

A high-resolution, single-molecule study directly assesses the prevalence and dynamics of DNA looping in gene regulation in live E. coli cells.
Abstract
DNA looping mediated by transcription factors plays critical roles in prokaryotic gene regulation. The “genetic switch” of bacteriophage λ determines whether a prophage stays incorporated in the E. coli chromosome or enters the lytic cycle of phage propagation and cell lysis. Past studies have shown that long-range DNA interactions between the operator sequences OR and OL (separated by 2.3 kb), mediated by the λ repressor CI (accession number P03034), play key roles in regulating the λ switch. In vitro, it was demonstrated that DNA segments harboring the operator sequences formed loops in the presence of CI, but CI-mediated DNA looping has not been directly visualized in vivo, hindering a deep understanding of the corresponding dynamics in realistic cellular environments. We report a high-resolution, single-molecule imaging method to probe CI-mediated DNA looping in live E. coli cells. We labeled two DNA loci with differently colored fluorescent fusion proteins and tracked their separations in real time with ∼40 nm accuracy, enabling the first direct analysis of transcription-factor-mediated DNA looping in live cells. Combining looping measurements with measurements of CI expression levels in different operator mutants, we show quantitatively that DNA looping activates transcription and enhances repression. Further, we estimated the upper bound of the rate of conformational change from the unlooped to the looped state, and discuss how chromosome compaction may impact looping kinetics. Our results provide insights into transcription-factor-mediated DNA looping in a variety of operator and CI mutant backgrounds in vivo, and our methodology can be applied to a broad range of questions regarding chromosome conformations in prokaryotes and higher organisms.
Author Summary
One mechanism cells use to regulate gene expression is DNA looping, whereby two distant DNA sites are brought together by regulatory proteins. The looping then either enhances interactions between other regulatory proteins bound at the separate sites or brings those regulatory proteins close to RNA polymerase at the promoter. Recent work in bacteriophage λ has suggested that DNA looping mediated by a transcription factor called λ repressor CI plays a critical role in regulating the expression of λ genes and consequently in determining the fate of the host E. coli bacterial cells. CI-mediated DNA looping has been directly demonstrated in vitro, but it has only been indirectly inferred in vivo. For the current study we developed a method to visualize CI-mediated DNA looping in individual live E. coli cells. We labeled two DNA sites—one each side of the proposed loop—with differently colored fluorescent fusion proteins, allowing us to measure their separation with an accuracy of a few tens of nanometers. Using this method, we directly analyzed CI-mediated DNA looping, providing insight into how transcription factor-mediated DNA looping influences gene regulation in live E. coli cells. Our methodology can be applied to a broad range of questions regarding chromosome conformation in prokaryotes and higher organisms.
Introduction
Looping between two DNA sites, mediated by transcription factors, is a ubiquitous mechanism in prokaryotic transcription regulation [1]. DNA looping brings two distal DNA sites into close proximity, enhancing interactions between transcription factors bound at separate sites or bringing transcription factors close to RNA polymerase at the promoter. Knowing when and how DNA loops in vivo is important to understand the role of DNA looping in gene regulation and cell decision-making; some studies found molecular details of gene regulation have little influence on gene expression [2]–[4], while others suggested that DNA looping could trigger cell phenotype switching [5] and influence fluctuations in transcription activity [6].
DNA looping was first suggested for the transcription factor AraC (accession number P0A9E0) in the E. coli arabinose operon. Disruption of an AraC binding site ∼280 bp upstream of the promoter reduced AraC-mediated repression nearly 10-fold, indicating a long-range interaction between the promoter and upstream DNA [7]. Subsequently, DNA looping mediated by transcription factors LacI [8] (accession number P03023), DeoR [9] (accession number P0ACK5), NtrC [10] (accession number P0AFB8), GalR [11] (accession number P03024), and bacteriophage λ repressor CI [12],[13] was reported. The length of the intervening DNA in these loops can be as short as 58 bp (lac operon [8]) or as long as ∼5 kilobases (deo operon [9]).
Biochemical, biophysical, and genetic studies have established important roles of DNA looping in transcription regulation. However, transcription-factor-mediated DNA looping on the length scale of a few kilobases in prokaryotic cells has not been directly visualized in vivo, and the in vivo dynamics of DNA looping are difficult to investigate. Chromosome conformation capture (3C) has been used to detect juxtaposition of DNA sites separated by hundreds of kilobases in both eukaryotic and prokaryotic cells [14],[15], but high background of interactions at the kilobase scale limits the utility of these methods in studying typical prokaryotic DNA loops [16]. An in vivo imaging method using fluorescent proteins fused to DNA-binding proteins bound to tandem arrays of hundreds of binding sites has been employed to visualize homologous chromosome pairing in yeast induced by double-strand breaks [17]; however, an array of several kilobases of binding sites makes this method unsuitable for studying DNA loops of only a few kilobases. In addition, the long array of tightly bound protein molecules may be detrimental to cells [18].
We developed a two-color, high-resolution imaging method to directly measure the end-to-end separation of two DNA sites 2.3 kb apart in live E. coli cells (Figure 1a). This method is based on the ability to precisely determine the location of a specific DNA site in vivo [19]. By expressing a fluorescent protein in fusion with a DNA-binding protein in a cell with only three tandem binding sites (spanning less than 100 bp), the resulting fluorescent spot is diffraction-limited, and the location of the binding site can be determined with sub-diffraction-limited precision by fitting its fluorescence profile to a two-dimensional Gaussian function [20]. By labeling two ends of a DNA segment with two unique sets of binding sequences and co-expressing corresponding fluorescent DNA-binding fusion proteins of different colors, the distance between the two DNA sites can be determined with a precision of a few tens of nanometers. An in vitro experiment employing the same principle measured intramolecular distances using organic dyes [21], but this approach has not been demonstrated in vivo with comparable resolution using fluorescent proteins.
Figure 1. Visualizing DNA looping in vivo by localizing OR and OL with fluorescent DNA-binding fusion proteins.

(a) λWT construct. Three tandem lacOsym and tetO sites, termed lacO3 and tetO3, were placed immediately next to OL and OR, respectively. Red and yellow fluorescent fusion proteins LacI-mCherry and TetR-EYFP bind lacO3 and tetO3, respectively. DNA looping mediated by a CI octamer (blue) or an additional CI tetramer (dashed) brings lacO3 and tetO3 together. Strains λOR3− and λOL3− harbor mutations (described in main text) to OR3 and OL3, respectively, that prevent CI dimers from binding these operator sites. (b) LacI-mCherry and TetR-EYFP are expressed co-transcriptionally from separate ribosome binding sites on a plasmid. (c) Illustration of 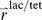 measurement. The observed distance between mCherry and EYFP spots indicates the distance between lacO3 and tetO3 projected onto the imaging plane. (d) Positive control λnull. The centers of lacO3 and tetO3 are separated by only 66 bp (see Figure S2a). (e) Negative control λΔOL. OL is deleted to eliminate CI-mediated DNA looping.
measurement. The observed distance between mCherry and EYFP spots indicates the distance between lacO3 and tetO3 projected onto the imaging plane. (d) Positive control λnull. The centers of lacO3 and tetO3 are separated by only 66 bp (see Figure S2a). (e) Negative control λΔOL. OL is deleted to eliminate CI-mediated DNA looping.
We used our method to probe the mechanisms and dynamics of DNA looping mediated by the bacteriophage λ repressor CI [22] in live E. coli cells and investigate its regulation of transcription from the CI promoter PRM. The λ repressor CI is an essential transcription factor in determining the fate of an E. coli cell infected by the bacteriophage λ. When CI is expressed, it represses lytic promoters to commit to an extraordinarily stable lysogenic state that persists for millions of generations [23]–[25]. However, upon induction by UV irradiation or other specific events, CI degradation can trigger an irreversible switch from lysogenic to lytic gene expression within one cell generation time [26].
The robustness of the λ regulatory circuit has been extensively studied. Among many important features of the system such as promoter-operator arrangement [27],[28], CI autoregulation [3],[29],[30], and cooperative binding [31]–[34], DNA looping between the homologous rightward and leftward operators OR and OL, separated by 2.3 kb, was shown to play significant, fate-determining roles in the λ lifecycle [13],[35]. Cooperative binding of CI dimers at the subsites OR1 and OR2 of OR represses the lytic promoter PR (reviewed in [36]) and simultaneously activates CI's own promoter, PRM, by accelerating transcription initiation [37]–[39]. At higher CI concentrations, an additional CI dimer binds to OR3 and represses PRM [40].
As illustrated in Figure 1a, an octameric CI complex (with or without an additional CI tetramer) can mediate DNA looping by bridging OR and OL. These higher-order complexes result from interactions between CI dimers bound to subsites at OR123 and OL123, and were first identified in vitro by ultracentrifugation [41] and later visualized by EM [12] and AFM [42]. Looping dynamics were investigated in vitro using tethered particle motion (TPM) [43]–[46].
To gain quantitative insight into the relationship between CI-mediated DNA looping and transcription regulation, thermodynamic models and numerical simulations were developed [33],[35],[44],[47]–[52]. Key parameters in these studies were the free energies of octameric and tetrameric CI interactions that mediate DNA looping [35]. These free energies specify the DNA looping probability at a given condition (temperature, CI concentration, etc.) and hence the extent to which distal DNA sites affect each other. To date, DNA-looping probabilities and free energies were either estimated indirectly in in vivo studies by measuring PRM and PR activities in various operator mutants with a priori assumptions of DNA looping states [35],[49],[51] or measured using purified components in vitro, where conditions differ from those in a cellular environment [42]–[46]. Consequently, these studies yielded varying estimates for the free energies of DNA looping and the degree to which DNA looping influences PRM activity. Hence, the roles of CI-mediated DNA looping in transcription regulation are still in debate [13],[35],[49],[51],[53].
In this study, we tracked the apparent separation between the OR and OL sites on a λ DNA segment (termed OR–OL DNA below) in real time in live E. coli cells, from which we obtained the first direct estimates of in vivo looping frequencies and kinetics for both wild-type DNA and for DNA carrying mutations in OR3 and OL3. We also measured corresponding CI expression levels in these strains by counting the number of CI transcripts in individual cells. Applying these independent, in vivo measurements to a thermodynamic model, we were able to obtain looping free energies and quantify the influence of DNA looping on PRM expression. Furthermore, we discuss how the compaction of the E. coli chromosome may impact DNA looping kinetics. The methodology established in this work can be extended to a broad range of questions regarding chromosomal DNA conformation and/or gene activities in prokaryotes and higher organisms.
Results
High-Resolution Imaging of Two DNA Sites
We inserted the construct shown in Figure 1a into the E. coli chromosome. It contains three tandem tetO sites (tetO3) [54] and three tandem lacOsym sites (lacO3) [55] flanking the wild-type λ lysogen sequence from OR to OL (including the PR, PRM and PL promoters and the cI, rexA (accession number P68924) and rexB (accession number P03759) genes). In this construct, called λWT, CI is expressed from PRM and regulates its own expression. The lacO-binding and tetO-binding proteins LacI and TetR (accession number P04483) were fused with red and yellow fluorescent proteins to generate LacI-mCherry and TetR-EYFP, and were expressed from an inducible plasmid (Figure 1b).
With the combination of strong induction, weak ribosome binding sites, and carefully controlled growth, we achieved sufficiently low LacI-mCherry and TetR-EYFP expression levels to detect distinct, diffraction-limited mCherry and EYFP spots in single cells. We then fit the fluorescence intensity profile of each individual spot with a two-dimensional Gaussian function to estimate its centroid position. The average localization precisions for individual spots of LacI-mCherry and TetR-EYFP were 17 and 14 nm, respectively (Figure S1a). Subsequently, we transformed EYFP coordinates into mCherry coordinates using fiducial data to calculate the vector between the mCherry and EYFP spots arising from LacI-mCherry and TetR-EYFP protein molecules bound to the same OR–OL DNA segment. We called this vector 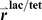 (Figure 1c). The magnitude of the vector,
(Figure 1c). The magnitude of the vector, 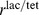 , is the two-dimensional projection of the distance between lacO3 and tetO3 onto the image plane; on average, it is proportional to the end-to-end distance between lacO3 and tetO3 in three dimensions. The total error for an
, is the two-dimensional projection of the distance between lacO3 and tetO3 onto the image plane; on average, it is proportional to the end-to-end distance between lacO3 and tetO3 in three dimensions. The total error for an 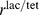 measurement, including fitting errors in determining centroid of individual spots (Figure S1a), registration errors in aligning EYFP and mCherry two-color images (∼10 nm based upon experiments using fluorescent beads), and contributions from local fluorescent background, was on average ∼40 nm (see below). With very low TetR-EYFP and LacI-mCherry expression, it was inevitable that not all lacO3 and tetO3 sites were bound by fusion protein molecules. Furthermore, not all fusion protein molecules were fluorescent due to stochastic chromophore maturation. Figure 2a contains typical data showing that a subset of cells was successfully labeled at both sites. We analyzed all cells having distinct fluorescent spots in both emission channels to calculate
measurement, including fitting errors in determining centroid of individual spots (Figure S1a), registration errors in aligning EYFP and mCherry two-color images (∼10 nm based upon experiments using fluorescent beads), and contributions from local fluorescent background, was on average ∼40 nm (see below). With very low TetR-EYFP and LacI-mCherry expression, it was inevitable that not all lacO3 and tetO3 sites were bound by fusion protein molecules. Furthermore, not all fusion protein molecules were fluorescent due to stochastic chromophore maturation. Figure 2a contains typical data showing that a subset of cells was successfully labeled at both sites. We analyzed all cells having distinct fluorescent spots in both emission channels to calculate 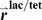 . We expected
. We expected 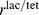 to decrease when DNA between lacO3 and tetO3 looped.
to decrease when DNA between lacO3 and tetO3 looped.
Figure 2. High-resolution imaging of lacO3 and tetO3 sites separated by 66 bp (λnull) or 2.3 kb (λΔOL).

(a) Fluorescent images of λnull. Arrows highlight molecules that exclusively appeared in mCherry (magenta, top) and EYFP (green, middle) channels, indicating a lack of significant crosstalk between the two channels. Squares show a spot that appeared in both channels. In the overlay image (bottom), fluorescence images were bandpass filtered and background was subtracted. Only cells having both mCherry and EYFP fluorescence were used in analysis. Scale bar, 2 µm. The image order and color scheme are repeated in (b–e). (b) Fluorescent images of λΔOL. Scale bar, 1 µm. (c–e) Timelapse images of spots acquired every 100 ms; (c) and (d) are spots in white squares in (a) and (b), respectively, and (e) shows an additional λΔOL spot, whose apparent separation can be easily detected by eye. Top and middle rows show mCherry and EYFP channels, respectively, and bottom rows show two-color overlays on brightfield images. (f–h) Trajectory 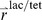 vectors from fitting fluorescence data for spots in (c–e). Coordinates are in nm. Vertices indicate the
vectors from fitting fluorescence data for spots in (c–e). Coordinates are in nm. Vertices indicate the 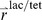 vector and subsequent time points are connected by lines that are colored to indicate elapsed time.
vector and subsequent time points are connected by lines that are colored to indicate elapsed time.
Distinguishing Between Looped and Unlooped States
To determine whether our two-color imaging method was sufficient to distinguish between looped and unlooped DNA in the crowded intracellular environment, we constructed two control strains (Table 1). In the positive control λnull, the centers of lacO3 and tetO3 sites are separated by 66 bp (Figure 1d). The outmost lacOsym and tetO sites are separated by less than 40 nm (Figure S2a). The close proximity of lacO3 and tetO3 mimicked permanently looped DNA. In the negative control λΔOL, we inserted the λ sequence from OR up to but not including OL between lacO3 and tetO3 (Figure 1e). The resulting λΔOL DNA has comparable length as the wild-type λ DNA, but CI-mediated DNA looping between OR and OL is abolished.
Table 1. Descriptions of strains and plasmids used in this study.
| Strain Name | Genotype |
| λnull | MG1655 ΔlacIzya::lacO3tetO3 |
| λWT | λnull lacO3tetO3::(OR–OL) |
| λΔOL | λWT ΔOL |
| λOR3− | λWT OR3r1 |
| λOL3− | λWT OL3–4 |
| λΔOLPRM−cI− | λΔOL PRM−cI− |
| λΔOLPRM−cI−/cItrans | λΔOL PRM−cI− (pACL18) |
| λCIG147D | λWT cI(G147D) |
| λCIG147D/cIG147D,trans | λCIG147D (pACL17) |
| pZH102R33Y29 | pLau53 [82] pBad-{lacI-mCherry}-{tetR-EYFP} |
| pZH102R33TD | pLau53 pBad-{lacI-mCherry-EYFP} |
| pACL18 | pACYC184-cI wt |
| pACL17 | pACYC184-cI G147D |
The 2.3-kb, wild-type phage λ sequence from OR to OL was incorporated into the E. coli chromosome in λWT. Strains λOR3−, λOL3−, and λWTG147D contain the r1 [80], OL3–4 [35], and CIG147D [62] mutations, respectively. Curly brackets indicate fusion products expressed from a single ribosome binding site. These are shorthand strain names; names used in our lab are listed in Table S4.
We first examined λnull and λΔOL in two-color fluorescence images to determine whether we could discriminate between looped and unlooped DNA by eye. We obtained at least sixty 20-frame movies (100 ms exposures; 2 s total) for each strain in each of three independent experiments. Typical fluorescence images are shown in Figure 2a and b. Crosstalk between the two emission channels was negligible, as bright mCherry and EYFP spots only appeared in the corresponding channel but not the other.
Figure 2c and d show 1 s of typical data for individual λnull and λΔOL spots. Representative movies for the two strains and others discussed below are included as Movies S1, S2, S3, S4, S5, S6. As expected for a permanently looped configuration, the positive control λnull exhibited overlapping EYFP and mCherry spots (Figure 2c). Generally, λΔOL molecules did not exhibit spot separation that was easily identifiable by eye (Figure 2d). However, some λΔOL molecules displayed large displacements between the LacI-mCherry and TetR-EYFP spots that were distinguishable by eye (Figure 2e); such images were not observed for λnull.
Visual inspection of the apparent separation between the LacI-mCherry and TetR-EYFP spots suggested that comparing the end-to-end separation in OR–OL DNAs required a more quantitative approach. We calculated 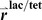 for all OR–OL DNA molecules in the λΔOL and λnull strains that exhibited fluorescent spots in both EYFP and mCherry images. Figure 2f–h shows
for all OR–OL DNA molecules in the λΔOL and λnull strains that exhibited fluorescent spots in both EYFP and mCherry images. Figure 2f–h shows 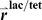 calculations for movies in Figure 2c–e, respectively, and Figure S3 shows
calculations for movies in Figure 2c–e, respectively, and Figure S3 shows 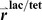 vectors for all movies lasting 0.8 s or longer. We then compiled the corresponding probability density distributions (PDF,
vectors for all movies lasting 0.8 s or longer. We then compiled the corresponding probability density distributions (PDF, 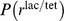 , Figure 3a) and cumulative density distributions (CDF,
, Figure 3a) and cumulative density distributions (CDF, 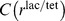 , Figure 3b) of the vector magnitude,
, Figure 3b) of the vector magnitude, 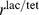 . The long-tailed PDF observed for λnull (Figure 3a) is consistent with the expected end-to-end distance distribution measured from two spots with a fixed separation when the localization of each spot is subject to Gaussian fitting error [56]. A simple numerical simulation of the end-to-end distance PDF for two sites separated by 22 nm and each subject to 22-nm localization error largely recapitulates the long-tailed distribution (Figure S2c).
. The long-tailed PDF observed for λnull (Figure 3a) is consistent with the expected end-to-end distance distribution measured from two spots with a fixed separation when the localization of each spot is subject to Gaussian fitting error [56]. A simple numerical simulation of the end-to-end distance PDF for two sites separated by 22 nm and each subject to 22-nm localization error largely recapitulates the long-tailed distribution (Figure S2c).
Figure 3. End-to-end distance (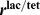 ) distributions and looping frequency fitting.
) distributions and looping frequency fitting.

(a) Probability density distribution (PDF) of the 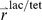 vector magnitude
vector magnitude 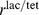 for the looped (λnull, red) and unlooped (λΔOL, blue) controls. The PDF is estimated for 10-nm bins as described in the main text. Light-colored areas indicate 1 s.e.m. calculated by bootstrapping. (b) Cumulative density (CDF) of
for the looped (λnull, red) and unlooped (λΔOL, blue) controls. The PDF is estimated for 10-nm bins as described in the main text. Light-colored areas indicate 1 s.e.m. calculated by bootstrapping. (b) Cumulative density (CDF) of 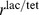 for the looped (λnull) and unlooped (λΔOL) controls. The CDF is estimated for 10-nm bins as described in the main text. Light-colored area indicates 1 s.e.m. calculated by bootstrapping. (c) The PDF is shown for strains λWT (green), λOR3− (orange), and λOL3− (purple), calculated as in (a), and PDFs for strains λnull and λΔOL are shown as dashed lines for comparison. (d) CDF estimates for three strains (dots; λWT, green; λOR3−, orange; λOL3−, purple) were fit as linear combinations of the positive (λnull) and negative (λΔOL) control CDFs to estimate looping frequency. Colored lines indicate CDF fits and CDFs for strains λnull and λΔOL are shown as dashed lines for comparison.
for the looped (λnull) and unlooped (λΔOL) controls. The CDF is estimated for 10-nm bins as described in the main text. Light-colored area indicates 1 s.e.m. calculated by bootstrapping. (c) The PDF is shown for strains λWT (green), λOR3− (orange), and λOL3− (purple), calculated as in (a), and PDFs for strains λnull and λΔOL are shown as dashed lines for comparison. (d) CDF estimates for three strains (dots; λWT, green; λOR3−, orange; λOL3−, purple) were fit as linear combinations of the positive (λnull) and negative (λΔOL) control CDFs to estimate looping frequency. Colored lines indicate CDF fits and CDFs for strains λnull and λΔOL are shown as dashed lines for comparison.
We found that the 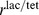 distribution for λΔOL was distinctly different from that of λnull (p<10−3); the difference was reproduced in three independent experiments (Figure S1b). The mean separations,
distribution for λΔOL was distinctly different from that of λnull (p<10−3); the difference was reproduced in three independent experiments (Figure S1b). The mean separations, 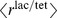 , were 47 (N = 1,153) and 71 nm (N = 979) for λnull and λΔOL respectively (results and measurement errors summarized in Table 2). Peaks in
, were 47 (N = 1,153) and 71 nm (N = 979) for λnull and λΔOL respectively (results and measurement errors summarized in Table 2). Peaks in 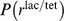 plots centered at ∼40 nm, reflecting our experimental precision in determining
plots centered at ∼40 nm, reflecting our experimental precision in determining 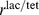 ; that is, OR–OL molecules with
; that is, OR–OL molecules with 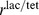 below 40 nm could not be distinguished from each other. Hence, it was more meaningful to compare distributions of
below 40 nm could not be distinguished from each other. Hence, it was more meaningful to compare distributions of 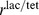 at large values where
at large values where 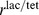 distributions differed most prominently. The cumulative probability of
distributions differed most prominently. The cumulative probability of 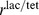 being 75 nm or more was ∼40% for λΔOL and only ∼15% for λnull (Figure 3b). Furthermore, two-dimensional distributions of
being 75 nm or more was ∼40% for λΔOL and only ∼15% for λnull (Figure 3b). Furthermore, two-dimensional distributions of 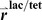 vectors (Figure S4) were clearly wider for λΔOL than for λnull. Thus, by examining
vectors (Figure S4) were clearly wider for λΔOL than for λnull. Thus, by examining 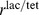 distributions, we could distinguish between the looped and unlooped control strains, suggesting that this approach could be used to probe CI-mediated DNA looping.
distributions, we could distinguish between the looped and unlooped control strains, suggesting that this approach could be used to probe CI-mediated DNA looping.
Table 2. Summary of measurements and sample statistics in this study.
| Strain | r lac/tet Measurements | Mean r lac/tet (nm) | Median r lac/tet (nm) | Looping Frequency | CI Expression Level (WLU) |
| λnull | 1,153 | 47±1 | 41±1 | N/A | N/A |
| λΔOL | 979 | 71±1 | 63±2 | N/A | 1.38±0.05 |
| λWT | 962 | 52±1 | 45±1 | 79±6% | 1.00±0.05 |
| λOR3− | 784 | 59±1 | 50±2 | 53±7% | 2.50±0.07 |
| λOL3− | 781 | 56±1 | 48±1 | 60±7% | 2.51±0.07 |
Errors are all 1 s.e.m. as estimated from 1,000 bootstrapped samples.
Compact Conformation of Unlooped DNA λΔOL Does Not Depend on Transcription or Nonspecific CI Binding
We measured the mean end-to-end separation 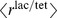 for λΔOL at 71-nm, much shorter than the ∼200-nm distance expected for B-form DNA with a typical 50-nm in vitro persistence length [57]. While such a result is expected given the many factors known to compact prokaryotic chromosomes [58], it is possible that nonspecifically bound CI on the λΔOL DNA and/or PRM transcription activity could influence the
for λΔOL at 71-nm, much shorter than the ∼200-nm distance expected for B-form DNA with a typical 50-nm in vitro persistence length [57]. While such a result is expected given the many factors known to compact prokaryotic chromosomes [58], it is possible that nonspecifically bound CI on the λΔOL DNA and/or PRM transcription activity could influence the 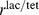 distribution, as indicated by a series of recent studies in vitro and in higher eukaryotic systems [46],[59],[60].
distribution, as indicated by a series of recent studies in vitro and in higher eukaryotic systems [46],[59],[60].
To examine these possibilities, we first compared the 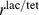 distribution of the λΔOL strain to that of a control strain λΔOLPRM−cI−/cItrans (Table 1, Figure S5a and b). In this control strain, promoter PRM was mutated to abolish transcription and the cI start codon was eliminated, but CI binding to OR was unaffected (Figure S5c, d, and e). In addition, we expressed CI from a plasmid at ∼9 times its level in λWT (Table S8). We found that the
distribution of the λΔOL strain to that of a control strain λΔOLPRM−cI−/cItrans (Table 1, Figure S5a and b). In this control strain, promoter PRM was mutated to abolish transcription and the cI start codon was eliminated, but CI binding to OR was unaffected (Figure S5c, d, and e). In addition, we expressed CI from a plasmid at ∼9 times its level in λWT (Table S8). We found that the 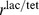 distributions of the λΔOL and λΔOLPRM−cI−/cItrans strains were indistinguishable (Figure S5a and b), demonstrating that the compact λΔOL distribution does not depend on PRM transcription. Furthermore,
distributions of the λΔOL and λΔOLPRM−cI−/cItrans strains were indistinguishable (Figure S5a and b), demonstrating that the compact λΔOL distribution does not depend on PRM transcription. Furthermore, 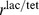 distributions for the same λΔOLPRM−cI− strain with or without the CI-expressing plasmid were indistinguishable (Figure S5a and b), suggesting that nonspecifically bound CI did not interact with specifically bound CI at OR operator sites to condense DNA in vivo [46].
distributions for the same λΔOLPRM−cI− strain with or without the CI-expressing plasmid were indistinguishable (Figure S5a and b), suggesting that nonspecifically bound CI did not interact with specifically bound CI at OR operator sites to condense DNA in vivo [46].
In Vivo Observations of DNA Looping
We next investigated DNA looping in the context of wild-type and mutant OR–OL DNAs. In λWT, the wild-type λ sequence from OR through OL was inserted between lacO3 and tetO3. CI could bind all OR and OL sites to mediate looping with both octameric and tetrameric CI complexes (Figure 1a). In λOR3− and λOL3−, mutations in OR3 and OL3 essentially eliminated CI binding to these operators at lysogenic CI concentrations (Table 1) [35],[61].
We measured 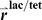 for these three strains and found that
for these three strains and found that 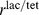 distributions differed significantly from those of the positive and negative controls λnull and λΔOL (p<10−3, except p = 0.004 for λWT and λnull), with
distributions differed significantly from those of the positive and negative controls λnull and λΔOL (p<10−3, except p = 0.004 for λWT and λnull), with 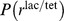 and
and 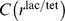 being intermediate to those of the controls (Figure 3c and d). Mean
being intermediate to those of the controls (Figure 3c and d). Mean 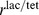 values for the three strains also fell in between those of λnull and λΔOL (Table 2). The wild-type strain had lower
values for the three strains also fell in between those of λnull and λΔOL (Table 2). The wild-type strain had lower 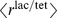 than λOR3− and λOL3−, and its distribution differed from those of the mutant strains with moderate to high significance (p = 0.001 and 0.048 for λOR3− and λOL3−, respectively);
than λOR3− and λOL3−, and its distribution differed from those of the mutant strains with moderate to high significance (p = 0.001 and 0.048 for λOR3− and λOL3−, respectively); 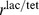 distributions for λOR3− and λOL3− were indistinguishable from each other (p = 0.493). The trend of λnull<λWT<λOR3−≈λOL3−<λΔOL for
distributions for λOR3− and λOL3− were indistinguishable from each other (p = 0.493). The trend of λnull<λWT<λOR3−≈λOL3−<λΔOL for 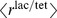 was reproduced in three independent experiments (Figure S1b). Assuming that a DNA molecule in the λWT, λOR3−, and λOL3− strains is in either a looped or unlooped state, the intermediate
was reproduced in three independent experiments (Figure S1b). Assuming that a DNA molecule in the λWT, λOR3−, and λOL3− strains is in either a looped or unlooped state, the intermediate 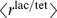 values of the three strains suggested that the fraction of looped DNA molecules (herein termed looping frequency) could be estimated by comparing
values of the three strains suggested that the fraction of looped DNA molecules (herein termed looping frequency) could be estimated by comparing 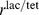 distributions of these strains to those of the looped and unlooped controls λnull and λΔOL.
distributions of these strains to those of the looped and unlooped controls λnull and λΔOL.
To further investigate whether the observed DNA looping in the λWT, λOR3−, and λOL3− strains could be abolished by eliminating CI cooperative binding rather than by deleting OL, we constructed a control strain λCIG147D (Table 1). This strain differs from λWT by a CI mutation G147D known to be defective in pairwise cooperative interaction [62],[63]. Structural evidence suggests that cooperative binding interfaces are shared for pairwise binding to adjacent operator sites and the formation of CI tetramers or octamers via DNA loops [64]. We found that the 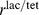 distribution of the λCIG147D strain was indistinguishable from that of λΔOL (Figure S5f and g,
Table S7). We note that this G147D mutant also diminishes PRM transcription because of its weakened ability to form a CI tetramer at the OR1 and OR2 sites; hence its expression level is lower than that with wild-type CI (Table S8). Therefore, we constructed another control strain (λCIG147D/cIG147D,trans), in which the CIG147D mutant protein was expressed constitutively at ∼11 times the CI expression level in λWT from a plasmid transformed into the λCIG147D strain (Table S8). We found that
distribution of the λCIG147D strain was indistinguishable from that of λΔOL (Figure S5f and g,
Table S7). We note that this G147D mutant also diminishes PRM transcription because of its weakened ability to form a CI tetramer at the OR1 and OR2 sites; hence its expression level is lower than that with wild-type CI (Table S8). Therefore, we constructed another control strain (λCIG147D/cIG147D,trans), in which the CIG147D mutant protein was expressed constitutively at ∼11 times the CI expression level in λWT from a plasmid transformed into the λCIG147D strain (Table S8). We found that 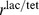 distribution of this strain was indistinguishable from that of the λΔOL and the λCIG147D strains, demonstrating that DNA looping could be abolished by eliminating CI cooperative binding.
distribution of this strain was indistinguishable from that of the λΔOL and the λCIG147D strains, demonstrating that DNA looping could be abolished by eliminating CI cooperative binding.
Estimating DNA Looping Frequency from 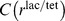
To quantitatively examine how operator mutations influence DNA looping, we estimated looping frequencies for λWT, λOR3−, and λOL3− by assuming a simple model. In this model, DNA molecule can only exist in one of two states, looped or unlooped, with 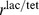 distributions for each state resembling those of the looped and unlooped controls, λnull and λΔOL, respectively. Therefore, the distribution
distributions for each state resembling those of the looped and unlooped controls, λnull and λΔOL, respectively. Therefore, the distribution 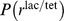 or
or 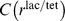 for one of the three strains is the linear combination of that of λnull and λΔOL, with their distributions weighted by the looping frequency,
for one of the three strains is the linear combination of that of λnull and λΔOL, with their distributions weighted by the looping frequency, 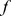 :
:
Using this model, we found that the looping frequency was 79% for λWT, and reduced to 53% for λOR3− and 60% for λOL3− (results with errors summarized in Table 2). The results were indistinguishable within error regardless of whether cumulative or probability density distributions were used, or whether data points from all frames or only the first frame of each molecule's movie were used (Table S1). The looping frequencies for λOR3− and λOL3− were indistinguishable from each other within error, suggesting a similar role of OR3 and OL3 in loop formation. Reduced looping frequencies of λOR3− and λOL3− compared to λWT suggest that while a CI octamer at OR12 and OL12 is sufficient to loop DNA, the resulting loop can be further stabilized by an additional CI tetramer only if both OR3 and OL3 are intact. To our knowledge, these measurements provide the first quantitative in vivo estimates of DNA looping frequencies that are independent of gene regulation models.
Estimating DNA Looping Kinetics
In the above analyses, we only utilized 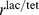 , the magnitude of the
, the magnitude of the 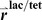 vector, and discarded information about the direction of
vector, and discarded information about the direction of 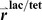 and its evolution in time. Looping frequencies estimated from
and its evolution in time. Looping frequencies estimated from 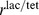 distributions are analogous to equilibrium constants and lack kinetic information. While many DNA molecules only exhibited fluorescent spots in both EYFP and mCherry channels for one or two consecutive frames due to photobleaching, some molecules had fluorescent spots lasting for several consecutive frames in both channels (Figure 2c–h; also see
distributions are analogous to equilibrium constants and lack kinetic information. While many DNA molecules only exhibited fluorescent spots in both EYFP and mCherry channels for one or two consecutive frames due to photobleaching, some molecules had fluorescent spots lasting for several consecutive frames in both channels (Figure 2c–h; also see 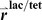 plots from molecules with many frames in Figure S3). By analyzing how
plots from molecules with many frames in Figure S3). By analyzing how 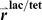 evolves in time, we can obtain additional information about DNA looping kinetics.
evolves in time, we can obtain additional information about DNA looping kinetics.
We calculated the autocorrelation of  (the average dot product of two
(the average dot product of two  vectors separated by a time lag) up to 0.5 s for each strain using all movies in which fluorescent spots in both channels lasted two or more frames (Figure 4a). The autocorrelation curves of all strains showed an initial drop of ∼2,500 nm2 at the first time lag, corresponding to uncorrelated errors in determining
vectors separated by a time lag) up to 0.5 s for each strain using all movies in which fluorescent spots in both channels lasted two or more frames (Figure 4a). The autocorrelation curves of all strains showed an initial drop of ∼2,500 nm2 at the first time lag, corresponding to uncorrelated errors in determining 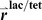 . After the initial drops, all autocorrelation curves showed positive correlation values that were approximately constant at time lags up to 0.5 s.
. After the initial drops, all autocorrelation curves showed positive correlation values that were approximately constant at time lags up to 0.5 s.
Figure 4. DNA looping kinetics and CI expression levels.
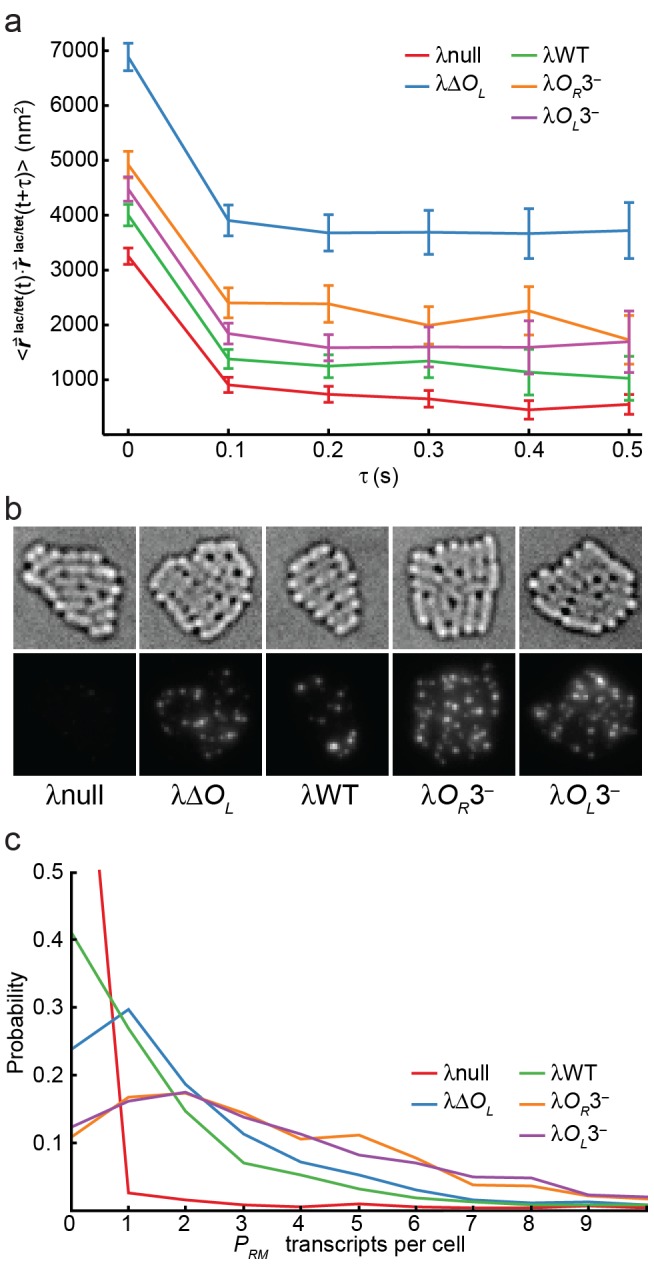
(a) Vector dot-product autocorrelation for 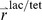 time trajectories for each strain. Plots show the average dot product of two
time trajectories for each strain. Plots show the average dot product of two 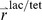 vectors separated by a given time lag. Error bars show 1 s.e.m. calculated by bootstrapping. (b) Typical smFISH images for λnull, which has no PRM transcripts, and for all other strains. Top, brightfield images showing a group of fixed cells for each strain. Bottom, maximum-intensity projections of fluorescence image stacks. Spots indicate one or more transcripts. (c) Distribution of PRM transcripts per cell determined by smFISH. The average expression level in wild-type λ units (WLU) is defined as the mean number of transcripts per cell in a given strain divided by the mean transcript number in λWT cells.
vectors separated by a given time lag. Error bars show 1 s.e.m. calculated by bootstrapping. (b) Typical smFISH images for λnull, which has no PRM transcripts, and for all other strains. Top, brightfield images showing a group of fixed cells for each strain. Bottom, maximum-intensity projections of fluorescence image stacks. Spots indicate one or more transcripts. (c) Distribution of PRM transcripts per cell determined by smFISH. The average expression level in wild-type λ units (WLU) is defined as the mean number of transcripts per cell in a given strain divided by the mean transcript number in λWT cells.
The observation of near constant autocorrelation values after the first time lag for all the strains indicated that the conformation of each DNA molecule, characterized by both the magnitude and orientation of 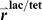 , persisted for at least 0.5 s. This provides a lower limit for the amount of time it takes for two DNA sites in the relaxed, unlooped state to move relative to each other and potentially form a DNA loop, and thus an upper limit of ∼2 s−1 for the rate of DNA looping. The plateau values are related to the averaged mean end-to-end separations—λΔOL has the highest autocorrelation plateau and λWT, λOR3−, and λOL3− have intermediate values because they contain a mixture of looped and unlooped DNA molecules.
, persisted for at least 0.5 s. This provides a lower limit for the amount of time it takes for two DNA sites in the relaxed, unlooped state to move relative to each other and potentially form a DNA loop, and thus an upper limit of ∼2 s−1 for the rate of DNA looping. The plateau values are related to the averaged mean end-to-end separations—λΔOL has the highest autocorrelation plateau and λWT, λOR3−, and λOL3− have intermediate values because they contain a mixture of looped and unlooped DNA molecules.
Single-Molecule Measurements of CI Expression Levels
Next, we measured average CI expression levels, 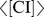 , in all strains in order to understand to what different extent DNA looping influences PRM regulation. We used single-molecule fluorescence in situ hybridization (smFISH, [2],[65],[66]), in which multiple fluorescently labeled oligonucleotides probe targeted nonoverlapping regions of cI mRNA, to count the number of PRM transcripts in individual cells (Figure 4b and c). Given the assumption that the average number of CI molecules translated per PRM transcript is the same in all strains and the observation of indistinguishable cell growth rates (Figure S6a and b), we expected average mRNA expression levels proportional to
, in all strains in order to understand to what different extent DNA looping influences PRM regulation. We used single-molecule fluorescence in situ hybridization (smFISH, [2],[65],[66]), in which multiple fluorescently labeled oligonucleotides probe targeted nonoverlapping regions of cI mRNA, to count the number of PRM transcripts in individual cells (Figure 4b and c). Given the assumption that the average number of CI molecules translated per PRM transcript is the same in all strains and the observation of indistinguishable cell growth rates (Figure S6a and b), we expected average mRNA expression levels proportional to 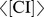 . The λnull strain does not contain the cI gene and was used as a negative control. All other strains were transcriptionally active. Under our experimental conditions, the false positive rate using the λnull strain was ∼1 transcript per 50 cells, two orders of magnitude below the levels of all other strains; false positives arise when nonspecifically bound probes occasionally co-localize to create a fluorescent spot above the detection threshold. Typical smFISH images of the five strains are shown in Figure 4b. We quantified the number of transcripts in each individual cell by dividing the total intensity of fluorescent spots in each cell by the average intensity of a single-transcript spot (Figure 4c). We then determined
. The λnull strain does not contain the cI gene and was used as a negative control. All other strains were transcriptionally active. Under our experimental conditions, the false positive rate using the λnull strain was ∼1 transcript per 50 cells, two orders of magnitude below the levels of all other strains; false positives arise when nonspecifically bound probes occasionally co-localize to create a fluorescent spot above the detection threshold. Typical smFISH images of the five strains are shown in Figure 4b. We quantified the number of transcripts in each individual cell by dividing the total intensity of fluorescent spots in each cell by the average intensity of a single-transcript spot (Figure 4c). We then determined 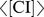 in wild-type λ units (WLU) by dividing the average number of transcripts in cells of a given strain by the average number of transcripts in λWT cells. We found that deleting OL increased
in wild-type λ units (WLU) by dividing the average number of transcripts in cells of a given strain by the average number of transcripts in λWT cells. We found that deleting OL increased 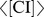 to ∼1.4 WLU (Table 2), indicating that the DNA loop formed between OL and OR in λWT enhances PRM repression. Mutating either OR3 or OL3 further increased
to ∼1.4 WLU (Table 2), indicating that the DNA loop formed between OL and OR in λWT enhances PRM repression. Mutating either OR3 or OL3 further increased 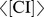 to ∼2.5 WLU. These observations are consistent with previous observations that although OL3 is 2.3 kb away from the PRM promoter, it has as important a role as OR3 in repressing PRM at lysogenic CI concentrations [13]. This suggests that PRM was not strongly repressed by CI binding to OR3 in the absence of a tetrameric interaction with an additional dimer at OL3. Finally, elevated
to ∼2.5 WLU. These observations are consistent with previous observations that although OL3 is 2.3 kb away from the PRM promoter, it has as important a role as OR3 in repressing PRM at lysogenic CI concentrations [13]. This suggests that PRM was not strongly repressed by CI binding to OR3 in the absence of a tetrameric interaction with an additional dimer at OL3. Finally, elevated 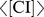 in λOL3− relative to λΔOL indicated that DNA looping could also activate PRM, which was likely mediated by the binding of a CI octamer at OL12 and OR12, and was consistent with recent in vivo [49],[51] and in vitro [53] experiments.
in λOL3− relative to λΔOL indicated that DNA looping could also activate PRM, which was likely mediated by the binding of a CI octamer at OL12 and OR12, and was consistent with recent in vivo [49],[51] and in vitro [53] experiments.
Evaluating Looping Free Energies and Transcription Activation Using a Thermodynamic Model
We have shown that reduced looping frequencies in λOL3− and λOR3− compared to that in λWT corresponded to increased expression levels of CI in the two strains, and that unlooped λΔOL has a higher expression level than the λWT strain. To establish a quantitative framework that explains all observed relationships between looping and CI expression levels, we refined a thermodynamic model, with which we estimated looping free energies and the degree to which DNA looping changes the activity of PRM. These parameters are important because free energies describe the likelihood of interaction between two distal DNA sites, and changes in promoter activity directly reflect the influence of DNA looping on gene regulation.
The thermodynamic approach was first applied to model repression and activation of PRM by CI bound to OR [52] and recently modified to address looping [35],[44],[49],[51]. Our modeling approach is unique in that we used two independent, in vivo measurements, looping frequencies, and corresponding CI expression levels, to refine parameters for DNA-looping free energies and transcription activities. In previous modeling work, DNA-looping free energies were either inferred from PRM and PR expression-level measurements [35],[49],[51] or estimated using in vitro data [44].
The thermodynamic model and fixed physical parameters from previous reports we used to estimate PRM expression levels and DNA looping frequencies are essentially identical to the one used to analyze in vivo gene expression experiments [35]. Briefly, we assume that DNA states can be enumerated, that steady-state, in vitro DNA-binding measurements are applicable in vivo, and that mean expression rate, 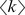 , equals the sum of all products
, equals the sum of all products 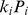 , where
, where 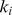 is the transcription rate in a particular state and
is the transcription rate in a particular state and 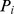 is the probability of the state at a given concentration of free CI dimers
is the probability of the state at a given concentration of free CI dimers 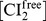 :
:
Each state is defined by its free energy, 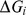 , the number of bound CI dimers,
, the number of bound CI dimers, 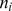 , and the degeneracy,
, and the degeneracy, 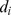 , which is the number of states with the same
, which is the number of states with the same 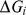 ,
, 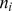 , and
, and 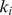 . The model is described in greater detail in the Materials and Methods section; all states considered are listed in Table S2.
. The model is described in greater detail in the Materials and Methods section; all states considered are listed in Table S2. 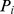 is normalized by the partition function,
is normalized by the partition function, 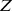 , so that the sum of all state probabilities is 1. Following earlier work [49] and considering that the CI-mediated loop is relatively long, we assumed looping free energies to be independent of parallel or antiparallel orientation. Note that loop orientation is important in shorter DNA loops such as those mediated by Gal repressor [67]. We approximated the average CI concentration,
, so that the sum of all state probabilities is 1. Following earlier work [49] and considering that the CI-mediated loop is relatively long, we assumed looping free energies to be independent of parallel or antiparallel orientation. Note that loop orientation is important in shorter DNA loops such as those mediated by Gal repressor [67]. We approximated the average CI concentration, 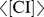 , as the concentration at which the degradation rate equaled the production rate.
, as the concentration at which the degradation rate equaled the production rate.
We refined our model to fit seven experimental observables: CI expression levels for λΔOL, λWT, λOR3−, and λOL3−, and the looping frequencies for λWT, λOR3−, and λOL3−. We varied four free parameters: the free energies of forming a CI octamer and tetramer in the DNA loop as defined by Dodd et al. [35], 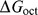 , and
, and 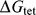 , and the PRM expression rates when OR12 is bound by CI and DNA is either looped (
, and the PRM expression rates when OR12 is bound by CI and DNA is either looped (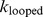 ) or unlooped (
) or unlooped (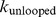 ).
). 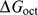 is the free energy of bringing together OR and OL when both are bound by two adjacent CI dimers to form a CI octamer, resulting in a looped conformation.
is the free energy of bringing together OR and OL when both are bound by two adjacent CI dimers to form a CI octamer, resulting in a looped conformation. 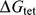 is the free energy of adding a CI tetramer to a loop already secured by a CI octamer. All other free energies and parameters such as specific and nonspecific DNA binding of CI were fixed at the values used by Dodd et al. [35]. The wild-type CI concentration was fixed to 220 nM (∼150 molecules/cell) based upon our previous experiment in which CI molecules were counted at the single-molecule level in a similar strain at similar growth conditions [3]. The CI degradation rate was fixed to give a half-life equal to the observed 2-h doubling time in our experiments.
is the free energy of adding a CI tetramer to a loop already secured by a CI octamer. All other free energies and parameters such as specific and nonspecific DNA binding of CI were fixed at the values used by Dodd et al. [35]. The wild-type CI concentration was fixed to 220 nM (∼150 molecules/cell) based upon our previous experiment in which CI molecules were counted at the single-molecule level in a similar strain at similar growth conditions [3]. The CI degradation rate was fixed to give a half-life equal to the observed 2-h doubling time in our experiments.
The four free parameters were adjusted to best fit our experimental measurements of looping frequencies and CI expression levels. Modeled looping frequencies and CI expression rates at different CI concentrations are shown in Figure 5a and b. The best fit estimated 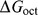 and
and 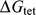 at 0.3 and −3.2 kcal/mol, respectively, and the CI expression rates at 1.9 nM/s and 4.5 nM/s for unlooped (
at 0.3 and −3.2 kcal/mol, respectively, and the CI expression rates at 1.9 nM/s and 4.5 nM/s for unlooped (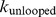 ) and looped (
) and looped (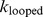 ) DNA when CI binds OR12. These results suggest that the DNA looping mediated by only a CI octamer is not strongly favored, while looping mediated by both an octamer and tetramer is the dominant configuration if all six binding sites are bound by CI dimers. Note that a small, positive
) DNA when CI binds OR12. These results suggest that the DNA looping mediated by only a CI octamer is not strongly favored, while looping mediated by both an octamer and tetramer is the dominant configuration if all six binding sites are bound by CI dimers. Note that a small, positive 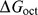 is consistent with measured looping frequencies greater than 50% for ΔλOL3− and ΔλOR3−, as one unlooped configuration could lead to multiple looped configurations (Table S2). The higher CI expression rate from the looped configuration suggests that, in the absence of OR3 binding, bringing the distal OL and OR sites together to form a DNA loop activates PRM to 2.4 times the unlooped level.
is consistent with measured looping frequencies greater than 50% for ΔλOL3− and ΔλOR3−, as one unlooped configuration could lead to multiple looped configurations (Table S2). The higher CI expression rate from the looped configuration suggests that, in the absence of OR3 binding, bringing the distal OL and OR sites together to form a DNA loop activates PRM to 2.4 times the unlooped level.
Figure 5. Thermodynamic modeling of PRM autoregulation by CI.

(a) Measured and modeled CI expression rates as a function of CI expression levels in wild-type λ units (WLU; the concentration of CI molecules in the λWT strain). Colored curves indicate the modeled dependence of CI expression rates on CI expression levels and dashed black curve indicates the CI degradation rate. Modeled, steady-state CI expression levels are indicated by white circles where the degradation curve intersects CI-expression-level curves. Vertical dashed lines indicate measured CI expression levels (Table 2). (b) Measured and modeled looping frequencies as a function of CI expression levels. Curves show the dependence of looping frequencies on CI expression levels; white circles indicate modeled CI expression levels and measured looping frequencies for λWT, λOR3−, and λOL3− with vertical lines indicating error in looping frequency estimates. (c, d) Fitting residual plots showing the uniqueness of best-fit model parameters. In each plot a parameter pair (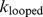 and
and 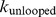 in c;
in c; 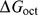 and
and 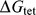 in d) is fixed, while the other parameter pair is varied and the corresponding minimum of the sum of squares of the difference between modeled and experimental parameters for all possible pairs was calculated. Parameter grids are colored according to the logarithm of the minimum sum of squares; well-defined minima indicate uniquely determined parameters.
in d) is fixed, while the other parameter pair is varied and the corresponding minimum of the sum of squares of the difference between modeled and experimental parameters for all possible pairs was calculated. Parameter grids are colored according to the logarithm of the minimum sum of squares; well-defined minima indicate uniquely determined parameters.
To test how sensitive the fitting results were to two fixed parameters that are poorly defined in previous work, we varied CI expression levels and nonspecific DNA binding affinity. We found that across the examined ranges, octameric looping energies, 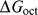 , were consistently near 0 and tetrameric looping energies,
, were consistently near 0 and tetrameric looping energies, 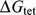 , were strongly favorable between −2.8 to −4.6 kcal/mol (Table S3). Similarly, CI expression rates
, were strongly favorable between −2.8 to −4.6 kcal/mol (Table S3). Similarly, CI expression rates 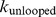 and
and 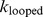 remained close to the original fit values, giving activation ratios between 1.7 and 2.5 (Table S3). We also verified that our fit parameters were unique—as shown in Figure 5c and d, the values of fit parameters corresponded to a well-defined minimum in the sum of squared residuals in the four-dimensional (two free energies and two expression rates) parameter space (Figure 5c and d). Hence we conclude that the four fit parameters resulted from the model were robust and well defined.
remained close to the original fit values, giving activation ratios between 1.7 and 2.5 (Table S3). We also verified that our fit parameters were unique—as shown in Figure 5c and d, the values of fit parameters corresponded to a well-defined minimum in the sum of squared residuals in the four-dimensional (two free energies and two expression rates) parameter space (Figure 5c and d). Hence we conclude that the four fit parameters resulted from the model were robust and well defined.
Discussion
In this work, we directly measure the end-to-end separation between two DNA sites separated by only 2.3 kb on the E. coli chromosome with high spatial resolution, and report the first estimates of CI-mediated DNA looping frequencies in live E. coli cells. We improved a thermodynamic model to estimate the free energies of DNA looping as well as the degree to which DNA looping enhances transcription regulation. Combining independent, single-molecule measurements of looping frequencies and CI expression levels increased confidence in this model. Our results provide insight into transcription-factor-mediated DNA looping in vivo, and the new method reported here also has the potential to address questions beyond DNA looping, including understanding of chromosome structure and dynamics in vivo. In the following, we compare our results with previous work, and discuss unique information provided by our new method.
Differences with in Vitro Looping Measurements
Our estimated looping frequencies of 79% for λWT and greater than 50% for λOR3− and λOL3− are larger than those observed in vitro by TPM and AFM, where looping frequencies at lysogenic CI concentrations were approximately 60% with wild-type operators and 10%–40% in the absence of OR3 and OL3 [42],[44],[46]. As looping frequency is directly linked to looping free energy, comparison of 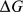 values showed the same trend:
values showed the same trend: 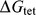 values estimated in these in vitro experiments were similar to our estimate of −3.2 kcal/mol, while in vitro
values estimated in these in vitro experiments were similar to our estimate of −3.2 kcal/mol, while in vitro 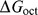 values were 1–2 kcal/mol higher than ours [44],[46].
values were 1–2 kcal/mol higher than ours [44],[46].
Significantly different 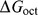 values likely resulted from differences between naked DNA in an in vitro environment and the compact, protein-decorated E. coli chromosome in the crowded cellular environment. Factors such as supercoiling and nonspecific, “histone-like” DNA-binding proteins could compact DNA and lead to more frequent encounters between OR and OL. Our observation that the unlooped λΔOL DNA was extremely compact (discussed in more detail below) was consistent with this view; this level of compaction (comparable to a polymer with a 3-nm rather than a 50-nm persistence length) could lead to a 50-fold increase in the rate at which OR and OL encounter each other [68]. The relatively unchanged
values likely resulted from differences between naked DNA in an in vitro environment and the compact, protein-decorated E. coli chromosome in the crowded cellular environment. Factors such as supercoiling and nonspecific, “histone-like” DNA-binding proteins could compact DNA and lead to more frequent encounters between OR and OL. Our observation that the unlooped λΔOL DNA was extremely compact (discussed in more detail below) was consistent with this view; this level of compaction (comparable to a polymer with a 3-nm rather than a 50-nm persistence length) could lead to a 50-fold increase in the rate at which OR and OL encounter each other [68]. The relatively unchanged 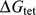 values could reflect the fact that the entropic and energetic costs of bringing OR and OL together are included in
values could reflect the fact that the entropic and energetic costs of bringing OR and OL together are included in 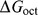 . Our looping frequency estimates confirm what were predicted by in vivo gene expression experiments—DNA was estimated to loop ∼72% of the time for wild-type OR–OL DNA and ∼69% for DNAs similar to our λOR3− and λOL3− constructs [35]. Correspondingly, the
. Our looping frequency estimates confirm what were predicted by in vivo gene expression experiments—DNA was estimated to loop ∼72% of the time for wild-type OR–OL DNA and ∼69% for DNAs similar to our λOR3− and λOL3− constructs [35]. Correspondingly, the 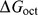 and
and 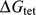 estimated in the in vivo work (−0.5 and −3.0 kcal/mol) [35] compared well to ours (0.3 and −3.2 kcal/mol).
estimated in the in vivo work (−0.5 and −3.0 kcal/mol) [35] compared well to ours (0.3 and −3.2 kcal/mol).
One important assumption we employed in calculating looping frequencies is that that looped and unlooped λWT, λOR3−, and λOL3− DNA molecules had similar 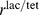 distributions to those of the looped control λnull and unlooped control λΔOL, respectively. It is possible that the unlooped states in the λWT, λOR3−, and λOL3− strains were more compact than that in λΔOL if after a DNA loop breaks OR–OL DNA does not always completely relax before it reforms again. In such a case, looping frequencies estimated using the linear-combination model would be upper limits on the true looping frequencies. Nevertheless, as we show above, our looping frequency estimates broadly agree with expectations from previous studies. Since this simple model only requires one free parameter and gives reasonable results, it is unnecessary to invoke more complicated models.
distributions to those of the looped control λnull and unlooped control λΔOL, respectively. It is possible that the unlooped states in the λWT, λOR3−, and λOL3− strains were more compact than that in λΔOL if after a DNA loop breaks OR–OL DNA does not always completely relax before it reforms again. In such a case, looping frequencies estimated using the linear-combination model would be upper limits on the true looping frequencies. Nevertheless, as we show above, our looping frequency estimates broadly agree with expectations from previous studies. Since this simple model only requires one free parameter and gives reasonable results, it is unnecessary to invoke more complicated models.
Effects of DNA Looping on Transcription Regulation
By comparing looping frequencies and corresponding CI expression levels in λWT, λΔOL, λOR3−, and λOL3−, we showed that loop stabilization by the CI tetramer between OR3 and OL3 is important for efficient PRM repression, and that looping mediated by a CI octamer at OR1 and OR2 is important for PRM activation. We note that while it is possible that the presence of tetO3 and lacO3 binding sites flanking OR–OL DNA may influence CI binding and/or transcription, this influence is negligible. This is because CI expression levels in these strains measured using smFISH are comparable to that of a wild-type λ lysogen (Table S8), and our results are consistent with previous observations [13],[49],[51],[53]. Furthermore, results are directly comparable as all strains used in this study are identical with respect to the presence and positioning of these binding sites.
Combining these results in our thermodynamic model, we estimated that CI-mediated DNA looping activates P
RM to 2.4 times its level when the DNA does not loop. This compares well to earlier estimates of 2–4 fold [49], and 1.6-fold for a high-expression PRM mutant [53]. Another study did not find looping activates transcription, modeling CI-concentration-dependent PR and PRM activities without invoking activation via looping (by assuming 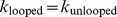 ) [35]. A later study indicated that this discrepancy may have resulted from different constructs used in the earlier study [49].
) [35]. A later study indicated that this discrepancy may have resulted from different constructs used in the earlier study [49].
The molecular basis for DNA loop-enhanced PRM activation is unclear. One possibility is that a CI dimer bound to OR2 interacts with RNA polymerase to a greater extent if it is part of a higher-order CI octamer [53]. Alternatively, a recent work showed that a DNA UP element proximal to OL [49],[69] enhances CI expression from PRM in looped DNA by contacting the α-C-terminal domain of RNA polymerase [51]. The activation mechanism could be clarified in future experiments measuring both looping frequency and PRM activity while varying operator and UP element sequences and introducing CI mutations affecting operator binding, oligomerization, and RNA polymerase interaction.
Kinetics of DNA Looping
We estimated the time scale a DNA molecule stays in a particular state by calculating the autocorrelation function of the 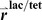 vector (Figure 4a). The
vector (Figure 4a). The 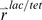 vector was strongly correlated for at least 0.5 s, suggesting that a particular DNA conformational state, either compact or extended, persisted for at least 0.5 s. This implies an upper limit of 2 s−1 for the rate of loop formation from the extended state. This upper bound of transition rate is in the range of what was observed in a previous TPM experiment, in which looped and unlooped states lasted for tens of seconds [44], and argues against a significantly faster rate used in a recent computer simulation (∼60 s−1) [50]. We note that although it is possible that transient CI unbinding does not necessarily lead to immediate and complete DNA conformational relaxation at our measurement time scale, the autocorrelation analysis puts an upper limit for the true transition rate between the looped and unlooped states. The same concern also applies to in vivo 3C and in vitro TPM experiments.
vector was strongly correlated for at least 0.5 s, suggesting that a particular DNA conformational state, either compact or extended, persisted for at least 0.5 s. This implies an upper limit of 2 s−1 for the rate of loop formation from the extended state. This upper bound of transition rate is in the range of what was observed in a previous TPM experiment, in which looped and unlooped states lasted for tens of seconds [44], and argues against a significantly faster rate used in a recent computer simulation (∼60 s−1) [50]. We note that although it is possible that transient CI unbinding does not necessarily lead to immediate and complete DNA conformational relaxation at our measurement time scale, the autocorrelation analysis puts an upper limit for the true transition rate between the looped and unlooped states. The same concern also applies to in vivo 3C and in vitro TPM experiments.
Slow transitions between looped and unlooped states imply that low or high expression states resulting from a particular DNA conformation could be long-lived, potentially committing a cell to a particular fate. Supporting this is a recent study that suggested that a single unlooping event could trigger induction of the lac operon [5]. We were unable to obtain time trajectories long enough to clearly identify looped/unlooped transitions for single DNA molecules. Development of brighter, faster maturing, and more photostable fluorescent proteins or in vivo labeling with synthetic fluorophores [70],[71] will help in increasing the number of measurements made on one DNA molecule, possibly enabling accurate measurement of DNA looping kinetics in vivo.
The Short End-to-End Separation of λΔOL Reflects the High Compactness of Chromosomal DNA
We observed very small end-to-end separation for the unlooped control (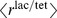 = 71 nm). This distance was shorter than expected from modeling the unlooped DNA as a noninteracting worm-chain with an in vitro persistence length of 50 nm [72], but consistent with the recently observed extreme bendability of short DNA molecules [73]. A noninteracting chain with an equivalent
= 71 nm). This distance was shorter than expected from modeling the unlooped DNA as a noninteracting worm-chain with an in vitro persistence length of 50 nm [72], but consistent with the recently observed extreme bendability of short DNA molecules [73]. A noninteracting chain with an equivalent 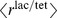 to that of λΔOL would have a persistence length of only 3 nm, which is physically infeasible. Our measurements of indistinguishable conformational distributions in the absence of PRM transcription and the presence of CI overexpression suggest that neither transcription nor nonspecifically bound CI played a major DNA-compacting role in our experiments. Furthermore, C. crescentus chromosomal DNA segments of ∼5 kb were found to be similarly compact and consistent with Brownian dynamics simulations of supercoiled DNA [74].
to that of λΔOL would have a persistence length of only 3 nm, which is physically infeasible. Our measurements of indistinguishable conformational distributions in the absence of PRM transcription and the presence of CI overexpression suggest that neither transcription nor nonspecifically bound CI played a major DNA-compacting role in our experiments. Furthermore, C. crescentus chromosomal DNA segments of ∼5 kb were found to be similarly compact and consistent with Brownian dynamics simulations of supercoiled DNA [74].
We attribute the small end-to-end separation observed for λΔOL to the high compaction of the E. coli chromosome in the crowded cellular environment. While the exact molecular mechanisms responsible for compaction remain unclear, previous studies found that in vitro binding of the histone-like HU proteins [75] (accession numbers P0ACF0, P0ACF4) and in vivo mammalian chromatin packing [76] reduced the apparent persistence length of DNA. Hence, it is possible that nucleoid-associated proteins such as HU may bring distal DNA sites together by protein–protein interactions and/or affect local DNA conformations by introducing bends and relieving torsional strain [77]. Another important factor could be negative supercoiling, which has been shown to compact the chromosomal DNA globally [78]. However, the exact effect of negative supercoiling on a 2.3-kb DNA segment is difficult to predict, because negative supercoiling could also introduce extended, plectonemic structures that promote large separations between DNA sites on relatively short length scales [78].
Potential Applications
Our two-color, high-resolution method can be applied to examine how chromosomal location, DNA length, genetic background, and growth conditions affect the distance between any two DNA sites on the E. coli chromosome. Furthermore, the spatial organization of the E. coli chromosome can be determined by systematically measuring 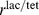 distributions between DNA sites throughout the chromosome. This method is similar to how chromosome conformation capture was used to generate a 3D model of the C. crescentus chromosome [79], but with significantly improved spatial resolution and without potential artifacts from fixation.
distributions between DNA sites throughout the chromosome. This method is similar to how chromosome conformation capture was used to generate a 3D model of the C. crescentus chromosome [79], but with significantly improved spatial resolution and without potential artifacts from fixation.
Materials and Methods
Strain and Plasmid Construction
A plasmid, pS2391, containing lacO3 and tetO3 (the tetO2 sequence [54] was used for each repeat in tetO3) sites was synthesized by Genewiz, Inc. Segments of λ DNA (OR through OL for λWT, OR up to but not including OL for λΔOL) from the wild-type lysogen JL5392 (a gift from John Little, University of Arizona) were amplified by PCR. This DNA was sequenced and inserted between lacO3 and tetO3 using the In-Fusion PCR cloning system (Clontech). A kanamycin-resistance cassette flanked by BamHI sites was amplified by PCR and inserted after lacO3. For strains with mutated operators, mutations r1 [80], OL3–4 [13], and cI G147D [62] were introduced to the λWT template via QuikChange (Agilent). A plasmid carrying the PRM−cI− mutations (Figure S5c) (λΔOLPRM − cI −) was constructed by overlapping PCR mutagenesis using complementary primers carrying the desired mutations, flanked by a forward primer that sits at the EcoRI site on the upstream end of the operon and a reverse primer at the ClaI site in the rexA gene downstream of cI. The 1.13 kb PCR product was introduced to the λΔOL plasmid by restriction ligation.
This procedure resulted in seven plasmids that were used as templates in subsequent chromosome insertion: pZH105 (λnull), pZH016 (λΔOL), pZH107 (λWT), pZH107r1 (λOR3−), pZH107OL3–4 (λOL3−), pACL006 (λWTG147D), and pACL007 (λΔOLPRM − cI −). Note that we use shorthand names such as λnull here for clarity; corresponding names used in our laboratory are listed in Table S4. The DNA sequence including lacO3, the λ DNA segment, tetO3, and the kanamycin resistance cassette was inserted into the chromosome of E. coli strain MG1655 by λ Red recombination [81], excising the lac operon, lacI, and all lacO sites.
To express the CI protein in trans from a plasmid, we constructed the plasmid pACL18 in which the wild-type cI ORF is driven by a constitutive promoter, PRMc, which has the wild-type −35 (TAGATA) and −10 (TAGATT) sequences, lacks OR2, and has a mutated OR1 sequence (CGCCTCGTGAGACCA) that eliminates binding by CI. The pRMc–cI fragment was then cloned to the ClaI site of the low-copy vector pACYC184. The plasmid pACL17 was generated similarly using a template containing the CIG147D mutation.
The two-color reporter plasmid pLau53, which expresses LacI-ECFP and TetR-EYFP polycistronically under the control of the PBAD promoter [82], was obtained from the Yale Coli Genetic Stock Center. Because the autofluorescence spectrum of live cells is generally strongest at wavelengths around 500 nm [83], single-molecule imaging of blue-shifted fluorescent proteins such as ECFP is difficult. The red fluorescent protein mCherry, which further benefits from a large Stokes shift, fast chromophore maturation rate, and high brightness relative to other monomeric RFPs [84], was inserted in place of ECFP. We also created a tandem LacI-mCherry-EYFP reporter, which was used as a fiducial marker, by inserting the linker sequence from the tandem-dimer fluorescent protein tdTomato [84] in between mCherry and EYFP.
To accurately localize a fluorescent spot arising from only a few fluorescent protein molecules above the background of unbound molecules within a cell, we reduced the reporter expression level by weakening the ribosome binding sites (RBSs). Weakened RBS sequences were designed using an online RBS calculator [85]. For example, the RBS for TetR-EYFP translation was the consensus AGGAGG Shine-Delgarno sequence in the parent plasmid pLau53. Our reporter plasmid had an ACCAGG Shine-Delgarno sequence, with a predicted ∼300-fold decrease in the TetR-EYFP translation rate. All sequences including chromosome insertions were verified by sequencing (Genewiz Inc). Reporter plasmids are described in Table 1.
Growth Condition
For all experiments reported in this study, cells were grown and imaged at room temperature (∼25°C) in M9 minimal media supplemented with MEM amino acids (Sigma). Cells were grown overnight with 0.4% glucose and 50 µg/ml carbenicillin to an optical density (OD600) of 0.4. After centrifugation at room temperature, cells were resuspended at OD600≈0.2 with 0.4% glycerol plus 0.2% L-arabinose and grown for 2 h (∼1 cell cycle) to induce LacI-mCherry and TetR-EYFP expression. Cells were again resuspended at OD600≈0.2 with 0.4% glucose and grown for another 2 h before immediate observation to allow time for fluorescent protein chromophores to mature.
We compared growth rates for the parent strain MG1655 to the experimental strain λnull to determine whether inserting the lacO3 and tetO3 construct into the chromosome and/or inducing expression from the reporter plasmid introduced a significant growth defect. Under induction growth conditions (∼27°C, M9 media with 0.4% glycerol and 1× MEM amino acids) starting at OD600≈0.1 and observing 8 h of growth, we measured doubling times of 2.7 h for MG1655 and 3.4 and 3.3 h for λnull harboring the reporter plasmid (in the absence and presence of 0.3% L-arabinose, respectively), indicating that there is no large growth defect associated with the insertion of the tandem operator sites into the chromosome and/or the expression of TetR-EYFP and LacI-mCherry fluorescent fusion proteins (Figure S6c).
Imaging Conditions
In each experiment, samples of all strains were placed on separate gel pads in the same growth chamber. Two sets of at least 30 movies were acquired for each strain, with the second set acquired in the reverse order to minimize any bias possibly introduced by observing some strains in a particular order. All images were acquired within less than one cell doubling time.
Cells were put on a gel pad made of 3% low-melting-temperature SeaPlaque agarose (Lonza) in M9 with glucose and imaged on an Olympus IX-81 inverted microscope with a 100× oil immersion objective (Olympus, PlanApo 100× NA 1.45) and additional 1.6× amplification. Images were split into red and yellow channels using an Optosplit II adaptor (Andor) and captured with an Ixon DU-895 (Andor) EM-CCD with a 13-µm pixel width using MetaMorph software (Molecular Devices). Laser illumination was provided at 514 nm by an argon ion laser (Coherent I-308), which also pumped a rhodamine dye laser (Coherent 599) tuned to ∼570 nm. A quarter-wave plate (Thorlabs) was used to circularly polarize excitation light. Emitted light was split by a long-pass filter, and the red and yellow images were filtered using HQ630/60 and ET540/30 bandpass filters (Chroma).
Measuring and Analyzing 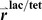
Images were inspected manually using a custom MATLAB script to identify spots that appeared in both EYFP and mCherry images. Images from all strains were displayed in random order without knowing the strain identify to avoid bias in spot selection. Pixel intensities within 3 pixels of the initial spot location were fitted with a symmetric, two-dimensional Gaussian distribution to estimate spot coordinates. The variance of the fit distribution was constrained to be less than 2 pixels. Spot-fitting error was estimated by scrambling residuals from a fit to the fluorescence data in 10 random permutations, adding them to the data, and fitting the resulting images; the reported error for a spot is the standard deviation of the distances between these fits and the initial fit to the raw data. Fitting error distributions are shown in Figure S1a.
The LacI-mCherry-EYFP tandem dimer (Figure S2b) in which the two fluorescent proteins were directly fused together was used to acquire fiducial control points to transform between the mCherry and EYFP coordinate systems. A projective transform was calculated from the control points using the cp2tform function in MATLAB. We found that relatively simple, global transformations were sufficient to transform coordinates of fluorescent beads (Tetraspeck, Invitrogen) with ∼10-nm registration error in our microscope setup, and did not see any further improvement with a locally weighted transformation used in in vitro two-color experiments [21]. This transformation was also used to generate the overlay images in Figure 2, Figure 3, and all supplemental movies. Fluorescent beads were not used as fiducial markers because the beads' emission spectra were different from those of the fluorescent proteins. Analysis was restricted to molecules in which mCherry and transformed EYFP coordinates were separated by less than 200 nm. Separations beyond this threshold were rare (∼1% of data, see two-dimensional distributions in Figure S4) and did not correlate with strain identity in any reasonable way. They possibly arose from data in which cells contained two labeled copies of OR–OL DNA.
After transformation into a uniform coordinate system, 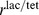 was calculated from the mCherry and EYFP coordinates and multiplied by an 81-nm pixel size (resulting from 160× magnification on a CCD with a 13-µm pixel width). Probability and cumulative distributions
was calculated from the mCherry and EYFP coordinates and multiplied by an 81-nm pixel size (resulting from 160× magnification on a CCD with a 13-µm pixel width). Probability and cumulative distributions 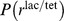 and
and 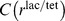 were calculated for 10-nm bins using the kernel smoothing probability density estimation (ksdensity) function in MATLAB, restricting the density to positive values and employing a uniform kernel width small enough to follow empirical cumulative density distributions without any systematic errors. Significant differences between
were calculated for 10-nm bins using the kernel smoothing probability density estimation (ksdensity) function in MATLAB, restricting the density to positive values and employing a uniform kernel width small enough to follow empirical cumulative density distributions without any systematic errors. Significant differences between 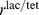 distributions were determined using a two-sample Kolmogorov–Smirnov test; two-tailed Student's t tests of sample means returned smaller, more significant p values. Errors in
distributions were determined using a two-sample Kolmogorov–Smirnov test; two-tailed Student's t tests of sample means returned smaller, more significant p values. Errors in 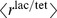 and
and 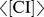 were determined by calculating the means of 1,000 bootstrapped samples; the reported error is the standard deviation of the calculated means. Looping frequencies were estimated by least squares fitting of 1,000 bootstrapped distributions (control distributions were also randomized on each iteration) and their error was calculated similarly.
were determined by calculating the means of 1,000 bootstrapped samples; the reported error is the standard deviation of the calculated means. Looping frequencies were estimated by least squares fitting of 1,000 bootstrapped distributions (control distributions were also randomized on each iteration) and their error was calculated similarly.
Single-Molecule Fluorescence in Situ Hybridization (smFISH)
Concentration measurements by smFISH followed a previously described protocol [66]. Transcripts from PRM were labeled with a mixture of 42 oligonucleotides labeled with CAL Fluor Red 610 (Biosearch Technologies), 31 of which hybridized to cI (11 targeted sequences not found in E. coli and did not cause a problematic level of false positives). Table S5 lists all 42 oligonucleotides. Labeled cells were imaged with 561-nm excitation at six imaging planes separated by 200 nm z-depth with negligible photobleaching. For each frame, fluorescent spots were automatically detected and fit to a Gaussian using a custom MATLAB routine. Nearly all molecules appeared in multiple image slices; the slice with the largest fit amplitude was kept. The integrated fluorescence of spots was observed to be quantized with one or a few molecules localized within one diffraction-limited spot. The intensity of one transcript was estimated from the distribution of spot intensities, and the number of molecules contributing to each spot was estimated from this quantization. The number of transcripts in each cell was estimated from the sum of the number of molecules in each spot within that cell. Alternatively, the number of molecules in one cell is proportional to its integrated fluorescence; this measurement provided the same average expression levels within error. The experiment was repeated to ensure that differences in labeling efficiency between samples were not responsible for differences in the number of detected molecules; combined data from both experiments were used for analysis.
Simulation of 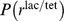
To generate simulated 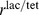 distributions, we first generated 10,000 random radial distances for a chain with a contour length
distributions, we first generated 10,000 random radial distances for a chain with a contour length  and persistence length
and persistence length  from a worm-like, noninteracting chain model using a Gaussian distribution with Daniels' approximation, which is accurate in the regime
from a worm-like, noninteracting chain model using a Gaussian distribution with Daniels' approximation, which is accurate in the regime  [72]:
[72]:
Each simulated  was projected onto the plane at a random angle to give a distance
was projected onto the plane at a random angle to give a distance  . Simulated spots were placed at
. Simulated spots were placed at  coordinates
coordinates  and
and 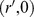 . The MATLAB function mvnrnd was then used to simulate normally distributed measurement error with a standard deviation of 22 nm to the coordinates of each simulated spot. This procedure was sufficient to simulate the λnull distribution (Figure S2c) using a fixed end-to-end distance of 22-nm (approximate distance between the centers of the lacO3 and lacO3 sites; Figure S2a). Note here that the simulation is simplified in that it assumes that each spot has the same 22-nm localization error. In reality, localization error varies between different spots (Figure S1a) and there are other sources of measurement error. These differences may explain the slight deviation of the simulated distribution from the experimental distribution. The same procedure was used to estimate the
. The MATLAB function mvnrnd was then used to simulate normally distributed measurement error with a standard deviation of 22 nm to the coordinates of each simulated spot. This procedure was sufficient to simulate the λnull distribution (Figure S2c) using a fixed end-to-end distance of 22-nm (approximate distance between the centers of the lacO3 and lacO3 sites; Figure S2a). Note here that the simulation is simplified in that it assumes that each spot has the same 22-nm localization error. In reality, localization error varies between different spots (Figure S1a) and there are other sources of measurement error. These differences may explain the slight deviation of the simulated distribution from the experimental distribution. The same procedure was used to estimate the 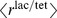 expected for 2.3-kb, B-form DNA with a 50-nm persistence (∼200 nm) as well as the apparently persistence length (3 nm) implied by the 71-nm
expected for 2.3-kb, B-form DNA with a 50-nm persistence (∼200 nm) as well as the apparently persistence length (3 nm) implied by the 71-nm 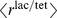 observed for λΔOL.
observed for λΔOL.
Thermodynamic Modeling
Additional descriptions of thermodynamic states are listed in Table S3. Parameter values were determined by first scoring a wide range of parameter values and iteratively searching narrower and more finely grained parameter ranges to manually minimize the sum of the squares of the differences between experimental and modeled values for looping frequency and CI expression level. We then refined this fit by least-squares minimization using MATLAB. This was done using a minimized model that only accounted for states likely to be populated near or above lysogenic CI concentrations (e.g., disregarding states in which OR1 and OR2 are unbound by CI). Using the same parameters and accounting for all 176 possible states (122 unique states accounting for degeneracy) did not significantly change the fit results. Fitting with this much more complex model gave octameric and tetrameric looping free energies of 0.6 and −3.3 kcal/mol and unlooped and looped expression rates of 2.1 and 5.3 nM/min. When determining parameters, rates were expressed in terms of changes in concentration per unit time; we followed earlier work in assuming that in a typical E. coli cell, a single molecule is at a concentration of ∼1.47 nM [35].
We do not report any estimate of fitting error; instead, we present only the parameters most consistent with our data and assumptions. Figure 5c and d shows that fit parameters were well-determined at a given combination of wild-type CI concentration and nonspecific binding parameters. As noted in the main text, varying these two parameters changed the absolute best-fit parameters, but did not dramatically change our conclusions. Furthermore, fixed parameters of previous studies were determined in a number of separate experiments employing different methods at temperatures other than 25°C; a rigorous estimate of modeling error would require knowing the error in the measurements of fixed parameters in our experimental conditions.
The basal CI expression rate, 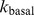 , was arbitrarily fixed at
, was arbitrarily fixed at 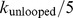 ; this did not have any significant impact on determining other parameters, as our measurements were all at or above lysogenic
; this did not have any significant impact on determining other parameters, as our measurements were all at or above lysogenic 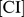 , where OR2 is almost always bound by a CI dimer. Additionally, the fraction of free CI dimers was fixed at its value for 150 CI molecules per cell at a given concentration of nonspecific binding sites and nonspecific binding affinity. Fixing the concentration of free CI dimers is a reasonable approximation if (1) nearly all CI molecules are in dimers and (2) the number of free nonspecific binding sites is not significantly changed by nonspecifically bound CI dimers.
, where OR2 is almost always bound by a CI dimer. Additionally, the fraction of free CI dimers was fixed at its value for 150 CI molecules per cell at a given concentration of nonspecific binding sites and nonspecific binding affinity. Fixing the concentration of free CI dimers is a reasonable approximation if (1) nearly all CI molecules are in dimers and (2) the number of free nonspecific binding sites is not significantly changed by nonspecifically bound CI dimers.
Image Representation in Figures
Figure 2a–e, Figure 4b, and Movies S1, S2, S3, S4, S5, S6 were prepared using NIH ImageJ [86]. Raw fluorescence image intensities were scaled linearly from the lowest to highest values in region shown. For EYFP/mCherry overlay images, brightfield images were inverted and converted to 8-bit RGB. Fluorescence images were bandpass filtered and background subtracted before being used to generate magenta (mCherry) and green (EYFP) 8-bit RGB images that were added to the brightfield image. The EYFP images were first transformed in MATLAB using the imtransform function and the same fiducial data that were used to transform EYFP spot locations into mCherry coordinates. For smFISH images (Figure 4b), the value of each pixel is the maximum value of that pixel in six images collected at different z-axis positions. Intensities for all images were scaled linearly from the minimum to the maximum of all pictures (117–4,840 counts in 16-bit images).
Supporting Information
Spot fitting and experimental error analysis. (a) Distribution of fitting errors for EYFP (green), mCherry (red) localizations, and 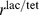 (black). Errors were estimated using a bootstrapping procedure by fitting raw data to a Gaussian distribution. The residuals from this fit were then randomly rearranged and added back to the data in 10 different permutations. The reported error is the standard deviation of the distance between these 10 locations and the initial fit location. Error in
(black). Errors were estimated using a bootstrapping procedure by fitting raw data to a Gaussian distribution. The residuals from this fit were then randomly rearranged and added back to the data in 10 different permutations. The reported error is the standard deviation of the distance between these 10 locations and the initial fit location. Error in 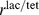 was determined similarly; from the 10 bootstrapped EYFP and mCherry fits, 100 distances were obtained and the error was estimated as the standard deviation of the difference between these distances and the distance determined from fitting the raw data. (b) A compilation of all data from three separate experiments was used for all analysis in the main text. Here,
was determined similarly; from the 10 bootstrapped EYFP and mCherry fits, 100 distances were obtained and the error was estimated as the standard deviation of the difference between these distances and the distance determined from fitting the raw data. (b) A compilation of all data from three separate experiments was used for all analysis in the main text. Here, 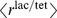 is shown for the individual experiments. Error was estimated as the standard deviation of the means of 1,000 bootstrapped distributions. Except for one sample (λOR3−, day 3), the estimated mean separations for all days followed the trend
is shown for the individual experiments. Error was estimated as the standard deviation of the means of 1,000 bootstrapped distributions. Except for one sample (λOR3−, day 3), the estimated mean separations for all days followed the trend 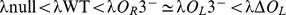 .
.
(TIF)
Estimate of positive control dimensions and apparent end-to-end distance distribution. (a) The maximum distance between TetR-EYFP and mCherry-LacI chromophores was approximated assuming straight DNA. All distances are in nm. Here, bound fusion proteins are shown on the same face of a DNA molecule, but this needs not be the case. Dimers of DNA-binding proteins were based on Protein Data Bank (PDB) entries for TetR (1QPI [87]) and LacI (1EFA [88]). Both fluorescent proteins are shown using the entry for GFP (1GFL [89]). Protein structures images generated using VMD [90]. (b) In an alternative positive control that was used to collect fiducial data for image registration, the plasmid pZH102R33TD encodes the tandem-dimer reporter LacI-mCherry-EYFP. (c) The 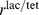 PDF for the λnull control (black line; 1 s.e.m. shown in red as in Figure 3a) is shown with the distribution of 10,000 numerically simulated end-to-end distances for two sites separated by 22 nm, randomly projected onto the 2D plane, and subjected to 22-nm localization error for both ends (dashed black line). PDFs were calculated using methods described in main text. See Materials and Methods for simulation details.
PDF for the λnull control (black line; 1 s.e.m. shown in red as in Figure 3a) is shown with the distribution of 10,000 numerically simulated end-to-end distances for two sites separated by 22 nm, randomly projected onto the 2D plane, and subjected to 22-nm localization error for both ends (dashed black line). PDFs were calculated using methods described in main text. See Materials and Methods for simulation details.
(TIF)
Plots showing trajectories of 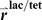 vectors for all data from all strains for every molecule that was fit in both the EYFP and mCherry images for at least 8 consecutive frames (800 ms). Green and magenta lines are single-color trajectories for TetR-EYFP and LacI-mCherry spots, respectively; the corresponding
vectors for all data from all strains for every molecule that was fit in both the EYFP and mCherry images for at least 8 consecutive frames (800 ms). Green and magenta lines are single-color trajectories for TetR-EYFP and LacI-mCherry spots, respectively; the corresponding 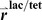 trajectory with time colored-coded from blue to red is plotted on top at the same length scale. Coordinates are in nm.
trajectory with time colored-coded from blue to red is plotted on top at the same length scale. Coordinates are in nm.
(PDF)
Two-dimensional distributions of the x and y components of 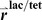 vectors. (a) A cartoon describes the calculation of the x and y components of the
vectors. (a) A cartoon describes the calculation of the x and y components of the 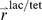 vector. In the projected image, the
vector. In the projected image, the 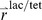 vector has two components determined by the arbitrary orientation of the detector. (b–f) Heat maps of the distribution of the x and y components of
vector has two components determined by the arbitrary orientation of the detector. (b–f) Heat maps of the distribution of the x and y components of 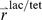 vectors of each strain. Plots were generated by binning the data for all
vectors of each strain. Plots were generated by binning the data for all 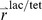 into 5 nm×5 nm bins. The resulting 2-dimensional distribution was then filtered with a Gaussian kernal (with a width similar to spot-localization precision) to approximate the smoothed distributions. Each image is colored by the probability of the
into 5 nm×5 nm bins. The resulting 2-dimensional distribution was then filtered with a Gaussian kernal (with a width similar to spot-localization precision) to approximate the smoothed distributions. Each image is colored by the probability of the 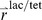 vector falling within a given bin according to the scale bar in (b).
vector falling within a given bin according to the scale bar in (b).
(TIF)
Experiments showing the effects of transcription, nonspecific CI binding and higher-ordered CI oligomer on DNA looping. (a) End-to-end distance (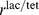 ) distributions (PDF) for λnull (red), λΔOL (blue), λΔOLPRM−cI− (purple), and λΔOLPRM−cI−/cItrans (green). The PDF is estimated for 10-nm bins. (b) Cumulative density of
) distributions (PDF) for λnull (red), λΔOL (blue), λΔOLPRM−cI− (purple), and λΔOLPRM−cI−/cItrans (green). The PDF is estimated for 10-nm bins. (b) Cumulative density of 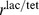 (CDF) for λnull (red), λΔOL (blue), λΔOLPRM−cI− (purple), and λΔOLPRM−cI−/cItrans (green). The CDF is estimated for 10-nm bins. (c) DNA sequence for the PRM−cI− mutant in comparison to the wild-type sequence. Mutated nucleotides are shown in red. (d) Gel shift assay monitoring the binding of wild-type CI protein. Lane 1–4, CI at concentrations of 0, 150, 300, and 600 nM binding to a 158-bp DNA fragment (20 nM) amplified from the plasmid pZH107 carrying the wild-type PRM DNA sequence. Lane 5–8, CI at concentrations of 150, 0, 300, and 600 nM (note loading order) binding to a 158-bp DNA fragment (20 nM) amplified from the plasmid pACL007 carrying the PRM−cI− sequence. Lane 9: empty. Lane 10–13, CI at concentrations of 0, 150, 300, and 600 nM binding to a 140-bp DNA fragment (20 nM) amplified from the E. coli hns promoter region, which CI does not bind specifically. Reaction mixtures were incubated in a buffer (10 mM Tris pH 8.0, 50 mM KCI, 1 mM MgCl2, 10% glycerol, 100 ug/ml BSA, 1 mM DTT) at room temperature for 10 min. Samples were electrophoresed in Bio-Rad 4–20% Gradient TBE gels (Bio-Rad, Hercules, CA) in a cold room and then stained with Ethidium Bromide for 30 min. (e) Fraction of bound DNA (intensity of low-weight band divided by intensity of lane over background) quantified using NIH ImageJ for the gel shown in (d). (f, g) Distributions of
(CDF) for λnull (red), λΔOL (blue), λΔOLPRM−cI− (purple), and λΔOLPRM−cI−/cItrans (green). The CDF is estimated for 10-nm bins. (c) DNA sequence for the PRM−cI− mutant in comparison to the wild-type sequence. Mutated nucleotides are shown in red. (d) Gel shift assay monitoring the binding of wild-type CI protein. Lane 1–4, CI at concentrations of 0, 150, 300, and 600 nM binding to a 158-bp DNA fragment (20 nM) amplified from the plasmid pZH107 carrying the wild-type PRM DNA sequence. Lane 5–8, CI at concentrations of 150, 0, 300, and 600 nM (note loading order) binding to a 158-bp DNA fragment (20 nM) amplified from the plasmid pACL007 carrying the PRM−cI− sequence. Lane 9: empty. Lane 10–13, CI at concentrations of 0, 150, 300, and 600 nM binding to a 140-bp DNA fragment (20 nM) amplified from the E. coli hns promoter region, which CI does not bind specifically. Reaction mixtures were incubated in a buffer (10 mM Tris pH 8.0, 50 mM KCI, 1 mM MgCl2, 10% glycerol, 100 ug/ml BSA, 1 mM DTT) at room temperature for 10 min. Samples were electrophoresed in Bio-Rad 4–20% Gradient TBE gels (Bio-Rad, Hercules, CA) in a cold room and then stained with Ethidium Bromide for 30 min. (e) Fraction of bound DNA (intensity of low-weight band divided by intensity of lane over background) quantified using NIH ImageJ for the gel shown in (d). (f, g) Distributions of 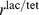 identical in description to those in (a, b) showing strains λnull (red), λΔOL (blue), λG147D (purple), and λG147D/cIG147D,trans (green).
identical in description to those in (a, b) showing strains λnull (red), λΔOL (blue), λG147D (purple), and λG147D/cIG147D,trans (green).
(TIF)
Growth rate comparisons. (a, b) Strains used in thermodynamic modeling were diluted from exponential growth to low optical densities in M9 minimal media supplemented with 0.4% glucose and carbenicillin as described in the main text. OD600 was measured over 10 h of growth for two replicate experiments. Strains are λΔOL (blue), λWT (red), λOR3− (green), and λOL3− (purple). Doubling times calculated using the Microsoft Excel LOGEST function range from 1.7 to 2.5 h. Two independent replicates are shown. (c) Growth rates for the parent E. coli strain MG1655 (blue) were compared to those of the control strain λnull in which the lac operon is replaced with a construct incorporating the lacO3 and tetO3 binding site arrays and which harbors the plasmid pZH102R33Y29 which expresses both TetR-EYFP and LacI-mCherry fluorescent fusion proteins upon arabinose induction. Strains were grown in M9 minimal media supplemented with 0.4% glycerol and λnull was grown in both the absence (red) and presence (green) of 0.3% L-arabinose. Doubling times were 2.7 h for MG1655 and 3.4 and 3.3 h for λnull in the absence and presence of L-arabinose, respectively.
(TIF)
Fluorescence movie montage for strain λnull corresponding to the data in Figure 2c. Single-color images for TetR-EYFP (top left) and LacI-mCherry (top right) data have intensities scaled linearly from the lowest to the highest pixel values in the first image in each time series. Before creating the overlay images (bottom), single-color images were background subtracted and bandpass filtered using the program ImageJ [91]. The overlay images are scaled to be twice as large as the single-color images. Scale bars correspond 4 µm in the small, single-color images and 2 µm in the overlay image. Ten consecutive image frames are shown in real time (10 frames per second); the movie is looped 5 times.
(MOV)
Fluorescence movie montage for strain λΔOL corresponding to the data in Figure 2d. Single-color images for TetR-EYFP (top left) and LacI-mCherry (top right) data have intensities scaled linearly from the lowest to the highest pixel values in the first image in each time series. Before creating the overlay images (bottom), single-color images were background subtracted and bandpass filtered using the program ImageJ [91]. The overlay images are scaled to be twice as large as the single-color images. Scale bars correspond 4 µm in the small, single-color images and 2 µm in the overlay image. Ten consecutive image frames are shown in real time (10 frames per second); the movie is looped 5 times.
(MOV)
Fluorescence movie montage for strain λΔOL corresponding to the data in Figure 2e. Single-color images for TetR-EYFP (top left) and LacI-mCherry (top right) data have intensities scaled linearly from the lowest to the highest pixel values in the first image in each time series. Before creating the overlay images (bottom), single-color images were background subtracted and bandpass filtered using the program ImageJ [91]. The overlay images are scaled to be twice as large as the single-color images. Scale bars correspond 4 µm in the small, single-color images and 2 µm in the overlay image. Thirteen consecutive image frames are shown in real time (10 frames per second); the movie is looped 5 times.
(MOV)
Fluorescence movie montage for strain λWT corresponding to a typical, long movie. Single-color images for TetR-EYFP (top left) and LacI-mCherry (top right) data have intensities scaled linearly from the lowest to the highest pixel values in the first image in each time series. Before creating the overlay images (bottom), single-color images were background subtracted and bandpass filtered using the program ImageJ [91]. The overlay images are scaled to be twice as large as the single-color images. Scale bars correspond 4 µm in the small, single-color images and 2 µm in the overlay image. Twelve consecutive image frames are shown in real time (10 frames per second); the movie is looped 5 times.
(MOV)
Fluorescence movie montage for strain λOR3− corresponding to a typical, long movie. Single-color images for TetR-EYFP (top left) and LacI-mCherry (top right) data have intensities scaled linearly from the lowest to the highest pixel values in the first image in each time series. Before creating the overlay images (bottom), single-color images were background subtracted and bandpass filtered using the program ImageJ [91]. The overlay images are scaled to be twice as large as the single-color images. Scale bars correspond 4 µm in the small, single-color images and 2 µm in the overlay image. Thirteen consecutive image frames are shown in real time (10 frames per second); the movie is looped 5 times.
(MOV)
Fluorescence movie montage for strain λOL3− corresponding to a typical, long movie. Single-color images for TetR-EYFP (top left) and LacI-mCherry (top right) data have intensities scaled linearly from the lowest to the highest pixel values in the first image in each time series. Before creating the overlay images (bottom), single-color images were background subtracted and bandpass filtered using the program ImageJ [91]. The overlay images are scaled to be twice as large as the single-color images. Scale bars correspond 4 µm in the small, single-color images and 2 µm in the overlay image. Twelve consecutive image frames are shown in real time (10 frames per second); the movie is looped 5 times.
(MOV)
Looping frequencies were estimated from alternate data sets using either all data or only the data from the first frames (for molecules appearing in more than one sequential frame) and fitting either probability (PDF) or cumulative (CDF) distributions. The first row results for each strain were reported in the main text.
(DOCX)
States used in thermodynamic modeling. We used free-energy parameters that were described by Dodd et al. [35]. States that will not be populated near lysogenic CI concentrations (e.g., those without OL1 or OL2 bound) are ignored; the reference state (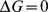 ) has CI dimers bound to OL1 and OL2. A state with OR free of CI is included to show activation in Figure 5a and b, but does not significantly change fit parameters; because OR1 and OR2 binding is highly cooperative, we do not model states with only one or the other operator bound. The degeneracy term indicates how many microstates exist with identical CI dimer binding patterns and free energies. A particular macrostate may have several microstates that differ in terms of parallel or antiparallel looping configurations or in the identity of binding sites participating in cooperative interactions (either through looping or through adjacent dimers). Here, we also list whether a state is looped (1 for looped; 2 for unlooped) as well as its transcription rate,
) has CI dimers bound to OL1 and OL2. A state with OR free of CI is included to show activation in Figure 5a and b, but does not significantly change fit parameters; because OR1 and OR2 binding is highly cooperative, we do not model states with only one or the other operator bound. The degeneracy term indicates how many microstates exist with identical CI dimer binding patterns and free energies. A particular macrostate may have several microstates that differ in terms of parallel or antiparallel looping configurations or in the identity of binding sites participating in cooperative interactions (either through looping or through adjacent dimers). Here, we also list whether a state is looped (1 for looped; 2 for unlooped) as well as its transcription rate,  (0; 1 for
(0; 1 for 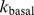 ; 2 for
; 2 for 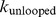 ; 3 for
; 3 for 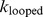 ). The free energy of state 2 is called
). The free energy of state 2 is called 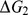 below.
below.
(DOCX)
Thermodynamic model fitting using alternative choices for wild-type CI concentration (expressed here in molecules/cell; in the model, 1 molecule per cell is equivalent to 1.47 nM) and the fraction of CI molecules that are in the form of free dimers. The approximation of a constant free-dimer fraction is reasonable if specifically bound CI dimers (up to 6 dimers composed of 12 monomers) do not make up a large fraction of total CI and if CI concentration is sufficiently high that almost all CI molecules are in dimeric complexes. The free-dimer fractions used here were calculated assuming the absence of specific binding sites using the parameters for nonspecific binding site affinity and concentration estimated by Dodd et al. [35]. Results in the first row are the same as those presented in the main text.
(DOCX)
Names of new strains used in this study (as used internally in our lab) and shorthand names used in the main text.
(DOCX)
Sequences of oligonucleotide probes for single-molecule fluorescence in situ hybridization (smFISH) experiment. Asterisks indicate probes that do not hybridize specifically with any E. coli sequence. All other probes hybridize nonoverlapping sequence in the cI coding region of the mRNA transcript from the PRM promoter.
(DOCX)
Measurement statistics for experiment comparing 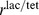 distributions for looped and unlooped control strains to
distributions for looped and unlooped control strains to 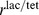 for strains lacking OL and having weakened PRM promoters with and without the overexpression of wild-type CI from a plasmid. Errors for the
for strains lacking OL and having weakened PRM promoters with and without the overexpression of wild-type CI from a plasmid. Errors for the 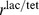 measurements are all 1 s.e.m. as estimated from 1,000 bootstrapped samples. Note that
measurements are all 1 s.e.m. as estimated from 1,000 bootstrapped samples. Note that 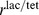 distributions display small, day-to-day variability between experiments (see Figure S1, this table, Table 2, Table S7), but the trend stays the same for a given set of experiments.
distributions display small, day-to-day variability between experiments (see Figure S1, this table, Table 2, Table S7), but the trend stays the same for a given set of experiments.
(DOCX)
Measurement statistics for experiment comparing 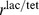 distributions for looped and unlooped control strains to
distributions for looped and unlooped control strains to 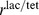 for strains in which CI harbors the G147D mutation with and without the overexpression of CIG147D from a plasmid. Errors for the
for strains in which CI harbors the G147D mutation with and without the overexpression of CIG147D from a plasmid. Errors for the 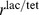 measurements are all 1 s.e.m. as estimated from 1,000 bootstrapped samples. Note that
measurements are all 1 s.e.m. as estimated from 1,000 bootstrapped samples. Note that 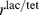 distributions display small, day-to-day variability between experiments (see Figure S1, this table, Table 2, Table S6), but the trends stays the same for a given set of experiments.
distributions display small, day-to-day variability between experiments (see Figure S1, this table, Table 2, Table S6), but the trends stays the same for a given set of experiments.
(DOCX)
CI expression levels measured by smFISH for wild-type phage lambda lysogen JL5392 and additional strains. For strains with replicate experiments (N, number of independent experiments), errors indicate standard deviation. The expression levels were normalized to wild-type units (WTUs) using the λWT strain.
(DOCX)
Acknowledgments
We thank Drs. Roger McMacken, Robert Schleif, and Cynthia Wolberger (Johns Hopkins University) for helpful discussions and critical comments on the manuscript. We thank Dr. Sankar Adhya (NIH) for the helpful suggestion of the G147D mutant experiment. We thank Dr. Henrik Flyvbjerg (DTU Nanotech) and Dr. Zan Luthey Schulten (UIUC) for helpful discussions.
Abbreviations
- 3C
chromosome conformation capture
- CDF
cumulative distribution function
probability density function
- RBS
ribosome binding site
- smFISH
single-molecule fluorescence in situ hybridization
- TPM
tethered partial motion
- WLU
Wild-type λ units
Funding Statement
This work was supported by NSF CAREER award 0746796 and March of Dimes Research grant 1-FY2011. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Schleif R (1992) DNA looping. Annu Rev Biochem 61: 199–223. [DOI] [PubMed] [Google Scholar]
- 2. So L, Ghosh A, Zong C, Sepulveda LA, Segev R, et al. (2011) General properties of transcriptional time series in Escherichia coli. Nat Genet 43: 554–560 doi:10.1038/ng.821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hensel Z, Feng H, Han B, Hatem C, Wang J, et al. (2012) Stochastic expression dynamics of a transcription factor revealed by single-molecule noise analysis. Nat Struct Mol Biol 19 (8) 797–802. [DOI] [PubMed] [Google Scholar]
- 4. Salman H, Brenner N, Tung C, Elyahu N, Stolovicki E, et al. (2012) Universal protein fluctuations in populations of microorganisms. Phys Rev Lett 108: 238105 doi:10.1103/PhysRevLett.108.238105 [DOI] [PubMed] [Google Scholar]
- 5. Choi PJ, Cai L, Frieda K, Xie XS (2008) A stochastic single-molecule event triggers phenotype switching of a bacterial cell. Science 322: 442–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vilar JM, Leibler S (2003) DNA looping and physical constraints on transcription regulation. J Mol Biol 331: 981–989. [DOI] [PubMed] [Google Scholar]
- 7. Dunn TM, Hahn S, Ogden S, Schleif RF (1984) An operator at −280 base pairs that is required for repression of araBAD operon promoter: addition of DNA helical turns between the operator and promoter cyclically hinders repression. Proceedings of the National Academy of Sciences 81: 5017–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Muller-Hill B (1998) The function of auxiliary operators. Mol Microbiol 29: 13–18. [DOI] [PubMed] [Google Scholar]
- 9. Dandanell G, Valentin-Hansen P, Larsen JE, Hammer K (1987) Long-range cooperativity between gene regulatory sequences in a prokaryote. Nature 325: 823–826 doi:10.1038/325823a0 [DOI] [PubMed] [Google Scholar]
- 10. Wyman C, Rombel I, North AK, Bustamante C, Kustu S (1997) Unusual oligomerization required for activity of NtrC, a bacterial enhancer-binding protein. Science 275: 1658–1661 doi:10.1126/science.275.5306.1658 [DOI] [PubMed] [Google Scholar]
- 11. Geanacopoulos M, Adhya S (2002) Genetic analysis of GalR tetramerization in DNA looping during repressosome assembly. J Biol Chem 277: 33148–33152 doi:10.1074/jbc.M202445200 [DOI] [PubMed] [Google Scholar]
- 12. Révet B, von Wilcken-Bergmann B, Bessert H, Barker A, Müller-Hill B (1999) Four dimers of lambda repressor bound to two suitably spaced pairs of lambda operators form octamers and DNA loops over large distances. Curr Biol 9: 151–154. [DOI] [PubMed] [Google Scholar]
- 13. Dodd IB, Perkins AJ, Tsemitsidis D, Egan JB (2001) Octamerization of lambda CI repressor is needed for effective repression of P(RM) and efficient switching from lysogeny. Genes Dev 15: 3013–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dekker J, Rippe K, Dekker M, Kleckner N (2002) Capturing chromosome conformation. Science 295: 1306–1311 doi:10.1126/science.1067799 [DOI] [PubMed] [Google Scholar]
- 15. Simonis M, Kooren J, Laat W de (2007) An evaluation of 3C-based methods to capture DNA interactions. Nature Methods 4: 895–901 doi:10.1038/nmeth1114 [DOI] [PubMed] [Google Scholar]
- 16. Dekker J (2006) The three “C” s of chromosome conformation capture: controls, controls, controls. Nature Methods 3: 17–21 doi:10.1038/nmeth823 [DOI] [PubMed] [Google Scholar]
- 17. Houston PL, Broach JR (2006) The dynamics of homologous pairing during mating type interconversion in budding yeast. PLoS Genet 2: e98 doi:10.1371/journal.pgen.0020098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Possoz C, Filipe SR, Grainge I, Sherratt DJ (2006) Tracking of controlled Escherichia coli replication fork stalling and restart at repressor-bound DNA in vivo. EMBO J 25: 2596–2604 doi:10.1038/sj.emboj.7601155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang W, Li G-W, Chen C, Xie XS, Zhuang X (2011) Chromosome organization by a nucleoid-associated protein in live bacteria. Science 333: 1445–1449 doi:10.1126/science.1204697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thompson RE, Larson DR, Webb WW (2002) Precise nanometer localization analysis for individual fluorescent probes. Biophysical Journal 82: 2775–2783 doi:10.1016/S0006-3495(02)75618-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Churchman LS, Ökten Z, Rock RS, Dawson JF, Spudich JA (2005) Single molecule high-resolution colocalization of Cy3 and Cy5 attached to macromolecules measures intramolecular distances through time. Proceedings of the National Academy of Sciences of the United States of America 102: 1419–1423 doi:10.1073/pnas.0409487102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ptashne M (2004) A genetic switch: phage lambda revisited. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 23. Rozanov DV, D'Ari R, Sineoky SP (1998) RecA-independent pathways of lambdoid prophage induction in Escherichia coli. J Bacteriol 180: 6306–6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Little JW, Shepley DP, Wert DW (1999) Robustness of a gene regulatory circuit. EMBO J 18: 4299–4307 doi:10.1093/emboj/18.15.4299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aurell E, Brown S, Johanson J, Sneppen K (2002) Stability puzzles in phage lambda. Phys Rev E Stat Nonlin Soft Matter Phys 65: 051914. [DOI] [PubMed] [Google Scholar]
- 26. Amir A, Kobiler O, Rokney A, Oppenheim AB, Stavans J (2007) Noise in timing and precision of gene activities in a genetic cascade. Mol Syst Biol 3: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Atsumi S, Little JW (2004) Regulatory circuit design and evolution using phage lambda. Genes Dev 18: 2086–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Michalowski CB, Short MD, Little JW (2004) Sequence tolerance of the phage lambda PRM promoter: implications for evolution of gene regulatory circuitry. J Bacteriol 186: 7988–7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Becskei A, Serrano L (2000) Engineering stability in gene networks by autoregulation. Nature 405: 590–593 doi:10.1038/35014651 [DOI] [PubMed] [Google Scholar]
- 30. Michalowski CB, Little JW (2005) Positive autoregulation of cI is a dispensable feature of the phage lambda gene regulatory circuitry. J Bacteriol 187: 6430–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson AD, Meyer BJ, Ptashne M (1979) Interactions between DNA-bound repressors govern regulation by the lambda phage repressor. Proc Natl Acad Sci U S A 76: 5061–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson AD, Poteete AR, Lauer G, Sauer RT, Ackers GK, et al. (1981) lambda repressor and cro–components of an efficient molecular switch. Nature 294: 217–223. [DOI] [PubMed] [Google Scholar]
- 33. Shea MA, Ackers GK (1985) The OR control system of bacteriophage lambda: a physical-chemical model for gene regulation. Journal of Molecular Biology 181: 211–230 doi:10.1016/0022-2836(85)90086-5 [DOI] [PubMed] [Google Scholar]
- 34. Babić AC, Little JW (2007) Cooperative DNA binding by CI repressor is dispensable in a phage λ variant. Proceedings of the National Academy of Sciences 104: 17741–17746 doi:10.1073/pnas.0602223104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dodd IB, Shearwin KE, Perkins AJ, Burr T, Hochschild A, et al. (2004) Cooperativity in long-range gene regulation by the lambda CI repressor. Genes Dev 18: 344–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ptashne M, Backman K, Humayun MZ, Jeffrey A, Maurer R, et al. (1976) Autoregulation and function of a repressor in bacteriophage lambda. Science 194: 156–161. [DOI] [PubMed] [Google Scholar]
- 37. Reichardt L, Kaiser AD (1971) Control of λ repressor synthesis. Proceedings of the National Academy of Sciences of the United States of America 68: 2185–2189 doi:VL - 68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meyer BJ, Ptashne M (1980) Gene regulation at the right operator (OR) of bacteriophage lambda. III. lambda repressor directly activates gene transcription. J Mol Biol 139: 195–205. [DOI] [PubMed] [Google Scholar]
- 39. Hawley DK, McClure WR (1982) Mechanism of activation of transcription initiation from the lambda PRM promoter. J Mol Biol 157: 493–525. [DOI] [PubMed] [Google Scholar]
- 40. Maurer R, Meyer BJ, Ptashne M (1980) Gene regulation at the right operator (OR) of bacteriophage [lambda]: I. OR3 and autogenous negative control by repressor. J Mol Biol 139: 147–161 doi:10.1016/0022-2836(80)90302-2 [DOI] [PubMed] [Google Scholar]
- 41. Senear DF, Laue TM, Ross JBA, Waxman E, Eaton S, et al. (1993) The primary self-assembly reaction of bacteriophage .lambda. cI repressor dimers is to octamer. Biochemistry 32: 6179–6189 doi:10.1021/bi00075a010 [DOI] [PubMed] [Google Scholar]
- 42. Wang H, Finzi L, Lewis DEA, Dunlap D (2009) AFM studies of repressor oligomers securing DNA loops. Current Pharmaceutical Biotechnology 10: 494–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zurla C, Franzini A, Galli G, Dunlap DD, Lewis DEA, et al. (2006) Novel tethered particle motion analysis of CI protein-mediated DNA looping in the regulation of bacteriophage lambda. Journal of Physics: Condensed Matter 18: S225–S234 doi:10.1088/0953-8984/18/14/S07 [Google Scholar]
- 44. Zurla C, Manzo C, Dunlap D, Lewis DEA, Adhya S, et al. (2009) Direct demonstration and quantification of long-range DNA looping by the λ bacteriophage repressor. Nucleic Acids Res 37: 2789–2795 doi:10.1093/nar/gkp134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Finzi L, Dunlap DD (2010) Single-molecule approaches to probe the structure, kinetics, and thermodynamics of nucleoprotein complexes that regulate transcription. Journal of Biological Chemistry 285: 18973–18978 doi:10.1074/jbc.R109.062612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Manzo C, Zurla C, Dunlap DD, Finzi L (2012) The effect of nonspecific binding of lambda repressor on DNA looping dynamics. Biophys J 103: 1753–1761 doi:10.1016/j.bpj.2012.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vilar JM, Saiz L (2005) DNA looping in gene regulation: from the assembly of macromolecular complexes to the control of transcriptional noise. Current Opinion in Genetics & Development 15: 136–144 doi:10.1016/j.gde.2005.02.005 [DOI] [PubMed] [Google Scholar]
- 48. Lou C, Yang X, Liu X, He B, Ouyang Q (2007) A quantitative study of lambda-phage SWITCH and its components. Biophys J 92: 2685–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Anderson LM, Yang H (2008) DNA looping can enhance lysogenic CI transcription in phage lambda. Proc Natl Acad Sci U S A 105: 5827–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Morelli MJ, ten Wolde PR, Allen RJ (2009) DNA looping provides stability and robustness to the bacteriophage λ switch. Proceedings of the National Academy of Sciences 106: 8101–8106 doi:10.1073/pnas.0810399106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cui L, Murchland I, Shearwin KE, Dodd IB (2013) Enhancer-like long-range transcriptional activation by λ CI-mediated DNA looping. Proc Natl Acad Sci USA 110: 2922–2927 doi:10.1073/pnas.1221322110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ackers GK, Johnson AD, Shea MA (1982) Quantitative model for gene regulation by lambda phage repressor. Proc Natl Acad Sci U S A 79: 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lewis D, Le P, Zurla C, Finzi L, Adhya S (2011) Multilevel autoregulation of λ repressor protein CI by DNA looping in vitro. Proc Natl Acad Sci USA 108: 14807–14812 doi:10.1073/pnas.1111221108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hillen W, Gatz C, Altschmied L, Schollmeier K, Meier I (1983) Control of expression of the Tn10-encoded tetracycline resistance genes: equilibrium and kinetic investigation of the regulatory reactions. Journal of Molecular Biology 169: 707–721 doi:10.1016/S0022-2836(83)80166-1 [DOI] [PubMed] [Google Scholar]
- 55. Sadler JR, Sasmor H, Betz JL (1983) A perfectly symmetric lac operator binds the lac repressor very tightly. Proc Natl Acad Sci USA 80: 6785–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Churchman LS, Flyvbjerg H, Spudich JA (2006) A non-Gaussian distribution quantifies distances measured with fluorescence localization techniques. Biophys J 90: 668–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lu Y, Weers B, Stellwagen NC (2001) DNA persistence length revisited. Biopolymers 61: 261–275 doi:10.1002/bip.10151 [DOI] [PubMed] [Google Scholar]
- 58. Thanbichler M, Shapiro L (2006) Chromosome organization and segregation in bacteria. Journal of Structural Biology 156: 292–303 doi:10.1016/j.jsb.2006.05.007 [DOI] [PubMed] [Google Scholar]
- 59. O'Sullivan JM, Tan-Wong SM, Morillon A, Lee B, Coles J, et al. (2004) Gene loops juxtapose promoters and terminators in yeast. Nat Genet 36: 1014–1018 doi:10.1038/ng1411 [DOI] [PubMed] [Google Scholar]
- 60. Tan-Wong SM, French JD, Proudfoot NJ, Brown MA (2008) Dynamic interactions between the promoter and terminator regions of the mammalian BRCA1 gene. Proc Natl Acad Sci USA 105: 5160–5165 doi:10.1073/pnas.0801048105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sarai A, Takeda Y (1989) Lambda repressor recognizes the approximately 2-fold symmetric half-operator sequences asymmetrically. Proc Natl Acad Sci USA 86: 6513–6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Beckett D, Burz DS, Ackers GK, Sauer RT (1993) Isolation of lambda repressor mutants with defects in cooperative operator binding. Biochemistry 32: 9073–9079. [DOI] [PubMed] [Google Scholar]
- 63. Burz DS, Ackers GK (1996) Cooperativity mutants of bacteriophage lambda cI repressor: temperature dependence of self-assembly. Biochemistry 35: 3341–3350 doi:10.1021/bi952055x [DOI] [PubMed] [Google Scholar]
- 64. Stayrook S, Jaru-Ampornpan P, Ni J, Hochschild A, Lewis M (2008) Crystal structure of the lambda repressor and a model for pairwise cooperative operator binding. Nature 452: 1022–1025 doi:10.1038/nature06831 [DOI] [PubMed] [Google Scholar]
- 65. Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S (2006) Stochastic mRNA synthesis in mammalian cells. PLoS Biol 4: e309 doi:10.1371/journal.pbio.0040309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zong C, So L, Sepúlveda LA, Skinner SO, Golding I (2010) Lysogen stability is determined by the frequency of activity bursts from the fate-determining gene. Mol Syst Biol 6: 440 doi:10.1038/msb.2010.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lia G, Semsey S, Lewis DEA, Adhya S, Bensimon D, et al. (2008) The antiparallel loops in gal DNA. Nucleic Acids Research 36: 4204–4210 doi:10.1093/nar/gkn389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shimada J, Yamakawa H (1984) Ring-closure probabilities for twisted wormlike chains. Application to DNA. Macromolecules 17: 689–698 doi:10.1021/ma00134a028 [Google Scholar]
- 69. Giladi H, Koby S, Prag G, Engelhorn M, Geiselmann J, et al. (1998) Participation of IHF and a distant UP element in the stimulation of the phage lambda PL promoter. Mol Microbiol 30: 443–451. [DOI] [PubMed] [Google Scholar]
- 70. Wombacher R, Heidbreder M, Linde S van de, Sheetz MP, Heilemann M, et al. (2010) Live-cell super-resolution imaging with trimethoprim conjugates. Nature Methods 7: 717–719 doi:10.1038/nmeth.1489 [DOI] [PubMed] [Google Scholar]
- 71. Klein T, Loschberger A, Proppert S, Wolter S, van de Linde S, et al. (2011) Live-cell dSTORM with SNAP-tag fusion proteins. Nat Meth 8: 7–9 doi:10.1038/nmeth0111-7b [DOI] [PubMed] [Google Scholar]
- 72. Becker NB, Rosa A, Everaers R (2010) The radial distribution function of worm-like chains. Eur Phys J E Soft Matter 32: 53–69 doi:10.1140/epje/i2010-10596-0 [DOI] [PubMed] [Google Scholar]
- 73. Vafabakhsh R, Ha T (2012) Extreme bendability of DNA less than 100 base pairs long revealed by single-molecule cyclization. Science 337: 1097–1101 doi:10.1126/science.1224139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hong S-H, Toro E, Mortensen KI, de la Rosa MAD, Doniach S, et al. (2013) Caulobacter chromosome in vivo configuration matches model predictions for a supercoiled polymer in a cell-like confinement. Proc Natl Acad Sci USA 110: 1674–1679 doi:10.1073/pnas.1220824110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Van Noort J, Verbrugge S, Goosen N, Dekker C, Dame RT (2004) Dual architectural roles of HU: formation of flexible hinges and rigid filaments. Proc Natl Acad Sci U S A 101: 6969–6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ringrose L, Chabanis S, Angrand P-O, Woodroofe C, Stewart AF (1999) Quantitative comparison of DNA looping in vitro and in vivo: chromatin increases effective DNA flexibility at short distances. EMBO J 18: 6630–6641 doi:10.1093/emboj/18.23.6630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Swinger KK, Rice PA (2004) IHF and HU: flexible architects of bent DNA. Current Opinion in Structural Biology 14: 28–35 doi:10.1016/j.sbi.2003.12.003 [DOI] [PubMed] [Google Scholar]
- 78. Vologodskii AV, Levene SD, Klenin KV, Frank-Kamenetskii M, Cozzarelli NR (1992) Conformational and thermodynamic properties of supercoiled DNA. Journal of Molecular Biology 227: 1224–1243 doi:10.1016/0022-2836(92)90533-P [DOI] [PubMed] [Google Scholar]
- 79. Umbarger MA, Toro E, Wright MA, Porreca GJ, Baù D, et al. (2011) The three-dimensional architecture of a bacterial genome and its alteration by genetic perturbation. Molecular Cell 44: 252–264 doi:10.1016/j.molcel.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Meyer BJ, Maurer R, Ptashne M (1980) Gene regulation at the right operator (OR) of bacteriophage lambda. II. OR1, OR2, and OR3: their roles in mediating the effects of repressor and cro. J Mol Biol 139: 163–194. [DOI] [PubMed] [Google Scholar]
- 81. Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proceedings of the National Academy of Sciences 97: 6640–6645 doi:10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lau IF, Filipe SR, Soballe B, Okstad OA, Barre FX, et al. (2003) Spatial and temporal organization of replicating Escherichia coli chromosomes. Mol Microbiol 49: 731–743. [DOI] [PubMed] [Google Scholar]
- 83. Benson RC, Meyer RA, Zaruba ME, McKhann GM (1979) Cellular autofluorescence—is it due to flavins? J Histochem Cytochem 27: 44–48. [DOI] [PubMed] [Google Scholar]
- 84. Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, et al. (2004) Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22: 1567–1572. [DOI] [PubMed] [Google Scholar]
- 85. Salis HM, Mirsky EA, Voigt CA (2009) Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotech 27: 946–950 doi:10.1038/nbt.1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Abramoff MD, Magalhães PJ, Ram SJ (2004) Image processing with ImageJ. Biophotonics International 11: 36–42. [Google Scholar]
- 87. Orth P, Schnappinger D, Hillen W, Saenger W, Hinrichs W (2000) Structural basis of gene regulation by the tetracycline inducible Tet repressor–operator system. Nature Structural & Molecular Biology 7: 215–219 doi:10.1038/73324 [DOI] [PubMed] [Google Scholar]
- 88. Bell CE, Lewis M (2000) A closer view of the conformation of the Lac repressor bound to operator. Nature Structural & Molecular Biology 7: 209–214 doi:10.1038/73317 [DOI] [PubMed] [Google Scholar]
- 89. Yang F, Moss LG, Phillips GN (1996) The molecular structure of green fluorescent protein. Nature Biotechnology 14: 1246–1251 doi:10.1038/nbt1096-1246 [DOI] [PubMed] [Google Scholar]
- 90. Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14: 33–28, 33-38, 27-28. [DOI] [PubMed] [Google Scholar]
- 91. Magelhaes PJ, Ram SJ, Abramoff MD (n.d.) Image processing with ImageJ. Biophotonics International 11: 36–42. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spot fitting and experimental error analysis. (a) Distribution of fitting errors for EYFP (green), mCherry (red) localizations, and (black). Errors were estimated using a bootstrapping procedure by fitting raw data to a Gaussian distribution. The residuals from this fit were then randomly rearranged and added back to the data in 10 different permutations. The reported error is the standard deviation of the distance between these 10 locations and the initial fit location. Error in was determined similarly; from the 10 bootstrapped EYFP and mCherry fits, 100 distances were obtained and the error was estimated as the standard deviation of the difference between these distances and the distance determined from fitting the raw data. (b) A compilation of all data from three separate experiments was used for all analysis in the main text. Here, is shown for the individual experiments. Error was estimated as the standard deviation of the means of 1,000 bootstrapped distributions. Except for one sample (λOR3−, day 3), the estimated mean separations for all days followed the trend .
(TIF)
Estimate of positive control dimensions and apparent end-to-end distance distribution. (a) The maximum distance between TetR-EYFP and mCherry-LacI chromophores was approximated assuming straight DNA. All distances are in nm. Here, bound fusion proteins are shown on the same face of a DNA molecule, but this needs not be the case. Dimers of DNA-binding proteins were based on Protein Data Bank (PDB) entries for TetR (1QPI [87]) and LacI (1EFA [88]). Both fluorescent proteins are shown using the entry for GFP (1GFL [89]). Protein structures images generated using VMD [90]. (b) In an alternative positive control that was used to collect fiducial data for image registration, the plasmid pZH102R33TD encodes the tandem-dimer reporter LacI-mCherry-EYFP. (c) The PDF for the λnull control (black line; 1 s.e.m. shown in red as in Figure 3a) is shown with the distribution of 10,000 numerically simulated end-to-end distances for two sites separated by 22 nm, randomly projected onto the 2D plane, and subjected to 22-nm localization error for both ends (dashed black line). PDFs were calculated using methods described in main text. See Materials and Methods for simulation details.
(TIF)
Plots showing trajectories of vectors for all data from all strains for every molecule that was fit in both the EYFP and mCherry images for at least 8 consecutive frames (800 ms). Green and magenta lines are single-color trajectories for TetR-EYFP and LacI-mCherry spots, respectively; the corresponding trajectory with time colored-coded from blue to red is plotted on top at the same length scale. Coordinates are in nm.
(PDF)
Two-dimensional distributions of the x and y components of vectors. (a) A cartoon describes the calculation of the x and y components of the vector. In the projected image, the vector has two components determined by the arbitrary orientation of the detector. (b–f) Heat maps of the distribution of the x and y components of vectors of each strain. Plots were generated by binning the data for all into 5 nm×5 nm bins. The resulting 2-dimensional distribution was then filtered with a Gaussian kernal (with a width similar to spot-localization precision) to approximate the smoothed distributions. Each image is colored by the probability of the vector falling within a given bin according to the scale bar in (b).
(TIF)
Experiments showing the effects of transcription, nonspecific CI binding and higher-ordered CI oligomer on DNA looping. (a) End-to-end distance () distributions (PDF) for λnull (red), λΔOL (blue), λΔOLPRM−cI− (purple), and λΔOLPRM−cI−/cItrans (green). The PDF is estimated for 10-nm bins. (b) Cumulative density of (CDF) for λnull (red), λΔOL (blue), λΔOLPRM−cI− (purple), and λΔOLPRM−cI−/cItrans (green). The CDF is estimated for 10-nm bins. (c) DNA sequence for the PRM−cI− mutant in comparison to the wild-type sequence. Mutated nucleotides are shown in red. (d) Gel shift assay monitoring the binding of wild-type CI protein. Lane 1–4, CI at concentrations of 0, 150, 300, and 600 nM binding to a 158-bp DNA fragment (20 nM) amplified from the plasmid pZH107 carrying the wild-type PRM DNA sequence. Lane 5–8, CI at concentrations of 150, 0, 300, and 600 nM (note loading order) binding to a 158-bp DNA fragment (20 nM) amplified from the plasmid pACL007 carrying the PRM−cI− sequence. Lane 9: empty. Lane 10–13, CI at concentrations of 0, 150, 300, and 600 nM binding to a 140-bp DNA fragment (20 nM) amplified from the E. coli hns promoter region, which CI does not bind specifically. Reaction mixtures were incubated in a buffer (10 mM Tris pH 8.0, 50 mM KCI, 1 mM MgCl2, 10% glycerol, 100 ug/ml BSA, 1 mM DTT) at room temperature for 10 min. Samples were electrophoresed in Bio-Rad 4–20% Gradient TBE gels (Bio-Rad, Hercules, CA) in a cold room and then stained with Ethidium Bromide for 30 min. (e) Fraction of bound DNA (intensity of low-weight band divided by intensity of lane over background) quantified using NIH ImageJ for the gel shown in (d). (f, g) Distributions of identical in description to those in (a, b) showing strains λnull (red), λΔOL (blue), λG147D (purple), and λG147D/cIG147D,trans (green).
(TIF)
Growth rate comparisons. (a, b) Strains used in thermodynamic modeling were diluted from exponential growth to low optical densities in M9 minimal media supplemented with 0.4% glucose and carbenicillin as described in the main text. OD600 was measured over 10 h of growth for two replicate experiments. Strains are λΔOL (blue), λWT (red), λOR3− (green), and λOL3− (purple). Doubling times calculated using the Microsoft Excel LOGEST function range from 1.7 to 2.5 h. Two independent replicates are shown. (c) Growth rates for the parent E. coli strain MG1655 (blue) were compared to those of the control strain λnull in which the lac operon is replaced with a construct incorporating the lacO3 and tetO3 binding site arrays and which harbors the plasmid pZH102R33Y29 which expresses both TetR-EYFP and LacI-mCherry fluorescent fusion proteins upon arabinose induction. Strains were grown in M9 minimal media supplemented with 0.4% glycerol and λnull was grown in both the absence (red) and presence (green) of 0.3% L-arabinose. Doubling times were 2.7 h for MG1655 and 3.4 and 3.3 h for λnull in the absence and presence of L-arabinose, respectively.
(TIF)
Fluorescence movie montage for strain λnull corresponding to the data in Figure 2c. Single-color images for TetR-EYFP (top left) and LacI-mCherry (top right) data have intensities scaled linearly from the lowest to the highest pixel values in the first image in each time series. Before creating the overlay images (bottom), single-color images were background subtracted and bandpass filtered using the program ImageJ [91]. The overlay images are scaled to be twice as large as the single-color images. Scale bars correspond 4 µm in the small, single-color images and 2 µm in the overlay image. Ten consecutive image frames are shown in real time (10 frames per second); the movie is looped 5 times.
(MOV)
Fluorescence movie montage for strain λΔOL corresponding to the data in Figure 2d. Single-color images for TetR-EYFP (top left) and LacI-mCherry (top right) data have intensities scaled linearly from the lowest to the highest pixel values in the first image in each time series. Before creating the overlay images (bottom), single-color images were background subtracted and bandpass filtered using the program ImageJ [91]. The overlay images are scaled to be twice as large as the single-color images. Scale bars correspond 4 µm in the small, single-color images and 2 µm in the overlay image. Ten consecutive image frames are shown in real time (10 frames per second); the movie is looped 5 times.
(MOV)
Fluorescence movie montage for strain λΔOL corresponding to the data in Figure 2e. Single-color images for TetR-EYFP (top left) and LacI-mCherry (top right) data have intensities scaled linearly from the lowest to the highest pixel values in the first image in each time series. Before creating the overlay images (bottom), single-color images were background subtracted and bandpass filtered using the program ImageJ [91]. The overlay images are scaled to be twice as large as the single-color images. Scale bars correspond 4 µm in the small, single-color images and 2 µm in the overlay image. Thirteen consecutive image frames are shown in real time (10 frames per second); the movie is looped 5 times.
(MOV)
Fluorescence movie montage for strain λWT corresponding to a typical, long movie. Single-color images for TetR-EYFP (top left) and LacI-mCherry (top right) data have intensities scaled linearly from the lowest to the highest pixel values in the first image in each time series. Before creating the overlay images (bottom), single-color images were background subtracted and bandpass filtered using the program ImageJ [91]. The overlay images are scaled to be twice as large as the single-color images. Scale bars correspond 4 µm in the small, single-color images and 2 µm in the overlay image. Twelve consecutive image frames are shown in real time (10 frames per second); the movie is looped 5 times.
(MOV)
Fluorescence movie montage for strain λOR3− corresponding to a typical, long movie. Single-color images for TetR-EYFP (top left) and LacI-mCherry (top right) data have intensities scaled linearly from the lowest to the highest pixel values in the first image in each time series. Before creating the overlay images (bottom), single-color images were background subtracted and bandpass filtered using the program ImageJ [91]. The overlay images are scaled to be twice as large as the single-color images. Scale bars correspond 4 µm in the small, single-color images and 2 µm in the overlay image. Thirteen consecutive image frames are shown in real time (10 frames per second); the movie is looped 5 times.
(MOV)
Fluorescence movie montage for strain λOL3− corresponding to a typical, long movie. Single-color images for TetR-EYFP (top left) and LacI-mCherry (top right) data have intensities scaled linearly from the lowest to the highest pixel values in the first image in each time series. Before creating the overlay images (bottom), single-color images were background subtracted and bandpass filtered using the program ImageJ [91]. The overlay images are scaled to be twice as large as the single-color images. Scale bars correspond 4 µm in the small, single-color images and 2 µm in the overlay image. Twelve consecutive image frames are shown in real time (10 frames per second); the movie is looped 5 times.
(MOV)
Looping frequencies were estimated from alternate data sets using either all data or only the data from the first frames (for molecules appearing in more than one sequential frame) and fitting either probability (PDF) or cumulative (CDF) distributions. The first row results for each strain were reported in the main text.
(DOCX)
States used in thermodynamic modeling. We used free-energy parameters that were described by Dodd et al. [35]. States that will not be populated near lysogenic CI concentrations (e.g., those without OL1 or OL2 bound) are ignored; the reference state () has CI dimers bound to OL1 and OL2. A state with OR free of CI is included to show activation in Figure 5a and b, but does not significantly change fit parameters; because OR1 and OR2 binding is highly cooperative, we do not model states with only one or the other operator bound. The degeneracy term indicates how many microstates exist with identical CI dimer binding patterns and free energies. A particular macrostate may have several microstates that differ in terms of parallel or antiparallel looping configurations or in the identity of binding sites participating in cooperative interactions (either through looping or through adjacent dimers). Here, we also list whether a state is looped (1 for looped; 2 for unlooped) as well as its transcription rate, (0; 1 for ; 2 for ; 3 for ). The free energy of state 2 is called below.
(DOCX)
Thermodynamic model fitting using alternative choices for wild-type CI concentration (expressed here in molecules/cell; in the model, 1 molecule per cell is equivalent to 1.47 nM) and the fraction of CI molecules that are in the form of free dimers. The approximation of a constant free-dimer fraction is reasonable if specifically bound CI dimers (up to 6 dimers composed of 12 monomers) do not make up a large fraction of total CI and if CI concentration is sufficiently high that almost all CI molecules are in dimeric complexes. The free-dimer fractions used here were calculated assuming the absence of specific binding sites using the parameters for nonspecific binding site affinity and concentration estimated by Dodd et al. [35]. Results in the first row are the same as those presented in the main text.
(DOCX)
Names of new strains used in this study (as used internally in our lab) and shorthand names used in the main text.
(DOCX)
Sequences of oligonucleotide probes for single-molecule fluorescence in situ hybridization (smFISH) experiment. Asterisks indicate probes that do not hybridize specifically with any E. coli sequence. All other probes hybridize nonoverlapping sequence in the cI coding region of the mRNA transcript from the PRM promoter.
(DOCX)
Measurement statistics for experiment comparing distributions for looped and unlooped control strains to for strains lacking OL and having weakened PRM promoters with and without the overexpression of wild-type CI from a plasmid. Errors for the measurements are all 1 s.e.m. as estimated from 1,000 bootstrapped samples. Note that distributions display small, day-to-day variability between experiments (see Figure S1, this table, Table 2, Table S7), but the trend stays the same for a given set of experiments.
(DOCX)
Measurement statistics for experiment comparing distributions for looped and unlooped control strains to for strains in which CI harbors the G147D mutation with and without the overexpression of CIG147D from a plasmid. Errors for the measurements are all 1 s.e.m. as estimated from 1,000 bootstrapped samples. Note that distributions display small, day-to-day variability between experiments (see Figure S1, this table, Table 2, Table S6), but the trends stays the same for a given set of experiments.
(DOCX)
CI expression levels measured by smFISH for wild-type phage lambda lysogen JL5392 and additional strains. For strains with replicate experiments (N, number of independent experiments), errors indicate standard deviation. The expression levels were normalized to wild-type units (WTUs) using the λWT strain.
(DOCX)