

Abstract
The opioid analgesic oxycodone is widely abused and increasingly associated with overdose deaths. A sensitive analytical method was developed for oxycodone and its metabolites, noroxycodone and oxymorphone, in human plasma, urine (±enzymatic hydrolysis at 50°C for 16 h) and liver microsomes (HLMs). Liquid–liquid extraction was followed by high-performance liquid chromatography–electrospray ionization-tandem mass spectrometry. The calibration range was 0.2–250 ng/mL for plasma and HLM and 10–5000 ng/mL for urine. Intra- and interrun accuracies were within 13.3% of target; precisions were within 12.8% for all matrices. Recoveries from plasma were: oxycodone, 75.6%; noroxycodone, 37.4% and oxymorphone, 18.2%. Analytes exhibited room temperature stability in plasma and urine up to 24 h, and freeze–thaw stability in plasma up to three cycles. In 24-h hydrolyzed urine from subjects administered intranasal oxycodone (30 mg/70 kg, n = 5), mean concentrations (ng/mL) and % daily doses excreted were: oxycodone, 1150, 6.53%; noroxycodone, 1330, 7.81% and oxymorphone, 3000, 17.1%. Oxycodone incubated with HLM produced more noroxycodone than oxymorphone. With a panel of recombinant human cytochrome P450s (CYPs), CYP2C18 and CYP3A4 produced the most noroxycodone, whereas CYP2D6 produced the most oxymorphone. These results demonstrate a new method suitable for both in vivo and in vitro metabolism and pharmacokinetic studies of oxycodone.
Introduction
Oxycodone (Figure 1) is a semisynthetic opioid analgesic derived from the opiate alkaloid thebaine (1). The clinical use of oxycodone dates back to the early 20th century. Widespread use in the USA, however, followed the introduction of oral immediate-release preparations and the controlled-release preparation, OxyContin® (1). After approval by the US Food and Drug Administration of OxyContin® for controlled-release pain relief in 1995, sales grew rapidly; by 2001, it became the most prescribed narcotic medication for treating moderate-to-severe pain in the USA (2, 3). In the meantime, widespread abuse and diversion of OxyContin® and other opioids grew substantially across the country (4–6). This prescription opioid epidemic has been linked to increased morbidity and mortality arising from abuse, misuse and dependence of opioids, including oxycodone (5–9).
Figure 1.
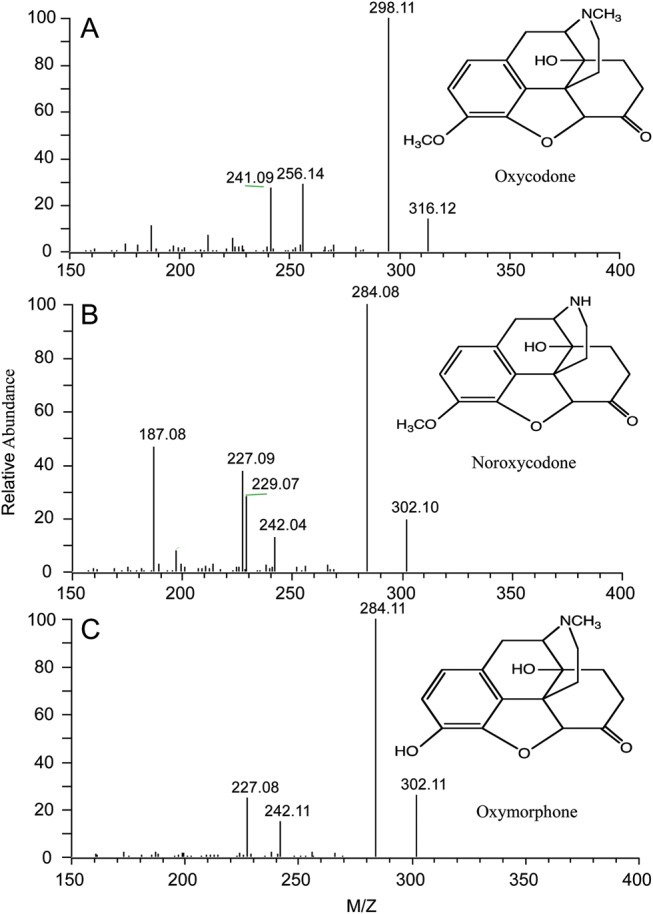
Full scan ESI mass spectrum of oxycodone, noroxycodone and oxymorphone obtained from a standard solution at 10 ng/μL.
Oxycodone is N-demethylated to noroxycodone and O-demethylated to oxymorphone (see Figure 1 for structures). Further metabolism involves combined oxidations to noroxymorphone, reduction in the keto group to a 6-hydroxyl, and N-oxidation and glucuronidation of the 3- and 6-hydroxyl groups (10–13). Products of the keto reduction and N-oxidation pathways have only been detected in urine samples from humans taking large doses. Limited information is available on the specific enzyme mediators of these reactions. A single study has shown that the N-demethylation to noroxycodone is carried out by cytochrome P450 (CYP) 3A4 (14); the same and an earlier study showed that CYP2D6 catalyzes the O-demethylation to oxymorphone (14, 15). A single study has shown that oxymorphone is glucuronidated by UDP-glucuronosyltransferase 2B7 (16). Oxymorphone and noroxymorphone share mu-opioid receptor activity with oxycodone, while noroxycodone does not. However, it is valuable to measure noroxycodone, because it is an index of CYP3A4 clearance. Noroxymorphone penetrates the blood–brain barrier poorly, and thus, oxycodone and oxymorphone are the active moieties (17, 18).
To help physicians, forensic scientists and other health-care providers better understand the clinical use and misuse of oxycodone, further studies on the pharmacokinetics and metabolism of the drug are needed. A sensitive and reliable method to measure oxycodone and its metabolites quantitatively in biological samples is needed to facilitate these studies.
There are several published methods for the determination of oxycodone and one or more of its metabolites in human biological fluids. Gas chromatography with mass spectrometry (MS) detection is the choice for most urine testing (19–21). Other methods include capillary electrophoresis–ion-trap multiple-stage MS (12), and high-performance liquid chromatography (HPLC) with ultraviolet or electrochemical detectors (22–25). HPLC with tandem MS (HPLC–MS-MS) has more recently been applied to measurements in plasma (26–28), urine (29, 30) and those plus other matrices (31), where it is usually a component of an opioid test panel. The reported lower limit of quantitation (LLOQ) for each of these methods varies across different matrices and metabolites. Our aim was to develop a simple, quick HPLC–MS-MS method suitable for multiple matrices that could detect oxycodone and its major metabolites, noroxycodone and oxymorphone. This novel method using electrospray ionization (ESI) as a source is described here. It was fully validated in human plasma with matrix cross-validation to human urine and human liver microsomes (HLMs). The application of this method for in vivo and in vitro metabolism studies of oxycodone to noroxycodone and oxymorphone in humans is also presented.
Experimental method
Materials
Oxycodone, noroxycodone, oxymorphone and their deuterated internal standards were purchased from Cerilliant Corporation (Round Rock, TX, USA). Concentrated formic acid (88%), glacial acetic acid and ammonium hydroxide were purchased from Fisher Scientific (Fair Lawn, NJ, USA). β-Glucuronidase (from Helix pomatia) and sodium acetate trihydrate were purchased from Sigma-Aldrich (St. Louis, MO, USA). Solvents were obtained from different sources, but all were, at a minimum, HPLC grade. Water used in the preparation of reagents, extraction and LC mobile phase was drawn from a Milli-Q filter apparatus (Millipore Corp., Billerica, MA, USA). Outdated human plasma was from the University of Utah blood bank. Silanized tubes were prepared by vapor-phase silanization using hexamethyldisilazane (Pierce, Rockford, IL, USA) in an oven under vacuum at 250°C for 2 h.
Extraction
Hydrolysis was performed on urine samples before extraction in order to obtain total (conjugated and nonconjugated) concentrations. Urine (200 µL) was incubated with 200 µL of 0.1 M sodium acetate buffer (with 5000 units/mL β-glucuronidase, pH 5) at 50°C for 16 h. Aliquots of 1.0 mL (for plasma and HLM) or 200 µL (for urine) of quality control (QC) samples, calibration standards and study samples were added to silanized tubes. Internal standard (50 µL of 0.1 ng/µL oxycodone-d6/noroxycodone-d3/oxymorphone-d3) was also added to each tube followed by 100 µL of concentrated ammonium hydroxide. All tubes were vortexed briefly, and 4 mL of 4:1 n-butyl chloride:acetonitrile was added. Tubes were then capped tightly and placed on a reciprocating tube rocker at low speed for 30 min. They were then centrifuged at 2600 rpm (830 g) for 10 min, and the organic layer was carefully transferred into 13 × 100 mm silanized culture tubes. The organic solvent was evaporated off under 10 psi air at 40°C in a Zymark Turbo Vap evaporator. The residues were reconstituted in 75 µL of 0.1% formic acid in Milli-Q water and transferred into autosampler vials.
HPLC–ESI-MS-MS analysis
HPLC–ESI-MS-MS analysis was performed on a Surveyor HPLC system and a TSQ Quantum triple quadrupole MS with ESI source (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The LC column was an YMC ODS-AQ 5 µm 2.0 × 100 mm (Waters Corporation, Milford, MA, USA). An isocratic program with 88% of 0.1% formic acid in Milli-Q water, and 12% of acetonitrile was used. Run time was 5 min with a flow rate of 0.2 mL/min. Autosampler temperature was set at 10°C, and column temperature was set at 22°C. Injection volume was 10 µL. The instrument was operated under a selective reaction monitoring mode. The capillary temperature was 280°C, and ESI spray voltage was set at 3.9 kV. High-purity N2 was used for both sheath and auxiliary gas. High-purity Ar was used for collision gas. The m/z 316 (MH+) to 298 and 322 to 304 transitions were used to analyze oxycodone and its internal standard oxycodone-d6. The m/z 302 (MH+) to 284 and 305 to 287 transitions were used to analyze noroxycodone, oxymorphone and their internal standards noroxycodone-d3 and oxymorphone-d3. Noroxycodone and oxymorphone have the same transition; their identifications were achieved by HPLC column separation. The concentration of oxycodone and its metabolites was determined by the peak area ratio of the analyte and its internal standard, with comparison to the calibration curve that was generated from the analysis of plasma or urine fortified with known concentrations of the analyte and its internal standard. All curves were quadratic with 1/X weighting.
Quality control
QC samples and calibrator stocks were prepared by different staff members. All calibrators were run in duplicate, one set at the beginning and one at the end of the run. For plasma and HLMs, they were 0.2, 0.5, 2, 5, 20, 50, 100 and 250 ng/mL. For urine, they were 10, 25, 100, 250, 500, 1250, 2500 and 5000 ng/mL. Calibrators were deleted from the curve if they deviated more than ±15% (±20% at the LLOQ) from the target. A valid run would have no >25% of calibrators deleted. QCs were run at a minimum of n = 2 for each QC concentration (low, medium and high). QC concentrations were selected based on the following criteria: low QC is no more than three times the LLOQ concentration, and the high QC is 70–80% of the upper limit of quantitation (ULOQ). For the medium QC, this study used the median of the curve. QCs were run at the beginning and the end of each sample batch. QCs were acceptable if they were within ±15% of target. For a run to pass, at least two-thirds of all QCs and at least one replicate at each QC concentration needed to pass.
Method validation
Method validation experiments were conducted to meet the guidelines for bioanalytical method validations specified by the US FDA (32). A full validation was performed in human plasma; only partial validation is required for alternative matrices. Partial validation experiments performed for urine included specificity, intra- and interrun precisions and accuracies, and room temperature matrix stability. For HLMs, only specificity and intrarun precision and accuracy experiments were performed.
Specificity was evaluated for all three matrices by using six different sources from each matrix. For each source, three replicates were spiked with internal standard only; one replicate was spiked with internal standard and analyte at the LLOQ (LLOQ sample). The primary evaluation was to compare the mean peak area ratio of any signal (often obtained by forced integration) at the retention time of analyte to its internal standard for each source with the mean peak area ratio of the six LLOQ samples. The mean peak area ratio of the three ‘internal standard only’ samples should not exceed 20% of the mean peak area ratio of the six LLOQ samples. The results of this experiment were used in part to determine the LLOQ. For each source, an additional replicate was also prepared with no fortifications. These were used to evaluate the potential contribution from the internal standard.
Recovery was determined at the three QC concentrations (n = 5 each). Analytes were fortified in one set of blank plasma and one set without matrix. The plasma set was extracted and then dried down along with the set without matrix. Both sets were then reconstituted and analyzed. Percent recovery was determined at each concentration by dividing the average peak area of extracted samples by the average peak area of unextracted samples and multiplying by 100.
Intrarun precision and accuracy were performed for all three matrices using the LLOQ and the three QC concentrations. LLOQ samples were prepared in the same six sources used for the specificity study. QCs were run at n = 5 for each concentration. Acceptance criteria for accuracy are mean results within 15% of the target value (20% for the LLOQ) and precision expressed as coefficient of variation (%CV) within 15% (20% for the LLOQ).
Interrun precision and accuracy were determined in plasma and urine using two additional runs of QCs at n = 5 for a total of 15 measurements for each QC concentration. Acceptance criteria for accuracy are mean results within 15% of the target value and precision for %CVs within 15%. %CV was calculated using analysis of variance (ANOVA) as previously described (33, 34).
Stability experiments were performed at low and high QC concentrations with n≥ 3. Room temperature stability evaluated QCs that were stored at room temperature for 24 h prior to extraction. Freeze–thaw stability evaluates QCs removed from the freezer to thaw unassisted, and then returned to the freezer for at least 12 h. This process was repeated twice, for a total of three cycles prior to analysis. Long-term stability was determined using QCs that had been stored at ∼ −20°C for the time specified. For processed sample stability, analyzed QCs were stored either on the autosampler or at −20°C until they were reanalyzed with freshly extracted calibrators and QCs. An acceptance criterion is mean results within 15% of target. To determine stock solution stability, stock solutions prepared at the beginning of the method validation were compared with that prepared at the conclusion of the validation experiments. Room temperature stock stability was also evaluated by comparing freshly made stock solution on the day of analysis with aliquots held at room temperature for a period of time. Stock stability was acceptable if the mean peak area counts of old stock were within 15% of freshly made stock.
In vivo specimens
Urine samples were collected during a study more fully described by Lofwall et al. (35). Subjects (n = 5) were adult volunteers who had experienced snorting opioids for recreational use, but were not physically dependent on opioids. Crushed OxyContin® was given by the intranasal route at 30 mg/70 kg. Blood was drawn into heparinized tubes at different time points over a 24-h dosing interval; plasma was collected and stored frozen until analysis. (Note: plasma data are not presented in this paper, but the area under the plasma concentration curve (AUC) is needed to calculate renal clearance.) Urine was collected during the entire 24 h, the volume measured and aliquots taken for frozen storage prior to analysis. All subjects provided informed consent and were compensated for participation in the studies. All protocols had received prior approval by the local institutional review board.
In vitro incubations
Incubations of oxycodone with HLMs and cDNA-expressed CYPs were performed using our previously described incubation conditions (36, 37). HLMs were prepared using the methods described by Nelson et al. (38) with the first centrifugation at 9,000 g, the homogenization buffer containing 0.25 M sucrose and 10 strokes of homogenization. The protein content was determined by the method of Lowry et al. (39). Three HLMs were used. Incubation was carried out using 0.5 mg/mL microsomal protein and 100 ng/mL oxycodone in an incubation buffer (0.1 M phosphate buffer, pH 7.4 with 1.0 mM ethylenediaminetetra-acetic acid and 5.0 mM MgCl2) with an NADPH-generating system (NADPH-GS) of 10 mM glucose-6-phosphate, 1.2 mM NADP and 1.2 units of glucose-6-phosphate dehydrogenase. The total volume was 0.5 mL. The reaction was initiated by adding the NADPH-GS and incubated at 37°C for 30, 60, 90 and 120 min in water bath shaker, then terminated by addition of 200 µL of ice-cold methanol. Oxycodone was incubated with microsomes prepared from insect cells transfected with cDNAs encoding for human CYPs 1A2, 2A6, 2B6, 2C8, 2C9*1, 2C18, 2C19, 2D6*1, 2E1, 3A4, 3A5 and 3A7 (BD Biosciences, Franklin Lakes, NJ, USA). Supersomes that coexpressed cytochrome b5 were used where available (not for 1A2, 2C18 and 3A5). Oxycodone (30 and 100 ng/mL) was incubated with 25 pmol of CYP at 37°C for 30 min in the incubation buffer (described above) with a final incubation volume of 200 µL. Further experiments were conducted with 2C18, 2D6 and 3A4 at an oxycodone concentration of 30 ng/mL with 5 and 10 min incubations. The reaction was initiated by adding a NADPH-GS and terminated by addition of 100 µL of ice-cold methanol. Human liver tissue was obtained from Tissue Transformation Tech (Edison, NJ, USA).
Results and discussion
Spectra and chromatography
The mass spectra of oxycodone, noroxycodone and oxymorphone are shown in Figure 1. The most abundant fragment that corresponds to a loss of water from all analytes was chosen as the quantitative fragment. This is a typical fragmentation as noted earlier (12, 40) for opioid structures that have a hydroxyl group at position 14. Other applications (26, 27) that monitor different transitions have comparable sensitivity to that reported herein. Selected ion chromatograms of blank plasma fortified with internal standards demonstrate that no significant peaks exist at the retention time for oxycodone and its metabolites (Figure 2A). Typical peak shapes and signal-to-noise ratios for extracts of plasma fortified with 0.2 ng/mL (LLOQ) oxycodone, noroxycodone and oxymorphone and their internal standard are shown in Figure 2B. All chromatograms show a Gaussian peak shape with sufficient signal-to-noise and good separation.
Figure 2.
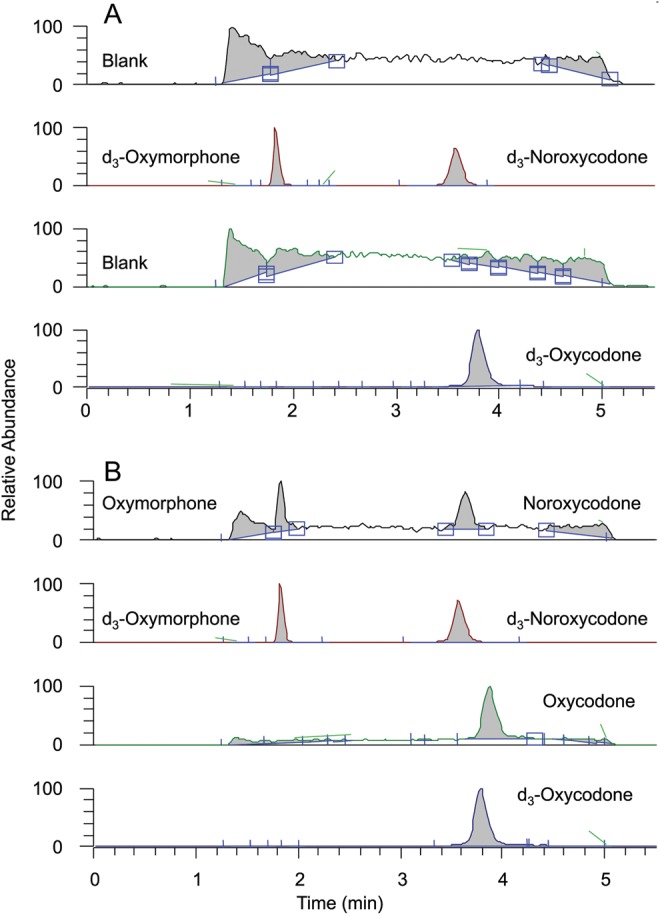
Representative chromatograms of extracted blank plasma with internal standards (A) and blank plasma fortified with 0.2 ng/mL oxycodone, noroxycodone and oxymorphone (B).
Wagner et al. (28) reported a LC–MS-MS column-switching method, which measures oxycodone and its metabolites in low pg/mL level using the following process: protein precipitation then neutralization, analyte trapping and sample clean-up using the Prospekt II system, followed lastly by HPLC–MS-MS analysis using the gradient mobile phase. The method also requires time-consuming adjusting sample conditions using acid, base and different solvents. Our method presented a simple, low-cost alternative (no additional costly equipment required), with a short and easy liquid/liquid extraction and a 5-min isocratic run time that makes it very efficient and suitable for high-throughput analysis.
Specificity
Specificities for oxycodone and its metabolites were determined in plasma, urine and HLM (Table I). Mean ratios relative to the mean LLOQ ranged from 1.04 to 4.51% with a mean of 1.92% for oxycodone, from 1.98 to 8.18% with a mean of 4.28% for oxymorphone and from 8.63 to 16.6% with a mean of 11.4% for noroxycodone in human plasma. In urine (both nonhydrolyzed and hydrolyzed), it ranged from 0.25 to 7.21% with a mean of 2.41% for oxycodone, from 1.73 to 12% with a mean of 4.75% for oxymorphone and from 0.93 to 6.80% with a mean of 2.43% for noroxycodone. In HLMs, it ranged from 3.18 to 5.53% with a mean of 3.81% for oxycodone, from 5.30 to 8.17% with a mean of 7.20% for oxymorphone and from 10.9 to 17.3% with a mean of 13.9% for noroxycodone. These experiments establish sufficient specificity for the use of an LLOQ at 0.2 ng/mL for oxycodone and its metabolites in plasma and HLM; and at 10 ng/mL in urine. In samples with no additions, peak heights at the retention time of the d0-analytes were not visible and similar to those with only internal standards added, excluding the contribution to the d0-analyte peak from the internal standard.
Table I.
Specificity for oxycodone, oxymorphone and noroxycodone in human plasma, urine and liver microsomes
| Matrix/analyte | Source number |
|||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| (mean peak area ratio as % of LLOQ) | ||||||
| Human plasma: LLOQ = 0.2 ng/mL | ||||||
| Oxycodone | 4.51 | 1.93 | 1.28 | 1.04 | 1.51 | 1.24 |
| Oxymorphone | 8.18 | 3.54 | 4.93 | 2.68 | 4.36 | 1.98 |
| Noroxycodone | 13.0 | 16.6 | 8.63 | 9.01 | 11.5 | 9.42 |
| Human urine: LLOQ = 10 ng/mL | ||||||
| Oxycodone | 1.02 | 0.86 | 0.37 | 0.54 | 0.25 | 0.42 |
| Oxymorphone | 3.76 | 5.52 | 2.94 | 2.47 | 1.73 | 1.90 |
| Noroxycodone | 1.90 | 1.72 | 1.75 | 1.45 | 0.93 | 0.98 |
| Hydrolyzed human urine: LLOQ = 10 ng/mL | ||||||
| Oxycodone | 7.21 | 5.92 | 3.61 | 3.32 | 1.73 | 3.69 |
| Oxymorphone | 12.0 | 10.5 | 5.59 | 5.78 | 2.00 | 2.79 |
| Noroxycodone | 6.80 | 3.94 | 3.45 | 2.08 | 1.99 | 2.12 |
| Human liver microsomes: LLOQ = 0.2 ng/mL | ||||||
| Oxycodone | 5.53 | 3.33 | 3.43 | 3.43 | 3.94 | 3.18 |
| Oxymorphone | 8.17 | 5.30 | 6.21 | 8.02 | 7.75 | 7.75 |
| Noroxycodone | 17.3 | 14.1 | 11.8 | 10.9 | 15.1 | 14.1 |
Values are from the mean of three replicates of the matrix source, which were fortified with internal standard and had peak areas determined for any signal within the retention time of respective d0-analyte peak width. Values were compared with the mean peak area ratio of 6 samples, one from each source, fortified with analyte at the LLOQ and internal standard. Source number is just a designation that varies from one matrix to another.
Specificity has only been reported for two other methods, both in plasma. One performed validation at the low QC instead of LLOQ concentration (28), and the other (27) reported no validation data. We determined specificity in all three matrices, which helped us substantiate the LLOQ.
These specificity experiments only show the selectivity of the method versus endogenous components in the matrices tested. They do not show selectivity from other drugs. These techniques, however, have been primarily used for the quantitation of clinical samples from pharmacokinetic or efficacy studies, where other drug use is purposefully limited. With HPLC separation and a mass transition monitoring, it is sufficient to select the right compound. Should this method be used for the determination of an unknown drug (i.e., forensic analyses), then additional transitions should always be monitored to ensure the correct ion identification. A related limitation of this assay is that it is designed for the precise and accurate determination of oxycodone and metabolites for research on metabolism and pharmacokinetics and is not designed or intended for use in forensics where polysubstance use is likely.
A qualitative ion suppression experiment was performed using continuous postcolumn infusion (10 µL/min) of a standard solution containing analytes of interest (250 ng/mL) using a syringe pump. After injecting the drug-free extracted blank samples from human plasma, urine and HLM in the LC system, a drop in the constant baseline at the retention time of the analytes would indicate ion suppression (41). No matrix-associated suppression was observed in the three matrices tested (data not shown).
Recovery
Recovery was evaluated at three QC concentrations. Mean recoveries of oxycodone, noroxycodone and oxymorphone were 75.6, 37.4 and 18.2%. No special effort was made to optimize recovery. The main focus of the assay is a simple, quick extraction procedure and use of common solvents. Other researchers reported improved recovery with either protein precipitation (26) or solid-phase extraction (27). Apparently, a higher recovery did not necessarily lead to a more sensitive method; we still achieved an LLOQ comparable with those other methods.
Precision and accuracy
Intrarun and interrun precision and accuracy experiments are summarized in Table II. The intrarun accuracies of the LLOQ in all matrices were within 12.5% of the target with intrarun precisions within 12.4%. The intrarun accuracies of other QCs were within 10.8% of the target with intrarun precision within 12.8%. Interrun accuracies of the QCs were within 5.0% of target with interrun precisions within 7.4%.
Table II.
Intrarun and Interrun (plasma only) accuracy and precision for the determination of oxycodone, oxymorphone and noroxycodone in human plasma, urine and liver microsomes
| Matrix/analyte | LLOQ | Low QC | Medium QC | High QC |
|---|---|---|---|---|
| 0.2 ng/mL | 0.6 ng/mL | 10.0 ng/mL | 200 ng/mL | |
| (% Target/%CV) | ||||
| Human plasma: intraruna | ||||
| Oxycodone | 87.5/4.0 | 107.8/12.8 | 104.0/1.0 | 102.5/1.5 |
| Oxymorphone | 105.5/6.2 | 98.3/6.1 | 100.0/2.0 | 101.0/2.5 |
| Noroxycodone | 100.5/2.5 | 89.2/9.9 | 99.5/2.1 | 99.0/1.0 |
| Human plasma: interrunb | ||||
| Oxycodone | 104.3/7.4 | 105.0/1.0 | 103.0/1.6 | |
| Oxymorphone | 101.5/5.2 | 101.0/1.0 | 101.5/0.1 | |
| Noroxycodone | 96.8/3.3 | 101.0/1.0 | 99.0/1.5 | |
| Human liver microsomes: intraruna | ||||
| Oxycodone | 100.0/5.5 | 102.3/1.6 | 96.5/1.0 | 95.0/2.1 |
| Oxymorphone | 102.5/11.7 | 96.0/11.8 | 96.4/3.2 | 94.0/4.3 |
| Noroxycodone | 95.0/9.5 | 100.7/4.6 | 103.0/1.9 | 101.0/3.0 |
| 10.0 ng/mL | 30.0 ng/mL | 350 ng/mL | 4,000 ng/mL | |
| Human urine: intraruna | ||||
| Oxycodone | 98.7/2.3 | 97.0/1.0 | 95.7/2.1 | 97.9/1.0 |
| Oxymorphone | 91.3/3.8 | 96.0/2.1 | 98.3/2.3 | 95.5/2.5 |
| Noroxycodone | 96.4/2.6 | 96.3/2.1 | 95.7/1.5 | 99.7/2.5 |
| Hydrolyzed human urine: intraruna | ||||
| Oxycodone | 97.2/1.2 | 93.7/1.8 | 92.9/1.2 | 95.2/2.0 |
| Oxymorphone | 101.0/4.0 | 95.3/3.8 | 94.0/3.3 | 93.7/3.3 |
| Noroxycodone | 105.0/12.4 | 101.3/4.9 | 103.1/3.0 | 96.7/2.0 |
aIntrarun values are from the mean of five replicates for QCs; for the LLOQ they are from the mean of six different sources of matrix each fortified at the LLOQ.
bValues are from the mean of 15 replicates, run as an n of 5 on 3 separate days. %CVs calculated from the use of one-way ANOVA.
Stability
We designed a series of studies to determine the stability of oxycodone and its metabolites in both human plasma and urine. The experiments emphasized the storage conditions for everyday analysis. Results (Table III) show that oxycodone and its metabolites are stable in human plasma for up to 24 h at room temperature, for 460 days at −20°C and after three freeze–thaw cycles. Similar experiments show that they are also stable in urine for up to 24 h at room temperature (Table III). Processed samples were stored at either −20°C for 7 days or for 4 days on the autosampler. Under both storage conditions, mean QC results were within 12.7% of the target concentration (Table III). Results also indicate that oxycodone and its metabolites are stable in methanol for up to 251 days at −20°C and 18 h at room temperature (Table III).
Table III.
Stability of oxycodone, oxymorphone and noroxycodone
| Matrix/storage conditions, concentration | Analyte |
||
|---|---|---|---|
| Oxycodone | Oxymorphone | Noroxycodone | |
| (% Target/%CV) | |||
| Plasma/room temperature for 24 h (n = 3), ng/mL | |||
| 0.2 | 101.2/1.2 | 97.5/1.5 | 99.3/3.4 |
| 200 | 103.0/0.5 | 99.5/1.5 | 100.0/2.0 |
| Plasma/−20°C for 460 days (n = 5), ng/mL | |||
| 0.2 | 99.8/2.2 | 91.8/6.9 | 93.8/6.4 |
| 200 | 97.5/0.5 | 95.0/1.6 | 95.0/1.6 |
| Plasma/three freeze–thaw cycles (n = 3), ng/mL | |||
| 0.2 | 101.0/1.0 | 97.7/1.9 | 93.7/7.5 |
| 200 | 103.5/2.9 | 97.5/2.1 | 100.5/3.0 |
| Processed Plasma/−20°C for 7 days (n = 5), ng/mL | |||
| 0.2 | 104.3/0.3 | 105.0/0.5 | 103.5/4.7 |
| 200 | 103.5/1.4 | 102.5/0.5 | 102.0/2.9 |
| Processed plasma/autosampler for 4 days (n = 5), ng/mL | |||
| 0.2 | 112.7/14.1 | 99.0/3.0 | 95.8/6.4 |
| 200 | 109.0/2.3 | 104.5/2.9 | 103.0/1.9 |
| Urine/room temperature for 24 h (n = 3), ng/mL | |||
| 30.0 | 97.7/1.7 | 94.7/2.5 | 93.3/1.1 |
| 4000 | 96.3/1.1 | 95.6/0.2 | 99.6/0.6 |
| Stock solutions, stored: (n = 5) | (% fresh stock/%CV) | ||
| −20°C for 251 days | 100.6/1.1 | 95.4/2.2 | 99.1/1.0 |
| Room temperature for 18 h | 100.7/4.4 | 103.4/12.9 | 98.4/2.3 |
Results are derived from the mean ± SD of n = 3 or 5 per concentration.
Neuvonen and Neuvonen (27) reported stability in human plasma. No significant degradation was observed during three freeze–thaw cycles, 5-h room temperature storage or 2-month −20°C storage. Our study extended findings for both short- and long-term stabilities
Optimization of urine sample hydrolysis time and urine excretion
A 200-µL aliquot of each urine sample was treated with 1000 units of β-glucuronidase in 200 µL of 0.1 M sodium acetate buffer (pH 5). The mixture was incubated at 50°C for 0, 1, 2, 4, 6, 16, 19 and 22 h. Figure 3 shows hydrolysis time versus mean oxycodone, noroxycodone and oxymorphone concentrations. Oxycodone and noroxycodone concentrations did not change appreciably during hydrolysis. This strongly suggests that they are not extensively conjugated. Oxymorphone concentrations increased substantially, over 10-fold, during hydrolysis. This experiment showed that hydrolysis at 50°C for 16 h was optimal.
Figure 3.

Hydrolysis time versus mean urine oxycodone (OC), noroxycodone (NO) and oxymorphone (OM) concentrations. Samples were incubated with β-glucuronidase at 50°C for the times shown. Results are the mean of five urine samples.
For these experiments, we used β-glucuronidase from H. pomatia, which contains both β-glucuronidase and arylsulfatase activity. This should allow for more thorough deconjugation, but does not permit one to specify that any conjugation is solely β-glucuronidation. Wey and Thurman (12) compared β-glucuronidase from H. pomatia and Escherichia coli at 24 versus 4 h incubation at 37°C and found more thorough deconjugation at 24 h for both enzymes, but the H. pomatia enzyme was much more effective at deconjugating oxymorphone, while no significant difference was noted for the deconjugation of oxycodone or noroxycodone. We have further optimized the time for hydrolysis. A limitation of our experiments is that we did not include controls with glucuronidated oxycodone, noroxycodone or oxymorphone; based on the results from Wey and Thormann, we used procedures that were comparable with those that describe the conjugation of oxycodone and noroxycodone (10–12).
These data were then used to calculate the percent of a daily dose of oxycodone that is excreted in urine over a 24-h period. About 6.5 and 7.8% of the daily dose was excreted as nonconjugated (free) oxycodone and noroxycodone; nonconjugated oxymorphone constituted only 1.3% of the dose, but total (conjugated and nonconjugated) oxymorphone made up 17.1% of the dose. The total of the three constitutes 31.4% of the daily dose. Using the plasma AUC of nonconjugated analytes for this subset of subjects from the previously published clinical study (35), renal clearance for nonconjugated analytes was 6.18 ± 1.97, 90.9 ± 42.7 and 14.2 ± 1.9 L/h.
In this study, we found that conjugated forms of oxycodone and noroxycodone comprised 8 and 3%, at most, of the respective compounds in urine. This is in sharp contrast to studies in laboratory animals and humans, where roughly one-third and one-fourth of oxycodone and noroxycodone in urine were conjugated (10, 11). The main difference among these studies was in the route of administration. Oxycodone was administered to the laboratory animals subcutaneously and to humans either orally or intramuscularly (10, 11); our samples were collected after intranasal administration. There is no immediate explanation for why intranasal administration would lead to reduced conjugation. We have, however, performed clinical studies where buprenorphine was given by the subcutaneous route (42) and the intranasal route (43). Here, buprenorphine, norbuprenorphine and their respective glucuronides were measured in plasma. If we compare the ratio of the Cmax (an average of conditions) of the glucuronide with that of buprenorphine after sublingual and intranasal administration, we find that, for buprenorphine-3-glucuronide, it went from 0.17 to 0.081 and for norbuprenorphine-3-glucuronide, it went from 0.49 to 0.098. These are 2.1- and 5.0-fold reductions, respectively. Thus, it appears that that reduced conjugation may result from intranasal opioid administration.
In vitro incubation of oxycodone with HLM and recombinant human P450s
Figure 4 shows the in vitro metabolism of oxycodone in HLM. Mean activities from three HLMs show that noroxycodone formation far exceeds that of oxymorphone concentration, as also observed by previous studies (14, 23). Their combined formation approximates the amount of oxycodone consumed (Figure 4A). The formation of noroxycodone (Figure 4B) and oxymorphone (Figure 4C) with incubation time in the three different HLMs shows that relative rates of N- and O-demethylation vary considerably. Noroxycodone formation was CO17 > CO18 > CO16, while oxymorphone formation was CO16 > CO17 > CO18. This variation may be caused by the difference in the amounts of specific CYP enzymes in each liver.
Figure 4.

Mean (n = 3) rates of oxycodone (OC) use and noroxycodone (NO) and oxymorphone (OM) formation (A), and NO (B) and OM (C) formation in individual HLM. HLM at 0.5 mg prot/mL was incubated with 100 ng/mL oxycodone; each time point per HLM was performed in duplicate.
Figure 5A shows the measured noroxycodone and oxymorphone formation and oxycodone utilization after incubation of oxycodone with cDNA-expressed CYPs. It can be seen that 2C18 and 3A4 are the major enzymes involved in the oxycodone N-demethylation. We did not find any involvement of 3A5 as reported by other researchers (23). CYP2D6 displayed highest activity for O-demethylation. CYP2C19 had barely detectable activity for both metabolites; CYPs 2B6 and 3A7 had barely detectable activity for noroxycodone formation (Figure 5A). Substrate use and product formation over time (Figure 5B) show that noroxycodone formation by CYP2C18 and oxymorphone formation by CYP2D6 are equivalent to substrate use. With CYP3A4, however, there is apparent greater oxycodone use than noroxycodone production. This suggests that CYP3A4 may be capable of catalyzing a reaction we did not monitor (e.g., carboxyl reduction).
Figure 5.

Oxycodone (OC) depletion and noroxycodone (NO) and oxymorphone (OM) formation in a panel of cDNA-expressed human liver CYPs incubated for a set time with 30 ng/mL oxycodone (A); OC depletion and NO and OM formation over time in CYP2C18, 2D6 and 3A4 incubated with 30 ng/mL oxycodone (B). Each result is from duplicate incubations.
Conclusion
We have developed a quick, simple and sensitive method using liquid–liquid extraction and HPLC–ESI-MS-MS to determine the concentration of oxycodone and its metabolites in various human matrices. One milliliter or less of the sample is used with acceptable precision and accuracy down to an LLOQ of 0.2 ng/mL. This is a very efficient method that can be used to follow the concentration of oxycodone and its metabolites in HLMs and supersomes after in vitro incubation. The stability of oxycodone and metabolites has been established under a number of conditions. We have optimized the conditions for the enzymatic hydrolysis time of urine; these results show that oxymorphone, but not oxycodone or noroxycodone, is highly conjugated. Approximately 31% of the daily dose is excreted in the urine with over half of that as oxymorphone glucuronide. The in vitro incubation experiments demonstrated that both N-demethylation and O-demethylation occur in HLMs, and the N-demethylation appeared to a greater degree than the O-demethylation, which may indicate that N-demethylation is the major oxidative pathway. CYP2C18 and CYP3A4 were identified to be the principal N-demethylases, and CYP2D6 made major contributions to O-demethylation. Thus, their relative expression and activity are expected to be major determinants of oxycodone clearance in vivo.
Funding
This work was supported in part by grants, a contract and a career development award from the National Institute on Drug Abuse, National Institutes of Health: R01 DA010100 (D.E.M.), R01 DA016718 (S.L.W.), N01-DA-9–7767 (D.E.M.) and K12 DA14040 (M.R.L.), and a grant from the National Institute of Justice 2011: DN-BX-K532 (D.E.M.).
References
- 1.Olkkola K.T., Hagelberg N.M. Oxycodone: new ‘old’ drug. Current Opinion in Anaesthesiology. 2009;22:459–462. doi: 10.1097/ACO.0b013e32832bc818. doi:10.1097/ACO.0b013e32832bc818. [DOI] [PubMed] [Google Scholar]
- 2.Rischitelli D.G., Karbowicz S.H. Safety and efficacy of controlled-release oxycodone: a systematic literature review. Pharmacotherapy. 2002;22:898–904. doi: 10.1592/phco.22.11.898.33628. doi:10.1592/phco.22.11.898.33628. [DOI] [PubMed] [Google Scholar]
- 3.Van Zee A. The promotion and marketing of oxycontin: commercial triumph, public health tragedy. American Journal of Public Health. 2009;99:221–227. doi: 10.2105/AJPH.2007.131714. doi:10.2105/AJPH.2007.131714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cicero T.J., Inciardi J.A., Munoz A. Trends in abuse of oxycontin and other opioid analgesics in the United States: 2002–2004. Journal of Pain. 2005;6:662–672. doi: 10.1016/j.jpain.2005.05.004. doi:10.1016/j.jpain.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Hall A.J., Logan J.E., Toblin R.L., Kaplan J.A., Bixler D., Crosby A.E., et al. Patterns of abuse among unintentional pharmaceutical overdose fatalities. Journal of the American Medical Association. 2008;300:2613–2620. doi: 10.1001/jama.2008.802. doi:10.1001/jama.2008.802. [DOI] [PubMed] [Google Scholar]
- 6.Okie S. A flood of opioids, a rising tide of deaths. New England Journal of Medicine. 2010;363:1981–1983. doi: 10.1056/NEJMp1011512. doi:10.1056/NEJMp1011512. [DOI] [PubMed] [Google Scholar]
- 7.Caravati E.M., Grey T., Nangle B., Rolfs R.T., Peterson-Poruceznik C.A. Increase in poisoning deaths caused by non-illicit drugs—Utah, 1991–2003. Morbidity and Mortality Weekly Report. 2005;54:33–36. [PubMed] [Google Scholar]
- 8.Paulozzi L.J., Ryan G.W. Opioid analgesics and rates of fatal drug poisoning in the United States. American Journal of Preventive Medicine. 2006;31:506–511. doi: 10.1016/j.amepre.2006.08.017. doi:10.1016/j.amepre.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 9.Cai R., Crane E., Poneleit K., Paulozzi L. Emergency department visits involving nonmedical use of selected prescription drugs in the United States, 2004–2008. Journal of Pain Pallitive Care and Pharmacotherapy. 2010;24:293–297. doi: 10.3109/15360288.2010.503730. doi:10.3109/15360288.2010.503730. [DOI] [PubMed] [Google Scholar]
- 10.Ishida T., Oguri K., Yoshimura H. Determination of oxycodone metabolites in urines and feces of several mammalian species. Journal of Pharmacobiodynamics. 1982;5:521–525. doi: 10.1248/bpb1978.5.521. doi:10.1248/bpb1978.5.521. [DOI] [PubMed] [Google Scholar]
- 11.Poyhia R., Seppala T., Olkkola K.T., Kalso E. The pharmacokinetics and metabolism of oxycodone after intramuscular and oral administration to healthy subjects. British Journal of Clinical Pharmacology. 1992;33:617–621. doi: 10.1111/j.1365-2125.1992.tb04090.x. doi:10.1111/j.1365-2125.1992.tb04090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wey A.B., Thormann W. Capillary electrophoresis-ion trap multiple-stage mass spectrometry for the differentiation and identification of oxycodone and its major metabolites in human urine. Journal of Chromatography B. 2002;770:191–205. doi: 10.1016/s1570-0232(01)00568-2. doi:10.1016/S1570-0232(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 13.Moore K.A., Ramcharitar V., Levine B., Fowler D. Tentative identification of novel oxycodone metabolites in human urine. Journal of Analytical Toxicology. 2003;27:346–352. doi: 10.1093/jat/27.6.346. doi:10.1093/jat/27.6.346. [DOI] [PubMed] [Google Scholar]
- 14.Lalovic B., Phillips B., Risler L.L., Howald W., Shen D.D. Quantitative contribution of CYP2D6 and CYP3A to oxycodone metabolism in human liver and intestinal microsomes. Drug Metabolism and Disposition. 2004;32:447–454. doi: 10.1124/dmd.32.4.447. doi:10.1124/dmd.32.4.447. [DOI] [PubMed] [Google Scholar]
- 15.Otton S.V., Wu D., Joffe R.T., Cheung S.W., Sellers E.M. Inhibition by fluoxetine of cytochrome P450 2D6 activity. Clinical Pharmacology and Therapeutics. 1993;53:401–409. doi: 10.1038/clpt.1993.43. doi:10.1038/clpt.1993.43. [DOI] [PubMed] [Google Scholar]
- 16.Coffman B.L., King C.D., Rios G.R., Tephly T.R. The glucuronidation of opioids, other xenobiotics and androgens by human UGT2B7Y(268) and UGT2B7H(268) Drug Metabolism and Disposition. 1998;26:73–77. [PubMed] [Google Scholar]
- 17.Chen Z.R., Irvine R.J., Somogyi A.A., Bocher F. Mu receptor binding of some commonly used opioids and their metabolites. Life Sciences. 1991;48:2165–2171. doi: 10.1016/0024-3205(91)90150-a. doi:10.1016/0024-3205(91)90150-A. [DOI] [PubMed] [Google Scholar]
- 18.Lalovic B., Kharasch E.D., Hoffer C., Risler L., Liu-Chen L.Y., Shen D.D. Pharmacokinetics and pharmacodynamics of oral oxycodone in healthy human subjects: role of circulating active metabolites. Clinical Pharmacology and Therapeutics. 2006;79:461–479. doi: 10.1016/j.clpt.2006.01.009. doi:10.1016/j.clpt.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 19.Meatherall R. GC-MS cofirmation of codeine, morphine, 6-acetylmorphine, hydrocone, hydromorphone, oxycodone, and oxymorphone in urine. Journal of Analytical Toxicology. 1999;23:177–186. doi: 10.1093/jat/23.3.177. doi:10.1093/jat/23.3.177. [DOI] [PubMed] [Google Scholar]
- 20.McKinley S., Snyder J.J., Welsh E., Kazarian C.M., Jamerson M.H., Klette K.L. Rapid quantitation of urinary oxycodone and oxymorphone using fast gas chromatography-mass spectrometry. Journal of Analytical Toxicology. 2007;31:434–441. doi: 10.1093/jat/31.8.434. doi:10.1093/jat/31.8.434. [DOI] [PubMed] [Google Scholar]
- 21.Goldberger B.A., Chronister C.W., Merves M.L. Quantitation of oxycodone in blood and urine using gas chromatography-mass spectrometry (GC-MS) Methods in Molecular Biology. 2010;603:453–460. doi: 10.1007/978-1-60761-459-3_44. doi:10.1007/978-1-60761-459-3_44. [DOI] [PubMed] [Google Scholar]
- 22.Wright A.W., Lawrence J.A., Iu M., Cramond T., Smith M.T. Solid-phase extraction method with high-performance liquid chromatography and electrochemical detection for quantitative analysis of oxycodone in human plasma. Journal of Chromatography B. 1998;712:169–175. doi: 10.1016/s0378-4347(98)00146-7. doi:10.1016/S0378-4347(98)00146-7. [DOI] [PubMed] [Google Scholar]
- 23.Menelaou A., Hutchinson M.R., Quinn I., Christensen A., Somogyi A.A. Quantification of the O- and N-demethylated metabolites of hydrocodone and oxycodone in human liver microsomes using liquid chromatography with ultraviolet absorbance detection. Journal of Chromatography B. 2003;785:81–88. doi: 10.1016/s1570-0232(02)00856-5. doi:10.1016/S1570-0232(02)00856-5. [DOI] [PubMed] [Google Scholar]
- 24.Kokubun H., Ouki M., Matoba M., Kubo H., Hoka S., Yago K. Determination of oxycodone and hydrocotarnine in cancer patient serum by high-performance liquid chromatography with electrochemical detection. Analytical Sciences. 2005;21:337–339. doi: 10.2116/analsci.21.337. doi:10.2116/analsci.21.337. [DOI] [PubMed] [Google Scholar]
- 25.Cheremina O., Bachmakov I., Neubert A., Brune K., Fromm M.F., Hinz B. Simultaneous determination of oxycodone and its major metabolite, noroxycodone, in human plasma by high-performance liquid chromatography. Biomedical Chromatography. 2005;19:777–782. doi: 10.1002/bmc.516. doi:10.1002/bmc.516. [DOI] [PubMed] [Google Scholar]
- 26.Edwards S.R., Smith M.T. Low-level quantitation of oxycodone and its oxidative metabolites, noroxycodone, and oxymorphone, in rat plasma by high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry. Journal of Chromatography B. 2007;848:264–270. doi: 10.1016/j.jchromb.2006.10.039. doi:10.1016/j.jchromb.2006.10.039. [DOI] [PubMed] [Google Scholar]
- 27.Neuvonen M., Neuvonen P.J. Determination of oxycodone, noroxycodone, oxymorphone, and noroxymorphone in human plasma by liquid chromatography-electrospray-tandem mass spectrometry. Therapeutic Drug Monitoring. 2008;30:333–340. doi: 10.1097/FTD.0b013e31816e2d4b. doi:10.1097/FTD.0b013e31816e2d4b. [DOI] [PubMed] [Google Scholar]
- 28.Wagner M., Bourgogne E., Varesio E., Hopfgartner G. Quantitation of polar analytes using column-switching: application to oxycodone and three metabolites in human plasma. Journal of Chromatography B. 2010;878:637–644. doi: 10.1016/j.jchromb.2010.01.014. doi:10.1016/j.jchromb.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 29.Edinboro L.E., Backer R.C., Poklis A. Direct analysis of opiates in urine by liquid chromatography-tandem mass spectrometry. Journal of Analytical Toxicology. 2005;29:704–710. doi: 10.1093/jat/29.7.704. doi:10.1093/jat/29.7.704. [DOI] [PubMed] [Google Scholar]
- 30.Cone E.J., Zichterman A., Heltsley R., Black D.L., Cawthon B., Robert T., et al. Urine testing for norcodeine, norhydrocodone, and noroxycodone facilitates interpretation and reduces false negatives. Forensic Science International. 2010;198:58–61. doi: 10.1016/j.forsciint.2009.12.005. doi:10.1016/j.forsciint.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 31.Coles R., Kushnir M.M., Nelson G.J., McMillin G.A., Urry F.M. Simultaneous determination of codeine, morphine, hydrocodone, hydromorphine, oxycodone, and 6-acetylmorphine in urine, serum, plasma, whole blood, and meconium by LC-MS-MS. Journal of Analytical Toxicology. 2007;31:1–14. doi: 10.1093/jat/31.1.1. doi:10.1093/jat/31.1.1. [DOI] [PubMed] [Google Scholar]
- 32.US FDA. Guidance for Industry—Bioanalytical Method Development. 2001 http://www.fda.gov/cder/guidance/index.htm. 4 January 2011, date last accessed. [Google Scholar]
- 33.Peters F.T. Method validation using LC-MS. In: Polettini A., editor. Applications of LC-MS in Toxicology. London: Pharmaceutical Press; 2006. pp. 71–95. [Google Scholar]
- 34.Fang W.B., Chang Y., McCance-Katz E.F., Moody D.E. Determination of naloxone and nornaloxone (noroxymorphone) by high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry. Journal of Analytical Toxicology. 2009;33:409–417. doi: 10.1093/jat/33.8.409. doi:10.1093/jat/33.8.409. [DOI] [PubMed] [Google Scholar]
- 35.Lofwall M.R., Moody D.E., Fang W.B., Nuzzo P.A., Walsh S.L. The pharmacokinetics of intranasal crushed OxyContin® and intravenous oxycodone in non-dependent prescription opioid abusers. Journal of Clinical Pharmacology. 2012;52:600–606. doi: 10.1177/0091270011401620. doi:10.1177/0091270011401620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neff J.A., Moody D.E. Differential N-demethylation of l-α-acetylmethadol (LAAM) and norLAAM by cytochrome P450s 2B6, 2C18, and 3A4. Biochemical and Biophysical Research Communications. 2001;284:751–756. doi: 10.1006/bbrc.2001.5054. doi:10.1006/bbrc.2001.5054. [DOI] [PubMed] [Google Scholar]
- 37.Chang Y., Moody D.E., McCance-Katz E.F. Novel metabolites of buprenorphine detected in human liver microsomes and human urine. Drug Metabolism and Disposition. 2006;34:440–448. doi: 10.1124/dmd.105.006148. [DOI] [PubMed] [Google Scholar]
- 38.Nelson A.C., Huang W., Moody D.E. Variables in human liver microsome preparation: impact on the kinetics of l-α-acetylmethadol (LAAM) N-demethylation and dextromethorphan O-demethylation. Drug Metabolism and Disposition. 2001;29:319–325. [PubMed] [Google Scholar]
- 39.Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.L. Protein measurement with the Folin phenol reagent. Journal of Biological Chemistry. 1951;193:265–275. [PubMed] [Google Scholar]
- 40.Pennanen K., Kotiaho T., Huikko J., Kostianen R. Identification of ozone-oxidation products of oxycodone by electrospray ion trap mass spectrometry. Journal of Mass Spectrometry. 2001;36:791–797. doi: 10.1002/jms.180. [DOI] [PubMed] [Google Scholar]
- 41.Annesley T.M. Ion suppression in mass spectrometry. Clinical Chemistry. 2003;49:1041–1044. doi: 10.1373/49.7.1041. doi:10.1373/49.7.1041. [DOI] [PubMed] [Google Scholar]
- 42.Moody D.E., Fang W.B., Morrison J., McCance-Katz E.F. Gender differences in pharmacokinetics of maintenance dosed buprenorphine. Drug and Alcohol Dependence. 2011;118:479–483. doi: 10.1016/j.drugalcdep.2011.03.024. doi:10.1016/j.drugalcdep.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Middleton L.S., Nuzzo P.A., Loftwall M.R., Moody D.E., Walsh S.L. The pharmacodynamic and pharmacokinetic profile of intranasal crushed buprenorphine and buprenorphine/naloxone tablets in opioid abusers. Addiction. 2011;106:1460–1473. doi: 10.1111/j.1360-0443.2011.03424.x. doi:10.1111/j.1360-0443.2011.03424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]