

Abstract
Background
Exercise causes physiological cardiac hypertrophy and benefits the diabetic heart. Mammalian switch-independent 3A (mSin3A) and histone deacetylases (HDACs) 1 and 2 regulate hypertrophic genes through associations with the DNA binding proteins repressor element-1 silencing transcription factor (REST) and O-linked β-N-acetylglucosamine transferase (OGT). O-linked β-N-acetylglucosamine (O-GlcNAc) is a glucose derivative that is chronically elevated in diabetic hearts, and a previous study showed that exercise reduces cardiac O-GlcNAc. We hypothesized that O-GlcNAc and OGT would physically associate with mSin3A/HDAC1/2 in the heart, and that this interaction would be altered by diabetes and exercise.
Methods
8-week-old type 2 diabetic db/db (db) and non-diabetic C57 mice were randomized to treadmill exercise or sedentary groups for 1 or 4 weeks.
Results
O-GlcNAc was significantly higher in db hearts and increased with exercise. Db hearts showed lower levels of mSin3A, HDAC1, and HDAC2 protein, but higher levels of HDAC2 mRNA and HDAC1/2 deacetylase activity. Elevated HDAC activity was associated with significantly blunted expression of α-actin and brain natriuretic peptide in db hearts. In sedentary db hearts, co-immunoprecipitation assays showed that mSin3A and OGT were less associated with HDAC1 and HDAC2, respectively, compared to sedentary C57 controls; however, exercise removed these differences.
Conclusions
These data indicate that diabetes and exercise oppositely affect interactions between pro-hypertrophic transcription factors, and suggest that an increase in total cardiac O-GlcNAc is a mechanism by which exercise benefits type 2 diabetic hearts.
Keywords: Exercise, Diabetes, Cardiac hypertrophy, O-GlcNAc, Fetal genes
Background
Cardiac hypertrophy is the adaptive enlargement of the myocardium in response to physical or neurohormonal stress. Type 2 diabetes is associated with a cardiac syndrome called diabetic cardiomyopathy, which is characterized by pathological hypertrophy, contractile dysfunction [1,2], and an intractable reliance on fatty acid oxidation [3-5]. By contrast, chronic endurance exercise training causes physiological hypertrophy that improves contractile mechanics [6,7] and myocardial metabolism [8,9]. Exercise benefits the type 2 diabetic heart [10-14], but the underlying mechanisms by which exercise and diabetes control cardiac hypertrophy are not well understood.
In non-diabetic hearts, activation of fetal genes is a protective mechanism [15-17] that accompanies pathological hypertrophy [17-21]. These fetal genes include fetal cytoskeletal proteins (skeletal α-actin, β-myosin heavy chain) and the atrial and brain natriuretic peptides (ANP and BNP) [22-24]. Importantly, exercise and diabetes moderate these genes differently. Exercise increases adult cardiac α-actin [25], but does not change fetal gene expression [26], whereas type 2 diabetes actually reduces circulating natriuretic peptides [27,28], and blocks the activation of fetal genes by hypertrophic stimuli in vitro [29]. This suggests that fetal gene regulation in diabetic hearts is different from that of exercised hearts and non-diabetic hearts, and therefore, may underlie the hypertrophic response to these conditions.
A potential mechanism for the effects of diabetes and exercise on fetal genes is through the post-translational modification of transcription factors by O-linked β-N-acetylglucosamine (O-GlcNAc). O-GlcNAc is a glucose derivative that post-translationally modifies serine/threonine residues [30,31]. O-GlcNAcylation modifies signal transduction in a manner analogous to phosphorylation; O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) add and remove the O-GlcNAc moiety, respectively. We have recently shown that O-GlcNAc modifies repressor element-1 silencing transcription factor (REST) [32], a transcription factor that represses fetal genes by recruiting the corepressors mammalian switch-independent 3A (mSin3A) and histone deacetylases (HDACs) 1 and 2 [33]. The HDAC enzymes deacetylate histone tails, thus condensing euchromatin and silencing gene expression; HDAC1 and HDAC2 specifically mediate fetal gene regulation by REST and mSin3A, and have been repeatedly linked to hypertrophic growth of the heart [34].
mSin3A and HDAC1 are O-GlcNAcylated in HepG2 liver carcinoma cells, and are recruited to gene loci by the OGT enzyme [35], which also O-GlcNAcylates itself [30]. The activity of OGT is regulated by cellular concentrations of UDP-GlcNAc substrate [36,37], which is increased in the diabetic heart [38]. Indeed, total O-GlcNAc and protein O-GlcNAcylation are elevated in the diabetic heart [4,39], but the functional implications of this finding are unclear as elevated cardiac O-GlcNAc is implicated in heart failure and cardiac dysfunction [39,40], but also cardioprotection [41,42]. Nevertheless, two previous studies showed that total protein O-GlcNAcylation and O-GlcNAcylation of the SP1 transcription factor are lowered by swimming exercise in both non-diabetic and streptozotocin-induced type 1 diabetic hearts [43,44], and OGA overexpression directly ameliorates the cardiovascular complications of type 2 diabetes [38].
We therefore hypothesized that a reduction in O-GlcNAc may be a mechanism by which exercise benefits the type 2 diabetic mouse heart. However, since these interactions have not been studied in the heart, the secondary purpose of this study was to characterize the effects of diabetes and moderate exercise on the mSin3A/HDAC1/2 complex. We used 4 weeks of moderate treadmill exercise training to investigate the early signaling mechanisms in the hypertrophic process. We show that exercise increases total protein O-GlcNAcylation in the type 2 diabetic db+/db+ mouse heart, and that exercise and diabetes have reciprocal effects on the association of HDAC1 and HDAC2 with fetal gene-regulating transcription factors.
Methods
Animal care and facilities
The procedures in this study followed the guidelines of the Washington State University Institutional Animal Care and Use Committee and the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH publication no. 85–23, revised 1996). 8-week-old type 2 diabetic mice (B6.BKS(D)-Leprdb/J, db+/db+ (db) and age-matched C57BL/6J non-diabetic lean db+/? background strain controls (C57) were purchased from Jackson Laboratories (Bar Harbor, ME). To control for activity, mice were singly housed without environmental enrichment in a climate-controlled vivarium on a 12:12 light:dark cycle. Mice consumed water and standard chow ad libitum, except for one overnight fast per week prior to blood glucose measurement.
Exercise training protocol
Mice ran on a 6-lane electric treadmill (Columbus Instruments, Columbus, OH) for 5 consecutive days a week with 2 days of rest. In week 0, all mice were acclimated to the treadmill by standing on the stationary belt for 10 min, then walking at 5 m/min for 20 min. Mice were then randomized to sedentary (n = 11) or exercise (n = 12) groups for 1 week, or sedentary (n = 16) or exercise (n = 15) groups for 4 weeks. Human patients with type 2 diabetes are commonly prescribed an exercise intensity of at least 40-60% of their aerobic capacity, but a higher intensity is recommended for maximum health benefits [45]. Therefore, we exercised mice at 10 m/min, which corresponds to approximately 70% of maximal oxygen uptake (VO2max) for the C57 strain [46]. Mice ran at this speed at 0% grade for 10, 20, 30, or 40 min in weeks 1, 2, 3, and 4, respectively. O-GlcNAc is a highly dynamic stress response; therefore, to remove confounding effects of stress, we kept the treadmill covered, and used gentle tactile stimuli rather than the electroshock apparatus to keep mice running. Sedentary groups were handled identically and spent equal time in the same treadmill environment on a stationary belt.
Blood glucose and body weight measurements
Blood glucose and body weight were measured weekly after an overnight fast. Blood glucose was measured with a glucometer (ACCU-CHEK® Aviva, Roche Diagnostics, Indianopolis, IN) in a small sample of tail blood. Glucose readings that exceeded the accuracy limit of the calibrated meter (33.3 mmol/L) were imputed this value for statistical analysis.
Tissue harvesting and morphological measurements
Following an overnight fast, mice were anesthetized rapidly using an isoflurane vaporizer chamber with 2-4% isoflurane gas in 100% oxygen. Mice were immediately decapitated and blood glucose was measured in neck blood. Whole hearts were excised and the atria were removed. Ventricular tissue was immediately wet-weighed, cut into four equal tissue aliquots, snap-frozen in liquid nitrogen, and stored at −80°C. The left tibia was dissected from each animal and measured from the tibial plateau to the lateral malleolus.
Western blotting
Ventricular tissue was homogenized in Tissue Protein Extraction Reagent (Sigma-Aldrich, St. Louis, MO); 20 mM sodium fluoride; 1 mM sodium orthovanadate; 3% protease inhibitor cocktail (Sigma); and 0.02% PUGNAc, an OGA inhibitor (Sigma), to inhibit O-GlcNAc removal from proteins. Total protein was quantified with a modified Lowry assay (BioRad, Hercules, CA). Proteins were separated by SDS-PAGE and transferred onto PVDF membranes, which were probed overnight at 4°C with primary antibodies (anti-HDAC1 and -HDAC2, Cell Signaling, Beverly, MA; anti-mSin3A, -REST, -OGT, and -calsequestrin, Abcam, Cambridge, MA; anti-NCOAT/OGA, Santa Cruz Biotechnology, Santa Cruz, CA; anti-phospho-HDAC1 (Ser 421/423), Millipore, Billerica, MA), then probed with the appropriate secondary antibodies for 1 hour at room temperature (see Table 1 for antibody details). Chemiluminescent substrates (Thermo Fisher Scientific, Rockford, IL) were used to detect horseradish peroxidase activity on a ChemiDoc (BioRad). Protein levels were quantified on duplicate blots with standard densitometry using ImageJ software (National Institutes of Health, Bethesda, MD), and normalized to the loading control calsequestrin.
Table 1.
Antibodies
| Target | Supplier | Cat # |
|---|---|---|
| CTD 110.6 (anti-O-GlcNAc) |
Gift from Mary-Ann Accavitti, University of Alabama at Birmingham |
n/a |
| RL2 (anti-O-GlcNAc) |
Abcam, Cambridge, MA |
2739 |
| anti-mSin3A |
Abcam |
3479 |
| anti-OGT |
Abcam |
50271 |
| anti-NCOAT/OGA |
Santa Cruz Biotechnology, Santa Cruz, CA |
sc-66612 |
| anti-HDAC1 |
Cell Signaling, Beverly, MA |
5356 |
| anti-HDAC2 |
Cell Signaling |
5113 |
| anti-Calsequestrin |
Abcam |
3516 |
| anti-REST |
Abcam |
21635 |
| anti-phospho-HDAC1 (ser421/423) | Millipore, Billerica, MA | 07-1575 |
O-GlcNAc Western blotting
Ventricular lysates were separated with SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked overnight at 4°C, probed with 1:5000 anti-O-GlcNAc antibody (CTD 110.6, generous gift of Mary-Ann Accavitti, University of Alabama at Birmingham), then probed with the appropriate secondary antibody. Horseradish peroxidase activity was detected on x-ray film with chemiluminescent substrate (Thermo Scientific) and quantified as described above for Western blotting.
Co-immunoprecipitation
Ventricular tissue was homogenized in Tissue Protein Extraction Reagent, 1% phosphatase inhibitor, 2% protease inhibitor (Sigma), and 0.02% PUGNAc. Lysates were assayed for total protein as described for Western blotting. Samples were diluted to equal protein concentrations and precleared over protein A/G agarose beads (Thermo Fisher Scientific) at 4°C for 4 hours. Precleared supernatants were then added to 25 ul of beads that had been incubated with primary antibody for 4 hours at 4°C. IP was performed overnight at 4°C; beads were then washed and eluted at 100°C for 5 min. The eluents were assayed for co-immunoprecipitated proteins using immunoblotting. The positive control was ventricular lysate; the negative control was ventricular lysate that was immunoprecipitated without antibody. Co-immunoprecipitated proteins were normalized to the level of captured target protein for analysis.
HDAC activity
HDAC enzyme activity was assayed with a colorimetric kit (Enzo Life Sciences, Farmingdale, NY) per the manufacturer’s instructions. The substrate for this assay is predominantly deacetylated by HDAC1/2 and sirtuin 1 (a class III HDAC), but not the class II HDACs. Optical density was read at 415 nm. Results are presented as fold changes from control absorbance.
RNA isolation and quantitative real-time PCR (qPCR)
RNA was isolated with an RNA isolation kit (Qiagen, Valencia, CA) and quantified on a NanoPhotometer™ spectrophotometer (Implen, Ontario, NY). Complementary DNA (cDNA) was generated using a cDNA synthesis kit (Thermo Fisher Scientific). qPCR was performed in triplicate with SYBR green fluorescence chemistry using a qPCR kit (Qiagen). The negative control contained water in place of cDNA template. Thermal cycling was performed on an iCycler iQTM Real Time PCR Detection System (Biorad) using the following cycle: 95°C for 10 min, and 40 cycles of 95°C for 30 sec and Tm for 10 sec. Primer specificity was confirmed by melting curve analysis. Amplification data were analyzed with the 2-ΔΔCt method for normalization to the housekeeping gene GAPDH as previously described [47]. See Table 2 for primer details.
Table 2.
Real-time PCR primers
|
Primer |
Forward |
Reverse |
Amplicon |
Annealing |
|---|---|---|---|---|
| size (kb) | temp (°C) | |||
| ANP |
TTCCGGTACCGAAGATAACAGCCA |
TGACACACCACAAGGGCTTAGGAT |
91 |
60 |
| BNP |
AGACAAGGGAGAACACGGCATCAT |
ACAGAATCATCTGGGACAGCACCT |
85 |
60 |
| HDAC1 |
TTCCTGCGTTCTATTCGCCCAGAT |
AACAAGCCATCAAACACCGGACAG |
98 |
60 |
| HDAC2 |
TACAACAGATCGCGTGATGACCGT |
TCCCTTTCCAGCACCAATATCCCT |
94 |
62 |
| α-skeletal actin |
TTGTGCACCGCAAATGCTTCTAGG |
GCAACCACAGCACGATTGTCGATT |
90 |
60 |
| α-cardiac actin |
TGTAGGTGATGAAGCCCAGAGCAA |
TGGTGCCAGATCTTCTCCATGTCA |
105 |
60 |
| β-myosin heavy chain |
TGGCTGGTGAGGTCATTGACAGAA |
TGGCTGGTGAGGTCATTGACAGAA |
104 |
60 |
| GAPDH | TGTGATGGGTGTGAACCACGAGAA | CATGAGCCCTTCCACAATGCCAAA | 133 | Per plate |
Statistics
Longitudinal effects of genotype and exercise on body weight and blood glucose were analyzed with two-factor repeated measures ANOVA followed by Bonferroni post-hoc tests. Non-normal data were log transformed prior to analysis. Two-factor ANOVA with Bonferroni post-hoc tests were used to describe genotype and exercise effects on protein levels, HDAC activity, and gene expression. Cardiac hypertrophy, tibia length, and wet heart weight data were resistant to transformations of normality and were analyzed with Kruskal-Wallis analysis of variance followed by Dunn’s post-hoc test. Values are presented as mean ± SEM and significance was accepted at P < 0.05.
Results
Db mice show obesity, hyperglycemia, and cardiac hypertrophy despite 4 weeks of exercise
Fasting blood glucose and body weight in db mice were significantly elevated relative to controls, and increased over the duration of the protocol (Figure 1) as the db mice developed overt diabetes. Exercise did not alter either variable in db mice; this is consistent with previously described work by others [27,48,49]. There were no differences in cardiac hypertrophy at 1 week; however, db mice showed overt cardiac hypertrophy (heart weight:tibia length) at the 4 week time point (Table 3). Tibia length was also significantly reduced in db mice at the 4 week time point, consistent with previous reports of reduced linear skeletal growth in db mice [50,51].
Figure 1.
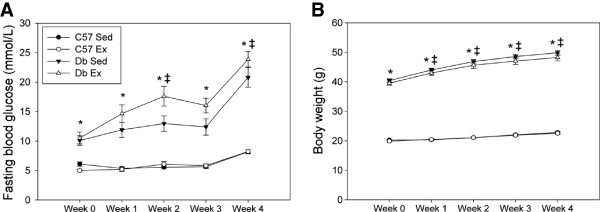
Effect of 4 weeks of treadmill exercise training on fasting blood glucose and body weight in C57BL/6J (C57) and db/db (db) mice. Ex = exercise (n = 15 per group); Sed = sedentary (n = 16 per group). * Significant effect of genotype within a time point, p < 0.05. ǂ Significant effect of time from previous consecutive time point, P < 0.05.
Table 3.
Morphological data
| 1 Week | ||||
|---|---|---|---|---|
| |
Wet heart (mg) |
Tibia (mm) |
Heart:tibia (mg:mm) |
N |
| C57 Sed |
115 ± 4 |
22.5 ± 0.2 |
5.13 ± 0.18 |
11 |
| C57 Ex |
111 ± 4 |
23.0 ± 0.2 |
4.83 ± 0.18 |
12 |
| Db Sed |
114 ± 3 |
22.4 ± 0.1 |
5.08 ± 0.14 |
11 |
| Db Ex |
116 ± 3 |
22.3 ± 0.2 |
5.20 ± 0.15 |
12 |
| |
4 Week |
|||
| |
Wet heart (mg) |
Tibia (mm) |
Heart:tibia (mg:mm) |
N |
| C57 Sed |
113 ± 2 |
23.5 ± 0.1 |
4.82 ± 0.09 |
16 |
| C57 Ex |
105 ± 2 |
23.4 ± 0.1 |
4.50 ± 0.10 |
15 |
| Db Sed |
123 ± 4* |
22.6 ± 0.1* |
5.45 ± 0.15* |
16 |
| Db Ex | 119 ± 3* | 22.4 ± 0.1* | 5.30 ± 0.13* | 15 |
*p < 0.05, significant effect of genotype.
Results are presented as mean ± SEM.
Total protein O-GlcNAcylation is increased by exercise in db mouse hearts
Exercise training increased cardiac O-GlcNAc in db mouse hearts at the 1 week time point; however, at this time point, protein levels of OGT and OGA were not different between groups (Figure 2). At 4 weeks, O-GlcNAc was elevated in db hearts relative to controls, and was significantly increased by exercise (Figure 3). As densitometry of the entire sample lane is dominated by the intense immunoreactive bands at 37 and 82 kDa, the analysis was also performed over the high, mid and low molecular weight ranges, which showed the same exercise-induced increases in db hearts. At this timepoint, levels of OGA and OGT were also significantly increased in db hearts relative to controls, independent of exercise.
Figure 2.

1 week of exercise increases total protein O-GlcNAcylation in db/db mouse hearts. (A) Western blot and (B-G) quantification of total protein O-GlcNAcylation in ventricular tissue from C57BL/6J (C57) and db/db (db) mice that were exercised (Ex) or sedentary (Sed) for 1 week. O-GlcNAc levels are normalized to the loading control calsequestrin (Csq). N = 6 per group. * Significant effect of genotype within an exercise group, P < 0.05. # Significant effect of exercise within a genotype group, P < 0.05. ‡ Significant interaction of exercise x genotype, P < 0.05.
Figure 3.

4 weeks of exercise increases total protein O-GlcNAcylation in db/db mouse hearts. (A) Western blot and (B-G) quantification of total protein O-GlcNAcylation in ventricular tissue from C57BL/6J (C57) and db/db (db) mice that were exercised (Ex) or sedentary (Sed) for 1 week. O-GlcNAc levels are normalized to the loading control calsequestrin (Csq). N = 6 per group. * Significant main effect of genotype, P < 0.05. # Significant effect of exercise within a genotype group, P < 0.05. ‡ Significant interaction of exercise x genotype, P < 0.05.
To analyze these effects further, we then analyzed O-GlcNAc by weight range within each genotype. Analysis of O-GlcNAc by weight range revealed a modest but significant decrease in total protein O-GlcNAcylation with exercise. Interestingly, however, we also observed an increase in O-GlcNAc on total and high molecular weight proteins over time in C57 hearts (Figure 4C). In db mice, however, total and high molecular weight O-GlcNAc increased over time (Figure 4G), and O-GlcNAcylation of mid and low molecular weight were increased by exercise (Figure 4I-J).
Figure 4.

Effects of 1 and 4 weeks of treadmill exercise on total protein O-GlcNAcylation in type 2 diabetic db/db and non-diabetic C57BL/6J hearts. (A) Western blot and (B-E) quantification of total protein O-GlcNAcylation in ventricular tissue from C57 mice that were treadmill exercised (Ex) or sedentary (Sed) for 1 or 4 weeks. (F) Western blot and (G-J) quantification of protein O-GlcNAcylation levels in ventricular tissue from Ex or Sed (db) mice. O-GlcNAc levels are normalized to the loading control calsequestrin (Csq). N=6 per group. * Significant effect of time within an exercise group, P<0.05. # Significant effect of exercise within a time point, P<0.05. ‡ Significant interaction of exercise group x time, P<0.05.
Diabetes reduces mSin3A/HDAC1/HDAC2 protein levels in the heart
REST protein levels were not different between groups at either time point (Figures 5 and 6). At the 1 week time point, mSin3A and HDAC1 were significantly lower in db hearts, independent of exercise (Figure 5). At the 4 week time point, mSin3A, HDAC1, as well as HDAC2 protein levels were significantly lower in db hearts, again independent of exercise (Figure 6). Exercise training reduced mSin3A levels in non-diabetic control hearts; however, mSin3A levels were even lower in db hearts and did not change with exercise (Figure 6B).
Figure 5.

Effects of diabetes and 1 week of exercise on protein levels of the mSin3A/HDAC1/2 transcription factor complex in type 2 diabetic db/db and non-diabetic C57BL/6J hearts. (A) Western blot and (B-E) quantification of members of the mSin3A/REST/HDAC1/2 complex in C57BL/6J (C57) and db/db (db) mice who were treadmill exercise trained (Ex) or sedentary (Sed) for 1 weeks. N = 6 per group. * Significant main effect of genotype, P < 0.05.
Figure 6.

Effects of diabetes and 4 weeks of exercise on protein levels of the mSin3A/HDAC1/2 transcription factor complex in type 2 diabetic db/db and non-diabetic C57BL/6J hearts. (A) Western blot and (B-E) quantification of members of the mSin3A/REST/HDAC1/2 complex in C57BL/6J (C57) and db/db (db) mice who were treadmill exercise trained (Ex) or sedentary (Sed) for 4 weeks. N=6 per group. * Significant main effect of genotype, P<0.05; # Significant effect of exercise within a genotype group, P<0.05.
Exercise rescues the mSin3A:HDAC1 association and the OGT:HDAC2 association in db mouse hearts
As stated above, cardiac hypertrophy as measured by heart weight:tibia length was not apparent at 1 week but was evident in db hearts at 4 weeks (Table 3); therefore, we analyzed the association of hypertrophy-regulating transcription factors in the 4 week group only. Co-immunoprecipitation of OGT showed that diabetes reduced the OGT:HDAC2 association (P < 0.05), but this difference was removed by exercise training (Figure 7). OGT association with mSin3A and HDAC1 was not different between groups. OGT was also modestly more associated with REST in sedentary db hearts compared to sedentary controls (P < 0.05).
Figure 7.

Effects of diabetes and 4 weeks of exercise on the association of mSin3A/HDAC1/2 complex with OGT in type 2 diabetic db/db and non-diabetic C57BL/6J hearts. (A) Immunoblot and (B-E) blot quantification showing the association of OGT with the mSin3A/REST/HDAC1/2 complex in C57BL/6J (C57) and db/db (db) mice who were treadmill exercise trained (Ex) or sedentary (Sed) for 4 weeks. N = 3 per group. * Significant effect of genotype within exercise group, P < 0.05.
Reciprocal co-immunoprecipitation of mSin3A showed that there were no differences in its association with OGT or HDAC2 (Figure 8). However, mSin3A was significantly more associated with REST in db hearts. Finally, the association of mSin3A with HDAC1 was significantly lower in sedentary db hearts compared to sedentary controls, and this difference was removed by exercise.
Figure 8.

Effects of diabetes and 4 weeks of exercise on the interaction of mSin3A/HDAC1/2 with OGT and O-GlcNAc in type 2 diabetic db/db and non-diabetic C57BL/6J hearts. (A) Immunoblot and (B-F) blot quantification showing the O-GlcNAc modification and the association of the mSin3A/REST/HDAC1/2 complex with OGT in C57BL/6J (C57) and db/db (db) mice who were treadmill exercise trained (Ex) or sedentary (Sed) for 4 weeks. N = 6 per group. * Significant effect of genotype within exercise group, P < 0.05. # Significant effect of exercise within a genotype group, P < 0.05. ‡ Significant main effect of exercise, P < 0.05.
It was recently reported that exercise reduces the O-GlcNAcylation of Specificity Protein 1 transcription factor (SP1) [43,44]. Therefore, we investigated whether exercise would alter the O-GlcNAc modification of mSin3A, a transcription factor that is directly involved in hypertrophic signaling. Although mSin3A immunoprecipitation appeared to show that the O-GlcNAcylation of mSin3A was increased by exercise (Figure 8A-B), reciprocal immunoprecipitation of O-GlcNAc did not confirm this effect (data not shown). Therefore, this effect of exercise should be viewed with caution.
Finally, we performed co-immunoprecipitation of HDAC1 to confirm the results of the mSin3A immunoprecipitation (Figure 9A). Immunoblotting for mSin3A showed the same effect of exercise on the HDAC1:mSin3A interaction; the association of mSin3A with HDAC1 was lower in sedentary db hearts relative to sedentary C57 controls, but exercise removed this difference.
Figure 9.
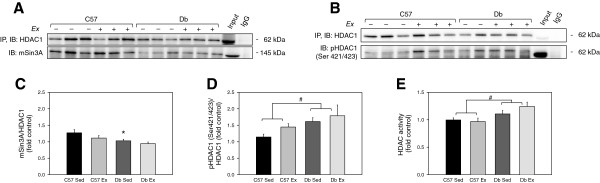
Effects of diabetes and 4 weeks of exercise on HDAC1 activation and the mSin3A/HDAC1 interaction. (A) Immunoprecipitation (IP) of HDAC1 and immunoblot for its association with mSin3A in ventricular tissue from C57BL/6J (C57) and db/db (db) mice that were treadmill exercised (Ex) or sedentary (Sed) for 4 weeks.(B) Immunoprecipitation (IP) of HDAC1 and immunoblot for phospho-serine 421/423, which activates HDAC1. (C-D) Blot quantification of (A) and (B). (E) Colorimetric assay quantification of class 1 HDAC activity in ventricular tissue. N = 6 per group for HDAC1 immunoprecipitation; N = 3 per group for HDAC1/2 activity assay. * Significant effect of genotype within an exercise group, P < 0.05. # Significant main effect of genotype, P < 0.05.
HDAC activity is increased in db mouse hearts
Phosphorylation of HDAC1 at serine 421 and 423 is specifically associated with HDAC1 activation [52]; therefore, we immunoprecipitated HDAC1 and immunoblotted for phospho-HDAC1 (Ser421/423). This showed that HDAC1 phosphorylation was significantly elevated in db hearts independent of exercise (P < 0.05) (Figure 9D). Colorimetric assay confirmed that class I HDAC activity was significantly higher in db mice independent of exercise (Figure 9E).
Diabetes blunts fetal gene expression in the heart
We investigated whether changes in these transcription factor interactions were associated with changes in the expression of fetal genes, such as ANP, BNP, and skeletal α-actin, which are regulated by REST and mSin3A [20,53]. Transcript levels of ANP and β-myosin heavy chain were not different between groups, but BNP and skeletal α-actin were significantly reduced in db hearts independent of exercise (Figure 10). HDAC2 gene expression was elevated in db hearts independent of exercise, and cardiac α-actin showed a prominent trend to be induced by exercise (P = 0.050) (Figure 10).
Figure 10.
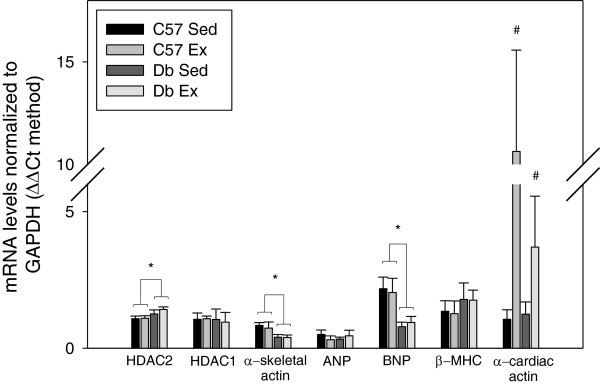
mRNA transcript levels in ventricular tissue from C57BL/6J (C57) or db/db (db) mice that were treadmill exercised (Ex) or sedentary (Sed) for 4 weeks. mRNA levels are normalized to the loading control GAPDH using the 2-∆∆Ct method. N = 8 per group for cardiac α-actin; N = 6 all other groups. * Significant main effect of genotype, P < 0.05. # Near-significant main effect of exercise, Ptrend = 0.050.
Discussion
While diabetes is a multifactorial cardiac insult, and diabetic cardiomyopathy is associated with multiple factors such as oxidative stress [54], lipotoxicity and mitochondrial dysfunction [55-57], and impaired calcium signaling [13], O-GlcNAc is emerging as an important signalling mechanism in the development of diabetic cardiomyopathy. Total protein O-GlcNAcylation is chronically elevated in the type 1 and 2 diabetic heart [4,39], and reducing protein O-GlcNAcylation by adenoviral overexpression of OGA [38] improves cardiac function. Similarly, lowering O-GlcNAc by intensive swim training [43,44] has been proposed as a mechanism by which exercise benefits the diabetic heart, and exercise lowers both the O-GlcNAc modification of the SP1 transcription factor and the OGT enzyme. O-GlcNAc directly mediates the expression of fetal genes in response to hypertrophic stimuli [29], and O-GlcNAc modifies mSin3A and HDAC1 [35], which regulate cardiac hypertrophy [20,58]. Previously, we have shown that exercise lowers the O-GlcNAc modification of the OGT enzyme [32], and others have shown that exercise lowers O-GlcNAcylation of the SP1 transcription factor [43]. Moderate exercise improves cardiac structure and function in humans with type 2 diabetes [59,60]; we therefore tested the hypothesis that moderate exercise would reduce O-GlcNAc in the type 2 diabetic heart, and would be associated with changes in the O-GlcNAc modification and activity of the mSin3A/HDAC1/2 transcription factor complex, which regulates hypertrophic genes.
Surprisingly, and in contrast with the previous studies, we found that 4 weeks of moderate treadmill exercise increased total O-GlcNAc in type 2 diabetic db mouse hearts. Also, while the previous studies showed that OGT was also reduced by exercise [43,44], we found that OGT and OGA expression was elevated in db hearts and did not change with exercise. Such parallel regulation of OGT and OGA expression has been previously reported [41], and may represent a compensatory relationship between these two opposing enzymes. The difference in our findings may be due to the use of type 2 db mice rather than streptozotocin-induced type 1 diabetic mice, and the use of moderate treadmill exercise rather than more intensive swimming exercise. However, other studies have shown that an upregulation of O-GlcNAc is essential in the cardiac stress response [61,62], is acutely cardioprotective [63,64], and is part of a constitutively active cardioprotection mechanism in the diabetic myocardium [42]. Therefore, these data suggest that an increase in cardiac O-GlcNAc in the type 2 diabetic heart may be a beneficial effect of exercise.
In our study, mSin3A immunoprecipitation revealed that exercise increased the O-GlcNAc modification of mSin3A; however, this was not supported by reciprocal O-GlcNAc immunoprecipitation. It is possible that the large amount of protein captured in the O-GlcNAc immunoprecipitation masked the changes in mSin3A O-GlcNAcylation, which we observed in the more specific mSin3A immunoprecipitation. However, these data underscore the importance of verifying changes in O-GlcNAcylation of individual proteins with reciprocal assays, and suggest that moderate changes in protein O-GlcNAcylation – including those in the present study – should be interpreted cautiously and confirmed by additional studies.
Nevertheless, our data do suggest an alternate mechanism for the beneficial effect of exercise on the diabetic heart. Db hearts showed lower protein levels of mSin3A, HDAC1, and HDAC2, and an increased association of mSin3A with REST, independent of exercise. Likewise, mRNA transcript levels of BNP and α-skeletal actin, which are typical markers of cardiac hypertrophy activated by HDAC1/2 [65] that are regulated via REST/mSin3A [33], were significantly lower in db hearts independent of exercise. The finding that blunted expression of fetal genes in diabetic hearts is not altered by exercise has been shown in previous studies [27,28]. Therefore, we suggest that the loss of HDAC1/2 and the increased association of the mSin3A corepressor with REST may underlie the blunted expression of fetal genes in the diabetic heart. Further, since the natriuretic peptides are both anti-hypertrophic and cardioprotective [15,17,28], we suggest that this mechanism may be responsible for the increased vulnerability of the diabetic heart to stress and heart failure [66,67].
Although we did not measure the structural or hemodynamic effects of the exercise protocol in db hearts, previous work has shown that the db heart shows similar cardiomyopathy to humans with type 2 diabetes [68,69], which are improved by exercise [13,14]. We show additionally that even the low intensity of exercise used in this protocol was sufficient to elevate the expression of cardiac α-actin in C57 hearts (P = 0.050). Cardiac α-actin is a marker of cardiomyocyte differentiation and hypertrophy [70], and is increased in physiologically hypertrophied hearts after chronic endurance exercise training [25]. Additional transcriptional changes were observed in db hearts, in which the exercise protocol significantly increased the association of mSin3A and OGT with HDAC1 and HDAC2, respectively. Therefore, although the exercise stimulus used in this study did not cause overt changes in cardiac mass, it induced transcriptional events consistent with the early stages of physiological cardiac remodelling.
Finally, these data show a potential interaction between HDAC1 and HDAC2 that has not previously been described in the heart. HDAC1 and HDAC2 regulate cardiac hypertrophy in a similar manner [58], and HDAC1 deficiency induces HDAC2 expression in embryonic stem cells [71]. In our study, the loss of HDAC1 protein preceded the loss of HDAC2 protein in db hearts, and was similarly associated with an increase in HDAC2 gene expression in db hearts. When HDAC2 deficiency was present at the 4 week time point, we observed an increase in the total activity of class I HDACs in db hearts, which was verified by an increase in the phosphorylation status of HDAC1 at Ser421/423. Phosphorylation at these residues is specifically associated with HDAC1 activity [52]. Therefore, these data suggest that the class I HDACs have compensatory effects on each other’s expression levels and activation by phosphorylation. Further, the reduction in HDAC2 protein levels in db mouse hearts did not occur until 4 weeks, and was associated with overt cardiac hypertrophy. Thus, the loss of HDAC2 in the diabetic heart is associated with the progression of hypertrophy in the diabetic heart, and may be more specifically involved in hypertrophy than HDAC1.
Conclusions
These data show that exercise increases O-GlcNAc in the type 2 diabetic db mouse heart, and that components of the mSin3A/HDAC1/2 chromatin-modifying complex interact with O-GlcNAc and OGT. Contrary to our hypothesis, exercise increased cardiac O-GlcNAc in the diabetic heart; this signalling mechanism may underlie the beneficial effect of exercise in the pathologically hypertrophied diabetic heart. Finally, we found that diabetes and exercise reciprocally affected the physical associations of mSin3A/HDAC1/2. The effects of exercise observed in this study were generally modest, which suggests that a moderate level of exercise, such as that prescribed for human patients with diabetes, does not have extreme effects on the mSin3A/HDAC1/2 complex. However, since this complex is a key regulator of cardiac hypertrophy, the results of this study suggest that exercise-induced changes in the association or activity of this complex may underlie the beneficial effect of moderate exercise in the diabetic heart.
Abbreviations
ANP: Atrial natriuretic peptide; BNP: Brain natriuretic peptide; C57: C57 background strain control mouse; Db: db/db type 2 diabetic mouse; HDAC: Histone deacetylase; mSin3A: Mammalian switch-independent 3A; O-GlcNAc: O-linked β-N-acetylglucosamine; OGT: O-GlcNAc transferase; OGA: O-GlcNAcase; REST: Repressor element-1 silencing transcription factor.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EJC performed the animal studies and laboratory experiments, and drafted the manuscript. SAM edited the manuscript and provided technical and intellectual guidance. Both authors read and approved the final manuscript.
Contributor Information
Emily J Cox, Email: emily.cox@email.wsu.edu.
Susan A Marsh, Email: susan.marsh@wsu.edu.
Acknowledgements
This work was supported by the National Institute of Health (HL-104549), WSU Office of Research and WSU College of Pharmacy. We are grateful for the help and attention of the vivarium staff and the intellectual assistance of Heidi M. Medford and Lindsey E. Miller.
References
- Falcao-Pires I, Palladini G, Goncalves N, van der Velden J, Moreira-Goncalves D, Miranda-Silva D, Salinaro F, Paulus WJ, Niessen HW, Perlini S, Leite-Moreira AF. Distinct mechanisms for diastolic dysfunction in diabetes mellitus and chronic pressure-overload. Basic Res Cardiol. 2011;106:801–814. doi: 10.1007/s00395-011-0184-x. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Yokoshiki H, Mitsuyama H, Mizukami K, Ono T, Tsutsui H. Conduction and refractory disorders in the diabetic atrium. Am J Physiol Heart Circ Physiol. 2012;303:H86–H95. doi: 10.1152/ajpheart.00010.2012. [DOI] [PubMed] [Google Scholar]
- Carley AN, Severson DL. Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochim Biophys Acta. 2005;1734:112–126. doi: 10.1016/j.bbalip.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Chess DJ, Stanley WC. Role of diet and fuel overabundance in the development and progression of heart failure. Cardiovasc Res. 2008;79:269–278. doi: 10.1093/cvr/cvn074. [DOI] [PubMed] [Google Scholar]
- Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord. 2010;11:31–39. doi: 10.1007/s11154-010-9131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggish AL, Yared K, Wang F, Weiner RB, Hutter AM, Picard MH, Wood MJ. The impact of endurance exercise training on left ventricular systolic mechanics. Am J Physiol Heart Circ Physiol. 2008;295:H1109–H1116. doi: 10.1152/ajpheart.00395.2008. [DOI] [PubMed] [Google Scholar]
- Vinereanu D, Florescu N, Sculthorpe N, Tweddel AC, Stephens MR, Fraser AG. Differentiation between pathologic and physiologic left ventricular hypertrophy by tissue Doppler assessment of long-axis function in patients with hypertrophic cardiomyopathy or systemic hypertension and in athletes. Am J Cardiol. 2001;88:53–58. doi: 10.1016/S0002-9149(01)01585-5. [DOI] [PubMed] [Google Scholar]
- Gertz EW, Wisneski JA, Stanley WC, Neese RA. Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J Clin Invest. 1988;82:2017–2025. doi: 10.1172/JCI113822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman BC, Tsvetkova T, Lowes B, Wolfel EE. Myocardial FFA metabolism during rest and atrial pacing in humans. Am J Physiol Endocrinol Metab. 2009;296:E358–E366. doi: 10.1152/ajpendo.90747.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loganathan R, Bilgen M, Al-Hafez B, Zhero SV, Alenezy MD, Smirnova IV. Exercise training improves cardiac performance in diabetes: in vivo demonstration with quantitative cine-MRI analyses. J Appl Physiol. 2007;102:665–672. doi: 10.1152/japplphysiol.00521.2006. [DOI] [PubMed] [Google Scholar]
- Broderick TL, Poirier P, Gillis M. Exercise training restores abnormal myocardial glucose utilization and cardiac function in diabetes. Diabetes Metab Res Rev. 2005;21:44–50. doi: 10.1002/dmrr.479. [DOI] [PubMed] [Google Scholar]
- Stolen KQ, Kemppainen J, Ukkonen H, Kalliokoski KK, Luotolahti M, Lehikoinen P, Hämäläinen H, Salo T, Airaksinen KE, Nuutila P, Knuuti J. Exercise training improves biventricular oxidative metabolism and left ventricular efficiency in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2003;41:460–467. doi: 10.1016/s0735-1097(02)02772-9. [DOI] [PubMed] [Google Scholar]
- Stolen TO, Hoydal MA, Kemi OJ, Catalucci D, Ceci M, Aasum E, Larsen T, Rolim N, Condorelli G, Smith GL, Wisloff U. Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ Res. 2009;105:527–536. doi: 10.1161/CIRCRESAHA.109.199810. [DOI] [PubMed] [Google Scholar]
- Hafstad AD, Lund J, Hadler-Olsen E, Hoper AC, Larsen TS, Aasum E. High- and Moderate-Intensity Training Normalizes Ventricular Function and Mechanoenergetics in Mice With Diet-Induced Obesity. Diabetes. 2013;62:2287–2294. doi: 10.2337/db12-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz AC, Hood SG, Woods RL, Dusting GJ, Ritchie RH. B-type natriuretic peptide prevents acute hypertrophic responses in the diabetic rat heart: importance of cyclic GMP. Diabetes. 2003;52:2389–2395. doi: 10.2337/diabetes.52.9.2389. [DOI] [PubMed] [Google Scholar]
- Gao XR, Tan YZ, Wang HJ. Overexpression of Csx/Nkx2.5 and GATA-4 enhances the efficacy of mesenchymal stem cell transplantation after myocardial infarction. Circ J. 2011;75:2683–2691. doi: 10.1253/circj.CJ-11-0238. [DOI] [PubMed] [Google Scholar]
- Franco V, Chen YF, Oparil S, Feng JA, Wang D, Hage F, Perry G. Atrial natriuretic peptide dose-dependently inhibits pressure overload-induced cardiac remodeling. Hypertension. 2004;44:746–750. doi: 10.1161/01.HYP.0000144801.09557.4c. [DOI] [PubMed] [Google Scholar]
- Brattelid T, Qvigstad E, Moltzau LR, Bekkevold SV, Sandnes DL, Birkeland JA, Skomedal T, Osnes JB, Sjaastad I, Levy FO. The cardiac ventricular 5-HT4 receptor is functional in late foetal development and is reactivated in heart failure. PLoS One. 2012;7:e45489. doi: 10.1371/journal.pone.0045489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azakie A, Fineman JR, He Y. Myocardial transcription factors are modulated during pathologic cardiac hypertrophy in vivo. J Thorac Cardiovasc Surg. 2006;132:1262–1271. doi: 10.1016/j.jtcvs.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Kuwahara K, Nishikimi T, Nakao K. Transcriptional regulation of the fetal cardiac gene program. J Pharmacol Sci. 2012;119:198–203. doi: 10.1254/jphs.12R04CP. [DOI] [PubMed] [Google Scholar]
- Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev. 2007;12:331–343. doi: 10.1007/s10741-007-9034-1. [DOI] [PubMed] [Google Scholar]
- Chang L, Kiriazis H, Gao XM, Du XJ, El-Osta A. Cardiac genes show contextual SWI/SNF interactions with distinguishable gene activities. Epigenetics. 2011;6:760–768. doi: 10.4161/epi.6.6.16007. [DOI] [PubMed] [Google Scholar]
- Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, Zhang D, Nakamura T, Takimoto E, Kass DA. Pivotal role of cardiomyocyte TGF-beta signaling in the murine pathological response to sustained pressure overload. J Clin Invest. 2011;121:2301–2312. doi: 10.1172/JCI44824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Port JD, Walker LA, Polk J, Nunley K, Buttrick PM, Sucharov CC. Temporal expression of miRNAs and mRNAs in a mouse model of myocardial infarction. Physiol Genomics. 2011;43:1087–1095. doi: 10.1152/physiolgenomics.00074.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burniston JG. Adaptation of the rat cardiac proteome in response to intensity-controlled endurance exercise. Proteomics. 2009;9:106–115. doi: 10.1002/pmic.200800268. [DOI] [PubMed] [Google Scholar]
- McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Kang PM, Izumo S. Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci USA. 2003;100:12355–12360. doi: 10.1073/pnas.1934654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutkowska J, Broderick TL, Bogdan D, Wang D, Lavoie JM, Jankowski M. Downregulation of oxytocin and natriuretic peptides in diabetes: possible implications in cardiomyopathy. J Physiol. 2009;587:4725–4736. doi: 10.1113/jphysiol.2009.176461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick TL, Jankowski M, Wang D, Danalache BA, Parrott CR, Gutkowska J. Downregulation in GATA4 and Downstream Structural and Contractile Genes in the db/db Mouse Heart. ISRN endocrinology. 2012;2012:736860. doi: 10.5402/2012/736860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh SA, Dell’Italia LJ, Chatham JC. Activation of the hexosamine biosynthesis pathway and protein O-GlcNAcylation modulate hypertrophic and cell signaling pathways in cardiomyocytes from diabetic mice. Amino Acids. 2011;40:819–828. doi: 10.1007/s00726-010-0699-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ande SR, Moulik S, Mishra S. Interaction between O-GlcNAc modification and tyrosine phosphorylation of prohibitin: implication for a novel binary switch. PLoS One. 2009;4:e4586. doi: 10.1371/journal.pone.0004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GW, Akimoto Y. In: Essentials of Glycobiology. 2. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editor. NY: Cold Spring Harbor; 2009. The O-GlcNAc Modification. [PubMed] [Google Scholar]
- Medford HM, Porter K, Marsh SA. Immediate effects of a single exercise bout on protein O-GlcNAcylation and chromatin regulation of cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2013;305(1):H114–H123. doi: 10.1152/ajpheart.00135.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roopra A, Sharling L, Wood IC, Briggs T, Bachfischer U, Paquette AJ, Buckley NJ. Transcriptional repression by neuron-restrictive silencer factor is mediated via the Sin3-histone deacetylase complex. Mol Cell Biol. 2000;20:2147–2157. doi: 10.1128/MCB.20.6.2147-2157.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee HJ, Kook H. Roles and targets of class I and IIa histone deacetylases in cardiac hypertrophy. J Biomed Biotechnol. 2011;2011:928326. doi: 10.1155/2011/928326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Zhang F, Kudlow JE. Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: coupling protein O-GlcNAcylation to transcriptional repression. Cell. 2002;110:69–80. doi: 10.1016/S0092-8674(02)00810-3. [DOI] [PubMed] [Google Scholar]
- Shafi R, Iyer SP, Ellies LG, O’Donnell N, Marek KW, Chui D, Hart GW, Marth JD. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci USA. 2000;97:5735–5739. doi: 10.1073/pnas.100471497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreppel LK, Hart GW. Regulation of a cytosolic and nuclear O-GlcNAc transferase. Role of the tetratricopeptide repeats. J Biol Chem. 1999;274:32015–32022. doi: 10.1074/jbc.274.45.32015. [DOI] [PubMed] [Google Scholar]
- Hu Y, Belke D, Suarez J, Swanson E, Clark R, Hoshijima M, Dillmann WH. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ Res. 2005;96:1006–1013. doi: 10.1161/01.RES.0000165478.06813.58. [DOI] [PubMed] [Google Scholar]
- Clark RJ, McDonough PM, Swanson E, Trost SU, Suzuki M, Fukuda M, Dillmann WH. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J Biol Chem. 2003;278:44230–44237. doi: 10.1074/jbc.M303810200. [DOI] [PubMed] [Google Scholar]
- Lunde IG, Aronsen JM, Kvaloy H, Qvigstad E, Sjaastad I, Tonnessen T, Christensen G, Gronning-Wang LM, Carlson CR. Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiol Genomics. 2012;44:162–172. doi: 10.1152/physiolgenomics.00016.2011. [DOI] [PubMed] [Google Scholar]
- Watson LJ, Facundo HT, Ngoh GA, Ameen M, Brainard RE, Lemma KM, Long BW, Prabhu SD, Xuan YT, Jones SP. O-linked beta-N-acetylglucosamine transferase is indispensable in the failing heart. Proc Natl Acad Sci USA. 2010;107:17797–17802. doi: 10.1073/pnas.1001907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RV, Zachara NE, Nielsen PH, Kimose HH, Kristiansen SB, Botker HE. Impact of O-GlcNAc on cardioprotection by remote ischaemic preconditioning in non-diabetic and diabetic patients. Cardiovasc Res. 2013;97(2):369–378. doi: 10.1093/cvr/cvs337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belke DD. Swim-exercised mice show a decreased level of protein O-GlcNAcylation and expression of O-GlcNAc transferase in heart. J Appl Physiol. 2011;111:157–162. doi: 10.1152/japplphysiol.00147.2011. [DOI] [PubMed] [Google Scholar]
- Bennett CE, Johnsen VL, Shearer J, Belke DD. Exercise training mitigates aberrant cardiac protein O-GlcNAcylation in streptozotocin-induced diabetic mice. Life Sci. 2013;92(11):657–663. doi: 10.1016/j.lfs.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Colberg SR, Sigal RJ, Fernhall B, Regensteiner JG, Blissmer BJ, Rubin RR, Chasan-Taber L, Albright AL, Braun B. American College of Sports M, American Diabetes A. Exercise and type 2 diabetes: the American College of Sports Medicine and the American Diabetes Association: joint position statement. Diabetes Care. 2010;33:e147–e167. doi: 10.2337/dc10-9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schefer V, Talan MI. Oxygen consumption in adult and AGED C57 mice during acute treadmill exercise of different intensity. Exp Gerontol. 1996;31:387–392. doi: 10.1016/0531-5565(95)02032-2. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Shearer J, Ross KD, Hughey CC, Johnsen VL, Hittel DS, Severson DL. Exercise training does not correct abnormal cardiac glycogen accumulation in the db/db mouse model of type 2 diabetes. Am J Physiol Endocrinol Metab. 2011;301:E31–E39. doi: 10.1152/ajpendo.00525.2010. [DOI] [PubMed] [Google Scholar]
- Lee S, Park Y, Zhang C. Exercise training prevents coronary endothelial dysfunction in type 2 diabetic mice. Am J Biomed Sci. 2011;3:241–252. doi: 10.5099/aj110400241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E. et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–859. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- Mozaffari MS, Baban B, Abdelsayed R, Liu JY, Wimborne H, Rodriguez N, Abebe W. Renal and glycemic effects of high-dose chromium picolinate in db/db mice: assessment of DNA damage. J Nutr Biochem. 2012;23:977–985. doi: 10.1016/j.jnutbio.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflum MK, Tong JK, Lane WS, Schreiber SL. Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J Biol Chem. 2001;276:47733–47741. doi: 10.1074/jbc.M105590200. [DOI] [PubMed] [Google Scholar]
- Kuwahara K, Saito Y, Takano M, Arai Y, Yasuno S, Nakagawa Y, Takahashi N, Adachi Y, Takemura G, Horie M. et al. NRSF regulates the fetal cardiac gene program and maintains normal cardiac structure and function. EMBO J. 2003;22:6310–6321. doi: 10.1093/emboj/cdg601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi T, Cheema Y, Tremble SM, Bell SP, Chen Z, Subramanian M, LeWinter MM, VanBuren P, Palmer BM. Zinc-induced cardiomyocyte relaxation in a rat model of hyperglycemia is independent of myosin isoform. Cardiovasc Diabetol. 2012;11:135. doi: 10.1186/1475-2840-11-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGavock JM, Lingvay I, Zib I, Tillery T, Salas N, Unger R, Levine BD, Raskin P, Victor RG, Szczepaniak LS. Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation. 2007;116:1170–1175. doi: 10.1161/CIRCULATIONAHA.106.645614. [DOI] [PubMed] [Google Scholar]
- Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- Rijzewijk LJ, van der Meer RW, Smit JW, Diamant M, Bax JJ, Hammer S, Romijn JA, de Roos A, Lamb HJ. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol. 2008;52:1793–1799. doi: 10.1016/j.jacc.2008.07.062. [DOI] [PubMed] [Google Scholar]
- Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PD, Buchner D, Pina IL, Balady GJ, Williams MA, Marcus BH, Berra K, Blair SN, Costa F, Franklin B. et al. Exercise and physical activity in the prevention and treatment of atherosclerotic cardiovascular disease: a statement from the Council on Clinical Cardiology (Subcommittee on Exercise, Rehabilitation, and Prevention) and the Council on Nutrition, Physical Activity, and Metabolism (Subcommittee on Physical Activity) Circulation. 2003;107:3109–3116. doi: 10.1161/01.CIR.0000075572.40158.77. [DOI] [PubMed] [Google Scholar]
- Hordern MD, Coombes JS, Cooney LM, Jeffriess L, Prins JB, Marwick TH. Effects of exercise intervention on myocardial function in type 2 diabetes. Heart. 2009;95:1343–1349. doi: 10.1136/hrt.2009.165571. [DOI] [PubMed] [Google Scholar]
- Kazemi Z, Chang H, Haserodt S, McKen C, Zachara NE. O-linked beta-N-acetylglucosamine (O-GlcNAc) regulates stress-induced heat shock protein expression in a GSK-3beta-dependent manner. J Biol Chem. 2010;285:39096–39107. doi: 10.1074/jbc.M110.131102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachara NE, O’Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem. 2004;279:30133–30142. doi: 10.1074/jbc.M403773200. [DOI] [PubMed] [Google Scholar]
- Balakumar P, Sharma NK. Healing the diabetic heart: does myocardial preconditioning work? Cell Signal. 2012;24:53–59. doi: 10.1016/j.cellsig.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Nawata T, Takahashi N, Ooie T, Kaneda K, Saikawa T, Sakata T. Cardioprotection by streptozotocin-induced diabetes and insulin against ischemia/reperfusion injury in rats. J Cardiovasc Pharmacol. 2002;40:491–500. doi: 10.1097/00005344-200210000-00001. [DOI] [PubMed] [Google Scholar]
- Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T, Floss T, Goettlicher M, Noppinger PR, Wurst W. et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat Med. 2007;13:324–331. doi: 10.1038/nm1552. [DOI] [PubMed] [Google Scholar]
- Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. 1974;34:29–34. doi: 10.1016/0002-9149(74)90089-7. [DOI] [PubMed] [Google Scholar]
- Capes SE, Hunt D, Malmberg K, Gerstein HC. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet. 2000;355:773–778. doi: 10.1016/S0140-6736(99)08415-9. [DOI] [PubMed] [Google Scholar]
- Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146:5341–5349. doi: 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- Demarco VG, Ford DA, Henriksen EJ, Aroor AR, Johnson MS, Habibi J, Ma L, Yang M, Albert CJ, Lally JW. et al. Obesity-related alterations in cardiac lipid profile and nondipping blood pressure pattern during transition to diastolic dysfunction in male db/db mice. Endocrinology. 2013;154:159–171. doi: 10.1210/en.2012-1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SS, Bernstein HS. Cardiac regeneration using human embryonic stem cells: producing cells for future therapy. Regen Med. 2010;5:763–775. doi: 10.2217/rme.10.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagger G, O’Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, Seiser C. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002;21:2672–2681. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]