses (Table 1). The AB−/− mice provides a unique in vivo model to study dual saposins (saposin A and saposin B) interaction and their major control points in GSLs degradation. The analysis of AB−/− model demonstrated that saposins play different role in different GSL synthetic rate environment (Fig. 9). Both saposin A and saposin B have unique functions in the degradation of GalCer and sulfatide in CNS and kidney, respectively. These saposins exhibit compensatory interactions with each other in hydrolysis of LacCer in the brain, whereas saposin A is dominant for liver LacCer metabolism. Understanding the essential roles of the saposins in specific tissues/cell is critical to the mechanistic bases of their physiological function. Importantly, the AB−/− mice model revealed the tissue-specific interactions of these saposins in GSL metabolism and homeostasis that cannot be achieved by in vitro assays and by individual saposin-deficient models.
MATERIALS AND METHODS
Materials
The following were from commercial sources: TopoTA cloning vector and Alexa Fluor® 488 conjugated goat anti-rabbit antibody (Invitrogen, Carlsbad, CA, USA), rat anti-mouse CD68 monoclonal antibodies (Serotec, Oxford, UK), Molecular Dynamics Storm 860 scanner (GE Healthcare, Chicago, IL, USA), Antifade/4′,6-diamidino-2-phenylindole (DAPI), ABC Vectastain and alkaline phosphatase kit II (Black) were used (Vector Laboratory, Burlingame, CA, USA). Qiagen Quick-change II XL kit (Qiagen Inc., Valencia, CA) was used. Restriction enzymes were from New England Labs (Beverly, MA, USA). Alcian blue was from Poly Scientific (Bay Shore, NY, USA). (Liquid chromatography) LC/MS-MS internal standards for GluCer, GalCer, LacCer, ceramide, galactoylsphingosine, glucosylsphingosine and sulfatide were from Avanti Polar lipids, Inc. (Alabaster, AL, USA). Sulfatide with C16:0 was obtained from Mytreya LLC (Pleasant Gap, PA, USA). Electrospray ionization (ESI)-LC-MS/MS using a Waters Quattro Micro API triple quadrupole mass spectrometer (Milford, MA, USA) was used.
Construction of knock-in targeting vector and generation of saposin A and B-deficient mice
Mutations encoding Cys→Phe were introduced to saposins A and B to disrupt one of the three disulfide bridges in each saposin to produce deficiencies of both saposins A and B. The same strategy has been used for generation of individual saposin-deficient mice (26,29). The Cys→Phe codon mutation in the saposin A domain was created in exon 4 of prosaposin gene using Qiagen Quick-change II XL kit with mismatched oligonucleotide primers: mAF (5′-TG TCG GCC TCG TTT AAA GAG GTG GTT GAC-3′), mAR (5′-GTC AAC CTC TTT AAA CGA GGC CGA CA-3′) and subclone 6–1 as a template. The mismatched nucleotides are underlined. The long arm Xho I-Cla I (4.0 kb) was generated by PCR using subclone 6-1 with the mutation on saposin A, and the product was cloned into the TA cloning vector. The Cys→Phe codon mutation in the saposin B domain was created in exon 7 using subclone 4-1 as described (29). The short arm BamH-Kpn1 (1.6 kb) containing the mutation in saposin B was generated by PCR and cloned in TopoTA cloning vector. The Cys→Phe mutation created a unique Dra1 restriction enzyme site for Southern blot analysis. The saposin AB targeting construct was assembled in OSdupdel containing the Polymer enhancer/herpes simplex virus thymidine kinase (TK) (MC1) promoter with the neomycin (neo) gene flanked by two loxP sites and the TK gene driven by the 3′ phosphoglycerate kinase (PGK) promoter. The short arm (1.6 kb) containing the mutation on saposin B was cloned into BamHI/KpnI sites downstream of MC1-neo in the vector to form the 3′ homologous region. The long arm (4.0 kb) containing the mutation of saposin A released with EcoRI from the TA cloning vector was blunt ligated into NheI sites upstream of MC1-neo resulting in the 5′ homologous region. Both long arm and short arm on targeting vector were validated by direct sequencing. The targeting vector was linearized with NotI and introduced into the 129/SvEv ES cell line. Recombinant ES clones were screened by PCR analysis using the primers inside of the short arm: ABR1 (5′-GCC AGA CCT GTC AGT TTG TGA TGA A-3′) or outside of the short arm: ABR2 (5′-CTG GAG GAG TTC TAT ACG TGC CCA-3′), together with the primer in the 3′ region of neo, NEO3′F2 (5′-GGT GTT GGG TCG TTT GTT CGG ATC A-3′). The recombinant and WT clones generated 1.4 and 1.6 kb PCR products, respectively. The recombinant ES clones were confirmed with Southern blot analyses. The clones were digested by DraI/BglII to release 11.7 kb for WT and 5.6 kb for recombinant allele, respectively, probed with the 3′ probe. The mutations on saposin A and B in the recombinant clone were verified by PCR and sequencing.
The correctly targeted clones were used to generate chimeric mice by microinjection into C57BL/6J blastocysts. Electroporation, ES cell culture, selection and chimera breeding were carried out by the mouse Gene-targeting service core at the University of Cincinnati. The neo gene in the heterozygous F1 mice was removed by crossbreeding with Zp3-Cre mice [C57BL/6-TgN(Zp3-Cre)93Knw; The Jackson Laboratory] as described (29) and resulted in one loxP site in the offspring. PCR genotyping of AB−/− mice was conducted using the primers for B−/− mice as described (29). The strain of background for AB−/− mice was C57BL/6J/129SvEV. A−/−, B−/− and CD−/− mice are in the same strain background as AB−/− mice. The mice were maintained in microisolators in accordance with institutional guidelines under IACUC approval at Cincinnati Children's Research Foundation.
Histological and immunohistochemistry analyses
The mice were subjected to CO2 narcosis and perfused with saline followed by 4% paraformaldehyde in 1× PBS. The tissues were collected and fixed in 10% formalin or 4% paraformaldehyde. Formalin-fixed tissues were embedded in paraffin, sectioned and stained with hematoxylin and eosin (H&E). Tissues for ultrastructural analyses were fixed in 3% glutaraldehyde. CD68 monoclonal antibody staining was carried out with paraformaldehyde-fixed frozen sections as described (51). GFAP staining on paraffin tissue sections with mouse anti-GFAP monoclonal antibody and Alcian blue staining on paraformaldehyde-fixed frozen sections were described previously (29). Paraformaldehyde-fixed frozen sections were stained with rabbit anti-Ubiquitin (abcam, ab7780), mouse anti-p62 (Abnova, Cat: H00008878), rabbit anti-LC3 (Novus Biologicals, NB100-2220) and rat anti-Lamp 2 (abcam, ab25339) antibodies and detected with fluorescence probe. The paraffin tissue sections were stained with rabbit anti-APP (abcam, ab25339), followed by AB vector kit for color development. Immunohistochemistry of frozen sections with anti-p62 (abcam, ab91526) was carried out in BenchMark XT IHC/ISH Staining Module (Ventana Medical System, Tucson, AZ, USA) at Pathology Core in Cincinnati Children's Hospital Medical Center. The tissues sections were counterstained with hematoxylin.
Immunoblot
Saposin protein expression in tissues and cells were analyzed by immunoblots (28) with modifications. Rabbit anti-mouse saposin A (1 of 500), anti-mouse saposin B (1 of 800), anti-mouse saposin C (1 of 400) and anti-mouse saposin D (1 of 1000) were diluted in 1% milk in 1× PBS buffer. To detect prosaposin and intermediated processing forms, HRP conjugated goat anti-rabbit IgG (ICN, 55667) was applied (1 of 10 000 dilution in 1.25% milk in 1× PBS). The signal was developed using ECL detection reagent (GE Healthcare, RPN2132) according to the manufacturer's instructions. Saposin proteins in tissues were detected using direct Infrared fluorescence detection. The blots were incubated with primary antibodies diluted (as describe above) in 1× Odyssey blocking buffer. After a series wash with 0.5% Tween 20 in 1× PBS, IRDye 800 CW conjugated goat anti-rabbit IgG (Li-COR, 926-32211) diluted in 1× Odyssey blocking buffer (1 of 5000) was applied. The signals were imaged on Odyssey Infrared Imaging System (Li-COR Biosciences) using 800 nm fluorescence channel.
MBP was detected using sheep anti-human MBP (1 of 5000), and β-actin in the tissues was detected by mouse anti-β-actin antibody (1 of 10 000) (30). Immunoblots of LC3 were performed using tissue lysate prepared in mammalian protein extraction reagent (Thermo Scientific. Rockford, IL, USA). The homogenate in loading buffer was resolved on 4–20% gradient gel (Invitrogen) at 100 mA for 2 h. The protein was transferred to Hybond ECL membrane (Amersham Biosciences) by a semidry system at 200 mA for 1 h. After blocking in 5% milk, LC3 was detected by rabbit anti LC3 (Novus Biologicals, NB100-2220) diluted to 1 of 500 in 1% milk (1× PBS). The signal was developed using ECL detection reagent.
GSL analyses
The GSLs in tissue samples were extracted (52), and relative proportions of lipids in the tissues were determined by TLC with borate impregnated plates (10 cm2 Merck high performance TLC silica gel 60, 200 μm). Plates were developed in chloroform/methanol/water (65:25:4, v/v/v) for Gb3 and LacCer and in chloroform/methanol/0.22% CaCl2 (65:35:8, v/v/v) for resolving gangliosides. GSLs were visualized with primulin (100 mg/l in 80% acetone) and blue fluorescence scanning (Storm 860, GE Healthcare).
GSLs, including NFA sulfatide, HFA sulfatide, LacCer, GluCer, GalCer, ceramide and galactosylsphingosine, were determined by LC/MS. Analysis was carried out by LC-ESI-MS/MS using a Waters Quattro Micro API triple quadrupole mass spectrometer (Milford, MA, USA) interfaced with Acquity ultra performance liquid chromatography system. Online chromatographic separation was achieved using a Supelcosil-LC-18-DB column (33 × 3.0 mm i.d. 3.0 µm). For quantification of ceramide and LacCer, the ESI-MS/MS was operated in the multiple reaction monitoring modes, with detection of the transition pair of the individual protonated parent ions and their common daughter ion m/z 264. Galactosylsphingosine or glucosylsphingosine was measured by monitoring of the mass transition m/z 462.3> 282.4. Sulfatides were analyzed in negative ion mode by detection of their parent ion and common daughter ion of m/z 97.
The resolution of GluCer and GalCer was achieved using the Alvarez-Vasquez method (53) with modifications. The two isomers were resolved on a silica column (Supelco 2.1 × 250 mm) running under the hydrophobic interaction liquid chromatography (HILC) mode with an isocratic mobile phase of acetonitrile/methanol/acetic acid (97/2/1, v/v/v) with 5 mm ammonium acetate for 8 min. The method to resolve galactosylsphingosine and glucosylsphingosine was developed using a Kinetex column (Phenomenex, 100 × 2.1 mm, 2.6 µm) running under the HILC. An isocratic elution with 95% mobile phase A [acetonitrile/methanol/acetic acid (97/2/1, v/v/v) with 5 mm ammonium acetate] and 5% mobile phase B (isopropanol) was run for 10 min to resolve two isomers.
Optimized parameters for GSLs were determined with individual standard compounds. Galactosylsphingosine and glucosylsphingosine were quantitated using external standards (galactosylsphingosine and glucosylsphingosine). For ceramide analysis, C16, C18 and C24 ceramides were used to construct calibration curves, and C17 ceramide was employed as the internal standard. For sulfatide, C16 and C24 sulfatide standards were used to construct calibration curves, and C12 sulfatide was used as an internal standard. Calibration curves for GluCer were built using C16, C18 and C24:1 GluCer. C12 GluCer was used as the internal standard. Quantification of GSL species with various fatty acid chain lengths was realized using the curve of each GSL species with the closest number of chain length. In the method for the resolution of GluCer and GalCer analysis, five standards (C16, C18, C24:1 GluCer and C16, C24:1 GalCer) were used, and C12 GluCer and C12 GalCer were used separately as internal standards. The extracted tissue samples were suspended in methanol containing internal standard and injected into the LC/MS. GSL levels in the tissues were normalized to wet tissue weight. Data were analyzed by one-way analysis of variance (ANOVA) with Tukey's post hoc test and Student's t-test (GraphPad Prism version 5.00, GraphPad Software, San Diego, CA, USA).
Behavioral studies
Narrow bridge tests
The male mice at 8 and 12 weeks of age were subjected to the tests on square beams (1 m in length) with cross sections of 12 and 5 mm2 and round beams (1 m in length) with diameters of 17 and 11 mm as described (29). Latency to traverse each beam and the number of times the hind feet slipped off the beam were recorded. A total of 8 A−/− male, 20 AB−/− male and 17 age-matched WT male mice were included in the test. The mice were tested at 8 weeks and repeated at terminal stage for A−/− mice at 10–12 weeks and AB−/− mice at 12 weeks. The significance was analyzed by one-way ANOVA with Tukey's post hoc test (GraphPad Prism version 5.00, GraphPad Software).
Locomotor activity
Locomotor activity measurements (28) were assessed with central and peripheral activities recorded in 5 min intervals. Each genotype of mice was alternated between each chamber to minimize time of day effects. The tests were conducted under normal fluorescent light conditions. The same mice tested for narrow bridges were assessed for activity at 8 and 12 weeks. Activity counts were analyzed by two-factor mixed linear ANOVA (SAS v9.2, SAS Institute, Cary, NC, USA) with genotype and interval. Genotype was a between-subject factor and interval a within-subject factor. Significant interactions were analyzed by slice-effect ANOVAs at each interval. Significant interval effects were further analyzed using pairwise group comparisons using the false discovery rate method.
Tremor tests
Tremor in the mice was monitored in the Tremor Monitor System (San Diego Instruments). An individual mouse was secured in the animal enclosure. Tremor in mice was recorded for 34 min at 128 Hz with bandpass filter ranging from 1 to 60 Hz setting. The recorded data were analyzed using the software provided by the manufacturer. Tremor in mutant mice was presented as the highest amplitude or fast Fourier transform magnitude at a certain frequency when compared with WT mice. The significance was analyzed by two-way ANOVA with Tukey's post hoc test (GraphPad Prism version 5.00, GraphPad Software).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants R01 NS/DK 36681 and DK 36729 to G.A.G.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Mary Moran, Kui Xu and Venette Inskeep for their technical assistance, Lisa McMillin, Georgianne Ciraolo, Betsy DiPasquale, Meredith Taylor and Chris Woods for skilled tissue preparation and photomicrographs and Joyce Life-Ishmael for her clerical expertise.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Rorman E.G., Grabowski G.A. Molecular cloning of a human co-beta-glucosidase cDNA: evidence that four sphingolipid hydrolase activator proteins are encoded by single genes in humans and rats. Genomics. 1989;5:486–492. doi: 10.1016/0888-7543(89)90014-1. [DOI] [PubMed] [Google Scholar]
- 2.O'Brien J.S., Kretz K.A., Dewji N., Wenger D.A., Esch F., Fluharty A.L. Coding of two sphingolipid activator proteins (SAP-1 and SAP-2) by same genetic locus. Science. 1988;241:1098–1101. doi: 10.1126/science.2842863. [DOI] [PubMed] [Google Scholar]
- 3.Collard M.W., Sylvester S.R., Tsuruta J.K., Griswold M.D. Biosynthesis and molecular cloning of sulfated glycoprotein 1 secreted by rat Sertoli cells: sequence similarity with the 70-kilodalton precursor to sulfatide/GM1 activator. Biochemistry. 1988;27:4557–4564. doi: 10.1021/bi00412a050. [DOI] [PubMed] [Google Scholar]
- 4.Leonova T., Qi X., Bencosme A., Ponce E., Sun Y., Grabowski G.A. Proteolytic processing patterns of prosaposin in insect and mammalian cells. J. Biol. Chem. 1996;271:17312–17320. doi: 10.1074/jbc.271.29.17312. [DOI] [PubMed] [Google Scholar]
- 5.Hiraiwa M., Martin B.M., Kishimoto Y., Conner G.E., Tsuji S., O'Brien J.S. Lysosomal proteolysis of prosaposin, the precursor of saposins (sphingolipid activator proteins): its mechanism and inhibition by ganglioside. Arch. Biochem. Biophys. 1997;341:17–24. doi: 10.1006/abbi.1997.9958. [DOI] [PubMed] [Google Scholar]
- 6.Kolter T., Sandhoff K. Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu. Rev. Cell Dev. Biol. 2005;21:81–103. doi: 10.1146/annurev.cellbio.21.122303.120013. [DOI] [PubMed] [Google Scholar]
- 7.Xu Y.H., Barnes S., Sun Y., Grabowski G.A. Multi-system disorders of glycosphingolipid and ganglioside metabolism. J. Lipid Res. 2010;51:1643–1675. doi: 10.1194/jlr.R003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harzer K., Paton B.C., Christomanou H., Chatelut M., Levade T., Hiraiwa M., O'Brien J.S. Saposins (sap) A and C activate the degradation of galactosylceramide in living cells. FEBS Lett. 1997;417:270–274. doi: 10.1016/s0014-5793(97)01302-1. [DOI] [PubMed] [Google Scholar]
- 9.Li S.C., Sonnino S., Tettamanti G., Li Y.T. Characterization of a nonspecific activator protein for the enzymatic hydrolysis of glycolipids. J. Biol. Chem. 1988;263:6588–6591. [PubMed] [Google Scholar]
- 10.Zschoche A., Furst W., Schwarzmann G., Sanhoff K. Hydrolysis of lactosylceramide by human galactosylceramidase and GM1-beta-galactosidase in a detergent-free system and its stimulation by sphingolipid activator proteins, sap-B and sap-C. Activator proteins stimulate lactosylceramide hydrolysis. Eur. J. Biochem. 1994;222:83–90. doi: 10.1111/j.1432-1033.1994.tb18844.x. [DOI] [PubMed] [Google Scholar]
- 11.Sun Y., Qi X., Grabowski G.A. Saposin C is required for normal resistance of acid beta-glucosidase to proteolytic degradation. J. Biol. Chem. 2003;278:31918–31923. doi: 10.1074/jbc.M302752200. [DOI] [PubMed] [Google Scholar]
- 12.Qi X., Grabowski G.A. Acid beta-glucosidase: intrinsic fluorescence and conformational changes induced by phospholipids and saposin C. Biochemistry. 1998;37:11544–11554. doi: 10.1021/bi980785+. [DOI] [PubMed] [Google Scholar]
- 13.Azuma N., O'Brien J.S., Moser H.W., Kishimoto Y. Stimulation of acid ceramidase activity by saposin D. Arch. Biochem. Biophys. 1994;311:354–357. doi: 10.1006/abbi.1994.1248. [DOI] [PubMed] [Google Scholar]
- 14.Hulkova H., Cervenkova M., Ledvinova J., Tochackova M., Hrebicek M., Poupetova H., Befekadu A., Berna L., Paton B.C., Harzer K., et al. A novel mutation in the coding region of the prosaposin gene leads to a complete deficiency of prosaposin and saposins, and is associated with a complex sphingolipidosis dominated by lactosylceramide accumulation. Hum. Mol. Genet. 2001;10:927–940. doi: 10.1093/hmg/10.9.927. [DOI] [PubMed] [Google Scholar]
- 15.Paton B.C., Schmid B., Kustermann-Kuhn B., Poulos A., Harzer K. Additional biochemical findings in a patient and fetal sibling with a genetic defect in the sphingolipid activator protein (SAP) precursor, prosaposin. Evidence for a deficiency in SAP-1 and for a normal lysosomal neuraminidase. Biochem. J. 1992;285:481–488. doi: 10.1042/bj2850481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuchar L., Ledvinova J., Hrebicek M., Myskova H., Dvorakova L., Berna L., Chrastina P., Asfaw B., Elleder M., Petermoller M., et al. Prosaposin deficiency and saposin B deficiency (activator-deficient metachromatic leukodystrophy): report on two patients detected by analysis of urinary sphingolipids and carrying novel PSAP gene mutations. Am. J. Med. Genet. A. 2009;149A:613–621. doi: 10.1002/ajmg.a.32712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elleder M., Jerabkova M., Befekadu A., Hrebicek M., Berna L., Ledvinova J., Hulkova H., Rosewich H., Schymik N., Paton B.C., et al. Prosaposin deficiency – a rarely diagnosed, rapidly progressing, neonatal neurovisceral lipid storage disease. Report of a further patient. Neuropediatrics. 2005;36:171–180. doi: 10.1055/s-2005-865608. [DOI] [PubMed] [Google Scholar]
- 18.Von Figura K., Gieselmann V., Jaeken J. Metachromatic leukodystrophy. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Basis of Inherited Disease. Vol. 3. New York: McGraw-Hill; 2001. pp. 3695–3724. [Google Scholar]
- 19.Zhang X.L., Rafi M.A., DeGala G., Wenger D.A. Insertion in the mRNA of a metachromatic leukodystrophy patient with sphingolipid activator protein-1 deficiency. Proc. Natl. Acad. Sci. USA. 1990;87:1426–1430. doi: 10.1073/pnas.87.4.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pampols T., Pineda M., Giros M.L., Ferrer I., Cusi V., Chabas A., Sanmarti F.X., Vanier M.T., Christomanou H. Neuronopathic juvenile glucosylceramidosis due to sap-C deficiency: clinical course, neuropathology and brain lipid composition in this Gaucher disease variant. Acta Neuropathol. (Berl) 1999;97:91–97. doi: 10.1007/s004010050960. [DOI] [PubMed] [Google Scholar]
- 21.Rafi M.A., de Gala G., Zhang X.L., Wenger D.A. Mutational analysis in a patient with a variant form of Gaucher disease caused by SAP-2 deficiency. Somat. Cell Mol. Genet. 1993;19:1–7. doi: 10.1007/BF01233949. [DOI] [PubMed] [Google Scholar]
- 22.Tylki-Szymanska A., Groener J.E., Kaminski M.L., Lugowska A., Jurkiewicz E., Czartoryska B. Gaucher disease due to saposin C deficiency, previously described as non-neuronopathic form–no positive effects after 2-years of miglustat therapy. Mol. Genet. Metab. 2011;104:627–630. doi: 10.1016/j.ymgme.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 23.Spiegel R., Bach G., Sury V., Mengistu G., Meidan B., Shalev S., Shneor Y., Mandel H., Zeigler M. A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: first report of saposin A deficiency in humans. Mol. Genet. Metab. 2005;84:160–166. doi: 10.1016/j.ymgme.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 24.Diaz-Font A., Cormand B., Santamaria R., Vilageliu L., Grinberg D., Chabas A. A mutation within the saposin D domain in a Gaucher disease patient with normal glucocerebrosidase activity. Hum. Genet. 2005;117:275–277. doi: 10.1007/s00439-005-1288-x. [DOI] [PubMed] [Google Scholar]
- 25.Fujita N., Suzuki K., Vanier M.T., Popko B., Maeda N., Klein A., Henseler M., Sandhoff K., Nakayasu H. Targeted disruption of the mouse sphingolipid activator protein gene: a complex phenotype, including severe leukodystrophy and wide-spread storage of multiple sphingolipids. Hum. Mol. Genet. 1996;5:711–725. doi: 10.1093/hmg/5.6.711. [DOI] [PubMed] [Google Scholar]
- 26.Matsuda J., Vanier M.T., Saito Y., Tohyama J., Suzuki K. A mutation in the saposin A domain of the sphingolipid activator protein (prosaposin) gene results in a late-onset, chronic form of globoid cell leukodystrophy in the mouse. Hum. Mol. Genet. 2001;10:1191–1199. doi: 10.1093/hmg/10.11.1191. [DOI] [PubMed] [Google Scholar]
- 27.Matsuda J., Kido M., Tadano-Aritomi K., Ishizuka I., Tominaga K., Toida K., Takeda E., Suzuki K., Kuroda Y. Mutation in saposin D domain of sphingolipid activator protein gene causes urinary system defects and cerebellar Purkinje cell degeneration with accumulation of hydroxy fatty acid-containing ceramide in mouse. Hum. Mol. Genet. 2004;13:2709–2723. doi: 10.1093/hmg/ddh281. [DOI] [PubMed] [Google Scholar]
- 28.Sun Y., Ran H., Zamzow M., Kitatani K., Skelton M.R., Williams M.T., Vorhees C.V., Witte D.P., Hannun Y.A., Grabowski G.A. Specific saposin C deficiency: CNS impairment and acid beta-glucosidase effects in the mouse. Hum. Mol. Genet. 2010;19:634–647. doi: 10.1093/hmg/ddp531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun Y., Witte D.P., Ran H., Zamzow M., Barnes S., Cheng H., Han X., Williams M.T., Skelton M.R., Vorhees C.V., et al. Neurological deficits and glycosphingolipid accumulation in saposin B deficient mice. Hum. Mol. Genet. 2008;17:2345–2356. doi: 10.1093/hmg/ddn135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun Y., Witte D.P., Zamzow M., Ran H., Quinn B., Matsuda J., Grabowski G.A. Combined saposin C and D deficiencies in mice lead to a neuronopathic phenotype, glucosylceramide and alpha-hydroxy ceramide accumulation, and altered prosaposin trafficking. Hum. Mol. Genet. 2007;16:957–971. doi: 10.1093/hmg/ddm040. [DOI] [PubMed] [Google Scholar]
- 31.Vaccaro A.M., Salvioli R., Barca A., Tatti M., Ciaffoni F., Maras B., Siciliano R., Zappacosta F., Amoresano A., Pucci P. Structural analysis of saposin C and B. Complete localization of disulfide bridges. J. Biol. Chem. 1995;270:9953–9960. doi: 10.1074/jbc.270.17.9953. [DOI] [PubMed] [Google Scholar]
- 32.Aggarwal S., Yurlova L., Snaidero N., Reetz C., Frey S., Zimmermann J., Pahler G., Janshoff A., Friedrichs J., Muller D.J., et al. A size barrier limits protein diffusion at the cell surface to generate lipid-rich myelin-membrane sheets. Dev. Cell. 2011;21:445–456. doi: 10.1016/j.devcel.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 33.Schochet S.S., Jr, McCormick W.F., Powell G.F. Krabbe's disease. A light and electron microscopic study. Acta Neuropathol. 1976;36:153–160. doi: 10.1007/BF00685277. [DOI] [PubMed] [Google Scholar]
- 34.Ohshima T., Murray G.J., Swaim W.D., Longenecker G., Quirk J.M., Cardarelli C.O., Sugimoto Y., Pastan I., Gottesman M.M., Brady R.O., et al. Alpha-galactosidase A deficient mice: a model of Fabry disease. Proc. Natl. Acad. Sci. USA. 1997;94:2540–2544. doi: 10.1073/pnas.94.6.2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandhoff K., Kolter T., Harzer K. Sphingolipids activator proteins. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Basis of Inherited Disease. Vol. 3. New York: McGraw-Hill; 2001. pp. 3371–3388. [Google Scholar]
- 36.Hess B., Saftig P., Hartmann D., Coenen R., Lullmann-Rauch R., Goebel H.H., Evers M., von Figura K., D'Hooge R., Nagels G., et al. Phenotype of arylsulfatase A-deficient mice: relationship to human metachromatic leukodystrophy. Proc. Natl. Acad. Sci. USA. 1996;93:14821–14826. doi: 10.1073/pnas.93.25.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orvisky E., Park J.K., LaMarca M.E., Ginns E.I., Martin B.M., Tayebi N., Sidransky E. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: correlation with phenotype and genotype. Mol. Genet. Metab. 2002;76:262–270. doi: 10.1016/s1096-7192(02)00117-8. [DOI] [PubMed] [Google Scholar]
- 38.Svennerholm L., Vanier M.T., Mansson J.E. Krabbe disease: a galactosylsphingosine (psychosine) lipidosis. J. Lipid Res. 1980;21:53–64. [PubMed] [Google Scholar]
- 39.Matsuda J., Yoneshige A., Suzuki K. The function of sphingolipids in the nervous system: lessons learnt from mouse models of specific sphingolipid activator protein deficiencies. J. Neurochem. 2007;103(Suppl. 1):32–38. doi: 10.1111/j.1471-4159.2007.04709.x. [DOI] [PubMed] [Google Scholar]
- 40.Igisu H., Suzuki K. Progressive accumulation of toxic metabolite in a genetic leukodystrophy. Science. 1984;224:753–755. doi: 10.1126/science.6719111. [DOI] [PubMed] [Google Scholar]
- 41.Tohyama J., Vanier M.T., Suzuki K., Ezoe T., Matsuda J. Paradoxical influence of acid beta-galactosidase gene dosage on phenotype of the twitcher mouse (genetic galactosylceramidase deficiency) Hum. Mol. Genet. 2000;9:1699–1707. doi: 10.1093/hmg/9.11.1699. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi T., Shinnoh N., Goto I., Kuroiwa Y. Hydrolysis of galactosylceramide is catalyzed by two genetically distinct acid beta-galactosidases. J. Biol. Chem. 1985;260:14982–14987. [PubMed] [Google Scholar]
- 43.Chen Y.Q., Rafi M.A., de Gala G., Wenger D.A. Cloning and expression of cDNA encoding human galactocerebrosidase, the enzyme deficient in globoid cell leukodystrophy. Hum. Mol. Genet. 1993;2:1841–1845. doi: 10.1093/hmg/2.11.1841. [DOI] [PubMed] [Google Scholar]
- 44.Sun Y., Liou B., Ran H., Skelton M.R., Williams M.T., Vorhees C.V., Kitatani K., Hannun Y.A., Witte D.P., Xu Y.H., et al. Neuronopathic Gaucher disease in the mouse: viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 2010;19:1088–1097. doi: 10.1093/hmg/ddp580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chevrier M., Brakch N., Celine L., Genty D., Ramdani Y., Moll S., Djavaheri-Mergny M., Brasse-Lagnel C., Annie Laquerriere A.L., Barbey F., et al. Autophagosome maturation is impaired in Fabry disease. Autophagy. 2010;6:589–599. doi: 10.4161/auto.6.5.11943. [DOI] [PubMed] [Google Scholar]
- 46.Pacheco C.D., Kunkel R., Lieberman A.P. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum. Mol. Genet. 2007;16:1495–1503. doi: 10.1093/hmg/ddm100. [DOI] [PubMed] [Google Scholar]
- 47.Settembre C., Fraldi A., Jahreiss L., Spampanato C., Venturi C., Medina D., de Pablo R., Tacchetti C., Rubinsztein D.C., Ballabio A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008;17:119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- 48.Tamboli I.Y., Hampel H., Tien N.T., Tolksdorf K., Breiden B., Mathews P.M., Saftig P., Sandhoff K., Walter J. Sphingolipid storage affects autophagic metabolism of the amyloid precursor protein and promotes Abeta generation. J. Neurosci. 2011;31:1837–1849. doi: 10.1523/JNEUROSCI.2954-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ihara Y., Morishima-Kawashima M., Nixon R. The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012;2 doi: 10.1101/cshperspect.a006361. pii: a006361. doi: 10.1101/cshperspect.a006361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palmieri M., Impey S., Kang H., di Ronza A., Pelz C., Sardiello M., Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011;20:3852–3866. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 51.Sun Y., Quinn B., Witte D.P., Grabowski G.A. Gaucher disease mouse models: point mutations at the acid beta-glucosidase locus combined with low-level prosaposin expression lead to disease variants. J. Lipid Res. 2005;46:2102–2113. doi: 10.1194/jlr.M500202-JLR200. [DOI] [PubMed] [Google Scholar]
- 52.Sun Y., Ran H., Liou B., Quinn B., Zamzow M., Zhang W., Bielawski J., Kitatani K., Setchell K.D., Hannun Y.A., et al. Isofagomine in vivo effects in a neuronopathic Gaucher disease mouse. PLoS One. 2011;6:e19037. doi: 10.1371/journal.pone.0019037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alvarez-Vasquez F., Sims K.J., Cowart L.A., Okamoto Y., Voit E.O., Hannun Y.A. Simulation and validation of modelled sphingolipid metabolism in Saccharomyces cerevisiae. Nature. 2005;433:425–430. doi: 10.1038/nature03232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.