

Using a previously characterized kinase ribozyme, the authors used a variety of selection and shuffling approaches to obtain a ribozyme that has 30-fold greater activity than any of the ribozymes used to build it.
Keywords: ribozyme, fitness landscape, in vitro selection, synthetic shuffling, synthetic recombination
Abstract
The relationship between genotype and phenotype is often described as an adaptive fitness landscape. In this study, we used a combination of recombination, in vitro selection, and comparative sequence analysis to characterize the fitness landscape of a previously isolated kinase ribozyme. Point mutations present in improved variants of this ribozyme were recombined in vitro in more than 1014 different arrangements using synthetic shuffling, and active variants were isolated by in vitro selection. Mutual information analysis of 65 recombinant ribozymes isolated in the selection revealed a rugged fitness landscape in which approximately one-third of the 91 pairs of positions analyzed showed evidence of correlation. Pairs of correlated positions overlapped to form densely connected networks, and groups of maximally connected nucleotides occurred significantly more often in these networks than they did in randomized control networks with the same number of links. The activity of the most efficient recombinant ribozyme isolated from the synthetically shuffled pool was 30-fold greater than that of any of the ribozymes used to build it, which indicates that synthetic shuffling can be a rich source of ribozyme variants with improved properties.
INTRODUCTION
An important goal in the field of molecular evolution is to better understand the structure and properties of adaptive fitness landscapes. Theoretical models suggest that many properties of such landscapes, such as the total number of fitness peaks and the average number of mutational steps to a local fitness peak, depend primarily on two factors: N, the number of building blocks in a macromolecule, and K, the extent to which these building blocks interact (Kauffman 1993). These models also indicate that if a macromolecule is built of relatively independent building blocks, it can be more efficiently optimized by recombination than by random mutagenesis alone (Holland 1992; Kauffman 1993).
It is well established that proteins can be built of smaller independent units. In many cases, these units correspond to independently folding structural domains, and they sometimes also represent functional units of the protein. Furthermore, effects of single amino acid substitutions in proteins are typically independent of one another, except when the mutated positions are close in the three-dimensional fold of the protein (Wells 1990). For example, when combined, the effects of the subtilisin mutations Q103R and N218S are multiplicative with respect to kcat/Km and additive with respect to the free energy of transition-state stabilization (Chen et al. 1991). More systematic studies suggest that, in at least some cases, the adaptive fitness landscapes of proteins can be relatively smooth (Gregoret and Sauer 1993; Hatley et al. 2003; Shulman et al. 2004; Lunzer et al. 2005; Pal et al. 2005). For instance, mutational effects in 164 mutants of isopropylmalate dehydrogenase (containing various combinations of amino acids at six positions in this protein) are typically independent of one another (Lunzer et al. 2005).
Functional RNA molecules such as ribozymes can also be built of smaller, relatively independent units. For example, the P4-P6 domain of the Group I intron can fold correctly when separated from the rest of the ribozyme (Murphy and Cech 1993; Cate et al. 1996; Doherty and Doudna 1997), and secondary structure elements that can fold and function by themselves are often identified in the context of larger ribozymes by comparative sequence analysis (Ekland and Bartel 1995; Curtis and Bartel 2005). Secondary structure elements can often be broken down into even smaller building blocks. For example, in many cases, the thermodynamic stability of an RNA helix can be predicted by assuming that the contribution of a particular base pair depends only on the identity of the flanking base pairs in the helix (Tinoco et al. 1973; Freier et al. 1986), and effects of both point mutations (Bare and Uhlenbeck 1986; Sampson et al. 1992; Putz et al. 1993) and functional group substitutions (Silverman and Cech 1999) can be independent in the context of larger structured RNAs. At the same time, the fundamental role played by base pairs in RNA structure ensures that mutational effects at some pairs of positions in a functional RNA will be coupled, and mutational effects at groups of positions involved in tertiary interactions such as base triples can also be linked (Tanner et al. 1997). Examples of coupled mutational effects at pairs of positions that are distant in the three-dimensional structure of an RNA have also been reported (Putz et al. 1993).
In this study, we used a method of in vitro recombination called synthetic shuffling (Ness et al. 2002), in combination with in vitro selection, comparative sequence analysis, and site-directed mutagenesis, to investigate two related questions about the adaptive fitness landscape of a previously isolated kinase ribozyme (Curtis and Bartel 2005). First, to what extent are mutational effects in this ribozyme independent of one another (Fig. 1)? Second, as has been demonstrated for proteins (Stemmer 1994a,b; Crameri et al. 1998), is recombination a useful way to search the fitness landscape of this ribozyme for variants with improved properties? Our results indicate that mutational changes at 13 of the 91 pairs of positions in this ribozyme analyzed are strongly correlated, and ∼30 pairs show some evidence of correlation. About half of the strongest correlations could be rationalized in a reference mutational background, and further analysis revealed that, for some pairs of positions, mutational effects were strongly coupled in one sequence background but modestly coupled or even independent in others. Our results also show that recombination can be an effective way to search the adaptive fitness landscape of a ribozyme for variants with improved catalytic properties: the activity of the most efficient ribozyme isolated from the synthetically shuffled pool was 30-fold greater than any of the ribozymes used to build it, with a second-order rate enhancement approaching 1010-fold.
FIGURE 1.

Types of adaptive fitness landscapes. On the left side of each panel, six positions in a hypothetical ribozyme are shown, with links indicating pairs of positions at which mutational effects are coupled. On the right side of each panel, a hypothetical data set is plotted, illustrating the type of relationship that might be observed between the product of the two single-mutant effects (x-axis) and the double-mutant effect (y-axis) on the catalytic rate of the ribozyme for each of the 15 possible pairs of positions. The line in each graph indicates the expected relationship between the product of two single-mutant effects and the double-mutant effect for independent positions. (A) Hypothetical illustration of a smooth adaptive fitness landscape. All 15 pairs of positions are independent, so that the product of the mutational effects at any two positions is always equal to the mutational effect of the double mutant. (B) Hypothetical illustration of an intermediate adaptive fitness landscape. Mutational effects at four pairs of positions are linked and at 11 pairs of positions are independent. (C) Hypothetical illustration of a rugged adaptive fitness landscape. All positions are linked to all other positions, so that the product of two single-mutant effects is rarely equal to the double-mutant effect.
RESULTS
In vitro recombination by synthetic shuffling
Our questions about adaptive fitness landscapes were addressed in the context of a previously described kinase ribozyme called 5-16 (Curtis and Bartel 2005). This ribozyme thiophosphorylates itself at an internal 2′ hydroxyl group using GTPγS as a substrate (Fig. 2A). Its secondary structure consists of a pseudoknotted hairpin containing two asymmetric internal loops, flanked by two less conserved helices that are not required for catalytic activity (Fig. 2B). In addition to the prototype sequence, 22 variants of this ribozyme were previously isolated by random mutagenesis and reselection, some of which had catalytic activities 40-fold greater than that of the parental kinase.
FIGURE 2.

Activity and secondary structure of kinase ribozyme 5-16. (A) The 5-16 ribozyme thiophosphorylates itself at an internal 2′ hydroxyl group using GTPγS as a substrate. (B) Secondary structure and minimized catalytic core of 5-16.
Analysis of these variants revealed several pairs of covarying positions that corresponded to base pairs in the secondary structure of the ribozyme but provided little information about other types of correlations (Curtis and Bartel 2005). This limitation was due in part to the properties of the pool from which these ribozyme variants were isolated: because positions were mutagenized at a relatively low rate of 20% per position, variants were much more likely to retain the nucleotide of the parental ribozyme at a given position than to contain a mutational change. This effect is especially pronounced for interacting positions. For example, consider a base pair at which A–U is present in the initial isolate of the ribozyme, at which a G–C mutational change is neutral, and at which all other mutational changes inactivate the ribozyme. In a pool mutagenized at a rate of 20% per position, 99.3% of the active variants will contain an A–U at this position, whereas only 0.7% will contain the neutral G–C change (Ekland and Bartel 1995). Because neutral changes at correlated positions will occur only rarely in a randomly mutagenized pool, a large number of sequences would be required to establish the significance of correlations, especially at pairs of positions that are only weakly correlated.
To enhance our ability to identify correlated positions in this ribozyme, a pool was synthesized using a method of in vitro recombination called synthetic shuffling (Ness et al. 2002). Variable positions in this pool corresponded to positions that varied among previously isolated ribozyme variants (Curtis and Bartel 2005). For example, at position 11 in the minimized catalytic core of the ribozyme, at which most of our previously isolated ribozyme variants contain a U but two contain a C, the pool was synthesized to encode 50% U and 50% C (Supplemental Fig. 1). A pool synthesized in such a way will contain an equal number of each of the possible recombinants at a given pair of variable positions. Furthermore, because other positions in the pool are variable, each of these recombinants will occur in many different sequence backgrounds. By analyzing the sequences of ribozyme variants isolated from such a pool, we hoped to obtain information about the extent to which mutational changes at different pairs of positions in the ribozyme were correlated in the context of a large number of sequence backgrounds. A second possible advantage of this approach was that, if different mutations were responsible for the improved catalytic activity of ribozyme variants used to design our synthetically shuffled pool, and if the effects of these mutations were independent of one another, combining them into single molecules would generate ribozymes with even greater activities.
For our purposes, synthetic shuffling had several advantages over the more common method of DNA shuffling, in which homologous DNA sequences are randomly fragmented using DNase I and reassembled by mutually primed annealing and PCR amplification (Stemmer 1994a,b; Crameri et al. 1998). First, even nucleotides that are close in the primary sequence of the ribozyme can be readily recombined by synthetic shuffling, whereas the recombination frequency between two positions in a pool generated by DNA shuffling is expected to be proportional to the distance between them. This was especially important considering the small size of the ribozyme used in these experiments, as well as the need to recombine nucleotides that, in some cases, occurred consecutively in the primary sequence. Second, because synthetic shuffling does not rely on DNA annealing, even blocks of sequence linked by highly variable joining regions can be efficiently recombined. Third, the method is extremely straightforward from a technical perspective. Although not a concern for our study, a potential disadvantage of synthetic shuffling relative to DNA shuffling is that ribozymes cannot be recombined unless their sequences are first known.
Pool synthesis and in vitro selection
Our synthetically shuffled pool consisted of two subpools. Subpool A incorporated sequence variation from both the catalytic core (P2-P4) and flanking region (P1 and P5) of six of the most efficient kinase variants previously isolated by random mutagenesis and reselection (Curtis and Bartel 2005). Subpool B incorporated mutations from the minimized catalytic core (P2-P4) of each of 22 previously described kinase variants (Curtis and Bartel 2005), as well as mutations from P1 and P5 of two of these variants. The two subpools were mixed in equal amounts to generate the starting pool, which contained, on the average, nine copies of every possible sequence encoded by each subpool.
To isolate kinase ribozymes from this pool, RNA was incubated with GTPγS (Lorsch and Szostak 1994; Curtis and Bartel 2005), and molecules that became thiophosphorylated during the incubation were isolated on N-acryloylaminophenylmercuric chloride (APM) polyacrylamide gels (Igloi 1988; Unrau and Bartel 1998). These molecules were then amplified by RT-PCR and transcribed to generate RNA for the next round of selection. After three rounds, the pool catalyzed self-thiophosphorylation as efficiently as the fastest kinase ribozymes used in its design, and after two or three more rounds of selection, molecules from the pool were cloned and sequenced. Most of the isolated sequences were from subpool A, and we restricted our comparative sequence analysis to members of this subpool.
Of the subpool A sequences tested, ∼70% were catalytically active, corresponding to 65 different ribozyme variants. These ribozymes typically differed from one another at 15 to 30 positions, and the most closely related variants differed from one another by at least five mutations, indicating that most or all were independently derived. Although the primary sequences of these ribozymes could be readily aligned (Supplemental Fig. 1), an unambiguous alignment of secondary structure elements could only be generated for portions of the sequence corresponding to the 50-nt minimized catalytic core of the ribozyme (Fig. 3). For this reason, we focused our analysis on the 14 variable positions, and (14 × 13)/2 = 91 possible pairs of positions in this portion of the sequence (Fig. 4A).
FIGURE 3.

Active variants of a synthetically shuffled kinase ribozyme. Only the catalytic core of the ribozyme (containing the 14 positions analyzed by mutual information) is shown. Colored bars indicate paired regions in the ribozyme secondary structure. Numbers at the top of the alignment indicate shuffled positions and correspond to those in Figure 4A. Based on their frequencies in the synthetically shuffled pool, substitutions shown in red were predicted to increase the efficiency of the initial ribozyme isolate, and substitutions shown in pink were predicted to decrease the efficiency of the initial ribozyme isolate. Boxes indicate substitutions that were not encoded in the pool but likely occurred during RT-PCR. Ribozymes are arranged in approximate order of activity when assayed at 30 µM GTPγS. See Supplemental Figure 1 for a full alignment of these sequences.
FIGURE 4.

Network of correlated positions in the kinase ribozyme. (A) Secondary structure of the minimized catalytic core of the ribozyme. Positions that were variable in the synthetically shuffled pool are numbered. (B) Number of correlated pairs of positions in the ribozyme variants observed at the indicated cutoffs for significance of mutual information values (O, red bars). For comparison, the expected number of correlations in a randomly shuffled sequence alignment is also plotted (E, blue bars), with the number of correlated pairs observed in excess over those expected by chance indicated (numbers above bars). (C) Heat map showing the significance of mutual information correlations for each of the 91 pairs of analyzed positions in the ribozyme alignment. (D) Number of correlated pairs of positions in the starting pool observed at the indicated cutoffs for significance of mutual information values. Otherwise, as in panel B. (E) Heat map showing the significance of mutual information correlations for each of the 91 pairs of analyzed positions in an alignment of sequences from the starting pool. (F) Network of correlated positions among the 14 synthetically shuffled nucleotides in the minimized catalytic core of the ribozyme. Lines indicate 13 pairs of positions with mutual information values expected to occur no more than once in every 100 randomized control alignments (P ≤ 0.01). (G) Same as panel F, except lines indicate 26 pairs of positions with mutual information values expected to occur no more than once in every 20 randomized control alignments (P ≤ 0.05). (H) Number of 3-nt modules in the ribozyme network observed at the indicated cutoffs for significance of mutual information values (O, red bars). For comparison, the expected number of 3-nt modules in a randomly shuffled network with the same number of links is also plotted (E, blue bars), with the number of modules observed in excess over those expected by chance indicated (numbers above bars).
Identification of correlated pairs of positions using mutual information
To identify correlated positions in our alignment of synthetically shuffled ribozyme variants, mutual information values were calculated for each of the 91 possible pairs of positions in the alignment. Mutual information indicates the extent to which the nucleotide identity at one position in a sequence alignment provides information about the nucleotide at a second position (Chiu and Kolodziejczak 1991; Gutell et al. 1992) and has been previously used to identify correlated positions in both functional RNAs (Gutell et al. 1986, 1992; Chiu and Kolodziejczak 1991; Gautheret et al. 1995; Brown et al. 1996) and proteins (Martin et al. 2005). To assess the significance of these mutual information values, 1000 control alignments were generated in which the order of nucleotides in each variable column of the ribozyme alignment was randomly shuffled while preserving nucleotide composition. Because average mutual information values derived from these control alignments varied by up to 12-fold at different pairs of positions, a separate cutoff for significance was determined for each pair of positions analyzed. To assess whether pairs of positions might be correlated due to biases during pool synthesis, mutual information values were also determined for these 91 pairs of positions in an alignment of 89 sequences from the starting pool.
This analysis revealed a high degree of correlation between different positions in the ribozyme. For example, using as a cutoff for significance a mutual information value that occurred only once in every 100 pairs from shuffled control alignments, our analysis revealed that mutational changes at 13 pairs of positions in the ribozyme were significantly correlated (Fig. 4B,C; Table 1; Supplemental Table 1). In contrast, parallel analysis of the 89 sequences of the starting pool indicated that none of these 91 pairs were correlated at this significance cutoff (Fig. 4D,E). Although using such a cutoff was expected to yield less than one false positive for 91 pairs of positions analyzed (Fig. 4B), it was also expected to underestimate the true number of correlated pairs of positions. By analyzing the data using a spectrum of cutoffs for significance of mutual information values and subtracting the expected number of false positives, we estimate that mutational changes at ∼30 out of 91 pairs of positions analyzed were correlated in at least some sequence backgrounds (Fig. 4B).
TABLE 1.
Expected and observed dinucleotide frequencies for the 13 most strongly correlated pairs of analyzed positions in the kinase ribozyme
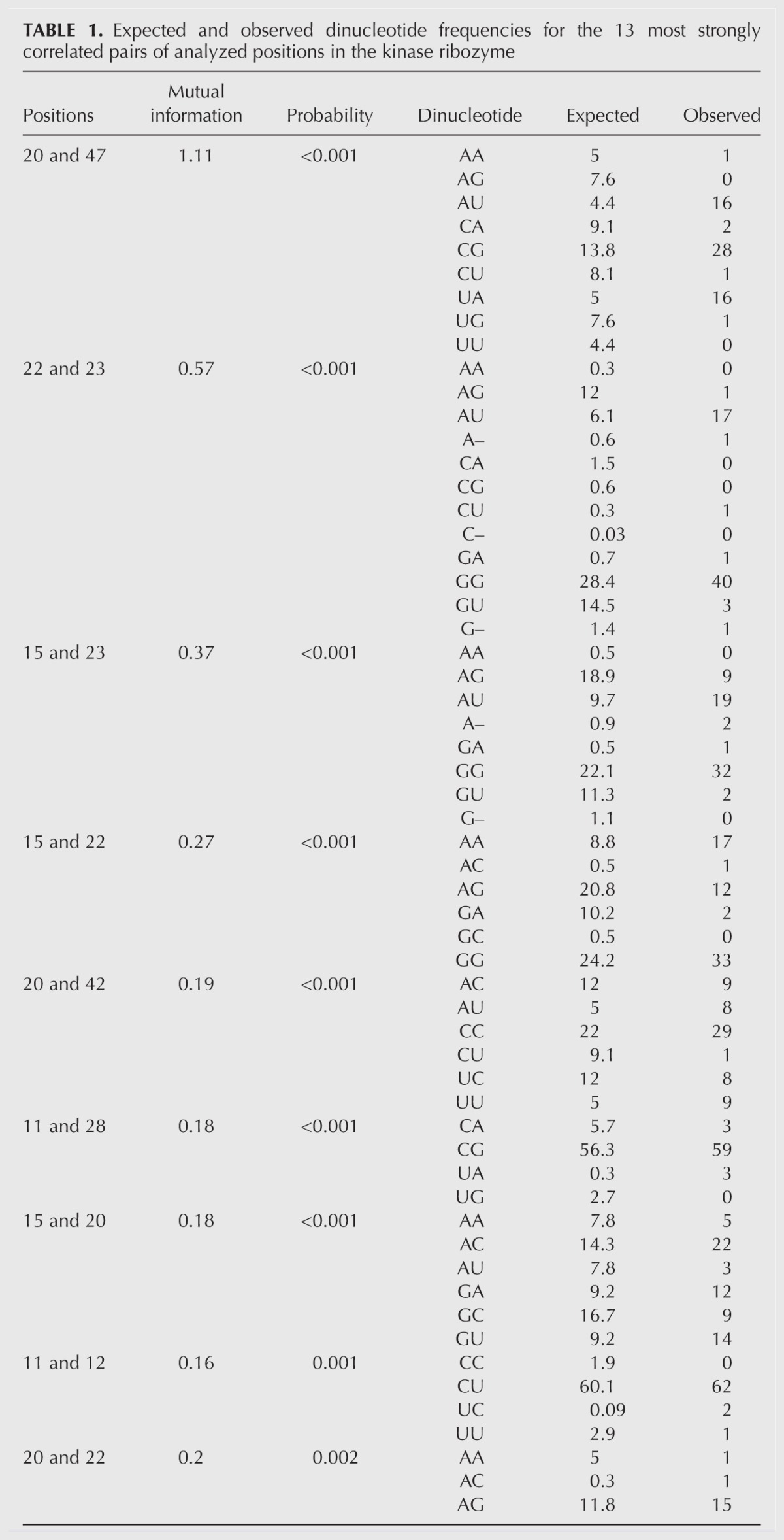
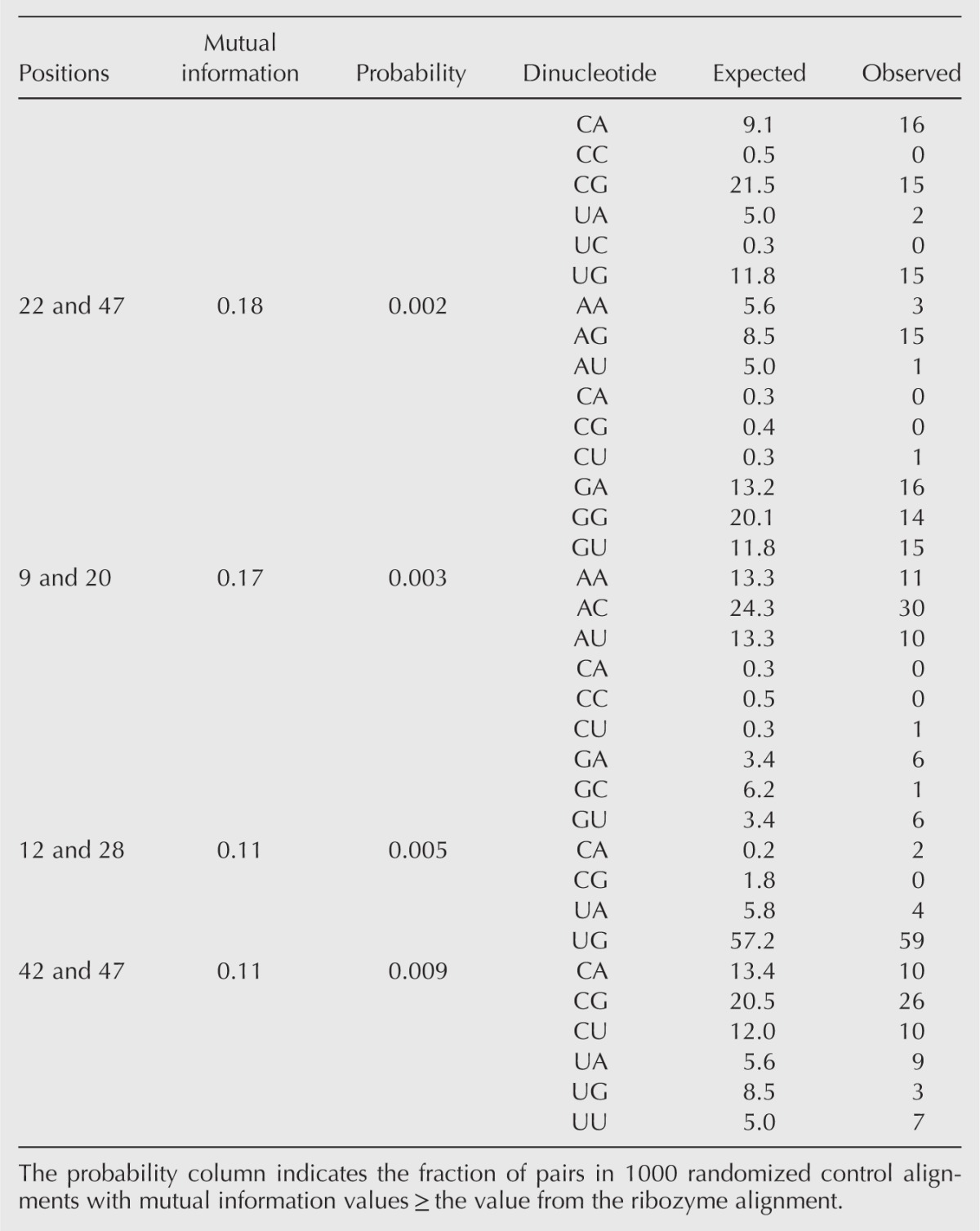
To search for higher-order relationships among correlated pairs of positions, we investigated the extent to which pairs overlapped to form networks of linked positions. Such overlapping pairs were observed at all mutual information significance cutoffs examined. For example, using a 0.01 cutoff for significance, seven of the 14 variable positions in the ribozyme catalytic core formed a network in which all positions were linked, either directly or indirectly, to all other positions (Fig. 4F). Increasing the significance cutoff to 0.05 increased the connectivity of this network such that it included 13 of the 14 variable positions in the ribozyme (Fig. 4G). Examination of these networks also revealed groups of positions (“modules”), in which all positions were directly linked to all other positions. Modules of 3 nt (i.e., 15–20–22) were most abundant, although at less stringent cutoffs modules of 4 (i.e., 9–20–42–47) and 5 nt (i.e., 15–20–22–23–47) were also observed (Fig. 4G). Modules were significantly more abundant in ribozyme networks than they were in randomized control networks with the same number of links. For example, five different 3-nt modules were observed in the ribozyme network generated using a 0.01 significance cutoff (Fig. 4F). In contrast, networks containing five or more 3-nt modules were never observed in 500 randomized control networks. By performing our analysis using a spectrum of cutoffs for significance of mutual information values and subtracting the expected number of false positives, we estimate that the correlation network of this ribozyme contains ∼20 modules of 3 nt (Fig. 4H).
Analysis of correlations by site-directed mutagenesis
To further assess the significance of these correlations, we investigated the extent to which they could be confirmed using site-directed mutagenesis. For each pair of positions tested, the effects of point mutations on self-thiophosphorylation activity of the ribozyme were determined at each individual position and compared to the double-mutant effect (Carter et al. 1984; Wells 1990; Mildvan et al. 1992; Horovitz 1996). Rates were calculated from reactions performed at subsaturating GTPγS concentrations (Fig. 5A), and thus the relative rates reflected relative reaction efficiencies (analogous to kcat/Km for multiturnover reactions). The departure from independence, D, was defined as the product of the two single-mutant effects divided by the double-mutant effect. For independent positions, the double-mutant effect should equal the product of the two single-mutant effects (D = 1), whereas for correlated positions the double-mutant effect could be either larger (D < 1) or smaller (D > 1) than that predicted from the single-mutant effects.
FIGURE 5.

Testing whether correlations indicate functionally coupled nucleotides. (A) Plot of observed rates of the reference ribozyme as a function of substrate concentration, showing that 30 µM GTPγS is below saturation for this construct. (B) APM polyacrylamide gel showing the effects of the 15A to 15G, 22A to 22G, and 15A-22A to 15G-22G mutations. Mutational effects at these positions were strongly coupled, with a D value of 170 in the mutational background used here. Reactions were performed in ribozyme selection buffer, in the presence of 1 µM ribozyme and 30 µM GTPγS. (C) The relationship between the product of the two single-mutant effects (x-axis) and the double-mutant effect (y-axis) for the 13 most strongly correlated pairs of positions (red diamonds; mutual information significance values ≤ 0.01) and 16 less strongly correlated pairs (blue diamonds; mutual information significance values > 0.01). The line indicates the expected relationship if the product of two single-mutant effects always equaled the double-mutant effect. Sequences and relative efficiencies of mutants used to calculate these D values are given in Supplemental Table 2.
Mutational effects were initially examined in the context of the reference ribozyme shown in Figure 4A. This construct lacked P1, P5, and both primer-binding sites, and it contained six mutations that occurred frequently among the synthetically shuffled ribozymes isolated in the selection. This ribozyme was chosen as a reference for three reasons. First, it was among the most efficient ribozyme variants generated in these experiments, which enabled characterization of mutants with efficiencies reduced by up to 104-fold. Second, using a construct that lacked P1 and P5, which are not essential for ribozyme function (Curtis and Bartel 2005) and could not be readily aligned in our alignment of synthetically shuffled kinase ribozymes, directed our focus to correlations in the most functionally important and well-understood part of the ribozyme. Third, we anticipate that the relatively small size of this construct might facilitate future structural studies that would enable interpretation of these correlations in the context of a three-dimensional fold.
Of the 13 most highly correlated pairs of positions identified by mutual information analysis, mutational effects of six were strongly coupled when analyzed in our reference mutational background, with D values ranging from ∼102 to 105 (Fig. 5B,C; Table 2). Two of these pairs (11–28 and 20–47) corresponded to base pairs in the secondary structure of the ribozyme, two (11–12 and 12–28) involved correlations between neighboring base pairs in a helix, and two (15–22 and 15–23) represented previously undetected interactions between opposite sides of a loop (Fig. 4A). Mutational effects at the remaining pairs tested in this mutational background were either modestly coupled (four pairs) or independent (three pairs) (Fig. 5C; Table 2). D values were also determined for 16 pairs of positions with mutual information values below our cutoff for significance. Although several of these pairs were modestly coupled, most were independent, and this independence spanned more than a 1000-fold range in self-thiophosphorylation efficiencies (Fig. 5C; Table 3).
TABLE 2.
D values for the 13 most strongly correlated pairs of analyzed positions in the kinase ribozyme
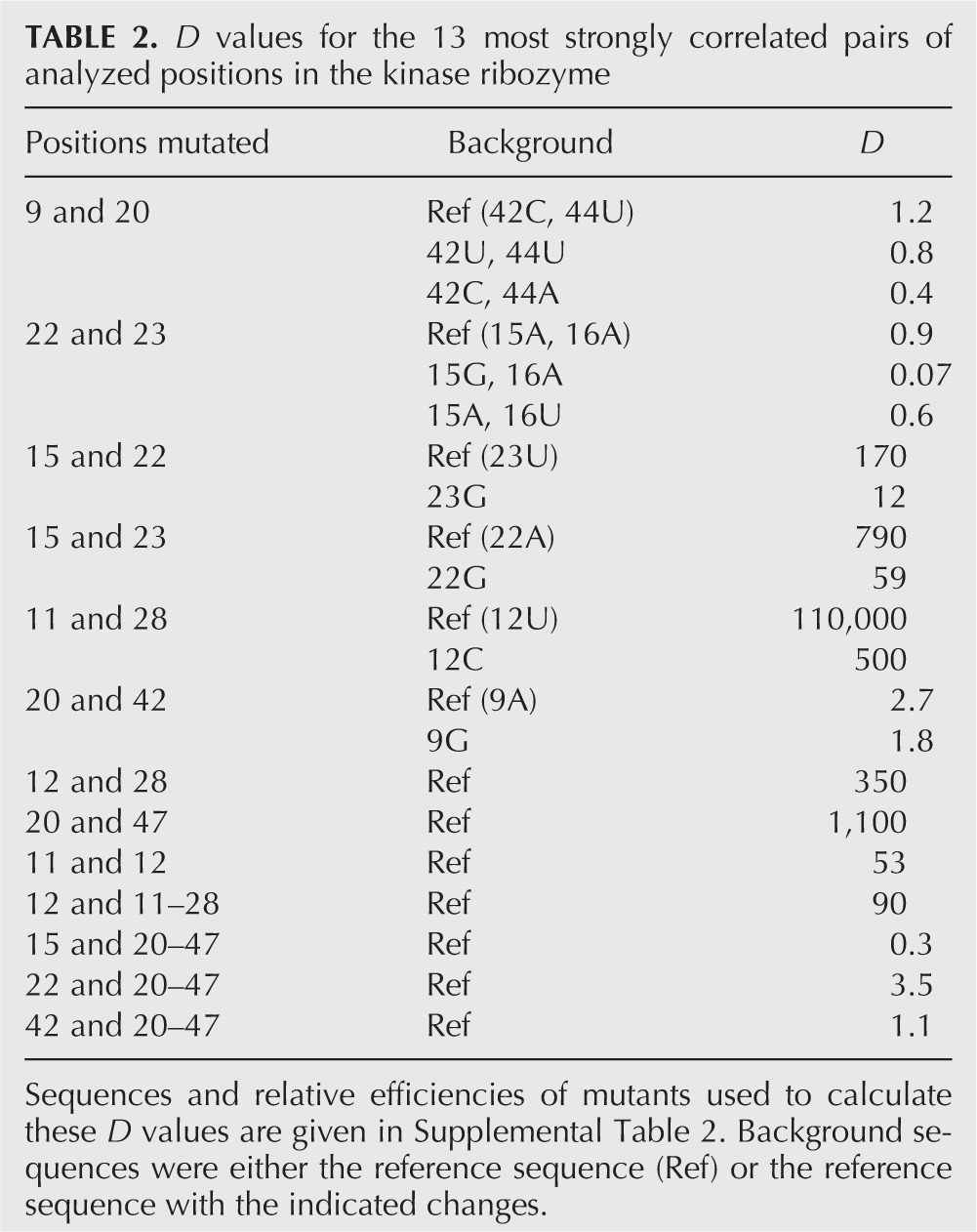
TABLE 3.
D values for 16 pairs of analyzed positions in the kinase ribozyme that were not correlated at a significant level
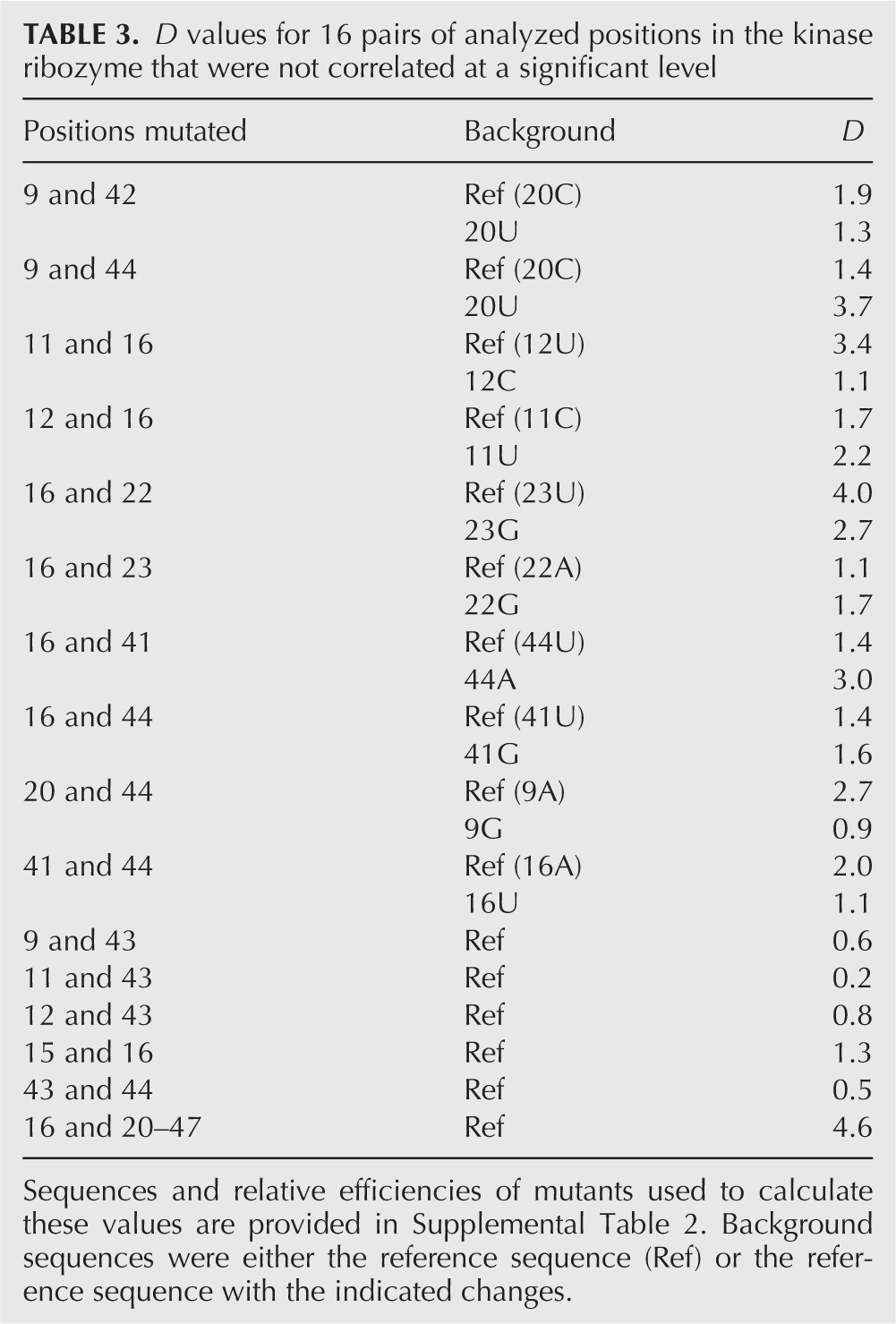
Although mutational effects at about half of the most highly correlated pairs of positions were strongly coupled, the number of correlations that could not be experimentally confirmed was higher than expected. Furthermore, D values predicted from the sequence alignment often differed considerably from those measured by mutagenesis. For example, at positions 15 and 23, AU occurred 19 times, GU occurred two times, AG occurred nine times, and GG occurred 32 times (Table 1). Based on these frequencies, the predicted D value for the 15–23 pair (using AU as the reference) was (19/2) × (19/9)/(19/32) = 34, but the value determined by site-directed mutagenesis was more than 20-fold higher (Table 2). One possible explanation for these observations is that D values for certain pairs can be strongly dependent on differences at additional positions in the ribozyme. To test this directly, D values for six of the most strongly correlated pairs were tested in alternative mutational backgrounds. In each case, the alternative background differed from the reference background at a single position that was correlated with at least one of the positions in the pair being tested. For example, because positions 15 and 22 were both correlated with position 23 according to mutual information analysis, the D value for the 15–22 pair was determined in both the reference 23U and alternative 23G backgrounds. In four of six cases, D values were strongly dependent on mutational background, and for one example, the D value changed by more than 100-fold (Table 2). This analysis also revealed several pairs of positions for which mutational effects were independent in the original background tested but either strongly coupled (22 and 23) or modestly coupled (9 and 20) in a second mutational background (Table 2). As a negative control, D values for six pairs of uncorrelated positions were also determined in a second mutational background. For these six examples, D values did not significantly change when measured in the alternative background (Table 3).
Combining advantageous mutations can produce more efficient ribozymes
Although the self-thiophosphorylation efficiencies of the ribozymes used to build our synthetically shuffled pool were up to 40 times greater than that of the initial isolate, the mutations responsible for the increased activity of these variants were not known, and the extent to which combining them into a single molecule would generate a more efficient ribozyme was not clear. To address these questions, we first investigated whether ribozymes isolated from the synthetically shuffled pool were more active than the ribozymes used to build it. This turned out to be the case: Improved ribozymes derived from both subpools were identified. The most efficient of these ribozymes (derived from subpool B) was 30 times more efficient than any of the ribozymes used to build the pool and 1300 times more efficient than the initial isolate (Fig. 6A), with a kcat/Km of 880 M−1 min−1 (adopting these multiple-turnover parameters for this single-turnover, self-thiophosphorylation reaction). Under optimized conditions, this ribozyme was even more efficient, with kcat of 0.4 min−1, Km of 30 μM, and kcat/Km of 1.3 × 104 M−1 min−1 (Fig. 6A,B).
FIGURE 6.

Isolation of improved ribozymes by synthetic shuffling. (A) APM polyacrylamide gel showing time course of 5-16 (the initial isolate of the kinase ribozyme), A-25 (one of the most efficient ribozymes used to build the synthetically shuffled pool), and Rec 7-5 (one of the most efficient ribozymes isolated from the synthetically shuffled pool). Reaction conditions were 0.2 µM ribozyme, 0.2 µM each blocking oligonucleotide (when necessary), 10 µM GTPγS, and ribozyme selection buffer. The reaction of Rec 7-5 in an optimized buffer (30 mM CaCl2, 200 mM KCl, 100 mM HEPES, pH 7.2) is also shown at the far right. (B) Michaelis-Menten parameters for the Rec 7-5 ribozyme. Reactions included 0.2 µM ribozyme and 0.2 µM each blocking oligonucleotide in optimized ribozyme selection buffer (30 mM CaCl2, 200 mM KCl, 100 mM HEPES, pH 7.2). (C) APM polyacrylamide gel showing the effects of the 11U-12C-28A to 11C-12U-28G and 16U-20A-47U to 16A-20C-47G substitution either individually or in combination. The 11C-12U-28G substitution was derived from the A-25 ribozyme, and the 16A-20C-47G substitution was derived from the A-24 ribozyme. Reactions were performed in ribozyme selection buffer, in the presence of 1 µM ribozyme and 30 µM GTPγS.
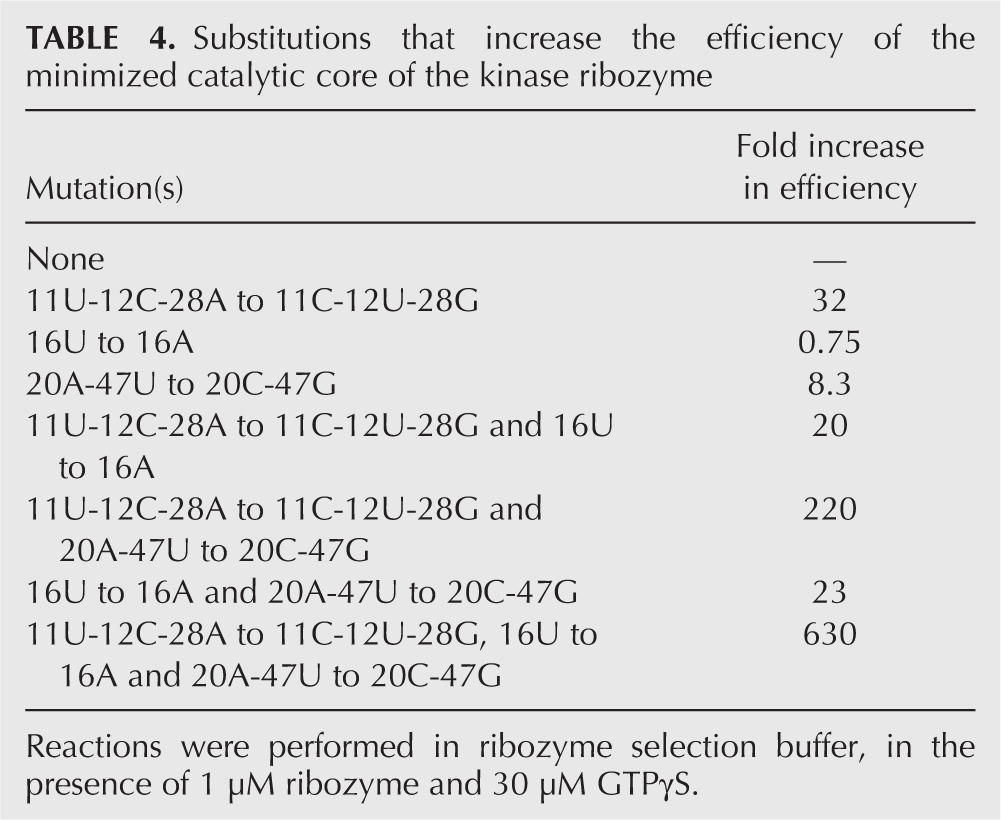
We next set out to identify some of the mutations responsible for the increased activity of the isolated ribozymes and to directly show that combining them would produce even more active ribozymes. At nine out of 14 variable positions in the minimized core of the ribozyme, mutational changes (relative to the sequence of the initial isolate) were identified that were more common than expected based on the composition of the starting pool (shaded in red in Fig. 3). The advantageous effects of six of these changes (derived from two different parental ribozymes used to build the pool) could be verified by site-directed mutagenesis. In the context of a minimized version of the initial isolate of the ribozyme, the 11U-12C-28A to 11C-12U-28G change (derived from the A-25 ribozyme used to build the pool) increased ribozyme efficiency 32-fold (Fig. 6C; Table 4). The 16U-20A-47U to 16A-20C-47G change (derived from the A-24 ribozyme used to build the pool) increased ribozyme efficiency 23-fold (Fig. 6C; Table 4). The mutational effects of the 11U-12C-28A to 11C-12U-28G and 16U-20A-47U to 16A-20C-47G changes were independent of one another: the efficiency of a ribozyme in which these changes were combined was more than 600-fold greater than that of the initial isolate of this ribozyme (Fig. 6C; Table 4). Mutational effects of other combinations of mutations at positions 11-12-28, 16, and 20-47 were also generally independent (Table 4).
TABLE 4.
Substitutions that increase the efficiency of the minimized catalytic core of the kinase ribozyme
Several other mutations that were predicted to increase the activity of the ribozyme could not be rationalized by site-directed mutagenesis. Out of 65 ribozymes selected from the synthetically shuffled pool, 31 contained the 15G-22G-23G sequence (Fig. 3), yet this change was deleterious in several different sequence backgrounds, as well as in the context of the full-length ribozyme. One possible explanation for this finding is that this mutational change only increases the activity of the ribozyme in the presence of additional substitutions in P1 and/or P5. Because this portion of the sequence could not be reliably aligned, such substitutions could not be identified. Alternatively, this mutational change might increase the fitness of ribozyme variants in the context of a selection (for example, by making them better templates for RT-PCR amplification) without increasing their thiophosphorylation efficiency.
Analysis of the sequence alignment of kinase ribozymes isolated in this selection also revealed several mutations likely to be deleterious based on their frequencies (shown in pink in Fig. 3), although the effects of these mutations were not tested by site-directed mutagenesis. Taken together, these results suggest that the ribozymes used to build the recombined pool, which were previously isolated from a randomly mutagenized pool (Curtis and Bartel 2005), contained both beneficial and deleterious mutations and that synthetic recombination followed by in vitro selection was a useful optimization strategy for at least two reasons: it combined beneficial mutations from different ribozyme variants into single molecules, and it removed deleterious mutations from these ribozymes.
DISCUSSION
One of the questions we set out to address in this study was whether the adaptive fitness landscape of a previously isolated kinase ribozyme was smooth or rugged (Fig. 1). Our results indicate that this fitness landscape is more rugged than might have been expected. Approximately 30 of the 91 pairs of positions examined showed some evidence of correlation as determined by mutual information analysis, and many of these pairs overlapped to form networks in which each position was correlated, either directly or indirectly, to all other positions in the network (Fig. 4). This number of correlations is significantly greater than either the two (11–28 and 20–47) expected based on the secondary structure alone, or the four (11–12, 11–28, 12–28 and 20–47) expected based on a nearest-neighbor model in which positions in a base pair are also correlated with positions in flanking base pairs (Tinoco et al. 1973; Freier et al. 1986). Correlated pairs of positions also occurred more frequently in this ribozyme than they typically do in proteins (Gregoret and Sauer 1993; Lunzer et al. 2005; Pal et al. 2005).
Of the 13 most strongly correlated pairs of positions identified by mutual information analysis, seven also displayed strongly coupled effects when mutated (Fig. 5C; Table 2). Our conclusions with regard to the remaining six correlations were less clear. Although correlations of this strength were unlikely to occur by chance and did not occur in the starting pool, mutational effects at these pairs were either weakly coupled (five pairs) or independent (one pair) and could not be differentiated from mutational effects at pairs of positions with lower mutual information values (Fig. 5C; Tables 2, 3). Although we cannot rule out the possibility that some of these correlations are unrelated to ribozyme function but instead represent interactions that contributed to the overall fitness of a ribozyme during the selection, we suggest that many reflect constraints that are important only in certain sequence backgrounds. In one case, we have shown this higher-order dependency directly: in the reference (15A-16A) and 15A-16U backgrounds, mutational effects at positions 22 and 23 were independent, whereas in the 15G-16A background, mutational effects were strongly coupled (Table 2). This idea is also consistent with our finding that D values can be extremely sensitive to mutational background (Table 2), as well as with results from several other studies. For example, in some cases, pairs of functional groups in RNA and DNA hairpins interact only in the presence of a functional group at a third site (Moody and Bevilacqua 2003; Moody et al. 2004), and mutations that are stabilizing in one sequence background can be destabilizing when tested in a different background (Guo et al. 2006). Background-specific effects have also been observed for RNA tertiary interactions such as base triples (Gautheret et al. 1995).
Mutational effects at pairs of positions in this ribozyme could be coupled for several reasons. The simplest interpretation is that each correlation reflects a direct physical interaction between two positions, although this does not necessarily imply an interaction in the catalytically active fold of the ribozyme. For example, correlations might represent favorable interactions that occur only during ribozyme folding. Correlations might also reflect selection against nonproductive interactions that would otherwise occur during or after folding. Of the seven correlated positions at which mutational effects were strongly coupled, four could be rationalized in terms of the existing secondary structure model of the ribozyme. These correlations corresponded to either base pairs (11–28 and 20–47) or interactions between neighboring base pairs (11–12 and 12–28). Three other pairs (15–22, 15–23, and 22–23) provided evidence for a previously undetected interaction between opposite sides of a loop. Of the remaining correlations, the 15–20, 15–47, 20–22, 22–47, 20–23, and 23–47 pairs were the most compelling. Each of these pairs provided evidence for a link between the two parts of the 15–20–22–23–47 module that are distant in the secondary structure of the ribozyme (Fig. 4A, G), and mutational effects at the four pairs that were tested were modestly coupled (Table 2).
A second goal of these experiments was to determine whether homologous recombination could be used to search the adaptive fitness landscape of this ribozyme for molecules with optimized properties. Although previous studies have shown that functional RNAs isolated from random sequences can typically be optimized using a combination of random mutagenesis and reselection (Ellington and Szostak 1990; Ekland and Bartel 1995), methods to further optimize these RNAs have not been extensively explored. In one approach, diversity was produced by fusing an RNA ligase ribozyme with weak polymerase activity to a random-sequence accessory domain. A more efficient and general polymerase was successfully isolated from the resulting pool (Johnston et al. 2001). In another study, a technique called nonhomologous random recombination was used to generate a pool of topological variants of a DNA aptamer to streptavidin (Bittker et al. 2002). The affinity of one of these variants toward streptavidin was 46-fold greater than that of the aptamer used to build the pool. Nonhomologous random recombination was also used to generate a pool of topological variants of a nucleotide synthase ribozyme (Wang and Unrau 2005). Although the ribozyme variants isolated from this pool were not faster than the ribozyme used to build it, some were considerably smaller. It is also known that recombination between different aptamer domains can generate molecules with the binding properties of both parental aptamers (Burke and Willis 1998), and recombination by random-priming was used in combination with several other approaches to improve the activity of an RNA polymerase ribozyme (Wochner et al. 2011). In this study, we found that recombination of point mutations by synthetic shuffling was an effective way to further improve a kinase ribozyme that had been previously improved by random mutagenesis and reselection: the activity of the most efficient kinase ribozyme isolated was 30-fold greater than that of the most efficient kinase used to build the synthetically shuffled pool and 1300-fold faster than that of the initial isolate (Fig. 6A). We anticipate that the method described here will provide a useful way to optimize other functional RNA molecules, and when used in combination with high-throughput sequencing (Pitt and Ferre-D’Amare 2010), mutual information analysis, and site-directed mutagenesis, it has the potential to provide additional information about RNA adaptive fitness landscapes.
MATERIALS AND METHODS
Pool synthesis
Our synthetically shuffled pool was generated from two smaller pools, called subpool A and subpool B. Subpool A incorporated mutations from kinase variants A-29, A-25, A-24, A-7, A-8, and A-1 (Curtis and Bartel 2005) and had the sequence 5′-GGAUGCCUGGUAAAGDRMGAKDAUACUACCCGRGYYGGRDGCAHGRKCACGRCAUAAUCGGUAGKYWHGCDUGCRYKSCHYDSKWYAHGBWGCAGVDDCYUCBBAAGUCAAUAGCCUAGGG-3′. Subpool B incorporated mutations from the minimized catalytic core (P2-P4) of each of 22 previously described kinase variants (Curtis and Bartel 2005), as well as mutations from P1 and P5 of A-29 and A-25, and had the sequence 5′-GGAUGCCUGGUAAAGKRMGAGKAUACUACCCGVGYYGGRNGSMNSDKCACGRCAUWRUCGGUAGDHHHRYDKRYRCKSCMYWSUUYAUGUAGCAGVWRCYUCBSAAGUCAAUAGCCUAGGG-3′ (R = A or G; Y = C or U; M = A or C; K = G or U; S = G or C; W = A or U; H = A, U or C; B = G, U or C; V = G, A or C; D = G, A or U; N = G, A, C or U; primer binding sites shown in bold, and connected to the pool by AA linkers). The two subpools were mixed in equal amounts to generate the starting pool, which contained, on the average, nine copies of every possible sequence encoded by each subpool.
In vitro selection
In vitro selection was as previously described (Curtis and Bartel 2005). Briefly, pool RNA was incubated with GTPγS, and molecules that became thiophosphorylated during the incubation were isolated on APM polyacrylamide gels, amplified by RT-PCR, and transcribed to generate RNA for the next round of selection. In round 1, the pool was incubated for 5 min at 100 µM GTPγS; in rounds 2 and 3, the pool was incubated for 1 min at 100 µM GTPγS; in rounds 4 and 5, the pool was incubated for 6 sec at 100 µM GTPγS; and in round 6, the pool was incubated for 6 sec at 10 µM GTPγS. As was the case in the reselection of 5-16 following random mutagenesis (Curtis and Bartel 2005), pool RNA was not dephosphorylated before each round of selection.
Ribozyme sequences
The sequences of active ribozymes isolated from subpool A are shown in Supplemental Figure 1. The sequence of Rec 7-5, which was isolated from subpool B, is GGAUGCCUGGUAAAGUACGAGGAUACUACCCGCGUUGGAUGCAUCAACACGGCAUUAUCCGAUAGGUUUAUUGAUACAGCACUGUUUAUGUAGCAGAUACUUCCGAAGUCAAUAGCCUAGGG.
Mutual information analysis
A sequence alignment containing 65 synthetically shuffled kinase ribozymes derived from subpool A was used for this analysis. Each of these ribozymes appeared to be independently derived (most sequences differed at 15 to 30 positions), and at 30 µM GTPγS, most had reaction rates within 50-fold of the fastest ribozyme isolated. An unambiguous alignment of secondary structure elements could be generated only for portions of the sequence corresponding to the minimized catalytic core of the ribozyme (P2-P4), and we focused our analysis on the 14 variable positions and 91 possible pairs of positions in this portion of the sequence. Mutual information values for each of these 91 pairs were calculated as previously described (Gorodkin et al. 1999). To determine the significance of mutual information, values from the ribozyme alignment were compared to values from 1000 control alignments, which were generated by randomly shuffling the order of nucleotides in each column in the ribozyme alignment. Because average mutual information values derived from these control alignments varied by up to 12-fold at different pairs of positions, a separate cutoff for significance of mutual information values was determined for each pair of positions analyzed.
Kinetic analysis
Ribozyme reactions were performed as previously described (Curtis and Bartel 2005). The 65 synthetically shuffled kinase ribozymes isolated in the selection were characterized at 1 µM ribozyme, 1 µM each blocking oligonucleotide, and 30 µM GTPγS, in ribozyme selection buffer (10 mM MgCl2, 5 mM CaCl2, 200 mM KCl, 100 mM HEPES, pH 7.2). Rates of the 49 mutants used to determine D values were measured at 1 µM ribozyme and 30 µM GTPγS. Pilot experiments indicated that 30 µM GTPγS was subsaturating under these conditions. Rates were typically determined from at least four points in the initial phase of the reaction using formula (1), where k is the observed rate constant and t is time.
Due to the slow rates of some mutants, rates were not normalized to the fraction of reacted ribozyme. On the average, rates changed by less than twofold when measured using different ribozyme preparations.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank U. Muller and M. Lawrence for comments on this manuscript, T. Jenkins for programming assistance, and members of the Bartel lab for helpful discussions. This work was supported by grant GM061835 from the National Institutes of Health.
REFERENCES
- Bare LA, Uhlenbeck OC 1986. Specific substitution into the anticodon loop of yeast tyrosine transfer RNA. Biochemistry 25: 5825–5830 [DOI] [PubMed] [Google Scholar]
- Bittker JA, Le BV, Liu DR 2002. Nucleic acid evolution and minimization by nonhomologous random recombination. Nat Biotechnol 20: 1024–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JW, Nolan JM, Haas ES, Rubio MA, Major F, Pace NR 1996. Comparative analysis of ribonuclease P RNA using gene sequences from natural microbial populations reveals tertiary structural elements. Proc Natl Acad Sci 93: 3001–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke DH, Willis JH 1998. Recombination, RNA evolution, and bifunctional RNA molecules isolated through chimeric SELEX. RNA 4: 1165–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter PJ, Winter G, Wilkinson AJ, Fersht AR 1984. The use of double mutants to detect structural changes in the active site of the tyrosyl-tRNA synthetase (Bacillus stearothermophilus). Cell 38: 835–840 [DOI] [PubMed] [Google Scholar]
- Cate JH, Gooding AR, Podell E, Zhou K, Golden BL, Kundrot CE, Cech TR, Doudna JA 1996. Crystal structure of a group I ribozyme domain: Principles of RNA packing. Science 273: 1678–1685 [DOI] [PubMed] [Google Scholar]
- Chen KQ, Robinson AC, Van Dam ME, Martinez P, Economou C, Arnold FH 1991. Enzyme engineering for nonaqueous solvents. II. Additive effects of mutations on the stability and activity of subtilisin E in polar organic media. Biotechnol Prog 7: 125–129 [DOI] [PubMed] [Google Scholar]
- Chiu DK, Kolodziejczak T 1991. Inferring consensus structure from nucleic acid sequences. Comput Appl Biosci 7: 347–352 [DOI] [PubMed] [Google Scholar]
- Crameri A, Raillard SA, Bermudez E, Stemmer WP 1998. DNA shuffling of a family of genes from diverse species accelerates directed evolution. Nature 391: 288–291 [DOI] [PubMed] [Google Scholar]
- Curtis EA, Bartel DP 2005. New catalytic structures from an existing ribozyme. Nat Struct Mol Biol 12: 994–1000 [DOI] [PubMed] [Google Scholar]
- Doherty EA, Doudna JA 1997. The P4–P6 domain directs higher order folding of the Tetrahymena ribozyme core. Biochemistry 36: 3159–3169 [DOI] [PubMed] [Google Scholar]
- Ekland EH, Bartel DP 1995. The secondary structure and sequence optimization of an RNA ligase ribozyme. Nucleic Acids Res 23: 3231–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington AD, Szostak JW 1990. In vitro selection of RNA molecules that bind specific ligands. Nature 346: 818–822 [DOI] [PubMed] [Google Scholar]
- Freier SM, Kierzek R, Jaeger JA, Sugimoto N, Caruthers MH, Neilson T, Turner DH 1986. Improved free-energy parameters for predictions of RNA duplex stability. Proc Natl Acad Sci 83: 9373–9377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautheret D, Damberger SH, Gutell RR 1995. Identification of base-triples in RNA using comparative sequence analysis. J Mol Biol 248: 27–43 [DOI] [PubMed] [Google Scholar]
- Gorodkin J, Staerfeldt HH, Lund O, Brunak S 1999. MatrixPlot: Visualizing sequence constraints. Bioinformatics 15: 769–770 [DOI] [PubMed] [Google Scholar]
- Gregoret LM, Sauer RT 1993. Additivity of mutant effects assessed by binomial mutagenesis. Proc Natl Acad Sci 90: 4246–4250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Gooding AR, Cech TR 2006. Comparison of crystal structure interactions and thermodynamics for stabilizing mutations in the Tetrahymena ribozyme. RNA 12: 387–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutell RR, Noller HF, Woese CR 1986. Higher order structure in ribosomal RNA. EMBO J 5: 1111–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutell RR, Power A, Hertz GZ, Putz EJ, Stormo GD 1992. Identifying constraints on the higher-order structure of RNA: Continued development and application of comparative sequence analysis methods. Nucleic Acids Res 20: 5785–5795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatley ME, Lockless SW, Gibson SK, Gilman AG, Ranganathan R 2003. Allosteric determinants in guanine nucleotide-binding proteins. Proc Natl Acad Sci 100: 14445–14450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland JH 1992. Genetic algorithms. Sci Am 267: 66–72 [Google Scholar]
- Horovitz A 1996. Double-mutant cycles: A powerful tool for analyzing protein structure and function. Fold Des 1: R121–R126 [DOI] [PubMed] [Google Scholar]
- Igloi GL 1988. Interaction of tRNAs and of phosphorothioate-substituted nucleic acids with an organomercurial. Probing the chemical environment of thiolated residues by affinity electrophoresis. Biochemistry 27: 3842–3849 [DOI] [PubMed] [Google Scholar]
- Johnston WK, Unrau PJ, Lawrence MS, Glasner ME, Bartel DP 2001. RNA-catalyzed RNA polymerization: Accurate and general RNA-templated primer extension. Science 292: 1319–1325 [DOI] [PubMed] [Google Scholar]
- Kauffman SA 1993. The origins of order. Oxford University Press, Oxford [Google Scholar]
- Lorsch JR, Szostak JW 1994. In vitro evolution of new ribozymes with polynucleotide kinase activity. Nature 371: 31–36 [DOI] [PubMed] [Google Scholar]
- Lunzer M, Miller SP, Felsheim R, Dean AM 2005. The biochemical architecture of an ancient adaptive landscape. Science 310: 499–501 [DOI] [PubMed] [Google Scholar]
- Martin LC, Gloor GB, Dunn SD, Wahl LM 2005. Using information theory to search for co-evolving residues in proteins. Bioinformatics 21: 4116–4124 [DOI] [PubMed] [Google Scholar]
- Mildvan AS, Weber DJ, Kuliopulos A 1992. Quantitative interpretations of double mutations of enzymes. Arch Biochem Biophys 294: 327–340 [DOI] [PubMed] [Google Scholar]
- Moody EM, Bevilacqua PC 2003. Folding of a stable DNA motif involves a highly cooperative network of interactions. J Am Chem Soc 125: 16285–16293 [DOI] [PubMed] [Google Scholar]
- Moody EM, Feerrar JC, Bevilacqua PC 2004. Evidence that folding of an RNA tetraloop hairpin is less cooperative than its DNA counterpart. Biochemistry 43: 7992–7998 [DOI] [PubMed] [Google Scholar]
- Murphy FL, Cech TR 1993. An independently folding domain of RNA tertiary structure within the Tetrahymena ribozyme. Biochemistry 32: 5291–5300 [DOI] [PubMed] [Google Scholar]
- Ness JE, Kim S, Gottman A, Pak R, Krebber A, Borchert TV, Govindarajan S, Mundorff EC, Minshull J 2002. Synthetic shuffling expands functional protein diversity by allowing amino acids to recombine independently. Nat Biotechnol 20: 1251–1255 [DOI] [PubMed] [Google Scholar]
- Pal G, Ultsch MH, Clark KP, Currell B, Kossiakoff AA, Sidhu SS 2005. Intramolecular cooperativity in a protein binding site assessed by combinatorial shotgun scanning mutagenesis. J Mol Biol 347: 489–494 [DOI] [PubMed] [Google Scholar]
- Pitt JN, Ferre-D'Amare AR 2010. Rapid construction of empirical RNA fitness landscapes. Science 330: 376–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putz J, Puglisi JD, Florentz C, Giege R 1993. Additive, cooperative and anti-cooperative effects between identity nucleotides of a tRNA. EMBO J 12: 2949–2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson JR, Behlen LS, DiRenzo AB, Uhlenbeck OC 1992. Recognition of yeast tRNAPhe by its cognate yeast phenylalanyl-tRNA synthetase: An analysis of specificity. Biochemistry 31: 4161–4167 [DOI] [PubMed] [Google Scholar]
- Shulman AI, Larson C, Mangelsdorf DJ, Ranganathan R 2004. Structural determinants of allosteric ligand activation in RXR heterodimers. Cell 116: 417–429 [DOI] [PubMed] [Google Scholar]
- Silverman SK, Cech TR 1999. Energetics and cooperativity of tertiary hydrogen bonds in RNA structure. Biochemistry 38: 8691–8702 [DOI] [PubMed] [Google Scholar]
- Stemmer WP 1994a. DNA shuffling by random fragmentation and reassembly: In vitro recombination for molecular evolution. Proc Natl Acad Sci 91: 10747–10751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer WP 1994b. Rapid evolution of a protein in vitro by DNA shuffling. Nature 370: 389–391 [DOI] [PubMed] [Google Scholar]
- Tanner MA, Anderson EM, Gutell RR, Cech TR 1997. Mutagenesis and comparative sequence analysis of a base triple joining the two domains of group I ribozymes. RNA 3: 1037–1051 [PMC free article] [PubMed] [Google Scholar]
- Tinoco I Jr, Borer PN, Dengler B, Levin MD, Uhlenbeck OC, Crothers DM, Bralla J 1973. Improved estimation of secondary structure in ribonucleic acids. Nat New Biol 246: 40–41 [DOI] [PubMed] [Google Scholar]
- Unrau PJ, Bartel DP 1998. RNA-catalysed nucleotide synthesis. Nature 395: 260–263 [DOI] [PubMed] [Google Scholar]
- Wang QS, Unrau PJ 2005. Ribozyme motif structure mapped using random recombination and selection. RNA 11: 404–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JA 1990. Additivity of mutational effects in proteins. Biochemistry 29: 8509–8517 [DOI] [PubMed] [Google Scholar]
- Wochner A, Attwater J, Coulson A, Holliger P 2011. Ribozyme-catalyzed transcription of an active ribozyme. Science 332: 209–212 [DOI] [PubMed] [Google Scholar]