

In the past decade, researchers have dramatically improved our understanding of the genetic basis of complex chronic diseases, such as Alzheimer’s disease and type 2 diabetes, through more than 1,000 genome-wide association studies (GWAS). These scan the genomes of thousands of people for known genetic variants, to find out which are associated with a particular condition.
Yet the findings from such studies are likely to have less relevance than was previously thought for the world’s population as a whole. Ninety-six per cent of subjects included in the GWAS conducted so far are people of European descent1 (see ‘Sampling bias’). And a recent Nature survey suggests that this bias is likely to persist in the upcoming efforts to sequence people’s entire genomes2.
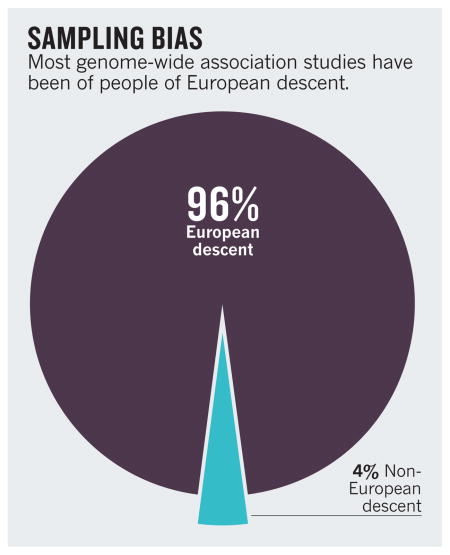
Geneticists worldwide must investigate a much broader ensemble of populations, including racial and ethnic minorities. If we do not, a biased picture will emerge of which variants are important, and genomic medicine will largely benefit a privileged few.
Since the 1970s, geneticists have known that most of the genetic variance between individuals stems from differences in DNA sequence (genetic variants). Of the millions of small sequence differences identified worldwide, some are ‘common variants’ — that is, they are found in more than 5% of people in many populations, and some of these occur widely, in people of different geographical and ethnic origins. Indeed, most GWAS have sought to find common variants associated with disease, in the hope that discoveries in one population will generalize to others.
GWAS have unearthed clear associations between common variants and many common diseases. But depending on the condition in question, these explain only between 5% and 50% of the diseases’ heritability. Many of the genetic factors thought to be responsible are still ‘missing’.
This suggests that ‘rare’ genetic variants (those that occur in less than 5% of the world’s population but which comprise the bulk of genetic variants) may be disproportionately important3 — both in determining a person’s risk of getting a complex disease and in predicting their response to a particular drug4. Rare variants tend to be population specific5 (see ‘Comparing the uncomparable’). So if they do play a key part in disease, the lack of diversity in genetic studies will be severely skewing our understanding of which are important.
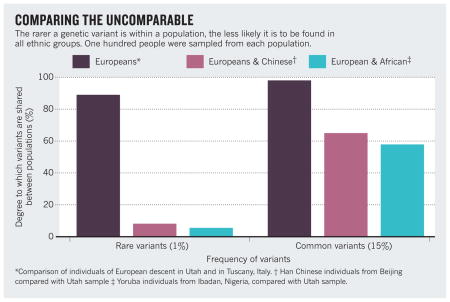
Several researchers have begun to assess the ability to generalize GWAS discoveries between different populations. Preliminary results suggest that findings from one population may not always easily translate to the rest of the world, although many more and larger comparisons are needed.
For example, in people with Native South American ancestry, a particular variant of a protein that transports cholesterol into cells is common and is strongly associated with low levels of high-density lipoprotein cholesterol, obesity and type 2 diabetes. European, Asian and African populations do not have this variant6.
Conversely, in dozens of studies in European populations, researchers have found 19 common single-nucleotide changes that are strongly associated with type 2 diabetes. In a further study of 6,000 people including European Americans, African Americans, Latinos, Japanese Americans and Native Hawaiians, 13 of these polymorphisms continue to be strongly associated with the disease7. Yet 5 of the 19 variants seem to have different effects in the different ethnic groups, and the role of one variant is unclear.
AVOIDING GENERALIZATIONS
There are several reasons why findings in one population might not generalize to another. Disease-associated versions (or alleles) of a gene may vary substantially in frequency in different ethnic groups. Also, GWAS identify genetic markers associated with a particular trait, not the mutations causing the disease. If a given marker is linked to a mixture of common and rare causal alleles8, some of the rare ones are likely to differ in frequency in different populations, or even be completely absent in some5.
The degree to which common genetic markers are linked to underlying causal mutations will also vary depending on the population being studied. For example, African populations are generally more genetically diverse than European, Asian or indigenous American populations, so one might expect to see weaker associations between markers and mutations in African and African-diaspora populations (such as African-Americans).
As well as genes and the environment differing between populations, the gene–environment interactions can vary, and these could significantly change a person’s likelihood of developing a disease.
Already there is evidence that measures of genetic ancestry can improve clinical care for people of mixed race. For example, physicians assessing the effects of lung disease compare measures of lung function (obtained by having the patient breathe into a spirometer) to a reference standard for healthy people of the same gender and racial group. Doctors make more accurate diagnoses when they use patients’ actual genetic ancestry to make comparisons, instead of self-selected or inferred categorizations of race or ethnicity9.
Likewise, researchers this year showed that Native American ancestry is associated with a greater risk of childhood acute lymphoblastic leukaemia returning after a remission, and that children with more than 10% Native American ancestry need an additional round of chemotherapy to respond to the treatment10.
So why are geneticists reluctant to undertake studies of people with diverse ancestries?
A key concern is that as more populations are included in a study, it becomes increasingly likely that a sequence variant will be associated with disease because of differences in race or ethnicity between cases and controls, rather than because of differences in people’s disease status. A lack of appropriate methods or access to the right data may make it difficult for many investigators to control for this. Certainly, it is hard to both collect samples from tens or hundreds of thousands of patients, and balance costs, statistical power and project deadlines with broad ethnic representation.
Such challenges, however, do not justify restricting the beneficiaries of medical genomic research to a small subset of humanity. Population-based studies must be carried out on a global scale. This means giving incentives to researchers in developed countries to increase the representation of minority populations in their studies and — crucially — empowering investigators in the developing world to undertake genomics research themselves.
COST EFFECTIVE
The ‘missing heritability problem’ has led many to become dismissive of GWAS. A danger of this GWAS fatigue is that it deters others from applying the approach to populations where it is likely to yield excellent results. GWAS has proved most successful in relatively small homogeneous populations — in Finland, Iceland and Costa Rica, say, where people generally stay put. Large families and limited migration are common among populations in Latin America, Africa and South Asia — suggesting that new and important associations between diseases and regionally common genetic variants may be found easily in these groups.
Moreover, large-scale GWAS are a feasible option for many research groups worldwide given that it now costs less than US$250 to obtain genetic data across millions of markers per person. (Whole-genome sequencing costs about 20 times more.) In fact, replicating an association study in a different ethnic group is often one-tenth the original cost. So at the very least, associations found in Europeans should be investigated in other ethnic groups.
Key to the success of global-scale GWAS are extensive and accurate catalogues of human genomic variation. The 1,000 Genomes Project is an excellent first step to providing a reference resource for researchers worldwide. It aims to catalogue genetic variants occurring in more than 1% of various populations throughout the world — including mixed-race North and South Americans as well as diverse populations from Africa, Europe, the Far East and South Asia.
Boosting genomic studies globally will also require initiatives that foster collaboration between countries, and enable the transfer of funding and technology beyond China, the United States and the European Union. The Human Heredity and Health in Africa Initiative, for example, is empowering local researchers. Supported by the US National Institutes of Health (NIH) and the UK Wellcome Trust in London, the initiative is the first major attempt to help African investigators study the genomic and environmental determinants of common diseases in African populations.
The private and philanthropic sector can also play an important part. The Slim Initiative for Genomic Medicine, a collaboration between the Mexican National Human Genome Research Institute and the Broad Institute of the United States, was launched last year and is enabling researchers to study type 2 diabetes and cancer in Latin American populations. This project is funded by the charitable foundation of the Mexican business magnate and philanthropist, Carlos Slim Helú.
For these efforts to be successful, researchers and physicians in the participating developing countries cannot simply provide samples. In addition, local expertise, resources and technology centres must be developed so that local populations benefit directly from home-grown research — as at the BGI (formerly the Beijing Genomics Institute) in Shenzhen, China. Local researchers will often better understand the history of local populations, such as whether they have recently switched from a rural to an urban lifestyle, and so will have deeper insights into likely environmental effects.
At the same time, medical geneticists working in wealthy nations must include their own minority and immigrant populations in their studies and develop the tools needed to compare results between populations. Many ‘exome’ sequencing projects (which sequence only the coding regions of the genome) are moving in this direction. For example, the US National Heart, Lung and Blood Institute aims to sequence the exomes of 7,000 people, roughly half of whom are African-Americans, to identify variants associated with cardiovascular and lung diseases.
PIECING TOGETHER THE MOSAIC
To make medical genomics truly global, geneticists need new statistical methods to dissect the contribution of genetic, sociocultural and environmental factors to both chronic and infectious disease.
Currently, ‘ancestry metrics’ are used to correct for the effect of shared ancestry on the results of association studies. But these methods do not work very well for groups whose genomes are a mosaic of fragments drawn from many different populations. Reference data from the relevant ancestral populations, including historically marginalized populations such as native Americans and Australian Aborigines, will help geneticists to separate spurious from real associations. And such understudied populations must be properly on board for this to happen. Researchers should gauge local values and concerns, and invest time and money into education and outreach to explain to the people they intend to study, as well as to the general public, why studying global (and local) health is so important.
The Center for Research on Genomics and Global Health headed by Charles Rotimi at the US National Human Genome Research Institute in Bethesda, Maryland, is beginning to gather the data and formulate the methods needed to understand the complex interplay that creates health disparities among ethnic groups for diseases such as type 2 diabetes, hypertension and obesity in Africa and elsewhere. Meanwhile, the Slim, the Wellcome Trust and the Bill & Melinda Gates Foundation based in Seattle, Washington, have begun to support research in understudied populations. Ultimately, however, global genomics needs the financial support of governments.
One way to encourage researchers to branch out may be for peer reviewers and granting bodies to stress the importance of racial and ethnic diversity in medical genetic studies. The NIH mandated the inclusion of diverse subjects in 1985. In the 26 years since, just 7% of GWAS have included minorities — perhaps because being more inclusive doesn’t win points for grant applicants.
It is tempting to focus on populations that are motivated, organized, medically compliant and otherwise easy to study. But by failing to develop resources, methodologies and incentives for underserved people, we risk perpetuating the health disparities that plague the medical system. Those most in need must not be the last to receive the benefits of genetic research.
SUMMARY.
Those most in need must not be the last to benefit from genetic research
Reviewers and granting bodies must demand racial and ethnic diversity in genome studies
Global genomics needs the financial support of governments and non-profits
Footnotes
The authors declare competing financial interests: for details see go.nature.com/gvtxn4
Contributor Information
Carlos D. Bustamante, Email: cdbustam@stanford.edu, Department of Genetics, Stanford University School of Medicine, Stanford, California 94305, USA
Esteban González Burchard, University of California, San Francisco, San Francisco, California 94143, USA.
Francisco M. De La Vega, Department of Genetics, Stanford University School of Medicine, Stanford, California 94305, USA
References
- 1.Need AC, Goldstein DB. Trends Genet. 2009;25:489–494. doi: 10.1016/j.tig.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 2.Nature. 2010;467:1026–1027. doi: 10.1038/4671026a. [DOI] [PubMed] [Google Scholar]
- 3.Manolio TA, et al. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McClellan J, King MC. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 5.Gravel S, et al. Proc Natl Acad USA. 2011 doi: 10.1073/pnas.1019276108. [DOI] [Google Scholar]
- 6.Acuna-Alonzo V, et al. Hum Mol Genet. 2010;19:2877–2885. doi: 10.1093/hmg/ddq173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waters KM, et al. PLoS Genet. 2010;6:e1001078. doi: 10.1371/journal.pgen.1001078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adeyemo A, Rotimi C. Public Health Genomics. 2010;13:72–79. doi: 10.1159/000218711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar R, et al. N Engl J Med. 2010;363:321–330. doi: 10.1056/NEJMoa0907897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang JJ, et al. Nature Genet. 2011;43:237–241. doi: 10.1038/ng.763. [DOI] [PMC free article] [PubMed] [Google Scholar]