

Abstract
C. Akay, K. A. Lindl, N. Shyam, B. Nabet, Y. Goenaga‐Vazquez, J. Ruzbarsky, Y. Wang, D. L. Kolson and K. L. Jordan‐Sciutto (2012) Neuropathology and Applied Neurobiology 38, 175–200
Activation status of integrated stress response pathways in neurones and astrocytes of HIV‐associated neurocognitive disorders (HAND) cortex
Aims: Combined anti‐retroviral therapy (cART) has led to a reduction in the incidence of HIV‐associated dementia (HAD), a severe motor/cognitive disorder afflicting HIV(+) patients. However, the prevalence of subtler forms of neurocognitive dysfunction, which together with HAD are termed HIV‐associated neurocognitive disorders (HAND), continues to escalate in the post‐cART era. The microgliosis, astrogliosis, dendritic damage, and synaptic and neuronal loss observed in autopsy cases suggest an underlying neuroinflammatory process, due to the neurotoxic factors released by HIV‐infected/activated macrophages/microglia in the brain, might underlie the pathogenesis of HAND in the post‐cART era. These factors are known to induce the integrated stress response (ISR) in several neurodegenerative diseases; we have previously shown that BiP, an indicator of general ISR activation, is upregulated in cortical autopsy tissue from HIV‐infected patients. The ISR is composed of three pathways, each with its own initiator protein: PERK, IRE1α and ATF6. Methods: To further elucidate the specific ISR pathways activated in the central nervous system of HAND patients, we examined the protein levels of several ISR proteins, including ATF6, peIF2α and ATF4, in cortical tissue from HIV‐infected patients. Results: The ISR does not respond in an all‐or‐none fashion in HAND, but rather demonstrates a nuanced activation pattern. Specifically, our studies implicate the ATF6 pathway of the ISR as a more likely candidate than the PERK pathway for increases in BiP levels in astrocytes. Conclusion: These findings begin to characterize the nature of the ISR response in HAND and provide potential targets for therapeutic intervention in this disease.
Keywords: astrocyte, ATF6, ER stress response, HAND, HIV, neurone
Introduction
HIV‐associated neurocognitive disorders (HAND) encompass a spectrum of clinical diagnoses ranging from minimal cognitive impairment to debilitating motor, cognitive and behavioural deficits. However, with the advent of combined anti‐retroviral therapy, the phenotype of HAND has been shifting towards the subtler forms and away from the more severe forms, such as HIV‐associated dementia (HAD), an AIDS‐defining disorder [1, 2]. In a recent study where a cohort of 200 aviraemic patients was evaluated for neurocognitive dysfunction, the prevalence was 27% for any degree of impairment; this number is estimated to be as high as 50% in sub‐Saharan Africa [3].
The neuropathological correlate of HAD, HIV encephalitis, exhibits astrocytosis, perivascular inflammatory macrophages, microglial nodules and multinucleated giant cells, all of which provide evidence that an inflammatory process underlies the pathogenesis of HAND disorders [4, 5, 6, 7]. The neuroinflammation in HAND can be tracked to HIV‐infected monocytes that differentiate into macrophages in the brain parenchyma after crossing the blood–brain barrier [8]. These infected macrophages constitute the viral reservoir in the brain; moreover, these infected macrophages are thought to lead to infection and/or activation of resident central nervous system (CNS) macrophages and microglia. While extensive studies have linked neuronal damage and death to the clinical presentation of HAND, neurones themselves lack the necessary co‐receptors for HIV binding and entry and are therefore not directly infected by the virus [9]. Rather, infected and/or activated macrophages, astrocytes and neurones release a wide range of soluble neurotoxic factors, including viral proteins, cytokines, chemokines, reactive oxygen species, and excitotoxins, into the extracellular milieu as part of the inflammatory response.
Several studies have shown that some of the soluble factors that are released as part of the HIV‐induced inflammatory response, as well as HIV viral proteins themselves, are able to induce the integrated stress response (ISR) [endoplasmic reticulum (ER) stress response, unfolded protein response], suggesting that the ISR may play a role in HAND [10]. The ISR orchestrates a multitude of cellular responses against a wide range of extra‐ and intracellular stresses by regulating intracellular Ca2+ levels; ER chaperone genes; protein synthesis, folding and degradation; and the endogenous antioxidant response [10, 11, 12, 13, 14, 15, 16, 17, 18]. A variety of insults can trigger the ISR, such as oxidative stress, hypoxia, toxins, nutrient deprivation and viral infection. In fact, many types of viruses actively regulate the ISR in their host cells, showing a great variety in the manners and mechanisms by which they do so, preventing components of ISR activity harmful to virus survival and replication and exploiting ISR components beneficial to the virus, and even showing cell‐type specificity [15, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34]. Further, a role for the ISR has been implicated in a number of neurodegenerative diseases, including Alzheimer disease and Parkinson disease, as well as in CNS injury, such as brain trauma and ischaemia [13, 35, 36, 37, 38, 39, 40]. Importantly, while the primary purpose of the ISR is to promote cellular survival in the short term, chronic ISR activation in response to sustained injury/insult or in cases of extreme insult can also lead to orchestrated cell death [15, 36, 38, 41, 42]. Thus, chronic ISR activation may provide a link between the inflammatory response to HIV infection in the CNS and neuronal dysfunction, damage and death in HAND.
The ISR is composed of three major pathways, each mediated by an upstream initiator protein: activating transcription factor 6 (ATF6), PKR‐like ER kinase (PERK) and inositol‐requiring enzyme 1α (IRE1α) [10]. All three initiator proteins are ER transmembrane proteins and each is maintained in its inactive state by interaction of its luminal domain with the ER resident chaperone protein, BiP/GRP78 (immunoglobulin heavy chain‐binding protein/glucose‐regulated protein 78), often considered to be the master regulator of the ISR [42]. However, conditions of cellular stress that activate the ISR disrupt the BiP:Initiator protein complexes, thereby freeing each of the initiators to activate its specific branch of the stress response.
Each initiator protein activates its path of the ISR via a distinct mechanism, allowing for a coordinated and well controlled three‐pronged cellular stress response [10]. ATF6 is a basic leucine zipper (bZIP) transcription factor that when freed from BiP translocates to the Golgi where it is cleaved into its active form, which then moves into the nucleus [43, 44, 45, 46]. Once in the nucleus, it binds the ER stress response element (ERSE) found in the upstream region of genes encoding proteins important for responding to the offending stresses, such as ER chaperone proteins and folding catalysts [47]. PERK and IRE1α are both kinases that, when released from BiP, homodimerize and activate via autophosphorylation [10]. PERK then activates of its branch of the ISR by phosphorylating the α‐subunit of the eukaryotic initiation factor 2 (eIF2α), causing a global inhibition of translation initiation, while simultaneously promoting the translation of certain stress response proteins, including activating transcription factor 4 (ATF4) [17]. Like its family member, ATF6, ATF4 is a bZIP transcription factor that binds the ERSE, thereby transactivating stress response genes [48]. Interestingly, eIF2α can be activated by other kinases in a stress‐specific manner: general control non‐depressible‐2 (GCN2), which senses amino acid starvation; the protein activated by dsRNA (PKR), which is important for the cellular response to viral infection; and haem‐regulated inhibitor, which senses haem deficiency in erythroid cells [49]. Autophosphorylation of the initiator protein IRE1α activates its endoribonuclease domains allowing it to cleave the mRNA of its one known target, X‐box binding protein 1 (XBP1). Once cleaved to its mature form, XBP1 mRNA encodes yet another bZIP transcription factor capable of binding the ERSE [50, 51].
While the three ISR pathways function via distinct mechanisms, they are all capable of activating the transcription of genes with the ERSE in their promoter regions, including the master regulator of the ISR, BiP, as well as others, such as CCAAT/enhancer binding protein (C/EBP) homologous protein (CHOP) and heat shock protein 70 (HSP70) [18]. This means that BiP is not only a critical overseer of the ISR, but also a key indicator of activity of any of the three ISR pathways. In a previous report, we showed that BiP is increased in the grey matter of mid‐frontal cortex of patients with HIV infection and with HIV‐induced neurocognitive impairment, indicating that the ISR is indeed activated in the CNS of these individuals [52]. However, as BiP can be upregulated by all branches of the ISR [18], our previous study did not determine which of the three pathways may be responding in this disease. It is important to note that, while BiP is implicated in the regulation of all three ISR pathways, the pathways do not necessarily exhibit concomitant, coordinated activation [53, 54, 55], allowing a more precise functionality of the ISR than an all‐or‐nothing response. This phenomenon is especially likely to occur during viral infection, as many viruses exploit specific ISR pathways, while inhibiting others, to enable persistence/latency [28, 30, 31, 33, 56, 57, 58]. Thus, in the study presented here, we sought to more specifically characterize the activation of the ISR in the CNS of HIV‐infected patients. Specifically, we used immunofluorescence (IFA) and immunoblotting to examine the levels of key players in the three prongs of the ISR in the grey matter of mid‐frontal cortical autopsy tissue from HAND cases. Here we report that ATF6β is increased in neurones and astrocytes in HIV(+) tissue and in HAND tissue over HIV(−) and neurocognitively normal control tissue, respectively. We also show via immunoblotting that total ATF6α, another isoform of ATF6, increases in HAND tissue. On the contrary, via immunoblotting, we saw no significant change in IRE1α across experimental groups, and that, if anything it showed a trend of decreasing in HIV(+) cases. Finally, in the PERK pathway of the ISR, we found that peIF2α increases in HIV(+) cases when compared with HIV(−) cases. Interestingly, although ATF4 increases in the cytoplasm of astrocytes in HAND cases over neurocognitively normal controls, total ATF4 as observed by IFA actually decreases. To expand our examination of the ISR further, we also examined two more ISR gene targets, CHOP and HSP70, via immunoblotting. Neither target changed significantly with either HIV infection or neurocognitive status, although CHOP showed a trend of decreasing in HIV(+) cases. Lastly, because of the significant increase in astrocytic BiP that we had observed previously [52], we determined the astrocytic nuclear levels of ATF6β and ATF4, which as transcription factors need to translocate to the nucleus to transactivate BiP. We found that ATF6β increases in the nucleus of astrocytes in both HAND and HIV(+) tissue with respect to control tissue. While nuclear astrocytic ATF4 does not change, total ATF4 colocalized with DAPI, which is predominantly neuronal nuclear ATF4, actually decreases in HAND cases. These findings (summarized in Table 1 and Figures 11 and 12) suggest that the increase in astrocytic BiP that we reported previously is more likely due to the ATF6 branch of the ISR than to the IRE1α or the PERK branches. Moreover, the data presented here demonstrate that the ISR activation that occurs in HAND and in HIV(+) tissue varies across cell types and is due to selective activation of the certain components of the ISR. Thus, further studies to uncover the nuances of ISR activation in this disease will be important to the development of potential therapies.
Table 1.
Summary of findings of protein levels for each ISR protein examined
| Total | Cytoplasmic | Nuclear | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Neuronal | Astrocytic | Total | Astrocytic | |||||||
| ATF6β | ↑ | ⇑ | ↑ | ⇑ | ↑ | ⇑ | ↔ | ⇔ | ↑ | ⇑ |
| ATF6α* | ↑* | ⇔* | ND | ND | ND | ND | ||||
| IRE1α* | ↔* | ⇔* | ND | ND | ND | ND | ||||
| peIF2α | ↑/↔* | ⇑ | ↑ | ⇑ | ND | ND | ND | |||
| ATF4 | ↓ | ⇔ | ↔ | ⇔ | ↑ | ⇑ | ↓ | ⇔ | ↔ | ⇔ |
| CHOP* | ↔* | ⇔* | ND | ND | ND | ND | ||||
| HSP70* | ↔* | ⇔* | ND | ND | ND | ND | ||||
| HAND vs. NcN | HIV(+) vs. HIV(−) | HAND vs. NcN | HIV(+) vs. HIV(−) | HAND vs. NcN | HIV(+) vs. HIV(−) | HAND vs. NcN | HIV(+) vs. HIV(−) | HAND vs. NcN | HIV(+) vs. HIV(−) | |
Closed arrows indicate changes observed in HAND tissue compared with neurocognitively normal tissue. Open arrows indicate changes observed in HIV(+) tissue compared with HIV(−) tissue. The symbols followed by * show results determined only by immunoblotting. For ATF6α, IRE1α, CHOP and HSP70 only immunoblotting and not IFA was conducted in this study. For peIF2α in HAND cases vs. neurocognitively normal cases, IFA and immunoblotting results disagreed with each other; IFA showed a significant but very slight increase (Figure 5A,B ) and immunoblotting showed no significant change (Figure 6).
ND, not determined; NcN, neurocognitively normal.
Materials and methods
Immunofluorescent analysis
Paraffin‐embedded tissue sections from the cortices of control and HIV(+) human autopsy cases were obtained from the tissue banks of National NeuroAIDS Tissue Consortium (NNTC). The age, sex, neurocognitive status and post‐mortem interval of each specimen were provided by NNTC and are depicted in Tables 2 and 3. The glass slides were prepared for immunofluorescent staining as described previously [52], except for ATF6β staining, where 10% normal horse serum was used as the blocking agent. Rabbit polyclonal antibodies to phosphorylated PERK (pPERK) (Thr980, 1:1000; Cell Signalling Inc., Beverly, MA, USA), peIF2α (pS52, 1:1000; Invitrogen, Carlsbad, CA, USA), ATF4 (C‐20, 1:600; Santa Cruz Biotechnology, Santa Cruz, CA, USA), glial fibrillary acidic protein (GFAP) (Z0334, 1:80; Dako, Carpinteria, CA, USA), goat polyclonal antibody raised against ATF6β (N‐17, 1:100; Santa Cruz Biotechnology) and mouse monoclonal antibody to microtubule‐associated protein 2 (MAP2) (SMI‐52, 1:200; Covance Research Products, Berkeley, CA, USA) were used at empirically defined dilutions. Antibodies are summarized in Table 4. The tyramide amplification system (New England Biolabs, Beverly, MA, USA) was used to detect the markers of ISR. To visualize staining, slides were incubated for 30 min with goat secondary antibodies (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) except for ATF6β, where a donkey anti‐goat secondary antibody (Jackson ImmunoResearch Laboratories) was utilized. The primary antibodies for ISR markers were detected by biotin‐conjugated, secondary antibodies (1:200) for use with the tyramide amplification system, and were subsequently visualized using Fluorescein isothiocyanate (FITC)‐conjugated Streptavidin (New England Biolabs). The GFAP and MAP2 primary antibodies were visualized by a Cy‐5‐conjugated, goat anti‐rabbit antibody (1:200) and a Cy‐3‐conjugated goat anti‐mouse antibody (1:200), respectively, and DAPI was used to visualize DNA (5 mM, Invitrogen). Slides were mounted in Citifluor AF1 (Citifluor, Ltd, London, UK) and laser confocal microscopy on a Biorad Radiance 2100 equipped with Argon, Green He/Ne, Red Diode, and Blue Diode lasers (BioRad, Hercules, CA, USA) was used to analyse the immunostaining, as described previously [15]. Five to ten high‐magnification (× 600) images with uniform threshold settings for each ISR marker were captured randomly from the areas of positive staining in the mid‐frontal cortical grey matter for each case.
Table 2.
For ATF6β and ATF4 the cases in Table 2 were used for immunofluorescent staining
| Neurocog | HIV status | Sex | Age | PMI (h) |
|---|---|---|---|---|
| HAND 1 | + | M | 35 | 9.25 |
| HAND 2 | + | F | 34 | 5 |
| HAND 3 | + | M | 57 | 5.5 |
| HAND 4 | + | M | 32 | 14.5 |
| HAND 5 | + | M | 38 | 5.5 |
| HAND 6 | + | M | 32 | 14 |
| HAND 7 | + | M | 36 | 2.5 |
| HAND 8 | + | M | 37 | 11.5 |
| HAND 9 | + | M | 31 | 8.83 |
| HAND 10 | + | M | 49 | 67.33 |
| HAND 11 | + | M | 42 | 27.33 |
| HAND 12 | + | M | 50 | 21 |
| HAND 13 | + | M | 43 | Unknown |
| NORMAL 1 | + | M | 50 | 18 |
| NORMAL 2 | + | M | 46 | 2.75 |
| CONTROL 1 | − | M | 46 | 27.65 |
| CONTROL 2 | − | F | 47 | 19.18 |
| CONTROL 3 | − | M | 40 | 14.66 |
| CONTROL 4 | − | F | 51 | 21.75 |
Neurocog, neurocognitive status; PMI, post‐mortem interval; M, male; F, female.
Table 3.
For peIF2α immunofluorescent analysis the cases listed in Table 3 were used
| Neurocog | HIV status | Sex | Age | PMI (h) |
|---|---|---|---|---|
| HAND 1 | + | M | 44 | 8.5 |
| HAND 2 | + | M | 31 | 20 |
| HAND 3 | + | M | 51 | 5 |
| HAND 4 | + | M | 33 | 6 |
| HAND 5 | + | N/A | 34 | 5 |
| HAND 6 | + | N/A | 32 | 14 |
| HAND 7 | + | N/A | 57 | 5.5 |
| NORMAL 1 | + | N/A | 52 | 15 |
| NORMAL 2 | + | N/A | 41 | N/A |
| NORMAL 3 | + | N/A | 44 | 21.25 |
| NORMAL 4 | + | N/A | 46 | 2.75 |
| NORMAL 5 | + | N/A | 45 | 13 |
| NORMAL 6 | + | N/A | 44 | 16.4 |
| CONTROL 1 | − | N/A | 58 | 7.42 |
| CONTROL 2 | − | N/A | 34 | 24.25 |
| CONTROL 3 | − | N/A | 51 | 21.75 |
| CONTROL 4 | − | N/A | 58 | 12.16 |
Neurocog, neurocognitive status; PMI, post‐mortem interval; M, male; N/A, not available.
Table 4.
Summary of antibodies used in this study
| Target | Species | Clone | Source | Dilution WB | Dilution IFA |
|---|---|---|---|---|---|
| ATF6α | Mouse | 70B1413.1 | Imgenex | 1:1000 | N/A |
| ATF6β | Goat | N‐17 | Santa Cruz | N/A | 1:100 with tyramide amplification |
| IRE1α | Rabbit | 14C10 | Cell Signaling | 1:1000 | N/A |
| peIF2α | Rabbit | PS52 | Invitrogen | 1:1000 | 1:1000 with tyramide amplification |
| eIF2α | Rabbit | N/A | Cell Signaling | 1:1000 | N/A |
| ATF4 | Rabbit | C20 | Santa Cruz | 1:1000 | 1:600 with tyramide amplification |
| CHOP | Mouse | 9C8 | Thermo Fisher | 1:500 | N/A |
| HSP70 | Rabbit | D69 | Cell Signaling | 1:1000 | N/A |
WB, western immunoblotting; IFA, immunofluorescent analysis; N/A, not available.
Post‐acquisition analysis for immunostaining was performed via a blinded fashion using MetaMorph 6.0 image analysis software (Universal Imaging, Inc., Downingtown, PA, USA). Total and phenotypic‐specific pixel intensity for each ISR marker was determined as described previously [52]. Additionally, astrocytic nuclear ATF4 and ATF6β pixel intensity was determined as follows: the astrocytic nuclei in each image were determined using GFAP and DAPI staining as guidance. These nuclei were then traced by hand and the integrated pixel intensity for the ISR marker was determined and then normalized for the area of the traced region. Averages are expressed at mean ± SEM. All data were analysed by GraphPad Prism 3.02 software (GraphPad Software, San Diego, CA, USA). For statistical analysis, Student's t‐test was used with post‐hoc Mann–Whitney U‐analysis. Values of P < 0.05 were considered significant for all statistical analyses performed.
Immunoblotting
Fresh‐frozen tissue samples from the cortices of control and HIV(+) human autopsy cases were obtained from the tissue banks of NNTC. The age, sex, neurocognitive status and post‐mortem interval of each specimen were provided by NNTC (Table 5). For protein extraction, tissue was homogenized in a Dounce tissue grinder (Kimble/Kontes, Vineland, NJ, USA) in lysis buffer [50 mM Tris‐HCl (pH 7.5), 0.5 M NaCl, 1% NP‐40, 0.5% sodium deoxycholate, 1% sodium dodecyl sulfate (SDS), 2 mM ethylene glycol tetraacetic acid (EGTA), 2 mM Ethylenediaminetetraacetic acid (EDTA), 5 mM NaF, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mg/ml leupeptin, 1 mM dithiothreitol (DTT), and protease inhibitor cocktail (Sigma, St. Louis, MO, USA)]. Homogenized tissue was sonicated three times in 6‐s cycles, and then centrifuged at 16 100 g for 30 min. The protein concentrations were determined by the Bradford method. For the immunoblotting of ATF6α, IRE1α, eIF2α, peIF2α, ATF4, CHOP and HSP70 (antibodies are summarized in Table 4), 20–100 mg of protein was loaded to each lane of a 10% Bis‐Tris gel. The samples were then transferred onto polyvinylidene difluoride (PVDF) membranes and blocked by tris‐buffered saline (TBS) with 1% Tween‐20 (TBS‐T) and 5% bovine serum albumin (BSA) for 1 h at room temperature. The membranes were incubated overnight at 4°C with the primary antibody raised against the protein of interest in TBS‐T with 5% BSA. The membranes were then washed three times with TBS‐T and incubated with secondary antibody conjugated to horseradish peroxidase (HRP) (Pierce Biotechnology, Rockford, IL, USA) in TBS‐T + 5% BSA for 1 h at room temperature. Following washes with TBS‐T three times, the membranes were developed using SuperSignal West Dura Extended Duration Substrate (Pierce Biotechnology) and exposed using Amersham Hyperfilm (GE Healthcare, Piscataway, NJ, USA). A broad‐range molecular weight ladder (BioRad) was run for each gel. For densitometric analysis, autographs were scanned into Adobe Photoshop (Adobe Systems, San Jose, CA, USA), and a fixed area centred over each band was assessed for pixel intensity by the ImageJ program (National Institutes of Health, Bethesda, MD, USA, V 1.36b) with normalization to the loading control. For loading control, the PVDF membranes were stained with the Coomassie Brilliant‐Blue R‐250 (BioRad) solution [0.1% Coomassie (w/v) in 50% Methanol] for 5 min, followed by destaining with 25% acetic acid in 50% methanol for 30 min. The membranes were then photographed by a Nikon Coolpix 995 camera (Nikon Inc., Melville, NY, USA), and further analysis was performed using NIH ImageJ as described above.
Table 5.
For all immunoblotting the cases listed in Table 5 were used
| Neurocog | HIV status | Sex | Age | PMI (h) |
|---|---|---|---|---|
| NcN | CONTROL 1 | M | 53 | 22.5 |
| NcN | CONTROL 2 | M | 48 | 9 |
| NcN | CONTROL 3 | M | 61 | 6.5 |
| NcN | CONTROL 4 | M | 50 | 16 |
| NcN | HIV 1 | N/A | N/A | N/A |
| NcN | HIV 2 | N/A | N/A | N/A |
| NcN | HIV 3 | N/A | N/A | N/A |
| NcN | HIV 4 | N/A | N/A | N/A |
| NcN | HIV 5 | N/A | N/A | N/A |
| NcN | HIV 6 | N/A | N/A | N/A |
| NcN | HIV 7 | N/A | N/A | N/A |
| HAND | HIV 8 | F | 54 | 7 |
| HAND | HIV 9 | M | 48 | 7.5 |
| HAND | HIV 10 | M | 62 | 4 |
| HAND | HIV 11 | N/A | 47 | N/A |
| HAND | HIV 12 | N/A | 44 | N/A |
| HAND | HIV 13 | N/A | 35 | N/A |
| HAND | HIV 14 | N/A | 34 | N/A |
| NcN | HIV 15 | M | 45 | N/A |
| NcN | HIV 16 | M | 46 | N/A |
| NcN | HIV 17 | M | 38 | N/A |
| NcN | HIV 18 | M | 49 | N/A |
| NcN | HIV 19 | M | 32 | N/A |
| NcN | HIV 20 | N/A | 39 | N/A |
| HAND | HIV 21 | N/A | 34 | N/A |
Neurocog, neurocognitive status; PMI, post‐mortem interval; NcN, neurocognitively normal; M, male; F, female; N/A, not available.
Results
ATF‐6β protein levels are increased in the mid‐frontal cortex of HAND cases and of HIV(+) cases
In our previous study, we had demonstrated via immunoblotting and IFA that BiP protein levels are increased in the grey matter of mid‐frontal cortex from HIV(+) autopsy cases and from HAND autopsy cases [52]. These findings indicated that the ISR is activated in the CNS of these patients, but demanded further study to determine more precisely the nature of ISR activation in this disease. Therefore, in the current study, we set out to determine which of the three ISR pathways are activated in the grey matter of mid‐frontal cortex of these patients. First, we examined the ATF6 pathway via quadruple‐label immunofluorescent confocal microscopy (IFA) for ATF6β protein levels. We had previously examined this branch of the ISR via immunoblotting for ATF6β[52]; however, the IFA method conducted in the current study allowed us to not only confirm the increases in total levels of ATF6β, but also examine its subcellular localization, as well as cell type‐specific changes in levels of this protein for a given disease state. In the current study, we found that total levels of ATF6β increase in the mid‐frontal cortex of HAND autopsy cases compared with levels in neurocognitively normal cases (Figure 1A,B ; summarized in Table 1, Figures 11 and 12). Additionally, total ATF6β protein levels increase in the mid‐frontal cortex of HIV(+) autopsy cases as compared with those seen in HIV(−) cases (Figure 1A,C ). These findings parallel our immunoblot data presented previously [52]. Next, we examined cell type‐specific changes in ATF6β expression by quantifying its colocalization with cell‐type markers, MAP2 and GFAP, which mark the cytoplasm of neurones and astrocytes, respectively. Importantly, protein levels of these two markers frequently change in HAND patients due to the reactive astrocytosis (increased GFAP) and neuronal damage and loss (MAP2 loss) associated with this disease [5, 7, 59, 60, 61, 62, 63, 64]. Thus, to ensure that any changes in cell type‐specific ATF6β levels were not due to an increase or decrease in the amount of the phenotypic marker present, we normalized colocalization values to total area of the phenotypic marker. Using this methodology, we found that ATF6β is increased in the cytoplasm of both neurones and astrocytes in HAND cases and in HIV(+) cases as compared with that in neurocognitively normal and HIV(−) cases, respectively (Figure 2A–D ). As a transcription factor, ATF6β must move into the nucleus to perform its function in cellular stress mitigation. We found that ATF6β was predominantly cytoplasmic in our staining, regardless of disease state; however, particularly in astrocytes, we did observe some nuclear staining, observed as colocalization with the nucleic acid marker, DAPI. Our initial analysis showed that total nuclear ATF6β did not change significantly (Figure 2E,F ). Because we had previously seen an increase in BiP protein levels that reached significance in astrocytes, but not in neurones [52], we sought to determine whether ATF6β might be increasing in the nuclei of astrocytes specifically, and is therefore able to transactivate BiP expressly in this cell type. As we have been unable to find a cell type‐specific nuclear marker that works for IFA, we had to manually determine astrocytic nuclei (see methods for details), a feasible task due to the relatively small number of astrocytes in each image. Our analysis revealed a statistically significant increase in astrocytic nuclear ATF6β in HAND cases as compared with neurocognitively normal cases, which was also true for HIV(+) cases when compared with HIV(−) cases (1, 2). Unfortunately, the overwhelmingly large number of neurones in our confocal images prohibited us from performing similar analysis to specifically quantify neuronal nuclear ATF6β. However, given that astrocytes comprise only a small percentage of cells captured in each image, it is likely that quantification of total nuclear ATF6β is indicative of neuronal nuclear ATF6β. Of note, while we detected the vast majority of ATF6β staining in either MAP2‐ or GFAP‐positive cells, we did observe small amounts of nuclear ATF6β staining in cells that were neither MAP2‐ nor GFAP‐positive.
Figure 1.
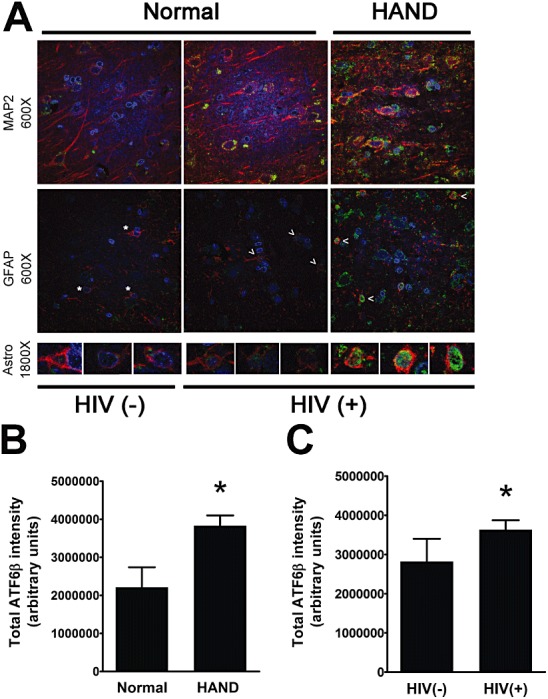
ATF6β increases in mid‐frontal cortical grey matter of patients with HAND and of patients infected with HIV. Formalin‐fixed, paraffin‐embedded mid‐frontal cortical autopsy tissue was quadruple labelled with antibodies for ATF6β and for the cell‐type markers for astrocytes (GFAP) and for neurones (MAP2), as well as the DNA‐binding compound, DAPI to label nuclei. (A) Grey matter in labelled slides was imaged using laser confocal microscopy: ATF6β is shown in green, GFAP and MAP2 are shown in red and are not both present in the same images, and DAPI is shown in blue. Overlap of ATF6β with a cell‐type marker appears yellow. ATF6β overlap with DAPI appears blue‐green. An example is shown for an HIV(−) uninfected control, an HIV(+) neurocognitively normal case, and a HAND case for each cell‐type marker. The third panel shows higher magnification images of astrocytes, which are denoted by asterisks [HIV(−) control] or arrowheads [HIV(+) cases]. (B and C) ATF6β integrated pixel intensity from confocal images (five per case) was quantified using Metamorph 6.0 software. Total ATF6β intensity increases in HAND cases (n = 12) over neurocognitively normal (NcN) cases (n = 5) (B) and in HIV(+) cases (n = 14) over HIV(−) (n = 3) controls (C). Values are shown as mean ± SEM. Student's t‐test and Mann–Whitney U post‐hoc analysis was used for statistical analysis to determine significance. *P < 0.05, compared with control.
Figure 2.
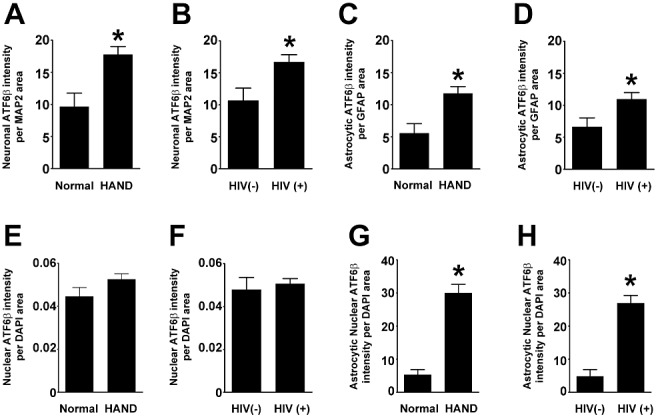
Neuronal and astrocytic ATF6β increases in HAND and in HIV(+) compared with neurocognitively normal and HIV(−) tissue, respectively, while nuclear ATF6β increases only in HAND tissue. Using Metamorph 6.0 software, total integrated pixel intensity of ATF6β was quantified and colocalization with cell‐type markers was determined and normalized to area of cell‐type marker to account for losses of cell‐type marker protein due to disease. (A and B) ATF6β increases in the cytoplasm of neurones in both HAND cases and HIV(+) cases when compared to neurocognitively normal (NcN) and HIV(−) cases, respectively. (C and D) ATF6β increases in the cytoplasm of astrocytes in both HAND cases and HIV(+) cases when compared to neurocognitively normal (NcN) and HIV(−) cases, respectively. (E and F) Total nuclear ATF6β levels do not increase in HAND cases over neurocognitively normal (NcN) cases or in HIV(+) cases over that seen in HIV(−). (G and H) Astrocytic nuclear ATF6β levels increase in both HAND cases and HIV(+) cases when compared to neurocognitively normal (NcN) and HIV(−) cases, respectively. Values are shown as mean ± SEM. Student's t‐test and Mann–Whitney U post‐hoc analysis was used for statistical analysis to determine significance. *P < 0.05, compared with control.
ATF6 has two forms, ATF6α and ATF6β, both of which are activated under stress conditions [65, 66, 67]. Thus, to further characterize activation of this branch of the ISR in HAND, we examined ATF6α levels via immunoblotting of lysates harvested from fresh‐frozen HAND cortical autopsy tissue. We found that total ATF6α protein levels increase in HAND tissue compared with neurocognitively normal control tissue (Figure 3A,B ). Interestingly, unlike our observations with ATF6β, we did not see a significant change in total ATF6α levels in HIV(+) tissue compared with HIV(−) controls (Figure 3A,C ). Finally, the number and types of cases available for immunoblot from each experimental group allowed analysis of the ATF6α immunoblot data according to each of the three experimental groups: (i) HIV(−)/neurocognitively normal; (ii) HIV(+)/neurocognitively normal; and (iii) HAND. When compared using one‐way anova, we found no significant differences between these three experimental groups; however, as expected, the HAND group trended towards an increase over the other two groups (Figure 3A,D ). These findings further support the conclusion that the ATF6 pathway is activated in HAND.
Figure 3.
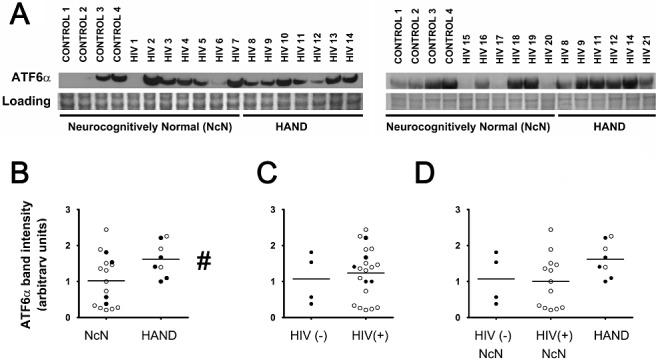
Total ATF6α increases in HAND tissue as observed via immunoblotting. Whole‐cell protein extracts were isolated from fresh‐frozen cortical autopsy tissue and were used for assessment of ATF6α protein levels. (A) Blots showing ATF6α levels in autopsy tissue, as well as a band from Coomassie staining of the PVDF membrane: HIV(−)/neurocognitively normal (control), HIV(+)/neurocognitively normal, and HAND. (B) ATF6α expression is increased in the mid‐frontal cortices of HAND cases (n = 8) compared with neurocognitively normal (NcN) cases (n = 17). Samples were run on two separate blots due to large number of sample sizes. Several cases were run on both blots to serve as technical replicates. For quantification, band intensities were first normalized to loading controls. Then, all samples on both blots were normalized to sample HIV 8 from their respective blot for comparison across blots. HIV 8 was used for normalization because it was run in the centre of the blot and was therefore unlikely to have any confounding factors due to proximity to the edge of the gel. For cases run on both gels, normalized values were averaged and are represented using closed circles. Cases run on only one gel are represented using open circles. (C) ATF6α levels did not significantly change in HIV(+) (n = 21) tissue compared with HIV(−) (control) tissue (n = 4). (D) Cases were divided into three groups: (i) HIV(−)/neurocognitively normal; (ii) HIV(+)/neurocognitively normal; and (iii) HAND. Groups were compared using one‐way anova and no significant differences were found. Coomassie staining of the PVDF membrane was used as a loading control for normalization of protein levels for quantification and statistical analysis as shown in B–D. #P < 0.05, compared with control, Student's t‐test for two‐way comparisons and one‐way anova for three‐way comparisons and Mann–Whitney U post‐hoc analysis.
IRE1α protein levels do not change in HAND or in HIV(+) mid‐frontal cortical autopsy tissue
Typically, IRE1α protein levels are not assessed to examine activation of this ISR pathway. Rather, to analyse activation of the IRE1α pathway, levels of spliced XBP1 mRNA are assessed. However, given the fragility of RNA and the nature of human autopsy tissue processing and handling, analysis of mRNA from such tissue samples obtained from tissue banks is not feasible. Further, the currently available XBP1 antibodies are not reliable for our assays. However, the E5 transforming protein of the high‐risk human papillomavirus type 16 (HPV‐16) has been shown to not only downregulate levels of XBP1 mRNA, but also downregulate IRE1α itself [24]. Thus, we were interested in whether a similar phenomenon might be occurring in cells from the cortex of HIV(+) individuals. Thus, via immunoblotting we examined levels of IRE1α in mid‐frontal cortical autopsy tissue from HIV(+) and HAND cases. We found that there were no significant changes in IRE1α levels across any of the experimental groups (Figure 4), although, interestingly, HIV(+) tissue, both neurocognitively normal and HAND, did trend towards a decrease compared with HIV(−) tissue.
Figure 4.
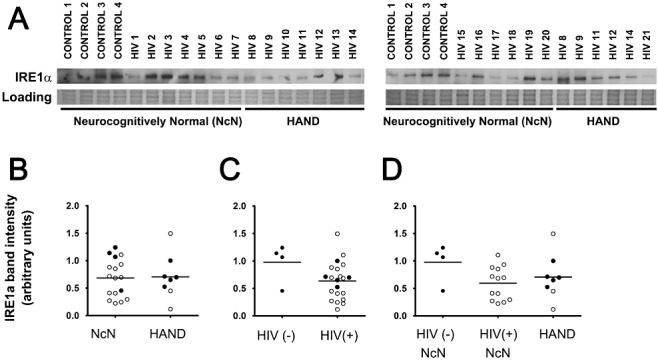
IRE1α levels do not change across experimental groups when examined via immunoblot analysis. Whole‐cell protein extracts were isolated from fresh‐frozen cortical autopsy tissue and were used for assessment of IRE1α protein levels. (A) Blots showing IRE1α levels in autopsy tissue, as well as a band from Coomassie staining of the PVDF membrane: HIV(−)/neurocognitively normal (control), HIV(+)/neurocognitively normal, and HAND. (B) IRE1α expression does not change in the mid‐frontal cortices of HAND cases (n = 8) compared with neurocognitively normal (NcN) cases (n = 17). Samples were run on two separate blots due to large number of sample sizes. Quantification was conducted as described for ATF6α in the Figure 3 legend. For cases run on both gels, normalized values were averaged and are represented using closed circles. Cases run on only one gel are represented using open circles. (C) IRE1α levels did not significantly change in HIV(+) (n = 21) tissue compared with HIV(−) (control) tissue (n = 4). (D) Cases were divided into three groups: (i) HIV(−)/neurocognitively normal; (ii) HIV(+)/neurocognitively normal; and (iii) HAND. Groups were compared using one‐way anova and no significant differences were found. Coomassie staining of the PVDF membrane was used as a loading control for normalization of protein levels for quantification and statistical analysis as shown in B–D. Student's t‐test for two‐way comparisons and one‐way anova for three‐way comparisons and Mann–Whitney U post‐hoc analysis.
peIF2α levels increase in HIV(+) mid‐frontal cortical autopsy tissue
The PERK arm of the ISR is activated by dimerization of PERK, which then activates via autophosphorylation and subsequently phosphorylates the alpha subunit of eIF2, thereby causing a global inhibition of protein translation [10]. Because there is no reliable antibody for pPERK, we conducted IFA and immunoblotting for peIF2α, as a downstream target of pPERK, to examine the activation of this branch of the ISR. We found that peIF2α increased in HIV(+) cases over HIV(−) cases, as observed by both IFA (Figure 5A,C ) and immunoblotting (Figure 6A,C ). Via immunoblotting, we also examined levels of unphosphorylated eIF2α (Figure 6), which when quantified showed no significant change across groups (data not shown). For quantification of peIF2α via immunoblotting, we determined values for peIF2α/total eIF2α per sample (Figure 6B–D ). We found that peIF2α increased in HIV(+) over HIV(−) tissue, but not in HAND tissue over neurocognitively normal tissue (Figure 6B–D ). We observed similar trends via IFA. However, in addition to the significant increase in peIF2α in HIV(+) tissue compared with HIV(−) tissue, via IFA we also observed a significant increase of peIF2α in HAND cases compared with neurocognitively normal cases (Figure 5A–C ), although this change was so small that it is questionable as to whether it is a meaningful change. Further, in our IFA study of peIF2α we had access to a larger cohort of cases than for the other IFA studies presented here. Thus, we were able to conduct one‐way anova between all three experimental groups. We observed a significant increase in the HIV(+) groups, both neurocognitively normal and HAND, when compared with HIV(−)/neurocognitively normal controls (Figure 5D ). When we examined peIF2α colocalized with MAP2 using IFA, we found that this neuronal peIF2α increased in HAND and in HIV(+) tissue over respective control groups (Figure 5E,F ). As with total peIF2α, neuronal peIF2α increases were more striking in HIV(+) tissue than they were in HAND tissue, and when the three experimental groups were compared using one‐way anova, both HIV(+) groups (neurocognitively normal and HAND) showed increased peIF2α compared with HIV(−) neurocognitively normal cases (Figure 5G ). These data suggest that increased peIF2α in mid‐frontal cortex is associated with HIV infection, rather than with HIV‐associated neurocognitive decline. Nuclear peIF2α was not determined because, as a post‐translational regulator, it is not expected to localize to the nucleus. Of note, we detected peIF2α staining exclusively in MAP2‐ or GFAP‐positive cells; however, unfortunately, GFAP staining was not of a quality and consistency to allow for quantification of colocalization using this marker. The cohort of tissue cases used for IFA of peIF2α was different from that used for the other ISR proteins examined in this study and was supplied by different tissue banks. Thus, it is probable that some aspect of tissue processing differed between these brain banks that affected the staining of this marker.
Figure 5.
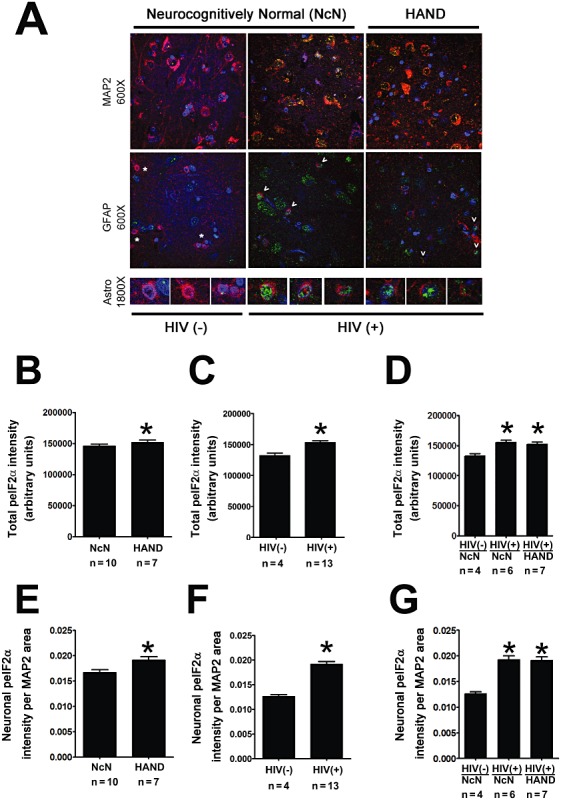
Total and neuronal peIF2α increase in HIV(+) tissue compared with HIV(−) tissue. Cortical autopsy tissue was quadruple labelled for peIF2α, GFAP, MAP2 and DAPI. (A) Grey matter in labelled slides was imaged using laser confocal microscopy: peIF2α is shown in green, GFAP and MAP2 are shown in red, and DAPI is shown in blue. Overlap of peIF2α with a cell‐type marker appears yellow and with DAPI appears blue‐green. An example is shown for an HIV(−) case, and HIV(+) neurocognitively normal case, and for a HAND case for each cell‐type marker. (B–E) peIF2α levels from confocal images (five per case) were quantified by determining integrated pixel intensity using Metamorph 6.0 software. Total and cytoplasmic neuronal peIF2α increases in HIV(+) cases (n = 13) over HIV(−) cases (n = 4) (C and F) and in HAND cases (n = 7) over neurocognitively normal cases (n = 10); however, the increase is quite small, despite reaching significance (B and E). (D and G) Cases were divided into three groups: (i) HIV(−)/neurocognitively normal; (ii) HIV(+)/neurocognitively normal; and (iii) HAND. Groups were compared using one‐way anova and HIV(+) cases, both neurocognitively normal (n = 6) and HAND (n = 7), were found to increase significantly over HIV(−) cases (n = 4). HAND cases, n = 7; neurocognitively normal cases, n = 10; HIV(+) cases, n = 13; HIV(−), n = 4. Values are shown as mean ± SEM. Student's t‐test was used for two‐way comparison and one‐way anova was used for three‐way comparisons and Mann–Whitney U post‐hoc analysis was used for statistical analysis to determine significance. *P < 0.05, compared with control.
Figure 6.
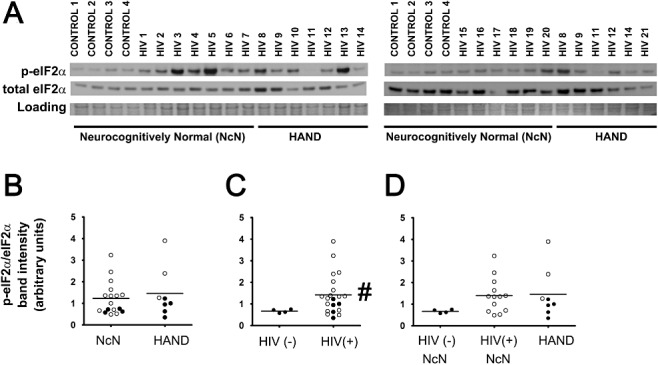
Total peIF2α increases in HIV(+) tissue as observed via immunoblotting. Whole‐cell protein extracts were isolated from fresh‐frozen cortical autopsy tissue and were used for assessment of peIF2α protein levels. (A) Blots showing peIF2α levels in autopsy tissue, as well as a band from Coomassie staining of the PVDF membrane: HIV(−)/neurocognitively normal (control), HIV(+)/neurocognitively normal, and HAND. (B) peIF2α expression does not change in the mid‐frontal cortices of HAND cases (n = 8) compared with neurocognitively normal (NcN) cases (n = 17). Samples were run on two separate blots due to large number of sample sizes. Quantification was conducted as described for ATF6α in the Figure 3 legend. For cases run on both gels, normalized values were averaged and are represented using closed circles. Cases run on only one gel are represented using open circles. (C) peIF2α levels increased significantly in HIV(+) (n = 21) tissue compared with HIV(−) (control) tissue (n = 4). (D) Cases were divided into three groups: (i) HIV(−)/neurocognitively normal; (ii) HIV(+)/neurocognitively normal; and (iii) HAND. Groups were compared using one‐way anova and no significant differences were found. Coomassie staining of the PVDF membrane was used as a loading control for normalization of protein levels for quantification and statistical analysis as shown in B–D. #P < 0.05, Student's t‐test for two‐way comparisons and one‐way anova for three‐way comparisons and Mann–Whitney U post‐hoc analysis.
Changes in ATF4 levels do not suggest functional activation as a transcription factor
While phosphorylation of eIF2α results in a global inhibition of translation, it also typically results in an increased rate of translation of certain ISR proteins, such as the transcription factor, ATF4 [10]. Thus, given the increases we observed in peIF2α levels, we sought to determine whether ATF4 levels were increased in mid‐frontal cortex from HAND autopsy cases. Interestingly, via IFA we found that total levels of ATF4 decreased in HAND cases as compared with neurocognitively normal cases (Figure 7A,B ). ATF4 staining was both nuclear and cytoplasmic and when we examined ATF4 that colocalized with DAPI we found that, like total ATF4 levels, nuclear ATF4 also decreased in HAND cases (Figure 8E ). However, we saw no change in total ATF4 (Figure 7C ) or total nuclear ATF4 levels (Figure 8F ) in HIV(+) cases compared with those seen in HIV(−) cases. Interestingly, when we examined cell type‐specific levels of cytoplasmic ATF4, we found that, while neuronal cytoplasmic ATF4 levels show no changes with either neurocognitive status or HIV status (Figure 8A,B ), astrocytic cytoplasmic ATF4 actually increased in HAND cases over neurocognitively normal cases and in HIV(+) cases over HIV(−) cases (Figure 8C,D ). However, as can be seen in the representative images (Figure 7A ), ATF4 staining was predominantly neuronal with a tendency towards strong nuclear staining in these cells, so it is not surprising that total ATF4 levels parallel total nuclear ATF4 levels, with cytoplasmic levels showing different trends. Interestingly, when we examined astrocytic nuclear ATF4, by isolating astrocytic nuclei manually as we did for ATF6β, we found that it did not change with either neurocognitive status or HIV infection status, suggesting that the increases in BiP levels we had observed previously in astrocytes [52] were not due to transactivation by ATF4. Of note, ATF4 staining was detected only in MAP2‐ or GFAP‐positive cells.
Figure 7.
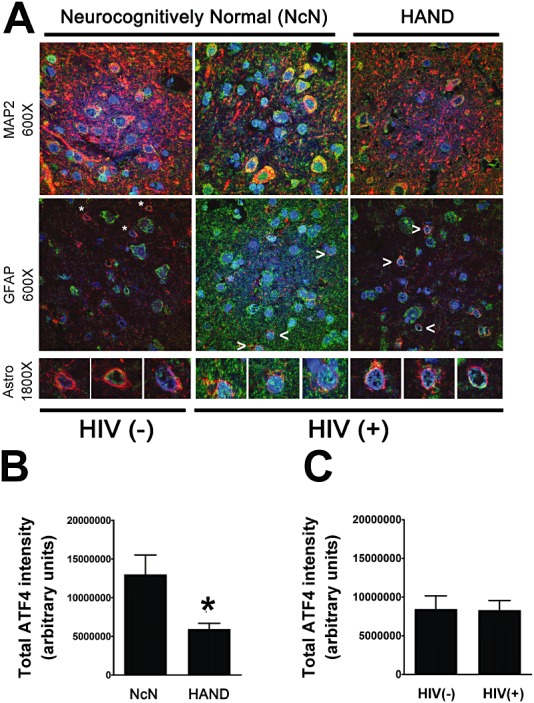
Total ATF4 decreases in HAND tissue compared with neurocognitively normal tissue. Paraffin‐embedded cortical autopsy tissue was quadruple labelled for ATF4, GFAP, MAP2 and DAPI. (A) Grey matter in labelled slides was imaged using laser confocal microscopy: ATF4 is shown in green, GFAP and MAP2 are shown in red, and DAPI is shown in blue. Overlap of ATF4 with a cell‐type marker appears yellow. ATF4 overlap with DAPI appears blue‐green. An example is shown for an HIV(−) uninfected control, an HIV(+) neurocognitively normal case, and a HAND case for each cell‐type marker. The third panel shows higher magnification images of astrocytes, which are denoted by asterisks [HIV(−) control] or arrowheads [HIV(+) cases]. (B and C) ATF4 protein levels from confocal images (five per case) were quantified by determining integrated pixel intensity using Metamorph 6.0 software. Total ATF4 decreases in HAND cases (n = 12) over neurocognitively normal cases (n = 5), but shows no change in HIV(+) cases (n = 14) compared with HIV(−) (n = 3) controls (C). Values are shown as mean ± SEM. Student's t‐test and Mann–Whitney U post‐hoc analysis was used for statistical analysis to determine significance. *P < 0.05, compared with control.
Figure 8.
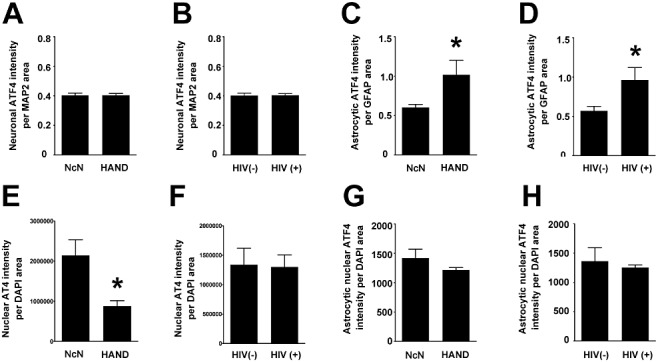
While total nuclear ATF4 protein levels parallel total ATF4 protein levels, cytoplasmic neuronal ATF4 and nuclear astrocytic ATF4 do not change with disease state and cytoplasmic astrocytic ATF4 increases in both HAND and HIV(+) tissue compared with neurocognitively normal and HIV(−) tissue, respectively. Using Metamorph 6.0 software, total integrated pixel intensity of ATF4 was quantified and colocalization with cell‐type markers was determined and normalized to area of cell‐type marker to account for losses of marker protein due to disease. (A and B) Neuronal cytoplasmic ATF4 does not change with disease state. (C and D) ATF4 in the cytoplasm of astrocytes increases in both HAND cases and HIV(+) cases when compared to neurocognitively normal and HIV(−) cases, respectively. (E and F) Total nuclear ATF4 protein levels decrease in HAND cases over neurocognitively normal cases, but do not change in HIV(+) cases over that seen in HIV(−) cases. (G and H) Astrocytic nuclear ATF4 levels do not change in either HAND or HIV(+) cases when compared to neurocognitively normal or HIV(−) cases, respectively. Values are shown as mean ± SEM. Student's t‐test and Mann–Whitney U post‐hoc analysis was used for statistical analysis to determine significance. *P < 0.05, compared with control.
When we examined ATF4 levels via immunoblotting, we saw no significant changes across experimental groups (Figure 9), although ATF4 levels trended towards a decrease in HIV(+) tissue compared with HIV(−) tissue (Figure 9B,C ). Given the variety of ATF4 responses in different cell types and subcellular compartments, the lack of significance and inconsistency in data collected via immunoblot is not unexpected, as the ratio of different types of cells is often different between fresh‐frozen tissue samples and paraffin‐embedded samples.
Figure 9.
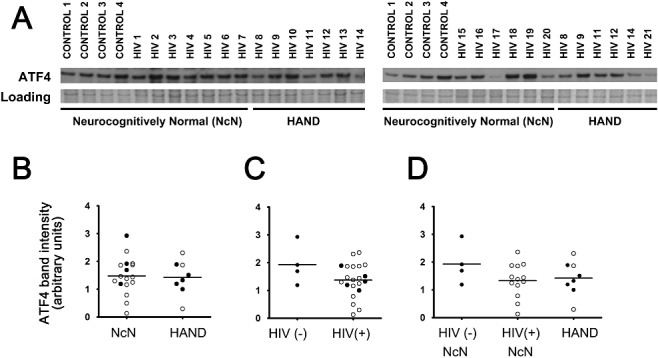
ATF4 levels do not change across experimental groups when examined via immunoblot analysis. Whole‐cell protein extracts were isolated from fresh‐frozen cortical autopsy tissue and were used for assessment of ATF4 protein levels. (A) Blots showing ATF4 levels in autopsy tissue, as well as a band from Coomassie staining of the PVDF membrane: HIV(−)/neurocognitively normal (control), HIV(+)/neurocognitively normal, and HAND. (B) ATF4 expression does not change in the mid‐frontal cortices of HAND cases (n = 8) compared with neurocognitively normal (NcN) cases (n = 17). Samples were run on two separate blots due to large number of sample sizes. Quantification was conducted as described for ATF6α in the Figure 3 legend. For cases run on both gels, normalized values were averaged and are represented using closed circles. Cases run on only one gel are represented using open circles. (C) ATF4 levels did not significantly change in HIV(+) (n = 21) tissue compared with HIV(−) (control) tissue (n = 4). (D) Cases were divided into three groups: (i) HIV(−)/neurocognitively normal; (ii) HIV(+)/neurocognitively normal; and (iii) HAND. Groups were compared using one‐way anova and no significant differences were found. Coomassie staining of the PVDF membrane was used as a loading control for normalization of protein levels for quantification and statistical analysis as shown in B–D. Student's t‐test for two‐way comparisons and one‐way anova for three‐way comparisons and Mann–Whitney U post‐hoc analysis.
CHOP and HSP70 protein levels do not change in HAND or in HIV(+) mid‐frontal cortical autopsy tissue
Often with ISR activation, not all target genes are equally transactivated [21, 22, 25, 28, 53, 54, 55, 57]. Thus, we sought to examine additional traditional ISR gene targets beyond BiP, which we had examined previously [52]. Specifically, we examined protein levels of CHOP and HSP70 via immunoblotting. We found no significant changes for either protein (Figure 10). However, CHOP levels unexpectedly appeared to trend towards a decrease in HIV(+) tissue, both neurocognitively normal and HAND, compared with HIV(−)/neurocognitively normal cases (Figure 10A,C,D ). These data suggest that different ISR gene targets are differentially transactivated in the cortex of HIV(+) patients.
Figure 10.
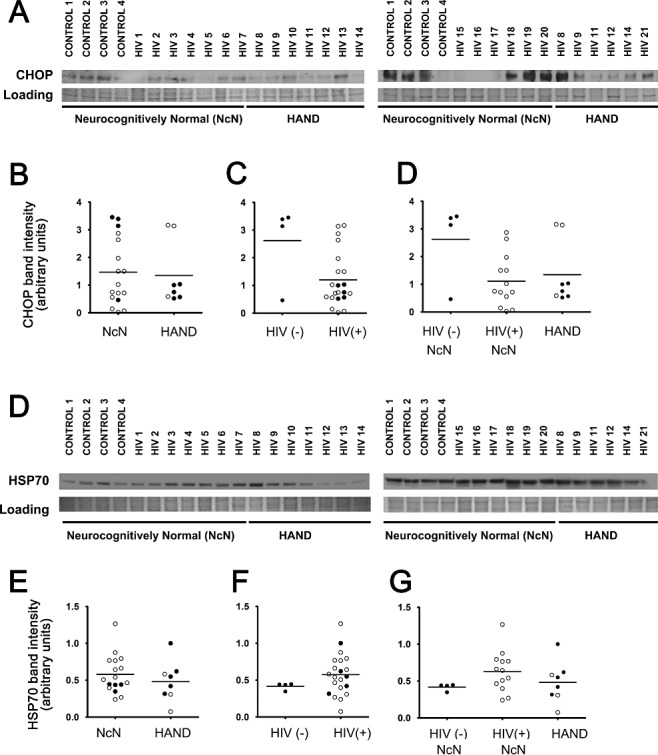
CHOP and HSP70 levels do not change across experimental groups when examined via immunoblot analysis. Whole‐cell protein extracts were isolated from fresh‐frozen cortical autopsy tissue and were used for assessment of CHOP and HSP70 protein levels. (A and D) Blots showing CHOP (A) and HSP70 (D) levels in autopsy tissue, as well as a band from Coomassie staining of the PVDF membrane: HIV(−)/neurocognitively normal (control), HIV(+)/neurocognitively normal, and HAND. (B and E) CHOP and HSP70 expression levels do not change in the mid‐frontal cortices of HAND cases (n = 8) compared with neurocognitively normal (NcN) cases (n = 17). Samples were run on two separate blots due to large number of sample sizes. Quantification was conducted as described for ATF6α in the Figure 3 legend. For cases run on both gels, normalized values were averaged and are represented using closed circles. Cases run on only one gel are represented using open circles. (C and F) CHOP and HSP70 levels did not significantly change in HIV(+) (n = 21) tissue compared with HIV(−) (control) tissue (n = 4). (D and G) Cases were divided into three groups: (i) HIV(−)/neurocognitively normal; (ii) HIV(+)/neurocognitively normal; and (iii) HAND. Groups were compared using one‐way anova and no significant differences were found for either CHOP or HSP70. Coomassie staining of the PVDF membrane was used as a loading control for normalization of protein levels for quantification and statistical analysis as shown in B–D. Student's t‐test for two‐way comparisons and one‐way anova for three‐way comparisons and Mann–Whitney U post‐hoc analysis.
Discussion
The direct cause and mechanism of neuronal degeneration and death in HAND is not currently understood. In this paper, we explore the nature of ISR activation in this disease, as we had previously reported an increase of BiP, an indicator of ISR activation, in the CNS of HIV(+) patients. While the ISR is largely a protective response, it can also culminate in a pro‐apoptotic response under certain conditions, and thus our observations of ISR activation in HAND required further investigation to determine whether the ISR ultimately provides protection or causes neuronal death or perhaps a combination of both in this disease. The findings presented here and summarized in 11, 12 and in Table 2 demonstrate the nuanced nature of the ISR, showing that it is not an all‐or‐nothing response and that it can vary across cell types.
Figure 11.
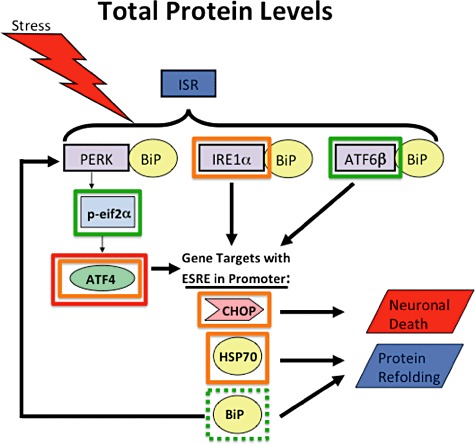
Diagram summarizing results of total protein levels for each protein examined. For ISR proteins outlined with green boxes, we observed increased protein levels. Specifically, for ATF6 and peIF2α we saw increases in both HAND and HIV(+) tissue compared with respective control groups when compared via IFA. When compared via immunoblot, ATF6 increased in HAND tissue over NcN tissue when compared via immunoblot, while peIF2α increased in HIV(+) tissue when compared with HIV(−) tissue. BiP (outlined in a dashed green box) was analysed previously [52] and increased via IFA and immunoblot. For proteins outlines with orange boxes, we saw no change. Specifically, IRE1α, CHOP and HSP70 showed no change across groups by immunoblot. ATF4 showed no change across groups by immunoblot, and showed no change in HIV(+) tissue over HIV(−) tissue via IFA. ATF is also outlined with a red box indicating that its levels decreased in HAND tissue compared with neurocognitively normal tissue via IFA.
Figure 12.
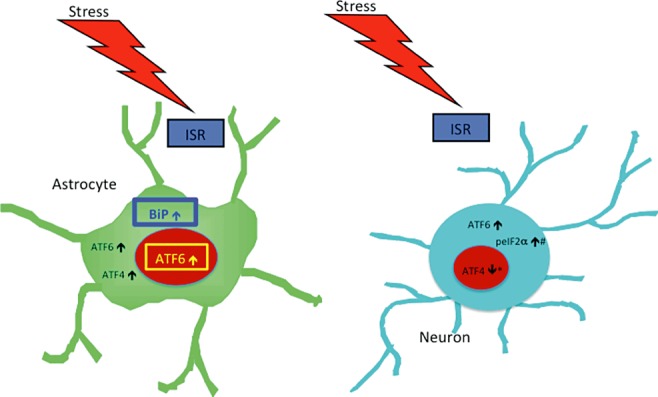
Diagram summarizing results of cell type‐specific protein levels for each protein examined. Changes observed in nuclear and cytoplasmic compartments are indicated in each cell‐type examined (neurones and astrocytes). Of particular interest, ATF6β in the only transcription we observed to increase in the nucleus of astrocytes, making it a particularly promising candidate as the ISR initiator protein responsible for our previously observed increase in astrocytic BiP levels [52]. Note, only proteins that exhibited a change in protein levels are indicated and not all proteins were examined for both subcellular compartments in both cell types (see chart 1 for reference). #: only determined for neurones because GFAP staining was not of a quality and consistency to allow for quantification of colocalization using this marker; *: indicates increase observed in total nuclear ATF4 levels, but because this staining was predominantly neuronal nuclei it was likely indicative of neuronal nuclear ATF4.
In our previous paper [52], we had shown that, in addition to an increase in BiP protein levels in HIV(+) and HAND tissue, ATF6β increased as seen by immunoblotting. This finding is in agreement with studies suggesting that ATF6 is upregulated at the transcriptional level by ISR activation in a positive feedback mechanism [68, 69]. Here we continued our examination of the ATF6β response in HIV(+) cortical tissue using IFA. We first confirmed our previous data, showing via IFA that total ATF6β increases in both HAND and HIV(+) cases when compared with neurocognitively normal and HIV(−) cases, respectively. Importantly, IFA allows examination of protein levels as they vary across subcellular compartments and across different cell types. ATF6 is a transcription factor that has two forms, ATF6α and ATF6β, that are both activated under stress conditions by translocation to the Golgi, followed by proteolytic cleavage, which allows its active, cleaved form to translocate to the nucleus and induce expression of target genes [65, 66, 67]. Manifestly, only nuclear ATF6 is able to perform its transcriptional function. In this report, we found that total nuclear ATF6β did not change in HAND or HIV(+) cases compared with controls, although astrocytic nuclear ATF6β did increase significantly in both HAND and HIV(+) tissue, suggesting that activation of ATF6β may be an important contributor to the increased BiP protein that we had previously observed in astrocytes in HIV(+) tissue. However, despite our findings in astrocytes, it is interesting to note that the majority of the total ATF6β staining was clearly non‐nuclear. These findings likely suggest that even though ATF6β is increased in HAND, it may not be efficiently moving into the nucleus to activate its target genes, particularly in neurones, although one must consider the caveat that some nuclear ATF6β may not be observable by IFA due to protein complexes formed during its nuclear activities.
ATF6 is cleaved to its active form by regulated intramembrane proteolysis by site‐1 protease (S1P) and site‐2 protease (S2P) working together consecutively [66, 67]. Several studies have shown that ATF6 translocates to the Golgi, but then fails to move to the nucleus in disease states or in models [70, 71]. One study found that pharmaceutical inhibition of the serine proteases prevented cleavage of ATF6 and thus prevented its nuclear localization [70]. This inhibition did not, however, inhibit ATF6 translocation to the Golgi [70]. Another study identified nucleobindin 1 as an endogenous inhibitor of S1P and found that overexpression of this inhibitor did not inhibit ATF6 localization in the Golgi, but prevented cleavage to its active, nuclear form [71]. Finally, mutant Chinese hamster ovary cells, M19 cells, are defective in induction of BiP and are deficient in S2P activity [72, 73]. Nadanaka et al. [74] found that some of the ATF6 in these cells is constitutively localized to the Golgi. Considering these findings, it is possible that ATF6β in HAND tissue is translocating to the Golgi but is then unable to move to the nucleus. It would be interesting to conduct IFA in HAND cortical autopsy tissue co‐labelling for ATF6β and a Golgi matrix marker, such as GM130, to determine if ATF6β is perhaps getting trapped in the Golgi in this disease. Alternatively, the hepatitis C virus NS4B protein is known to physically interact with ATF6β[34], indicating the potential for this virus to modulate ATF6‐meditated transcriptional activity and possibly to limit the subcellular mobility of this protein. This is of interest as HIV patients, particularly at the end of life, often suffer from many comorbidities, commonly including hepatitis C infection [75, 76, 77, 78, 79, 80].
The implications of a lack of nuclear localization of ATF6β in neurones are not clear. As mentioned previously, ATF6 has two forms, both of which are activated by stressors and are proteolytically cleaved to their active forms in the Golgi. The active form of ATF6α is a stronger, yet much shorter‐lived transcriptional activator than is ATF6β[81, 82]. The roles of these two isoforms of ATF6 remain unclear. Some studies have suggested that ATF6β, when localized to the nucleus, is an inhibitor of ATF6α transcriptional function [82], suggesting that ATF6β may function to regulate the strength and duration of ATF6α activity [81]. Interestingly, ATF6β undergoes N‐glycosylation, which is necessary for its proteolytic cleavage, but is not necessary for its translocation to the Golgi [83]. This may allow for control in the timing of ATF6β transcriptional repression of ATF6α activity. As we see increased ATF6β protein levels, but not high levels of nuclear ATF6β, particularly in neurones, it is possible that its N‐glycosylation is being prevented to allow continued transcriptional activation of ATF6α target genes.
In contrast to studies suggesting a transcriptional repressor function for ATF6β, other studies have found that, although ATF6β is a much less potent inducer of transcription, it is still able to provide some protection against stress by its induction of target ER stress genes [84]. Taken together with the fact that the transcriptionally active fragment of ATF6α is rapidly degraded, while that of ATF6β is longer‐lived, these findings that ATF6β may not be a transcriptional repressor of ATF6α, but rather an activator of ER stress genes, suggest the possibility that ATF6α may act as a strong immediate, but temporally limited, response to stress, while ATF6β would be responsible for a lower‐level, longer‐lived response to chronic stress, such as that observed in HAND [81, 85]. The nature of the coordination between theses two isoforms of ATF6 is a highly interesting and widely applicable topic that warrants future investigations across numerous disease states.
Finally, we did observe a significant increase in ATF6β in the nuclei of astrocytes, while this does not appear to be the case with neurones. These findings suggest that this pathway of the ISR shows definite differences between cell types in this disease. However, it is unclear whether its nuclear localization is prevented in neurones as a protective measure to avoid the apoptosis that can be associated with ISR activation, or whether nuclear localization of ATF6β in neurones would be beneficial, but is failing in the CNS of HAND patients. Overexpression of a cleaved, and therefore nuclear and active, form of ATF6β in neurones and astrocytes individually in in vitro and in vivo models of HIV‐associated neurodegeneration would provide useful information in this regard. Interestingly, of the ISR proteins examined here, only ATF6β appeared to localize to cells not positive for MAP2 or GFAP. Specifically, it appeared to show small amounts of colocalization with DAPI in cells whose nuclear morphology resembled that of oligodendrocytes, further implicating the ATF6 pathway as a contributor to BiP increases in HAND cortex [52]. Because the IFA protocols in this study were optimized to detect ISR markers in astrocytes and neurones as a follow‐up to our previous report [52], it is not improbable that other cell types express these proteins, but that they were simply not detectable by protocols used here. Future in vivo and in vitro studies designed to examine the ISR in other cell types in HAND cortex would be enlightening.
Viral activation and utilization of the IRE1α/XBP1 pathway of the ISR is well documented, in particular among the neurotropic RNA flaviviruses, such as West Nile virus, Japanese encephalitis virus, and dengue virus [28, 86, 87], as well as the RNA alphavirus, Semliki Forest virus [88]. Here, unable to analyse XBP1 mRNA levels due to the nature of human autopsy tissue processing, we report that total IRE1α protein levels do not change significantly in HIV(+) cortex or in HAND cortex compared with respective control groups. However, unexpectedly, it seems to trend towards a decrease in HIV(+) tissue. Interestingly, several viruses, including the severe acute respiratory syndrome (SARS) coronavirus, the hepatitis C virus, and the human cytomegalovirus, known to activate certain components of the ISR, have also been found to either not activate the IRE1α/XBP1 arm [33] or actively inhibit it at some level [30, 31, 32]. Further, HPV‐16 E5 protein has been shown to downregulate not only spliced XBP1, but also IRE1α itself [24]. In vitro models of HIV infection of the CNS, as well as in vivo animal models, are critical to further elucidate the role of the IRE1α/XBP1 pathway in the brain in this disease, particularly because analysis of XBP1 splicing is key to fully understanding this pathway, and thus systems which allow harvest of mRNA are needed.
In this study, we found that peIF2α increases in HIV(+) tissue compared with HIV(−) control tissue and by IFA shows a slight but significant increase in HAND tissue compared with neurocognitively normal controls. Phosphorylation of eIF2α inhibits its function in the initiation of translation causing a general suppression of the translation of new proteins [89, 90]. However, the phosphorylation of eIF2α actually increases the translation of a subset of proteins that include an internal ribosome entry site (IRES) in their 5′ untranslated region [91], such as ATF4. Importantly, eIF2α is the target of several other kinases in addition to PERK, including double‐stranded RNA‐activated protein kinase (PKR) and GCN2 [49]. As mentioned previously, the ISR possesses both pro‐survival and pro‐apoptotic roles [92], and peIF2α is capable of both [93]. In particular, one study suggested that the timing of phosphorylation of eIF2α may determine whether it is pro‐survival or pro‐apoptotic in its function, and further, this study suggested that the nature of the role of peIF2α in a given stress response may be influenced by the kinase that phosphorylates it under any given stress [93]. While all kinases known to target peIF2α do phosphorylate it on its serine 51 (S51), these kinases themselves are differentially regulated and perhaps the timing of their activation or the identities of their other target proteins affect whether peIF2α will play a pro‐survival or a pro‐apoptotic role. Interestingly, while we examined eIF2α as a part of the PERK pathway of the ISR, one must consider that another kinase could potentially phosphorylate eIF2α in the cortex of these patients. Of particular interest is PKR, given that it responds to double‐stranded RNA, as well as to interferon [49], and also considering that the increase of peIF2α was more striking in HIV(+) tissue over HIV(−) tissue, than in HAND tissue over neurocognitively normal controls. Muaddi et al. [93] show data suggesting a pro‐survival role for PERK and PKR, and a pro‐apoptotic role for GCN2, although other studies have suggested that PKR often has pro‐death function.
We next examined ATF4, which is a downstream target of the PERK/eIF2α pathway of the ISR. Interestingly, given our finding that peIF2α increases in HIV(+) tissue, we found that total and nuclear ATF4 decreases in HAND tissue, although it shows no change in HIV(+) tissue, compared with controls. In contrast, we found that ATF4 levels do not change in the cytoplasm of neurones and actually increase in the cytoplasm of astrocytes. These apparently contradictory findings are reconcilable when one takes into account that ATF4 in the cytoplasm of astrocytes makes up a very small amount of the total observed ATF4 and also that several neuronal cells show very strong ATF4 nuclear staining, which is not surprising given that ATF4 is a transcription factor. Interestingly, we did not see any change in nuclear ATF4 in astrocytes, even though we observed an increase in cytoplasmic ATF4 in these cells. This finding strongly suggests that ATF4 is not contributing to the increased BiP levels we had observed previously in HAND [52].
One possible explanation for the general overall decrease in ATF4 levels despite an increase in peIF2α levels relates back to the multiple functionality of peIF2α. Conceivably, in HIV(+) tissue and in HAND, eIF2α may function mostly to inhibit global protein translation and may not promote the specific expression of ATF4 protein. Additionally, the increases of peIF2α staining were far more apparent in HIV(+) tissue than they were in HAND tissue compared with respective controls, while total and nuclear ATF4 decreases are seen in HAND tissue, but not in HIV(+) tissue, suggesting an alternate method of regulation of ATF4 expression in this case. Interestingly, although ATF4 is usually discussed as a translationally regulated protein, there is evidence that it is transcriptionally regulated as well. In particular, another study found that phosphorylation of eIF2α failed to increase expression of ATF4 in their model, and that, rather, ATF4 was regulated at the transcriptional level by TIR‐domain‐containing adapter‐inducing interferon‐β (TRIF)‐mediated signalling through toll‐like receptors 3 and 4 [94]. Further, a recent study suggests a stimulus‐dependent transcriptional regulation of ATF4 that alters the amount of ATF4 mRNA available for translational upregulation by peIF2α[95]. Finally, another study showed that, under conditions of oxidative stress, retinal pigment epithelial ARPE‐19/HPV‐16 cells demonstrate transcriptional regulation of ATF4 [96]. This study is particularly interesting in the context of viral infection of the brain because the cells studied in this paper are transformed with the HPV‐16 DH5 protein; another study has shown an HPV‐16 transforming protein, E5, capable of downregulating components of the ISR [24]. Whether the alteration in ATF4 expression we observed in the study presented here represents a failure of the ISR to protect in HAND or whether it is actually a regulatory mechanism functioning to protect cells from the pro‐apoptotic side of the ISR is unclear, but could be examined in an in vitro model of HIV‐induced neuronal degeneration.
Finally, to expand our study of classic ISR gene targets beyond BiP, we examined levels of CHOP and HSP70 via immunoblotting. We saw no significant change in either of these proteins, and surprisingly, CHOP even trended towards a decrease. As the tissue used in these studies is from the end stage of disease, it is certainly possible that most cells that expressed CHOP have already died, and thus are not contributing protein to lysates harvested from the cortical autopsy tissue. Alternatively, in ISR activation associated with viral infection, not all target genes are equally activated. Specifically, hepatitis C virus, an RNA virus, increases BiP, but not CHOP [29]. Additionally, African swine fever virus, a DNA virus, actively inhibits induction of CHOP [23]. While this property of active inhibition of apoptosis seems to be more common of DNA viruses than of RNA viruses, it is not inconceivable that HIV could employ a similar mechanism. That HIV does not productively infect neurones and astrocytes makes this scenario less appealing, but it still may warrant future investigation.
Here we present the initial studies designed to characterize the nature of the ISR response in HAND. Our findings clearly show that the ISR does not respond in an all‐or‐none fashion in HAND, but rather demonstrates a nuanced activation pattern in this disease, as summarized in Figure 11 and in Table 1. In particular, our finding that ATF6 increases in the nuclei of astrocytes while ATF4 does not, implicates the ATF6 pathway of the ISR as a more likely candidate than the PERK pathway for the increases in BiP levels in astrocytes that we had observed previously [52] (summarized in Figure 12). Of note, the neurovirulent Moloney murine leukemia virus, which causes a syndrome in mice similar to the immunodeficiency and neurodegeneration seen in HIV‐infected patients, also induces ISR activation in astrocytes [97]. That ISR activation is increased in astrocytes in these viral infections is interesting. While increases in protective ISR proteins such as BiP are likely beneficial to astrocytes, chronic astrocytic activation of other components of the ISR might eventually contribute to neuronal toxicity in this disease. Specifically, the xCT subunit of the cysteine/glutamate exchange transporter (xC) is a downstream gene target of the ISR through transactivation by the bZIP transcription factor Nrf2 [98, 99]. xC plays a critical role in the production of glutathione, an important component of the cellular antioxidant response; however, to do so, it exports astrocytic glutamate into the extracellular milieu, potentially contributing to an excitotoxic environment and ultimately to neuronal death [100, 101]. The data presented in this paper compose an important novel foray into the characterization of the ISR response in HAND, which will be necessary if we are to understand whether this catch‐all stress pathway is merely unsuccessful in its attempts to protect neurones from death in the toxic environment of the HIV‐infected CNS and should thus be supported in its function via therapeutic intervention or whether the ISR is actively destroying neurones in this disease and should therefore be inhibited by any potential therapeutic interventions. Alternatively, it may be the case and is likely that some aspects of the ISR are protective in this disease while others are apoptotic. Mechanistic in vitro studies and animal models of HIV infection, such as SIV‐infected macaques, will be invaluable in gaining a full understanding into the nuances of ISR activation following HIV‐infection, which is vital to the development of therapies to prevent progression to HAND and to treat patients already suffering from this devastating disease.
Cagla Akay and Kathryn A. Lindl contributed equally.
Grants: This work was supported by the following grants: NS056885 (KJS), NS043994 (DLK).
References
- 1. Akay L, Coye MJ. Implementing an employee portal. Taking steps toward a ‘paperless’ future. Healthplan 2001; 42: 66–9 [PubMed] [Google Scholar]
- 2. McArthur JC, Haughey N, Gartner S, Conant K, Pardo C, Nath A, Sacktor N. Human immunodeficiency virus‐associated dementia: an evolving disease. J Neurovirol 2003; 9: 205–21 [DOI] [PubMed] [Google Scholar]
- 3. Simioni S, Cavassini M, Annoni JM, Rimbault Abraham A, Bourquin I, Schiffer V, Calmy A, Chave JP, Giacobini E, Hirschel B, Du Pasquier RA. Cognitive dysfunction in HIV patients despite long‐standing suppression of viremia. AIDS 2010; 24: 1243–50 [DOI] [PubMed] [Google Scholar]
- 4. Gorry PR, Ong C, Thorpe J, Bannwarth S, Thompson KA, Gatignol A, Vesselingh SL, Purcell DF. Astrocyte infection by HIV‐1: mechanisms of restricted virus replication, and role in the pathogenesis of HIV‐1‐associated dementia. Curr HIV Res 2003; 1: 463–73 [DOI] [PubMed] [Google Scholar]
- 5. Kaul M, Zheng J, Okamoto S, Gendelman HE, Lipton SA. HIV‐1 infection and AIDS: consequences for the central nervous system. Cell Death Differ 2005; 12 (Suppl. 1): 878–92 [DOI] [PubMed] [Google Scholar]
- 6. Ozdener H. Molecular mechanisms of HIV‐1 associated neurodegeneration. J Biosci 2005; 30: 391–405 [DOI] [PubMed] [Google Scholar]
- 7. Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol 2008; 40: 14–21 [DOI] [PubMed] [Google Scholar]
- 8. Albright AV, Soldan SS, Gonzalez‐Scarano F. Pathogenesis of human immunodeficiency virus‐induced neurological disease. J Neurovirol 2003; 9: 222–7 [DOI] [PubMed] [Google Scholar]
- 9. Gonzalez‐Scarano F, Martin‐Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol 2005; 5: 69–81 [DOI] [PubMed] [Google Scholar]
- 10. Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest 2002; 110: 1389–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK‐dependent cell survival. Mol Cell Biol 2003; 23: 7198–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hammond C, Helenius A. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J Cell Biol 1994; 126: 41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paschen W. Disturbances of calcium homeostasis within the endoplasmic reticulum may contribute to the development of ischemic‐cell damage. [Review][57 refs]. Med Hypotheses 1996; 47: 283–8 [DOI] [PubMed] [Google Scholar]
- 14. Schroder M, Kaufman RJ. ER stress and the unfolded protein response. [Review][328 refs]. Mutat Res 2005; 569: 29–63 [DOI] [PubMed] [Google Scholar]
- 15. Williams BL, Lipkin WI. Endoplasmic reticulum stress and neurodegeneration in rats neonatally infected with borna disease virus. J Virol 2006; 80: 8613–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang K, Kaufman RJ. Signaling the unfolded protein response from the endoplasmic reticulum. [Review][60 refs]. J Biol Chem 2004; 279: 25935–8 [DOI] [PubMed] [Google Scholar]
- 17. Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the Mammalian unfolded protein response. [Review][95 refs]. Annu Rev Cell Dev Biol 2002; 18: 575–99 [DOI] [PubMed] [Google Scholar]
- 18. Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. [Review][186 refs]. Genes Dev 1999; 13: 1211–33 [DOI] [PubMed] [Google Scholar]
- 19. Buchkovich NJ, Yu Y, Pierciey FJ, Alwine JC. human cytomegalovirus induces the endoplasmic reticulum chaperone BiP through increased transcription and activation of translation by using the BiP internal ribosome entry site. J Virol 2010; 84: 11479–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng G, Feng Z, He B. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF‐2 dephosphorylation by the 134.5 protein. J Virol 2005; 79: 1379–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jheng J‐R, Lau KS, Tang W‐F, Wu M‐S, Horng J‐T. Endoplasmic reticulum stress is induced and modulated by enterovirus 71. Cell Microbiol 2010; 12: 796–813 [DOI] [PubMed] [Google Scholar]
- 22. Mulvey M, Arias C, Mohr I. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1‐infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J Virol 2007; 81: 3377–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Netherton CL, Parsley JC, Wileman T. African swine fever virus inhibits induction of the stress‐induced proapoptotic transcription factor CHOP/GADD153. J Virol 2004; 78: 10825–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sudarshan SR, Schlegel R, Liu X. The HPV‐16 E5 protein represses expression of stress pathway genes XBP‐1 and COX‐2 in genital keratinocytes. Biochem Biophys Res Commun 2010; 399: 617–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tardif KD, Mori K, Siddiqui A, Hepatitis C. Virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J Virol 2002; 76: 7453–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xuan B, Qian Z, Torigoi E, Yu D. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress‐induced cell death. J Virol 2009; 83: 3463–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang HM, Ye X, Su Y, Yuan J, Liu Z, Stein DA, Yang D. Coxsackievirus B3 infection activates the unfolded protein response and induces apoptosis through downregulation of p58IPK and activation of CHOP and SREBP1. J Virol 2010; 84: 8446–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ambrose RL, Mackenzie JM. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J Virol 2010; 85: 2723–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joyce MA, Walters KA, Lamb SE, Yeh MM, Zhu LF, Kneteman N, Doyle JS, Katze MG, Tyrrell DL. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb‐uPA mice. PLoS Pathog 2009; 5: e1000291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Isler JA, Skalet AH, Alwine JC. Human cytomegalovirus infection activates and regulates the unfolded protein response. J Virol 2005; 79: 6890–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tardif KD. Hepatitis C virus suppresses the IRE1‐XBP1 Pathway of the unfolded protein response. J Biol Chem 2004; 279: 17158–64 [DOI] [PubMed] [Google Scholar]
- 32. Zheng Y, Gao B, Ye L, Kong L, Jing W, Yang X, Wu Z. Hepatitis C virus non‐structural protein NS4B can modulate an unfolded protein response. J Microbiol 2005; 43: 529–36 [PubMed] [Google Scholar]
- 33. Chan CP, Siu KL, Chin KT, Yuen KY, Zheng B, Jin DY. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J Virol 2006; 80: 9279–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tong WY, Nagano‐Fujii M, Hidajat R, Deng L, Takigawa Y, Hotta H. Physical interaction between hepatitis C virus NS4B protein and CREB‐RP/ATF6beta. Biochem Biophys Res Commun 2002; 299: 366–72 [DOI] [PubMed] [Google Scholar]
- 35. Paschen W, Yatsiv I, Shoham S, Shohami E. Brain trauma induces X‐box protein 1 processing indicative of activation of the endoplasmic reticulum unfolded protein response. J Neurochem 2004; 88: 983–92 [DOI] [PubMed] [Google Scholar]
- 36. Benavides A, Pastor D, Santos P, Tranque P, Calvo S. CHOP plays a pivotal role in the astrocyte death induced by oxygen and glucose deprivation. Glia 2005; 52: 261–75 [DOI] [PubMed] [Google Scholar]
- 37. Rissanen A, Sivenius J, Jolkkonen J. Prolonged bihemispheric alterations in unfolded protein response related gene expression after experimental stroke. Brain Res 2006; 1087: 60–6 [DOI] [PubMed] [Google Scholar]
- 38. Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase‐12 mediates endoplasmic‐reticulum‐specific apoptosis and cytotoxicity by amyloid‐beta. Nature 2000; 403: 98–103 [DOI] [PubMed] [Google Scholar]
- 39. Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 2001; 105: 891–902 [DOI] [PubMed] [Google Scholar]
- 40. Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci 2002; 22: 10690–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McCullough KD, Martindale JL, Klotz L‐O, Aw T‐Y, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down‐regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 2001; 21: 1249–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rao RV, Peel A, Logvinova A, del Rio G, Hermel E, Yokota T, Goldsmith PC, Ellerby LM, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett 2002; 514: 122–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Coye MJ, Jacks G, Everett WE, Akay L. Medical group adoption of Internet services. J Ambul Care Manage 2001; 24: 67–75 [DOI] [PubMed] [Google Scholar]
- 44. Akay C, Lindl KA, Wang Y, White MG, Isaacman‐Beck J, Kolson DL, Jordan‐Sciutto KL. Site‐specific hyperphosphorylation of pRb in HIV‐induced neurotoxicity. Mol Cell Neurosci 2011; 47: 154–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. White MG, Wang Y, Akay C, Lindl KA, Kolson DL, Jordan‐Sciutto KL. Parallel high throughput neuronal toxicity assays demonstrate uncoupling between loss of mitochondrial membrane potential and neuronal damage in a model of HIV‐induced neurodegeneration. Neurosci Res 2011; 70: 220–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang Y, Shyam N, Ting JH, Akay C, Lindl KA, Jordan‐Sciutto KL. E2F1 localizes predominantly to neuronal cytoplasm and fails to induce expression of its transcriptional targets in human immunodeficiency virus‐induced neuronal damage. Neurosci Lett 2010; 479: 97–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kohno K. How transmembrane proteins sense endoplasmic reticulum stress. Antioxid Redox Signal 2007; 9: 2295–303 [DOI] [PubMed] [Google Scholar]
- 48. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 1999; 10: 3787–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans 2006; 34: 7–11 [DOI] [PubMed] [Google Scholar]
- 50. Wang Y, White MG, Akay C, Chodroff RA, Robinson J, Lindl KA, Dichter MA, Qian Y, Mao Z, Kolson DL, Jordan‐Sciutto KL. Activation of cyclin‐dependent kinase 5 by calpains contributes to human immunodeficiency virus‐induced neurotoxicity. J Neurochem 2007; 103: 439–55 [DOI] [PubMed] [Google Scholar]
- 51. McArthur JC. HIV dementia: an evolving disease. J Neuroimmunol 2004; 157: 3–10 [DOI] [PubMed] [Google Scholar]
- 52. Lindl KA, Akay C, Wang Y, White MG, Jordan‐Sciutto KL. Expression of the endoplasmic reticulum stress response marker, BiP, in the central nervous system of HIV‐positive individuals. Neuropathol Appl Neurobiol 2007; 33: 658–69 [DOI] [PubMed] [Google Scholar]
- 53. Lee D, Singaravelu G, Park BJ, Ahnn J. Differential requirement of unfolded protein response pathway for calreticulin expression in Caenorhabditis elegans. J Mol Biol 2007; 372: 331–40 [DOI] [PubMed] [Google Scholar]
- 54. Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress‐responsive cis‐acting elements ERSE, UPRE and ERSE‐II. J Biochem 2004; 136: 343–50 [DOI] [PubMed] [Google Scholar]
- 55. Gjymishka A, Su N, Kilberg MS. Transcriptional induction of the human asparagine synthetase gene during the unfolded protein response does not require the ATF6 and IRE1/XBP1 arms of the pathway. Biochem J 2009; 417: 695–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cho HK, Cheong KJ, Kim HY, Cheong J. Endoplasmic Reticulum stress induced by Hepatitis B virus X protein enhances cyclooxygenase 2 expression via activating transcription factor‐4. Biochem J 2011; 435: 431–9 [DOI] [PubMed] [Google Scholar]
- 57. Johnson JS, Gentzsch M, Zhang L, Ribeiro CM, Kantor B, Kafri T, Pickles RJ, Samulski RJ. AAV exploits subcellular stress associated with inflammation, endoplasmic reticulum expansion, and misfolded proteins in models of cystic fibrosis. PLoS Pathog 2011; 7: e1002053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu Y‐P, Chang C‐M, Hung C‐Y, Tsai M‐C, Schuyler SC, Wang R. Japanese encephalitis virus co‐opts the ER‐stress response protein GRP78 for viral infectivity. Virol J 2011; 8: 128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nikawa J, Yamashita S. IRE1 encodes a putative protein kinase containing a membrane‐spanning domain and is required for inositol phototrophy in Saccharomyces cerevisiae. Mol Microbiol 1992; 6: 1441–6 [DOI] [PubMed] [Google Scholar]
- 60. Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993; 73: 1197–206 [DOI] [PubMed] [Google Scholar]
- 61. Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem 2000; 275: 27013–20 [DOI] [PubMed] [Google Scholar]
- 62. Li M, Baumeister P, Roy B, Phan T, Foti D, Luo S, Lee AS. ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress‐induced changes and synergistic interactions with NF‐Y and YY1. Mol Cell Biol 2000; 20: 5096–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ. IRE1‐mediated unconventional mRNA splicing and S2P‐mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev 2002; 16: 452–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Masliah E, Roberts ES, Langford D, Everall I, Crews L, Adame A, Rockenstein E, Fox HS. Patterns of gene dysregulation in the frontal cortex of patients with HIV encephalitis. J Neuroimmunol 2004; 157: 163–75 [DOI] [PubMed] [Google Scholar]
- 65. Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell 2002; 3: 99–111 [DOI] [PubMed] [Google Scholar]
- 66. Chen X, Shen J, Prywes R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J Biol Chem 2002; 277: 13045–52 [DOI] [PubMed] [Google Scholar]
- 67. Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane‐bound ATF6 by the same proteases that process SREBPs. Mol Cell 2000; 6: 1355–64 [DOI] [PubMed] [Google Scholar]
- 68. Asselah T, Bièche I, Mansouri A, Laurendeau I, Cazals‐Hatem D, Feldmann G, Bedossa P, Paradis V, Martinot‐Peignoux M, Lebrec D, Guichard C, Ogier‐Denis E, Vidaud M, Tellier Z, Soumelis V, Marcellin P, Moreau R. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J Pathol 2010; 221: 264–74 [DOI] [PubMed] [Google Scholar]
- 69. von dem Bussche A, Machida R, Li K, Loevinsohn G, Khander A, Wang J, Wakita T, Wands JR, Li J. Hepatitis C virus NS2 protein triggers endoplasmic reticulum stress and suppresses its own viral replication. J Hepatol 2010; 53: 797–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Okada T, Haze K, Nadanaka S, Yoshida H, Seidah NG, Hirano Y, Sato R, Negishi M, Mori K. A serine protease inhibitor prevents endoplasmic reticulum stress‐induced cleavage but not transport of the membrane‐bound transcription factor ATF6. J Biol Chem 2003; 278: 31024–32 [DOI] [PubMed] [Google Scholar]
- 71. Tsukumo Y, Tomida A, Kitahara O, Nakamura Y, Asada S, Mori K, Tsuruo T. Nucleobindin 1 controls the unfolded protein response by inhibiting ATF6 activation. J Biol Chem 2007; 282: 29264–72 [DOI] [PubMed] [Google Scholar]
- 72. Hasan MT, Chang CC, Chang TY. Somatic cell genetic and biochemical characterization of cell lines resulting from human genomic DNA transfections of Chinese hamster ovary cell mutants defective in sterol‐dependent activation of sterol synthesis and LDL receptor expression. Somat Cell Mol Genet 1994; 20: 183–94 [DOI] [PubMed] [Google Scholar]
- 73. Rawson RB, Zelenski NG, Nijhawan D, Ye J, Sakai J, Hasan MT, Chang TY, Brown MS, Goldstein JL. Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol Cell 1997; 1: 47–57 [DOI] [PubMed] [Google Scholar]
- 74. Nadanaka S, Yoshida H, Sato R, Mori K. Analysis of ATF6 activation in Site‐2 protease‐deficient Chinese hamster ovary cells. Cell Struct Funct 2006; 31: 109–16 [DOI] [PubMed] [Google Scholar]
- 75. Boesecke C, Rockstroh JK. Treatment of acute hepatitis C infection in HIV‐infected patients. Curr Opin HIV AIDS 2011; 6: 278–84 [DOI] [PubMed] [Google Scholar]
- 76. Dominguez S, Ghosn J, Valantin MA, Schruniger A, Simon A, Bonnard P, Caumes E, Pialoux G, Benhamou Y, Thibault V, Katlama C. Efficacy of early treatment of acute hepatitis C infection with pegylated interferon and ribavirin in HIV‐infected patients. AIDS 2006; 20: 1157–61 [DOI] [PubMed] [Google Scholar]
- 77. Kumar R, Singla V, Kacharya S. Impact and management of hepatitis B and hepatitis C virus co‐infection in HIV patients. Trop Gastroenterol 2008; 29: 136–47 [PubMed] [Google Scholar]
- 78. Lincoln D, Petoumenos K, Dore GJ. HIV/HBV and HIV/HCV coinfection, and outcomes following highly active antiretroviral therapy. HIV Med 2003; 4: 241–9 [DOI] [PubMed] [Google Scholar]
- 79. Matthews GV, Dore GJ. HIV and hepatitis C coinfection. J Gastroenterol Hepatol 2008; 23: 1000–8 [DOI] [PubMed] [Google Scholar]
- 80. Vogel M, Bieniek B, Jessen H, Schewe CK, Hoffmann C, Baumgarten A, Kroidl A, Bogner JR, Spengler U, Rockstroh JK. Treatment of acute hepatitis C infection in HIV‐infected patients: a retrospective analysis of eleven cases. J Viral Hepat 2005; 12: 207–11 [DOI] [PubMed] [Google Scholar]
- 81. Thuerauf DJ, Marcinko M, Belmont PJ, Glembotski CC. Effects of the isoform‐specific characteristics of ATF6 alpha and ATF6 beta on endoplasmic reticulum stress response gene expression and cell viability. J Biol Chem 2007; 282: 22865–78 [DOI] [PubMed] [Google Scholar]
- 82. Thuerauf DJ, Morrison L, Glembotski CC. Opposing roles for ATF6alpha and ATF6beta in endoplasmic reticulum stress response gene induction. J Biol Chem 2004; 279: 21078–84 [DOI] [PubMed] [Google Scholar]
- 83. Guan D, Wang H, Li VE, Xu Y, Yang M, Shen Z. N‐glycosylation of ATF6beta is essential for its proteolytic cleavage and transcriptional repressor function to ATF6alpha. J Cell Biochem 2009; 108: 825–31 [DOI] [PubMed] [Google Scholar]
- 84. Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell 2007; 13: 365–76 [DOI] [PubMed] [Google Scholar]
- 85. Hong M, Li M, Mao C, Lee AS. Endoplasmic reticulum stress triggers an acute proteasome‐dependent degradation of ATF6. J Cell Biochem 2004; 92: 723–32 [DOI] [PubMed] [Google Scholar]
- 86. Medigeshi GR, Lancaster AM, Hirsch AJ, Briese T, Lipkin WI, DeFilippis V, Fruh K, Mason PW, Nikolich‐Zugich J, Nelson JA. West nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J Virol 2007; 81: 10849–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yu CY, Hsu YW, Liao CL, Lin YL. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J Virol 2006; 80: 11868–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Barry G, Fragkoudis R, Ferguson MC, Lulla A, Merits A, Kohl A, Fazakerley JK. Semliki forest virus‐induced endoplasmic reticulum stress accelerates apoptotic death of mammalian cells. J Virol 2010; 84: 7369–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 2009; 136: 731–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I. Dendritic injury is a pathological substrate for human immunodeficiency virus‐related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann Neurol 1997; 42: 963–72 [DOI] [PubMed] [Google Scholar]
- 91. Komar AA, Hatzoglou M. Internal ribosome entry sites in cellular mRNAs: mystery of their existence. J Biol Chem 2005; 280: 23425–8 [DOI] [PubMed] [Google Scholar]
- 92. Masliah E, Achim CL, Ge N, De Teresa R, Wiley CA. Cellular neuropathology in HIV encephalitis. Res Publ Assoc Res Nerv Ment Dis 1994; 72: 119–31 [PubMed] [Google Scholar]
- 93. Muaddi H, Majumder M, Peidis P, Papadakis AI, Holcik M, Scheuner D, Kaufman RJ, Hatzoglou M, Koromilas AE. Phosphorylation of eIF2{alpha} at serine 51 is an important determinant of cell survival and adaptation to glucose deficiency. Mol Biol Cell 2010; 21: 3220–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Woo CW, Cui D, Arellano J, Dorweiler B, Harding H, Fitzgerald KA, Ron D, Tabas I. Adaptive suppression of the ATF4‐CHOP branch of the unfolded protein response by toll‐like receptor signalling. Nat Cell Biol 2009; 11: 1473–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Dey S, Baird TD, Zhou D, Palam LR, Spandau DF, Wek RC. Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J Biol Chem 2010; 285: 33165–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Miyamoto N, Izumi H, Miyamoto R, Bin H, Kondo H, Tawara A, Sasaguri Y, Kohno K. Transcriptional regulation of activating transcription factor 4 under oxidative stress in retinal pigment epithelial ARPE‐19/HPV‐16 cells. Invest Ophthalmol Vis Sci 2010; 52: 1226–34 [DOI] [PubMed] [Google Scholar]
- 97. Kim H‐T, Qiang W, Liu N, Scofield VL, Wong PKY, Stoica G. Up‐regulation of astrocyte cyclooxygenase‐2, CCAAT/enhancer‐binding protein, glucose‐related protein 78, eukaryotic initiation factor 2α, and c‐Jun N‐terminal kinase by a neurovirulent murine retrovirus. J Neurovirol 2005; 11: 166–79 [DOI] [PubMed] [Google Scholar]
- 98. Sasaki H, Sato H, Kuriyama‐Matsumura K, Sato K, Maebara K, Wang H, Tamba M, Itoh K, Yamamoto M, Bannai S. Electrophile response element‐mediated induction of the cystine/glutamate exchange trans porter gene expression. J Biol Chem 2002; 277: 44765–71 [DOI] [PubMed] [Google Scholar]
- 99. Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH. Coordinate regulation of glutathione biosynthesis and release by Nrf2‐expressing glia potently protects neurons from oxidative stress. J Neurosci 2003; 23: 3394–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Bannai S, Kitamura E. Transport interaction of L‐cystine and L‐glutamate in human diploid fibroblasts in culture. J Biol Chem 1980; 255: 2372–6 [PubMed] [Google Scholar]
- 101. Watanabe H, Bannai S. Induction of cystine transport activity in mouse peritoneal macrophages. J Exp Med 1987; 165: 628–40 [DOI] [PMC free article] [PubMed] [Google Scholar]