

Abstract
Signaling levels within sensory neurons must be tightly regulated to allow cells to integrate information from multiple signaling inputs and to respond to new stimuli. Herein we report a new role for the cGMP-dependent protein kinase EGL-4 in the negative regulation of G protein-coupled nociceptive chemosensory signaling. C. elegans lacking EGL-4 function are hypersensitive in their behavioral response to low concentrations of the bitter tastant quinine and exhibit an elevated calcium flux in the ASH sensory neurons in response to quinine. We provide the first direct evidence for cGMP/PKG function in ASH and propose that ODR-1, GCY-27, GCY-33 and GCY-34 act in a non-cell-autonomous manner to provide cGMP for EGL-4 function in ASH. Our data suggest that activated EGL-4 dampens quinine sensitivity via phosphorylation and activation of the regulator of G protein signaling (RGS) proteins RGS-2 and RGS-3, which in turn downregulate Gα signaling and behavioral sensitivity.
Author Summary
All animals rely on their ability to sense and respond to their constantly changing environments to survive. C. elegans (small roundworms) depend heavily upon their ability to taste and smell chemical information in their soil environment to find food and avoid danger. While similar signal transduction pathways are implicated in both C. elegans and vertebrate chemosensation, there are still large gaps in our understanding of the mechanisms used to regulate signaling in these systems. We have identified a new role for the C. elegans cGMP-dependent protein kinase (PKG) EGL-4 in the negative regulation of nociceptive chemosensory signaling. Our data suggest that EGL-4 negatively regulates signaling and behavior by activating known inhibitors of G protein-coupled signal transduction, RGS proteins. Using C. elegans behavioral response to aversive stimuli as the readout for neuronal activity, we provide the first evidence for PKG regulation of RGS function in sensory neurons in any system.
Introduction
The ability to detect and avoid noxious stimuli in the environment is critical to an organism's survival. Nociceptive sensory systems mediate detection of harmful stimuli, allowing rapid initiation of protective behavioral responses. In the nematode Caenorhabditis elegans, the pair of polymodal nociceptive head sensory neurons termed ASH respond to a broad range of aversive stimuli, including soluble chemicals (e.g. the bitter tastant quinine, heavy metals and SDS), odorants (e.g. octanol), ions (e.g. Na+), osmotic stress and mechanosensory stimulation (nose touch) [1]–[9]. Because the ASH sensory neurons synapse onto command interneurons that drive backward locomotion via their connections with motor neurons, ASH activation elicits reversal and stimulus avoidance.
In general, olfaction and some forms of taste (including bitter) are mediated by G protein-coupled signal transduction pathways [10]–[12]. Signaling is initiated when the chemosensory ligand binds to a seven-transmembrane G protein-coupled receptor (GPCR). This induces a conformational change in the receptor that activates the associated heterotrimeric G proteins. Gα becomes active upon exchange of GDP for GTP. Once dissociated, the Gα-GTP and Gβγ subunits can activate distinct signaling cascades within the cell [13]. Although the C. elegans genome encodes >500 predicted functional chemosensory GPCRs [14], only one aversive chemical stimulus, dihydrocaffeic acid, has been functionally coupled to a receptor, DCAR-1 [15]. However, the C. elegans stimulatory Gα subunits ODR-3 and GPA-3 (both most similar to Gαi/o) are used by ASH to mediate avoidance of a variety of stimuli [7], [8], [16]–[18].
Regulator of G protein signaling (RGS) proteins are important negative regulators of G protein-coupled signal transduction. They bind to Gα-GTP and accelerate the intrinsic GTPase activity of the Gα subunits. Once GTP is hydrolyzed (back to GDP), signaling via Gα is attenuated [19], [20]. By dampening Gα signaling, RGS proteins help to protect cells from overstimulation. Mammalian RGS proteins have been implicated in the regulation of sensory signaling. For example, RGS9-1 plays an important role in regulating the light response of rod photoreceptor cells. Retinas isolated from knock-out mice lacking RGS9-1 function displayed a prolonged dim flash response [21], while overexpression of the RGS9-1 containing complex resulted in a faster light response recovery in the retina rod cells of transgenic mice [22]. In addition, RGS21 is coexpressed with T2R bitter receptors and T1R2 and T1R3 sweet receptors in rat taste bud cells [23]. RGS21 also coprecipitates with α-gustducin, the Gαi protein that is coupled to T2R bitter receptors [23]–[27]. Taken together, these observations suggest a potential role for RGS21 in the regulation of taste transduction.
C. elegans lacking RGS-3 function are defective in their response to a subset of strong sensory stimuli detected by the ASH sensory neurons [28]. Interestingly, the behavioral defects appear to be due to increased signaling in the sensory neurons that in turns leads to decreased synaptic transmission [28]. Although our previous study did not identify chemosensory hypersensitivity (e.g. enhanced sensitivity to dilute quinine) in rgs-3 mutant animals, we note that feeding status and, consequently, biogenic amine (e.g. dopamine and serotonin) levels modulate rgs-3 behavioral responses [28]. For example, rgs-3 animals responded to 100% octanol (odorant) and 10 mM quinine (tastant) when they were assayed in the presence of food (E. coli bacterial lawn), and were only defective when assayed after a short (10 minute) period of starvation [28]. Taken together, the sensitivity of C. elegans to an environmental stimulus is ultimately coordinated by proteins (e.g. RGS-3) that directly regulate the sensory G protein-coupled signaling cascade, in conjunction with signals modulated by nutritional status. All of the tastant avoidance experiments presented herein were performed 30 minutes after animals were removed from food, in contrast to the previous study focused on rgs-3, where “off food” assays were performed 10 minutes after animals were removed from the bacterial food source [28].
cGMP-dependent protein kinases (PKGs) are serine/threonine kinases that are activated upon the binding of cGMP [29], [30] and their function is implicated in a variety of cellular contexts. PKGI-null mice have altered physiological processes including impaired vasorelaxation [31]–[33] and increased platelet aggregation and activation [34] via the nitric oxide/cGMP/PKGI signaling pathway [35]. They also display impaired nociceptive flexion reflexes [36] and reduced inflammatory hyperalgesia [37]. Mice lacking PKGII function exhibit enhanced anxiety and alcohol consumption [38]. In addition, mammalian PKG can act as an indirect negative regulator of G protein-coupled signal transduction. Upon activation, PKGI phosphorylates serine/threonine residues on RGS2 in mouse vascular smooth muscle cells [39] and Rat1 fibroblast cells [40], RGS3 and RGS4 in rat diencephalic astrocytes [41] and RGS4 in rabbit gastric smooth muscle cells [42]. Phosphorylation by PKG causes each RGS protein to translocate from the cytosol to the plasma membrane, stimulating its binding to Gαq and consequently enhancing GTPase activity and the dampening of Gα signaling [39]–[42].
In C. elegans, EGL-4 is a cGMP-dependent protein kinase with physiological roles including egg laying, life span, cell growth, quiescence and dauer formation [43]–[46]. Additionally, EGL-4 function contributes to C. elegans sensory responses. For example, egl-4(lof) animals are strongly defective for chemotaxis to the AWA-detected odorant diacetyl [47]. In the AWC olfactory neurons, EGL-4 functions in the cytoplasm and nucleus to regulate short term and long term olfactory adaptation, respectively [48], [49]. EGL-4 also acts with KIN-29, a salt-inducible kinase, to regulate chemoreceptor gene expression [50], [51], contributing to behavioral plasticity and chemosensory signal-dependent development [52].
In mammalian systems, cGMP binding activates PKG [53]–[55] by relieving an inhibitory interaction that blocks the kinase domain [56]. C. elegans EGL-4 has two allosteric cGMP-binding domains in its amino-terminal half [46] and altering key residues within either or both cGMP-binding domains renders animals completely defective for AWC-mediated odor adaptation [49]. Interestingly, while EGL-4 requires intact cGMP binding domains to accumulate in the AWC nucleus [49], it requires reduction in cGMP levels to enter the AWC nucleus and promote adaptation [57]. In uterine epithelial cells, nuclear EGL-4 regulates gene expression [58]. Guanylyl cyclases (GCYs) produce cGMP, and 27 receptor-type GCYs and 7 soluble GCYs are encoded by the C. elegans genome [59]. Although several GCYs have been shown to function in C. elegans sensory neurons, no GCYs are known to be expressed in or function in ASH.
Herein we describe a new role for the C. elegans cGMP-dependent protein kinase EGL-4 as a negative regulator of nociceptive chemosensory signaling. Animals lacking EGL-4 function respond better than wild-type animals to dilute concentrations of several ASH-detected chemical stimuli, including the bitter tastant quinine. We provide evidence that a subset of GCYs function non-cell-autonomously to generate cGMP to stimulate EGL-4 phosphorylation and activation of RGS proteins in ASH to dampen G protein-coupled chemosensory signal transduction and behavioral sensitivity.
Results
EGL-4 Regulates Behavioral Sensitivity to Quinine
EGL-4 has not previously been implicated in nociceptive chemosensory signaling in C. elegans and egl-4(n479) loss-of-function (lof) animals effectively avoid the bitter tastant 10 mM quinine (Figure 1A). However, we noticed that egl-4(lof) animals appeared to respond to 10 mM quinine slightly better than wild-type animals (p<0.01). To determine whether EGL-4 might contribute to chemosensory signaling in a way that might not be fully revealed by this high concentration of quinine, which elicits a robust behavioral response even in wild-type animals, we challenged both genotypes with dilute (1 mM) quinine. At this lower concentration, egl-4(lof) animals were clearly hypersensitive, with significantly more egl-4(lof) animals responding to the dilute quinine than wild-type animals (Figure 1A). As loss of EGL-4 function led to quinine hypersensitivity, we reasoned that excess EGL-4 function could result in diminished quinine sensitivity. ad450 is a gain-of-function (gof) allele of egl-4. Consistent with our prediction, egl-4(gof) animals were no longer hypersensitive to quinine and responded worse than wild-type animals to 10 mM quinine (Figure 1A).
Figure 1. C. elegans EGL-4 regulates quinine sensitivity in ASH.

(A) egl-4(lof) animals respond better than wild-type animals to dilute (1 mM) quinine, while egl-4(gof) animals show a decreased sensitivity to 10 mM quinine, when compared to wild-type animals. p<0.0001 for each. (B) The ASH sensory neurons are the primary neurons used to detect quinine, but the ASK neurons also contribute [7]. The osm-10 [3], srb-6 [61] and srbc-66 [62] promoters were used to drive expression of wild-type egl-4 in egl-4(lof) animals. The osm-10 promoter expresses in ASH, ASI, PHA and PHB, while the srb-6 promoter drives expression in ASH, ADL, ADF, PHA and PHB. ASH is the only head sensory neuron common to both promoters. The srbc-66 promoter expresses in ASK. While egl-4(lof) animals respond better than wild-type animals to 1 mM quinine, restoring EGL-4 function in ASH significantly diminished this hypersensitivity (p<0.0001 for both). EGL-4 expression in ASK had no effect (p>0.5). (C) RNAi knock-down of egl-4 in the ASH sensory neurons of otherwise wild-type animals, using the osm-10 [3] or srb-6 [61] promoter resulted in behavioral hypersensitivity to dilute (1 mM) quinine, similar to egl-4(lof) animals (p<0.0001 when compared to N2 animals for both transgenes). The percentage of animals responding is shown. The combined data of ≥3 independent lines, n≥120 transgenic animals, is shown. Error bars represent the standard error of the mean (SEM). Alleles used: egl-4(n479) loss-of-function and egl-4(ad450) gain-of-function. WT = the N2 wild-type strain. lof = loss-of-function. gof = gain-of-function.
The ASH sensory neurons are the main cells used to detect quinine in C. elegans, but the ASK neurons also contribute [7]. EGL-4 is broadly expressed throughout the animal, including these sensory neurons [60]. To determine whether EGL-4 function in either the ASHs or ASKs is sufficient to regulate quinine response, the cell-selective promoters osm-10 (ASH) [3], srb-6 (ASH) [61] and srbc-66 (ASK) [62] were used to restore EGL-4 function in each neuron pair and animals were assayed for response to 1 mM quinine. Expression in ASH, but not ASK, returned the egl-4(lof) response to wild-type levels so that animals were no longer hypersensitive to the dilute stimulus (Figure 1B). To assess whether selective loss of EGL-4 function in the ASH sensory neurons could also lead to quinine hypersensitivity, we used the cell-specific RNAi approach of Esposito et al. [63]. The osm-10 [3] and srb-6 [61] promoters were used to co-express a non-coding fragment of egl-4 in both the sense and antisense orientations in the ASH neurons of otherwise wild-type animals. egl-4 knock-down using either promoter resulted in hypersensitivity to 1 mM quinine, similar to egl-4(lof) animals (Figure 1C). Taken together, our data suggest a role for EGL-4 in the negative regulation of quinine avoidance in the ASH sensory neurons.
EGL-4 Does Not Regulate ASH Sensitivity in General
In addition to the ASH-mediated avoidance of quinine, C. elegans avoid several additional bitter compounds [7]. We therefore tested whether EGL-4 selectively regulates quinine avoidance (Figure 1) or whether it could regulate bitter taste responses generally. We assayed the response of egl-4(lof) animals to the bitter tastants amodiaquine and primaquine (Figures 2A and 2B). egl-4(lof) animals were hypersensitive to amodiaquine, with more animals responding than wild-type across a range of concentrations (Figure 2A). However, egl-4(lof) animals responded to primaquine similarly to wild-type animals and did not appear hypersensitive (Figure 2B).
Figure 2. EGL-4 does not regulate ASH sensitivity in general.

C. elegans respond to bitter stimuli in addition to quinine. (A) Animals lacking EGL-4 function are hypersensitive to dilute amodiaquine (p≤0.01 when compared to wild-type animals), but not dilute primaquine (B) (p≥0.05). The percentage of animals responding is shown. The ASH sensory neurons also detect the volatile odorant octanol, the heavy metal copper and the detergent SDS. (C) egl-4(lof) mutant animals are moderately hypersensitive to dilute octanol (p<0.02). Time to respond is shown. (D–E) egl-4(lof) animals respond similarly to wild-type animals to both copper and SDS, across a range of concentrations (p>0.1 for each concentration, except p = 0.03 for 1 mM copper). The percentage of animals responding is shown. n>40 for each. All tastants were dissolved in M13 buffer, pH 7.4. Error bars represent the standard error of the mean (SEM). Allele used: egl-4(n479) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function.
In C. elegans, as in mammals, bitter compounds signal through G protein-coupled receptor pathways [7], [10], [11]. The aversive odorant 1-octanol, which like quinine is detected primarily by the ASH sensory neurons [61], [64], also activates G protein-coupled signaling [16], [65]. To determine whether EGL-4 regulates octanol avoidance, animals were assayed for their time to reverse when presented with 100%, 30% or 10% octanol. At each concentration egl-4(lof) animals responded better than wild-type animals (Figure 2C), suggesting that EGL-4 normally dampens this olfactory response.
The ASH sensory neurons also detect soluble stimuli (copper and SDS) that are thought to signal in a G protein-independent manner [4], [6], [8]. To assess whether EGL-4 regulation of sensory signaling extends to these compounds, animals were tested for their avoidance response across a range of concentrations for each. In all cases, the response of egl-4(lof) animals was similar to that of wild-type animals (Figures 2D and 2E). We conclude that EGL-4 regulates response to a subset of G protein-coupled chemosensory responses, including the bitter tastants quinine and amodiaquine and the aversive odorant octanol, but does not regulate ASH sensitivity in general.
EGL-4 Functions in the Cytoplasm of the ASH Sensory Neurons to Regulate Quinine Sensitivity and Calcium Signaling
Upon prolonged exposure to attractive odorants, EGL-4 translocates from the cytoplasm into the nucleus of the AWC olfactory neurons to regulate long-term adaptation in a manner downstream of primary sensory transduction [49]. To determine where within the ASH sensory neurons EGL-4 might function to regulate quinine sensitivity, we used the osm-10 promoter [3] to express a functional GFP−EGL-4 fusion protein [49] and visualized its subcellular localization via GFP fluorescence. As was previously reported for naïve AWC neurons [49], [66], GFP−EGL-4 was distributed throughout ASH in wild-type animals, with expression seen in both the cytoplasm and the nucleus (Figure 3A).
Figure 3. EGL-4 functions in the cytoplasm to regulate calcium signaling.

(A) EGL-4 functions in the cytoplasm to regulate behavioral sensitivity to quinine. The osm-10 promoter [3] was used to express wild-type EGL-4, EGL-4 lacking its endogenous nuclear localization sequence (NLS) or EGL-4 with an additional NLS in the ASH sensory neurons of egl-4(lof) animals. In each case, the EGL-4 was expressed as a fusion with the green fluorescent protein (GFP). Wild-type GFP−EGL-4 was localized throughout the cell and rescued the quinine hypersensitivity of egl-4(lof) animals. GFP−EGL-4(ΔNLS) was restricted to the cytoplasm and rescued the hypersensitivity of egl-4(lof) animals as well as wild-type GFP−EGL-4 (p>0.05). NLS−GFP−EGL-4 was sequestered to the nucleus and had only a small, but statistically significant, rescuing effect (p<0.001). (B) Stimulus-evoked calcium transients in the ASH neurons are enhanced in egl-4(lof) animals. The sra-6 promoter [61] was used to express the genetically encoded calcium indicator G-CaMP3 [69] in the ASH sensory neurons, kyEx2865 (sra-6p::G-CaMP3;ofm-1p::gfp) [70]. Using a microfluidic device, an adult animal was restrained while quinine was delivered to its nose for 10 seconds (black horizontal bar), and the change in fluorescence intensity was recorded. The average ratio change ± the standard error of the mean (SEM) is indicated on each trace. n = 10 animals for each condition. (C) The averaged maximum evoked calcium change for 10, 1 and 0 mM quinine is shown. egl-4(lof) animals showed an elevated ASH calcium flux upon exposure to 1 mM quinine when compared to wild-type animals (p<0.05) that is rescued by osm-10p::egl-4 expression in ASH (p>0.1 when compared to wild-type animals). Error bars represent the SEM. Alleles used: egl-4(n479) loss-of-function and egl-4(ad450) gain-of-function. WT = the N2 wild-type strain. lof = loss-of-function, gof = gain-of-function. s = seconds.
To ascertain whether the cytoplasmic or nuclear pools of EGL-4 contribute to the regulation of quinine avoidance, we used the osm-10 promoter [3] to express two modified forms of GFP−EGL-4 in egl-4(lof) animals. Deletion of the endogenous nuclear localization sequence, GFP−EGL-4(ΔNLS), lead to cytoplasmic accumulation and had no effect on the ability of EGL-4 to rescue the quinine hypersensitivity of egl-4(lof) animals (Figure 3A). Conversely, egl-4(lof) animals expressing the constitutively nuclear NLS−GFP−EGL-4 remained hypersensitive in their response to 1 mM quinine (Figure 3A). Combined, these results suggest a primarily cytoplasmic role for EGL-4 in the negative regulation of quinine signaling.
Genetically encoded calcium indicators such as G-CaMP [67] and cameleon [68] can be used to monitor the activity of C. elegans neurons. In response to quinine, a transient rise in intracellular calcium levels has been seen in the ASHs of wild-type animals [8], [17], [65]. We imaged quinine-evoked calcium fluxes in wild-type, egl-4(lof) and egl-4(gof) animals expressing G-CaMP3 in ASH (sra-6p::G-CaMP3) [69], [70]. Consistent with their observed behavioral hypersensitivity (Figures 1 and 3A), egl-4(lof) animals displayed quinine-evoked ASH calcium fluxes of greater amplitude in response to 1 mM quinine, while expression of wild-type egl-4 in ASH, using the osm-10 promoter [3], rescued this elevated calcium flux (Figures 3B and 3C). Conversely, egl-4(gof) animals showed a diminished calcium flux in response to 10 mM quinine (Figures 3B and 3C), consistent with their decreased behavioral sensitivity to this concentration (Figure 1). Taken together, we suggest that the elevated calcium fluxes seen in the absence of EGL-4 function underlie the enhanced sensitivity to quinine.
RGS Proteins Are Targets of EGL-4
PKGs have been shown to down-regulate Gαq signaling by directly phosphorylating and activating RGS proteins in mouse vascular smooth muscle cells, Rat1 fibroblast cells, rat diencephalic astrocytes, and rabbit gastric smooth muscle cells [39]–[42]. The C. elegans genome encodes twelve predicted RGS proteins [71]. To determine if the loss of RGS function also results in quinine hypersensitivity, similar to loss of EGL-4, we tested animals lacking each of the eight neuronally expressed RGS proteins for response to 1 mM quinine (Figure 4A). rgs-2(lof) and rgs-3(lof) animals displayed a statistically significant hypersensitive response, with more animals than wild-type responding to the dilute stimulus. Although an RGS-2 reporter construct did not show expression in ASH [72], RGS-3 is known to be expressed in and function in the quinine-detecting ASH sensory neurons [28]. We used the osm-10 promoter [3] to conduct cell-selective knock-down of rgs-2 and rgs-3 in the ASH sensory neurons of wild-type animals and in both cases, knock-down also resulted in hypersensitivity to dilute quinine (Figure 4B). Furthermore, consistent with a role in dampening sensory sensitivity, overexpression of RGS-2 or RGS-3 in the ASHs of wild-type animals decreased quinine response (Figure 4C).
Figure 4. RGS proteins are targets of EGL-4.

(A) Animals lacking each of the 8 neuronally expressed RGS proteins were tested for response to 1 mM quinine. rgs-2(lof) and rgs-3(lof) animals respond better than wild-type animals to dilute (1 mM) quinine (p<0.001). (B) RNAi knock-down of rgs-2 or rgs-3 in the quinine-detecting ASH sensory neurons of otherwise wild-type animals, using the osm-10 promoter [3], resulted in behavioral hypersensitivity to dilute (1 mM) quinine, similar to rgs-2(lof) and rgs-3(lof) animals, respectively (p>0.05 for both transgenes when compared to the respective rgs loss-of-function animals). The srb-6 promoter [61] was also used for ASH knock-down of rgs-3, and similarly resulted in hypersensitivity to 1 mM quinine (data not shown). (C) Wild-type animals overexpressing ectopic rgs-2 or rgs-3 cDNA displayed diminished response to 10 mM quinine (p<0.0001 when compared to wild-type animals). (D) rgs-2(lof);egl-4(lof) and rgs-3(lof);egl-4(lof) double mutant animals responded to dilute (1 mM) quinine similarly to egl-4(lof) animals (p>0.1 for each). (E) egl-4(gof) animals lacking either RGS-2 or RGS-3 function responded to 10 mM quinine similarly to the rgs-2(lof) and rgs-3(lof) animals, respectively (p>0.5) and (F) were hypersensitive to 1 mM quinine (p<0.001 when compared to wild-type animals). (G) rgs-2(lof) and rgs-3(lof) animals are hypersensitive to dilute (1 mM) quinine. ASH expression of wild-type RGS-2 or RGS-3, using the osm-10 promoter [3], rescued quinine hypersensitivity in the respective loss-of-function animals. The predicted PKG phosphorylation target site in each was mutated (ΔP), making RGS-2(S126A) and RGS-3(S154A). rgs-2(lof) animals expressing RGS-2(ΔP) and rgs-3(lof) animals expressing RGS-3(ΔP) remained hypersensitive to dilute quinine (p>0.05 for each). The percentage of animals responding is shown. The combined data of ≥3 independent lines, n≥120 transgenic animals, is shown. Error bars represent the standard error of the mean (SEM). Alleles used: egl-4(n479), rgs-1(nr2017), rgs-2(vs17), rgs-3(vs19), rgs-6(vs62), rgs-10(ok1039), rgs-10/11(vs109), egl-10(md176) and eat-16(tm761) loss-of-function and egl-4(ad450) gain-of-function. WT = the N2 wild-type strain. lof = loss-of-function. gof = gain-of-function.
If EGL-4 functions in the same pathway as RGS-2 and RGS-3 to regulate chemosensory signaling, then egl-4(lof);rgs-2(lof) and egl-4(lof);rgs-3(lof) double mutant animals should display a behavioral phenotype similar to egl-4(lof) animals and the hypersensitivity should not be additive. We found that the double mutant animals' response to dilute (1 mM) quinine was indistinguishable from animals lacking only EGL-4 function (Figure 4D), suggesting that they do function in the same regulatory pathway. To determine whether EGL-4 might function upstream of the RGS proteins, we utilized the egl-4(gof) animals, which display reduced sensitivity to quinine (Figure 1A). If RGS-2 and RGS-3 function downstream of EGL-4, then loss of either in combination with the egl-4(gof) allele should relieve the dampened sensitivity of egl-4(gof) mutants in response to 10 mM quinine. Indeed, egl-4(gof);rgs-2(lof) and egl-4(gof);rgs-3(lof) double mutants both responded to 10 mM quinine similarly to the rgs single mutants (Figure 4E). In addition, the double-mutant animals were hypersensitive to 1 mM quinine, similar to the rgs single mutants (Figure 4F).
The characterized consensus sequence of mammalian PKG, seen in 75% of targets surveyed, consists of (R/K)2–3-X-S*/T* [73]–[76]. A search for putative PKG phosphorylation sites in RGS-2 and RGS-3 using NetPhosK 1.0 server [77] revealed one site in each protein, serine 126 in RGS2 and serine 154 in RGS-3. Both of the predicted target serines were changed to alanine and the mutated constructs, expressed under the control of the osm-10 promoter [3], were compared to the wild-type cDNAs for their ability to rescue quinine hypersensitivity. If EGL-4 phosphorylates RGS-2 and RGS-3 to stimulate their activity, loss of the target phosphorylation site(s) in each RGS protein should preclude activation. This would result in an RGS protein that cannot rescue the hypersensitivity of the animals lacking the corresponding RGS protein. As shown in Figure 4G, ASH expression of wild-type RGS-2 or RGS-3 in the respective loss-of-function animals, using the osm-10 promoter [3], returned quinine sensitivity to the levels of wild-type animals. However, rgs-2(lof) animals expressing RGS-2(S126A) and rgs-3(lof) animals expressing RGS-3(S154A) remained hypersensitive to quinine, suggesting that these putative PKG phosphorylation sites are required for RGS-2 and RGS-3 function. We conclude that EGL-4 functions upstream of RGS-2 and RGS-3, and propose that phosphorylation by EGL-4 may be required for the function of these regulatory proteins in ASH.
The Guanylyl Cyclases ODR-1, GCY-27, GCY-33 and GCY-34 Act Upstream of EGL-4
A single point mutation (T276A) within the cGMP-binding domain of EGL-4 abolished its function in AWC-mediated adaptation [49]. Although no guanylyl cyclase (GCY) has been reported to be expressed in ASH, we reasoned that because EGL-4 is a PKG, cGMP binding is likely required for its function in the regulation of quinine avoidance. Although ASH expression of wild-type EGL-4 significantly rescued the egl-4(lof) quinine hypersensitivity, egl-4(lof) animals expressing EGL-4(T276A) remained hypersensitive to dilute quinine (Figure 5A). This suggests that cGMP binding is required for EGL-4 function in ASH-mediated avoidance behaviors. It also suggests that one or more GCYs may provide cGMP to regulate ASH function.
Figure 5. Guanylyl cyclases act upstream of EGL-4.

(A) The osm-10 promoter [3] was used to express wild-type EGL-4 or the cGMP binding mutant EGL-4(T276A) in the ASH sensory neurons of egl-4(lof) animals. While wild-type EGL-4 significantly rescued the egl-4(lof) quinine hypersensitivity (p<0.001), egl-4(lof) animals expressing EGL-4(T276A) remained hypersensitive. (B) Loss-of-function mutations in the guanylyl cyclase genes odr-1, gcy-27, gcy-33 and gcy-34 resulted in behavioral hypersensitivity to dilute (1 mM) quinine (p<0.01 for each). egl-4(gof) animals lacking ODR-1, GCY-27, GCY-33 or GCY-34 function showed diminished sensitivity to both (C) 10 mM quinine and (D) 1 mM quinine, similar to egl-4(gof) single mutant animals (p>0.1 for each double mutant when compared to egl-4(gof) animals). The percentage of animals responding is shown. (E) The osm-10 promoter [3] (ASH, ASI, PHA and PHB), srb-6 promoter [62] (ASH, ADL, ADF, PHA and PHB), and srbc-66 [62] (ASK) promoters were used in cell-selective rescue experiments. The quinine hypersensitivity of odr-1(lof) animals was rescued by srb-6p::odr-1 expression (p<0.001), but not osm-10p::odr-1 expression (p>0.5). gcy-27(lof) hypersensitivity was rescued by all three promoters (p<0.001 for each). Neither gcy-33(lof) nor gcy-34(lof) hypersensitivity was rescued using the osm-10 or srb-6 promoters (p>0.05 for each). The combined data of ≥3 independent lines, n≥120 transgenic animals, is shown. Error bars represent the standard error of the mean (SEM). Alleles used: egl-4(n479), odr-1(n1936), gcy-27(ok3653), gcy-33(ok232) and gcy-34(ok2953) loss-of-function and egl-4(ad450) gain-of-function. WT = the N2 wild-type strain. lof = loss-of-function. gof = gain-of-function.
We assayed animals with loss-of-function alleles for 19 of the 34 GCYs encoded by the C. elegans genome [59] for response to dilute (1 mM) quinine (data not shown). Four GCY mutants, odr-1, gcy-27, gcy-33 and gcy-34, responded better than wild-type animals (Figure 5B). To determine whether the guanylyl cyclases ODR-1, GCY-27, GCY-33 and GCY-34 function upstream of EGL-4, we again utilized the egl-4(gof) animals, which display reduced sensitivity to 10 mM quinine (Figure 1A). A loss-of-function allele of each of the 4 GCYs was combined with the egl-4(gof) mutation and double mutant animals were assayed for quinine avoidance. Each double mutant remained less sensitive to both 10 mM and 1 mM quinine, similar to the egl-4(gof) single mutant animals (Figures 5C and 5D), supporting a role for ODR-1, GCY-27, GCY-33 and GCY-34 upstream of EGL-4 in the regulation of quinine sensitivity.
GCY expression has not previously been reported in ASH, consistent with our own analysis of odr-1, gcy-27, gcy-33 and gcy-34 GFP reporter constructs (data not shown). However, gcy-27 is expressed in the ASK head sensory neurons [59] that also contribute to quinine response [7]. To determine whether expression of any of these GCYs in ASH was sufficient to rescue the quinine hypersensitivity of the respective loss-of-function animals, we used the osm-10 [3] and srb-6 [61] promoters in cell-selective rescue experiments, as their expression overlaps in ASH. The hypersensitivity of odr-1(lof) animals was rescued by srb-6p::odr-1, but not osm-10p::odr-1 expression, and expression using neither promoter was sufficient to rescue gcy-33(lof) or gcy-34(lof) hypersensitivity (Figure 5E). Interestingly, the hypersensitivity of gcy-27(lof) animals was rescued by both osm-10p::gcy-27 and srb-6p::gcy-27, as well as by ASK-selective expression using the srbc-66 promoter [62] (Figure 5E). However, as these constructs contain gcy-27 genomic sequence, we cannot rule out the possibility that regulatory information within the introns could direct expression in addition cells beyond those predicted by the cell-selective promoters used. Taken together, and consistent with the reported expression patterns, these results suggest that the primary site of GCY function is in cells other than the ASHs, and that the cyclases may function in a non-cell-autonomous manner to provide cGMP to regulate EGL-4 function in ASH.
Discussion
EGL-4 Functions as a Negative Regulator of Quinine Sensitivity
PKGs regulate the physiological responses of a variety of cell types, and have a wide range of known and predicted protein targets [35], [78]. As the C. elegans genome encodes only two PKGs, EGL-4 (also known as PKG-1) and PKG-2, it provides an excellent model environment in which to further elucidate the unique physiological roles of PKGs in different cellular contexts. In the AWC olfactory neurons, EGL-4 has been shown to regulate both short-term and long-term adaption. At the cellular level, adaptation is thought to serve as a protective measure against prolonged stimulation, as in the case of photoreceptor signaling for long-term light adaptation [79]. In C. elegans, short-term olfactory adaptation (<30 minutes) diminishes odorant sensitivity for up to 60 minutes and utilizes cytoplasmic EGL-4, whereas long-term adaptation to prolonged odor exposure (>80 minutes) requires the translocation of EGL-4 to the nucleus, where it can alter gene expression and decrease sensitivity to attractive scents for the lifetime of the animal (until the animal is refed) [48], [49], [66]. The cellular localization of EGL-4 depends on the levels of cGMP, as increased cGMP resulting from the loss of phosphodiesterases blocks odor-induced nuclear accumulation of EGL-4 [57], and loss of ODR-1 function leads to constitutively nuclear EGL-4 [57]. In addition, ectopic expression of constitutively nuclear EGL-4 mimicked the adapted state and decreased sensitivity to AWC-detected attractive odors [49]. However, a majority of mammalian PKG substrates are signaling proteins, suggesting an important role for PKGs in cytoplasmic cellular responses that may be closer to the initial stimulus signaling event [35], [78].
Similar to PKGI and PKGII in mammalian systems, whose targets include components of G protein-coupled signaling [39]–[42], the c-Jun N-terminal kinases (JNK) pathway [80] and the anti-apoptotic pathway that includes Bad and Akt [81], EGL-4 appears to function in or regulate multiple signal transduction pathways in C. elegans. For example, EGL-4 likely functions upstream of the TGF-β pathway SMAD transcription factors DAF-3 and DAF-5, as mutations in DAF-3 and DAF-5 suppress several egl-4 mutant phenotypes, including chemosensory, dauer formation, egg laying and body size defects [47]. In addition, EGL-4 has been implicated in C. elegans Notch signaling in sensory neurons, acting directly or indirectly downstream of Notch receptor activation. Animals lacking the DOS-motif Notch co-ligand OSM-11 do not effectively avoid 100% octanol, and this defect is partially suppressed by the gain-of-function egl-4(ad450) allele [82]. We now provide the first direct evidence for EGL-4 function in the ASH nociceptive neurons, and show that it acts to negatively regulate G protein-coupled signal transduction and dampen behavioral sensitivity to the bitter tastant quinine.
We found that egl-4(lof) animals respond better than wild-type animals to 10 mM quinine and also avoid dilute levels of quinine (1 mM) that wild-type animals do not respond to (Figure 1A). EGL-4 function in the two bilaterally symmetric ASHs was both necessary and sufficient to regulate quinine sensitivity (Figures 1B and 1C). Given the short time period that animals are given to initiate the avoidance response (four seconds), we reasoned that EGL-4's primary function in modulating quinine sensitivity is likely the regulation of signal transduction; nuclear translocation and transcriptional changes are unlikely to occur on this timescale. Indeed, in contrast to the necessary nuclear localization of EGL-4 to mediate long-term odor adaptation in AWC [49], cytoplasmic EGL-4 expression in the ASHs was sufficient to rescue the quinine hypersensitivity of egl-4(lof) animals (Figure 3A).
Calcium influx is essential for neuronal function and increased intracellular calcium levels trigger exocytosis of neurotransmitter-containing synaptic vesicles [83]. Consistent with their observed enhanced behavioral sensitivity, upon exposure to quinine egl-4(lof) animals exhibited an elevated ASH calcium flux, when compared to wild-type animals (Figures 3B and 3C). Combined, our data suggest that EGL-4 normally dampens ASH signaling. Interestingly, this is in contrast to the role of mouse PKGI in centrally located nociceptors in mice. In these cells, PKGI phosphorylation of IP3R(Ser1755) potentiates IP3R-mediated calcium release from internal stores [84]–[86]. This promotes stimulus-induced synaptic transmission between nociceptors and spinal-periaqueductal grey projection neurons, ultimately leading to the withdrawal reflex (to applied pressure, intrathecally administered NMDA or thermal stimuli) and development of hyperalgesia [84]–[88]. Thus, PKGI promotes an aversive behavioral response in mice, while EGL-4 dampens behavioral sensitivity to a nociceptive stimulus in C. elegans. This difference highlights the diversity of mechanisms by which PKGs can regulate signaling in different cellular contexts.
EGL-4 Negatively Regulates Quinine Sensitivity through RGS-2 and RGS-3
As the C. elegans response to quinine utilizes the Gα subunits ODR-3 and GPA-3 [7], [8], our results suggest a role for EGL-4 in the negative regulation of a G protein-coupled signal transduction pathway. We found that when C. elegans rgs-2(lof) and rgs-3(lof) animals were assayed 30 minutes after being removed from their bacterial food source, they also displayed a significant hypersensitivity phenotype (Figure 4), and RGS-2 and RGS-3 function in ASH was both necessary and sufficient to regulate quinine sensitivity. When the predicted PKG phosphorylation sites in RGS-2 and RGS-3 were changed to alanines, the mutated constructs failed to rescue quinine hypersensitivity. These findings support previous studies using mammalian smooth muscle, astrocyte and fibroblast cells [39]–[42], wherein PKGs can function as activators of RGS proteins, and provide the first evidence of this mechanism in nociceptive sensory neurons. It is also interesting to speculate that EGL-4 may target RGS proteins to regulate additional physiological processes, such as C. elegans egg laying [47], [72], [89]–[92]. We examined the amino acid sequences of all twelve C. elegans RGS proteins and identified predicted PKG phosphorylation sites in RGS-1, RGS-6, RGS-7, EGL-10 and EAT-16.
The sensitivity of C. elegans to environmental stimuli is dramatically and dynamically regulated by an animal's nutritional status, which influences signaling levels of biogenic amines such as serotonin and dopamine [14], [93]. In particular, the responses of wild-type animals to nociceptive stimuli diminish upon food deprivation [28], [64], [94]–[96]. rgs-3(lof) animals are defective in their avoidance of 10 mM quinine when assayed just 10 minutes after removal from food (Figure S1) [28]. This defective response is the result of elevated signaling in the ASHs in the absence of the negative regulator, which ultimately leads to decreased synaptic transmission [28]. We see rgs-3(lof) animals responding better than wild-type animals to quinine (10 mM and 1 mM) when they are assayed 30 minutes after removal from food (Figure 4). Similarly, rgs-2(lof) animals are defective in response to 10 mM quinine when assayed after only the short (10 minute) period of starvation (Figure S1), but are hypersensitive when assayed after being off food for 30 minutes (Figure 4). Interestingly, it is not until 45 minutes off food that rgs-2(lof);rgs-3(lof) double mutant animals reach the level of hypersensitivity seen in the single mutants at 30 minutes off food (Figure S1). It is possible that at the intermediate time off food (30 minutes) there is still “too much signaling” in the absence of both RGS-2 and RGS-3 to allow response to 1 mM quinine, similar to the elevated signaling that blocks the response of rgs-3(lof) single mutant animals to 10 mM quinine at 10 minutes off food [28]. The longer (45 minute) period off food may allow the increased signaling due to loss of both RGSs to attenuate over time, bringing it into a range that allows for the sensitized behavioral response. Because the time-course for quinine sensitivity differs between egl-4(lof) animals and rgs-2(lof);rgs-3(lof) animals, EGL-4 may have additional targets within the ASH sensory neurons. Alternatively, RGS-2 and RGS-3 may retain residual function in egl-4(lof) animals, such that loss of EGL-4 function does not affect cellular signaling as adversely as loss of the RGS proteins themselves.
Modulation of Quinine Sensitivity Upstream of EGL-4
PKGs require cGMP for their activation, and the levels of cGMP within a cell are modulated by production by guanlyl cyclases and breakdown by phosphodiesterases. Guanylyl cyclases are widely expressed in mammalian tissues and exist in two forms: soluble and transmembrane [97]. Mammalian soluble GCYs have been well studied in smooth muscle cells, where they are activated by nitric oxide (NO) to produce cGMP [98], [99]. Transmembrane GCYs have been well characterized for their role in natriuresis and phototransduction [100], [101]. While 34 GCYs are encoded by the C. elegans genome, the physiological roles of most are unknown. Our analysis revealed that EGL-4 requires cGMP binding in order to negatively regulate quinine sensitivity (Figure 5A), suggesting that a pool of cGMP is available to activate EGL-4 in the ASH nociceptive neurons. Moreover, animals lacking the function of the transmembrane guanylyl cyclases ODR-1 and GCY-27, or the soluble guanylyl cyclases GCY-33 and GCY-34, are hypersensitive in their response to dilute quinine (Figure 5B). However, these GCYs do not appear to function directly in the ASHs (Figure 6), suggesting that other neurons in the circuit may provide the cGMP that regulates ASH function, perhaps via GAP junctions between cGMP-generating neurons and ASH. For example, gcy-27 is expressed in ASK [59], which forms GAP junctions directly with ASH. Continued studies in C. elegans may yield new insights into nociceptive signaling in mammalian systems, where PKGI is known to function in central nociceptors [84]–[86].
Figure 6. Model for EGL-4 regulation of nociceptive signaling.
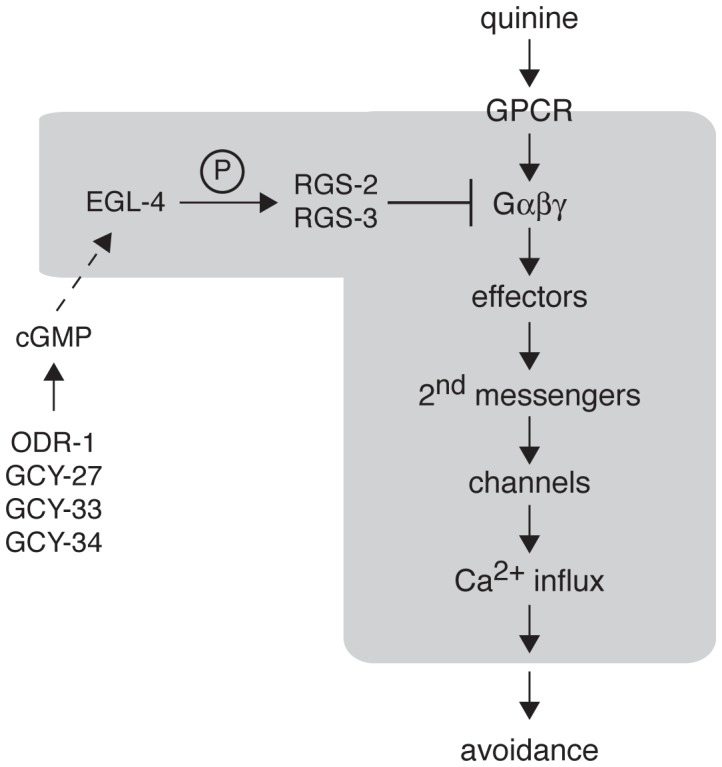
The cGMP-dependent protein kinase EGL-4 regulates C. elegans behavioral sensitivity to the bitter tastants quinine and amodiaquine and the volatile odorant octanol. Wild-type chemosensory signaling is initiated in the ASH sensory neurons when a ligand (such as quinine) binds to a GPCR to activate the associated heterotrimeric G proteins. The activated G proteins (Gα-GTP and Gβγ) interact with downstream effectors to generate second messengers that can activate channels in the plasma membrane, allowing Ca2+ influx. Through connections with downstream interneurons and motor neurons, ASH activation is ultimately translated into behavioral avoidance (backward locomotion). Signaling is terminated in part by regulator of G protein signaling (RGS) proteins, which promote the hydrolysis of GTP to GDP by the Gα subunit. EGL-4 phosphorylation of RGS-2 and RGS-3 stimulates their activity. The guanylyl cyclases ODR-1, GCY-27, GCY-33 and GCY-34 may function in alternate neurons to provide the cGMP that is required for EGL-4 function in ASH. In the absence of EGL-4 function, RGS-2 and RGS-3 do not efficiently downregulate Gα signaling, leading to increased Ca2+ levels in response to receptor activation. This increased signaling in the ASH sensory neurons leads behavioral hypersensitivity to weak stimuli. The molecular events believed to be happening within the ASHs themselves are included within the grayed area.
Materials and Methods
C. elegans Culture
Strains were maintained under standard conditions on NGM agar plates seeded with OP50 E. coli bacteria [102].
Strains
Strains used in this study include: N2 Bristol wild-type, MT1074 egl-4(n479), DA521 egl-4(ad450), CX10979 kyEx2865 (sra-6p::G-CaMP3;ofm-1p::gfp), FG414 (kyEx2865 sra-6p::G-CaMP3;ofm-1p::gfp), FG417 egl-4(n479);kyEx2865 (sra-6p::G-CaMP3;ofm-1p::gfp), FG413 egl-4(ad450);kyEx2865 (sra-6p::G-CaMP3;ofm-1p::gfp), FG454 egl-4(n479);udEx208 (osm-10p::egl-4;myo-3p::mCherry);kyEx2865 (sra-6p::G-CaMP3;ofm-1p::gfp), LX147 rgs-1(nr2017), LX160 rgs-2(vs17), LX242 rgs-3(vs19), LX533 rgs-6(vs62), FG105 rgs-10(ok1039), FG108 rgs-10/11(vs109), MT8504 egl-10(md176), LX1226 eat-16(tm761), FG329 egl-4(n479);rgs-2(vs17), FG330 rgs-3(vs19);egl-4(n479), FG275 egl-4(ad450);rgs-2(vs17), FG276 rgs-3(vs19);egl-4(ad450), FG269 rgs-3(vs19);rgs-2(vs17), FG376 rgs-3(vs19);egl-4(n479);rgs-2(vs17), VC2796 gcy-3(gk1154), RB1010 gcy-5(ok930), IK800 gcy-8(oy44), VC2675 gcy-15(gk1102), VC2450 gcy-17(gk1155), VC2321 gcy-18(ok3047);nT1[q1351], RB1909 gcy-19(ok2472), RB1935 gcy-20(ok2538), RB924 gcy-23(ok797), VC2375 gcy-25(gk1187), CZ3714 gcy-31(ok296), RB1048 gcy-32(ok995), AX1295 gcy-35(ok769), RB626 gcy-37(ok384), IK597 gcy-23(nj37);gcy-8(oy44);gcy-18(nj38), FG290 odr-1(n1936), FG280 gcy-27(ok3653), FG278 gcy-33(ok232), FG279 gcy-34(ok2953), FG294 egl-4(ad450);odr-1(n1936), FG288 gcy-27(ok3653);egl-4(ad450), FG289 egl-4(ad450);gcy-33(ok232) and FG285 egl-4(ad450);gcy-34(ok2953).
Transgenic Strains
Germline transformations were performed as previously described [103]. For egl-4, rgs-2, rgs-3, odr-1, gcy-27, gcy-33, and gcy-34 rescue experiments, 50 ng/µl of pJM67 elt-2::gfp plasmid [104] was used as the co-injection marker, along with 50 ng/µl of the rescuing plasmid. For rgs-2 and rgs-3 overexpression, 250 ng/µl of the rgs plasmid was co-injected with 50 ng/µl of pJM67 elt-2::gfp plasmid [104]. For egl-4 rescue with G-CaMP3 expression, 5 ng/µl of myo-3p::mCherry plasmid (Yamamoto Lab) was used as the co-injection marker, along with 50 ng/µl of the osm-10p::egl-4 rescuing plasmid. Animals expressing the egl-4 rescuing array (udEx208) were then crossed with kyEx2865-expressing (sra-6p::G-CaMP3;ofm-1p::gfp) animals, and egl-4(n479) animals co-expressing both arrays were isolated. Cell-specific RNAi knock-down experiments were performed as previously described [63]. 25 ng/µl of pJM67 elt-2::gfp plasmid [104] was co-injected with 50 ng/µl of each PCR fusion product [63].
Plasmid Construction
pFG1
The ∼900 bp osm-10 promoter was isolated from CR142 [105] using PstI/BamHI and inserted into these sites of Fire vector pPD49.26 (Fire Lab C. elegans Vector Kit, Addgene).
pFG50
The ∼2.1 kb srbc-66 promoter was isolated from srbc-66::gfp [62] using HindIII/BamHI and inserted into these sites of Fire vector pPD49.26 (Fire Lab C. elegans Vector Kit, Addgene).
pFG34 osm-10p::egl-4
The egl-4 cDNA (isoform 2a.1) was PCR amplified from odr-3::egl-4 [48], incorporating a 5′ XbaI site and a 3′ NcoI, and subcloned into the NheI and NcoI sites of pFG1.
pFG99 srb-6p::egl-4
The ∼1.3 kb srb-6 promoter was isolated from pFG10 [106] using SphI/BamHI and inserted into these sites of pFG34 (replacing the osm-10 promoter).
pFG53 srbc-66p::egl-4
The egl-4 cDNA (isoform 2a.1) was PCR amplified from odr-3::egl-4 [48], incorporating a 5′ XbaI site and a 3′ NcoI, and subcloned into the NheI and NcoI sites of pFG50.
pFG97 osm-10p::rgs-2
The rgs-2 cDNA was PCR amplified from pMD6 [72], incorporating a 5′ SalI site and a 3′ NcoI site, and subcloned into these sites of pFG1.
pHA446 osm-10p::rgs-3
cDNA encoding the “A” isoform of RGS-3, including the initiator methionine, was amplified from yk252h2 (kind gift of Y. Kohara), incorporating a 5′ NheI and a 3′ NcoI site. This fragment was inserted into the Fire vector pPD49.26 (Fire Lab C. elegans Vector Kit, Addgene) to create pHA443. The ∼900 bp osm-10 promoter was isolated from CR142 [105] using Pst-1/Xma-1 and inserted into these sites of pHA443.
pFG98 osm-10p::rgs-2(S126A)
Site-directed mutagenesis (QuikChange, Stratagene) was used to incorporate the S126A change into pMD6 [72]. The rgs-2(S126A) cDNA was then PCR amplified, incorporating a 5′ SalI site and a 3′ NcoI site, and subcloned into these sites of pFG1.
pFG93 osm-10p::rgs-3(S154A)
Site-directed mutagenesis (QuikChange, Stratagene) was used to incorporate the S154A change into pHA443 (A isoform of RGS-3, above). The rgs-3(S154A) cDNA was then PCR amplified, incorporating a 5′ NheI site and a 3′ NcoI site, and subcloned into these sites of pFG1.
pFG80 osm-10p::gfp::egl-4
SphI/NgoMIV digest was used to remove 2496 bp of the 2678 bp odr-3 promoter from odr-3::gfp::egl-4 [49]. The osm-10 promoter was isolated from pFG1 using SphI/XmaI and inserted into these sites upstream of gfp::egl-4.
pFG111 osm-10p::gfp::egl-4(ΔNLS)
SphI/NgoMIV digest was used to remove 2496 bp of the 2678 bp odr-3 promoter from odr-3::gfp::egl-4(ΔNLS) [49]. The osm-10 promoter was isolated from pFG1 using SphI/XmaI and inserted into these sites upstream of gfp::egl-4(ΔNLS).
pFG82 osm-10p::NLS::gfp::egl-4
SphI/NgoMIV digest was used to remove 2496 bp of the 2678 bp odr-3 promoter from odr-3::NLS::gfp::egl-4 [49]. The osm-10 promoter was isolated from pFG1 using SphI/XmaI and inserted into these sites upstream of NLS::gfp::egl-4.
pFG113 osm-10p::gfp::egl-4(T276A)
SphI/NgoMIV digest was used to remove 2496 bp of the 2678 bp odr-3 promoter from odr-3::gfp::egl-4(T276A) [49]. The osm-10 promoter was isolated from pFG1 using SphI/XmaI and inserted into these sites upstream of gfp::egl-4(T276A).
pFG133 osm-10p::gcy-33
cDNA encoding the “A” isoform of GCY-33, including the initiator methionine, was isolated from gcy-31p::gcy-33 [107] using NheI/Asp718I and subcloned into these sites of pFG1.
pFG134 srb-6p::gcy-33
cDNA encoding the “A” isoform of GCY-33, including the initiator methionine, was isolated from gcy-31p::gcy-33 [107] using NheI/Asp718I and subcloned into these sites of pFG10 [106].
pFG138 osm-10p::gcy-34
Genomic gcy-34 sequence was PCR amplified from N2 lysate, incorporating a 5′ NheI site and a 3′ Asp718I site, and subcloned into these sites of pFG1.
pFG139 srb-6p::gcy-34
The gcy-34 genomic sequence was isolated from pFG138 using NheI/Asp718I and subcloned into these sites of pFG10 [106].
KC3 odr-1p::odr-1b::gfp
R01E6.1SC [108] contains the odr-1 genomic region from 2.6 kb upstream of the start site for odr-1b to 79 bp of the last exon (22nd) of odr-1b. This construct is missing the last 46 nucleotides of the 22nd exon of odr-1b and the 3′ UTR, as predicted by PACdb ID: R01E6.3_216. We added back the last 46 nt and all of the 3′ UTR (449 nt) by first amplifying the 3′ end of odr-1b and all of the 3′ UTR from N2 genomic DNA, adding a KpnI site on the 3′ end of the amplicon. This was Topo cloned and cut from the Topo cloning vector with KpnI. This KpnI fragment was inserted into the Kpn1 site of R01E6.1SC [108] to yield KC1. In order to tag ODR-1b with GFP, site-directed mutagenesis was used to add adjacent AgeI and NruI sites at the very last codon of odr-1b (before stop) so that gfp could be inserted in frame. This construct was called KC2. Finally, odr-1p::odr-1b::gfp (KC3) was generated by cutting KC2 with AgeI/NruI and inserting gfp (Fire vector pPD95.77 cut with EcoRI, then blunted, then cut with AgeI).
pFG140 osm-10p::odr-1
Genomic sequence encoding ODR-1 was PCR amplified from KC3 (above), incorporating a 5′ NheI site and a 3′ NcoI site, and subcloned into these sites of pFG1.
pFG141 srb-6p::odr-1
Genomic odr-1 sequence was isolated from pFG140 using NheI/NcoI and subcloned into these sites of pFG10 [106].
pFG142 osm-10p::gcy-27
Genomic gcy-27 sequence was PCR amplified from N2 lysate, incorporating a 5′ NheI site and a 3′ BsrGI site, and subcloned into the NheI/Asp718 sites of pFG1.
pFG143 srb-6p::gcy-27
The gcy-27 genomic sequence was isolated from pFG142 using NheI/ApaI and subcloned into these sites of pFG10 [106].
pFG144 srbc-66p::gcy-27
The gcy-27 genomic sequence was isolated from pFG142 using NheI/ApaI and subcloned into these sites of pFG50.
All constructs were verified by sequencing when appropriate.
Calcium Imaging
Neuronal calcium changes were recorded using the GFP-based fluorescent calcium reporter G-CaMP3 [69], [70] and based on described methods [109], [110]. Briefly, a microfluidic device similar to that used by [110] was fabricated by the Stanford Microfluidics Foundry and used to immobilize a kyEx2865 (sra-6p::G-CaMP3;ofm-1p::gfp)-expressing worm for imaging while streams of buffer or quinine (or buffer for controls) under laminar flow were alternatively presented to the nose of the worm. To reduce the influence of neuronal activation by blue light, we pre-exposed the worm to blue light for 5–8 seconds. The worm was imaged one minute later using a 40X air objective on an inverted Axiovert 200 microscope (Zeiss, Oberkochen, Germany). Images were captured with an exposure time of 20 milliseconds every 500 milliseconds with an ORCA-Flash 2.8 camera (Hamamatsu, Shizuoka Pref., Japan) and recorded over 15 seconds with µManager software [111]. The movies were analyzed using ImageJ (Rasband, W.S., ImageJ, US NIH, Bethesda, MD, USA). The mean signal intensities of the region of interest (ROI) and background were determined and the background corrected value taken for further analysis. The percent change in fluorescence intensity based on the average intensity of the first 3 frames (delta F/F0) was calculated. The ROI was centered on the cell body of the ASH neuron. After 5 seconds of imaging with M13 buffer (pH 7.4), the quinine stimulus was given for the last 10 seconds of imaging. Quinine was dissolved as a 1 or 10 mM solution in M13 and the pH was adjusted to 7.4 after dissolving. Ten worms for each condition were tested and averaged. A one-tailed unpaired Student's t-Test for each point in all conditions was performed. The maximum change in fluorescence before and after exposure to quinine was determined and used for statistics.
Behavioral Assays
Well-fed young adults were used for analysis, and all behavioral assays were performed on at least three separate days, in parallel with controls. The number of transgenic animals assayed in each experiment is indicated within the figure legends, and in all cases n≥58 for non-transgenic animals. Response to the soluble tastant quinine was scored as the percentage of animals that initiated backward locomotion within 4 seconds of encountering a quinine drop placed on the agar plate in front of a forward moving animal [6], [7], [65]. Quinine was dissolved in M13, pH 7.4 [112]. For quinine avoidance assays, animals were tested 30 minutes after transfer to NGM plates lacking bacteria (“off food”). Response to octanol was scored as the amount of time it took an animal to initiate backward locomotion when presented with a hair dipped in octanol [3], [61]. For octanol avoidance assays, animals were tested 10–20 minutes after transfer to NGM plates lacking bacteria and assays were stopped at 20 seconds. All data is presented as ± standard error of the mean (SEM). The Student's two-tailed t-Test was used for statistical analysis, except for panels 4A and 5B, in which the one-way Anova with Tukey's Honestly Significant Difference (HSD) statistical analysis was used.
Supporting Information
Length of starvation alters C. elegans sensitivity to quinine. Animals lacking EGL-4, RGS-2, RGS-3 or RGS-2 and RGS-3 function were tested for response to both (A) 10 mM quinine and (B) 1 mM quinine when starved for increasing lengths of time. (A) rgs-2(lof) and rgs-3(lof) animals responded to 10 mM similarly to wild-type animals when assayed 30 minutes after removal from their bacterial food source (p>0.5 and p>0.1, respectively). rgs-2(lof);rgs-3(lof) double mutant animals did not respond similarly to wild-type animals until 60 minutes after removal from food (p>0.5). (B) The behavioral sensitivity of rgs-2(lof), rgs-3(lof) and rgs-2(lof);rgs-3(lof) animals to dilute quinine increased with longer periods off food. The percentage of animals responding is shown. The combined data of n≥40 animals is shown. Error bars represent the standard error of the mean (SEM). Alleles used: egl-4(n479), rgs-2(vs17), and rgs-3(vs19) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function, min = minutes.
(PDF)
Acknowledgments
We thank Cori Bargmann and Doug Portman for valuable discussions and are grateful to Cori Bargmann, Anne Hart, Michael Koelle and Piali Sengupta for reagents.
Funding Statement
This work was supported by the National Science Foundation (MCB-0917896, DMF). Some strains used in this study were obtained from the Caenorhabditis Genetics Center, which is funded in part by the National Institutes of Health - National Center for Research Resources. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bargmann CI, Thomas JH, Horvitz HR (1990) Chemosensory cell function in the behavior and development of Caenorhabditis elegans . Cold Spring Harb Symp Quant Biol 55: 529–538. [DOI] [PubMed] [Google Scholar]
- 2. Kaplan JM, Horvitz HR (1993) A dual mechanosensory and chemosensory neuron in Caenorhabditis elegans . Proc Natl Acad Sci U S A 90: 2227–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hart AC, Kass J, Shapiro JE, Kaplan JM (1999) Distinct signaling pathways mediate touch and osmosensory responses in a polymodal sensory neuron. J Neurosci 19: 1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sambongi Y, Nagae T, Liu Y, Yoshimizu T, Takeda K, et al. (1999) Sensing of cadmium and copper ions by externally exposed ADL, ASE, and ASH neurons elicits avoidance response in Caenorhabditis elegans . Neuroreport 10: 753–757. [DOI] [PubMed] [Google Scholar]
- 5. Troemel ER (1999) Chemosensory signaling in C. elegans . Bioessays 21: 1011–1020. [DOI] [PubMed] [Google Scholar]
- 6. Hilliard MA, Bargmann CI, Bazzicalupo P (2002) C. elegans responds to chemical repellents by integrating sensory inputs from the head and the tail. Curr Biol 12: 730–734. [DOI] [PubMed] [Google Scholar]
- 7. Hilliard MA, Bergamasco C, Arbucci S, Plasterk RH, Bazzicalupo P (2004) Worms taste bitter: ASH neurons, QUI-1, GPA-3 and ODR-3 mediate quinine avoidance in Caenorhabditis elegans . EMBO J 23: 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hilliard MA, Apicella AJ, Kerr R, Suzuki H, Bazzicalupo P, et al. (2005) In vivo imaging of C. elegans ASH neurons: cellular response and adaptation to chemical repellents. EMBO J 24: 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chatzigeorgiou M, Bang S, Hwang SW, Schafer WR (2013) tmc-1 encodes a sodium-sensitive channel required for salt chemosensation in C. elegans . Nature 494: 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chandrashekar J, Hoon MA, Ryba NJ, Zuker CS (2006) The receptors and cells for mammalian taste. Nature 444: 288–294. [DOI] [PubMed] [Google Scholar]
- 11. Palmer RK (2007) The pharmacology and signaling of bitter, sweet, and umami taste sensing. Mol Interv 7: 87–98. [DOI] [PubMed] [Google Scholar]
- 12. Dryer L, Berghard A (1999) Odorant receptors: a plethora of G-protein-coupled receptors. Trends Pharmacol Sci 20: 413–417. [DOI] [PubMed] [Google Scholar]
- 13. McCudden CR, Hains MD, Kimple RJ, Siderovski DP, Willard FS (2005) G-protein signaling: back to the future. Cell Mol Life Sci 62: 551–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bargmann CI (2006) Chemosensation in C. elegans. In: Community TCeR, editor. WormBook: WormBook. [DOI] [PMC free article] [PubMed]
- 15. Aoki R, Yagami T, Sasakura H, Ogura K, Kajihara Y, et al. (2011) A seven-transmembrane receptor that mediates avoidance response to dihydrocaffeic acid, a water-soluble repellent in Caenorhabditis elegans . J Neurosci 31: 16603–16610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roayaie K, Crump JG, Sagasti A, Bargmann CI (1998) The Gα protein ODR-3 mediates olfactory and nociceptive function and controls cilium morphogenesis in C. elegans olfactory neurons. Neuron 20: 55–67. [DOI] [PubMed] [Google Scholar]
- 17. Esposito G, Amoroso MR, Bergamasco C, Di Schiavi E, Bazzicalupo P (2010) The G protein regulators EGL-10 and EAT-16, the Giα GOA-1 and the Gqα EGL-30 modulate the response of the C. elegans ASH polymodal nociceptive sensory neurons to repellents. BMC Biol 8: 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jansen G, Thijssen KL, Werner P, van der Horst M, Hazendonk E, et al. (1999) The complete family of genes encoding G proteins of Caenorhabditis elegans . Nat Genet 21: 414–419. [DOI] [PubMed] [Google Scholar]
- 19. Ross EM, Wilkie TM (2000) GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem 69: 795–827. [DOI] [PubMed] [Google Scholar]
- 20. Hollinger S, Hepler JR (2002) Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev 54: 527–559. [DOI] [PubMed] [Google Scholar]
- 21. Chen CK, Burns ME, He W, Wensel TG, Baylor DA, et al. (2000) Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9-1. Nature 403: 557–560. [DOI] [PubMed] [Google Scholar]
- 22. Krispel CM, Chen D, Melling N, Chen YJ, Martemyanov KA, et al. (2006) RGS expression rate-limits recovery of rod photoresponses. Neuron 51: 409–416. [DOI] [PubMed] [Google Scholar]
- 23. von Buchholtz L, Elischer A, Tareilus E, Gouka R, Kaiser C, et al. (2004) RGS21 is a novel regulator of G protein signalling selectively expressed in subpopulations of taste bud cells. European Journal of Neuroscience 19: 1535–1544. [DOI] [PubMed] [Google Scholar]
- 24. Mclaughlin SK, Mckinnon PJ, Margolskee RF (1992) Gustducin is a taste-cell-specific G-protein closely related to the transducins. Nature 357: 563–569. [DOI] [PubMed] [Google Scholar]
- 25. Ming D, Ruiz-Avila L, Margolskee RF (1998) Charcterization and solubilization of bitter-responsive receptors that couple to gustducin. Proc Natl Acad Sci U S A 95: 8933–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wong GT, Gannon KS, Margolskee RF (1996) Transduction of bitter and sweet taste by gustducin. Nature 381: 796–800. [DOI] [PubMed] [Google Scholar]
- 27. Huang L, Shanker YG, Dubauskaite J, Zheng JZ, Yan W, et al. (1999) Gγ13 colocalized with gustducin in taste receptor cells and mediates IP3 responses to bitter denatonium. Nat Neurosci 2: 1055–1062. [DOI] [PubMed] [Google Scholar]
- 28. Ferkey DM, Hyde R, Haspel G, Dionne HM, Hess HA, et al. (2007) C. elegans G protein regulator RGS-3 controls sensitivity to sensory stimuli. Neuron 53: 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hofmann F (2005) The biology of cyclic GMP-dependent protein kinases. J Biol Chem 280: 1–4. [DOI] [PubMed] [Google Scholar]
- 30. Lincoln TM, Dey N, Sellak H (2001) Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: from the regulation of tone to gene expression. J Appl Physiol 91: 1421–1430. [DOI] [PubMed] [Google Scholar]
- 31. Koeppen M, Feil R, Siegl D, Feil S, Hofmann F, et al. (2004) cGMP-dependent protein kinase mediates NO- but not acetylcholine-induced dilations in resistance vessels in vivo. Hypertension 44: 952–955. [DOI] [PubMed] [Google Scholar]
- 32. Pfeifer A, Klatt P, Massberg S, Ny L, Sausbier M, et al. (1998) Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J 17: 3045–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sausbier M, Schubert R, Voigt V, Hirneiss C, Pfeifer A, et al. (2000) Mechanisms of NO/cGMP-dependent vasorelaxation. Circ Res 87: 825–830. [DOI] [PubMed] [Google Scholar]
- 34. Massberg S, Sausbier M, Klatt P, Bauer M, Pfeifer A, et al. (1999) Increased adhesion and aggregation of platelets lacking cyclic guanosine 3′,5′-monophosphate kinase I. J Exp Med 189: 1255–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Francis SH, Busch JL, Corbin JD, Sibley D (2010) cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev 62: 525–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmidt H, Werner M, Heppenstall PA, Henning M, Moré MI, et al. (2002) cGMP-mediated signaling via cGKIα is required for the guidance and connectivity of sensory axons. J Cell Biol 159: 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tegeder I, Del Turco D, Schmidtko A, Sausbier M, Feil R, et al. (2004) Reduced inflammatory hyperalgesia with preservation of acute thermal nociception in mice lacking cGMP-dependent protein kinase I. Proc Natl Acad Sci U S A 101: 3253–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Werner C, Raivich G, Cowen M, Strekalova T, Sillaber I, et al. (2004) Importance of NO/cGMP signalling via cGMP-dependent protein kinase II for controlling emotionality and neurobehavioural effects of alcohol. Eur J Neurosci 20: 3498–3506. [DOI] [PubMed] [Google Scholar]
- 39. Osei-Owusu P, Sun X, Drenan RM, Steinberg TH, Blumer KJ (2007) Regulation of RGS2 and second messenger signaling in vascular smooth muscle cells by cGMP-dependent protein kinase. J Biol Chem 282: 31656–31665. [DOI] [PubMed] [Google Scholar]
- 40. Tang KM, Wang GR, Lu P, Karas RH, Aronovitz M, et al. (2003) Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat Med 9: 1506–1512. [DOI] [PubMed] [Google Scholar]
- 41. Pedram A, Razandi M, Kehrl J, Levin ER (2000) Natriuretic peptides inhibit G protein activation. Mediation through cross-talk betwen cyclic cGMP-dependent protein kinase and regulators of G protein-signaling proteins. Journal of Biological Chemistry 275: 8. [DOI] [PubMed] [Google Scholar]
- 42. Huang J, Zhou H, S M, Sriwai W, Murthy KS (2006) Inhibition of Gαq-dependent PLC-β activity by PKG and PKA is mediated by phosphorylation of RGS4 and GRK2. American Journal of Physiology Cell Physiology 292: 9. [DOI] [PubMed] [Google Scholar]
- 43.Hu PJ (2007) Dauer. In: Community TCeR, editor. WormBook: WormBook.
- 44.Savage-Dunn C (2005) TGF-β signaling. In: Community TCeR, editor. WormBook: WormBook.
- 45. Raizen DM, Cullison KM, Pack AI, Sundaram MV (2006) A novel gain-of-function mutant of the cyclic GMP-dependent protein kinase egl-4 affects multiple physiological processes in Caenorhabditis elegans . Genetics 173: 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stansberry J, Baude EJ, Taylor MK, Chen PJ, Jin SW, et al. (2001) A cGMP-dependent protein kinase is implicated in wild-type motility in C. elegans . Journal of Neurochemistry 76: 1177–1187. [DOI] [PubMed] [Google Scholar]
- 47. Daniels SA, Ailion M, Thomas JH, Sengupta P (2000) egl-4 acts through a transforming growth factor-β/SMAD pathway in Caenorhabditis elegans to regulate multiple neuronal circuits in response to sensory cues. Genetics 156: 123–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. L'Etoile ND, Coburn CM, Eastham J, Kistler A, Gallegos G, et al. (2002) The cyclic GMP-dependent protein kinase EGL-4 regulates olfactory adaptation in C. elegans . Neuron 36: 1079–1089. [DOI] [PubMed] [Google Scholar]
- 49. Lee JI, O'Halloran DM, Eastham-Anderson J, Juang BT, Kaye JA, et al. (2010) Nuclear entry of a cGMP-dependent kinase converts transient into long-lasting olfactory adaptation. Proc Natl Acad Sci U S A 107: 6016–6021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. van der Linden AM, Nolan KM, Sengupta P (2007) KIN-29 SIK regulates chemoreceptor gene expression via an MEF2 transcription factor and a class II HDAC. EMBO J 26: 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van der Linden AM, Wiener S, You YJ, Kim K, Avery L, et al. (2008) The EGL-4 PKG acts with KIN-29 salt-inducible kinase and protein kinase A to regulate chemoreceptor gene expression and sensory behaviors in Caenorhabditis elegans . Genetics 180: 1475–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lanjuin A, Sengupta P (2002) Regulation of chemosensory receptor expression and sensory signaling by the KIN-29 Ser/Thr kinase. Neuron 33: 369–381. [DOI] [PubMed] [Google Scholar]
- 53. Kuo JF (1974) Guanosine 3′:5′-monophosphate-dependent protein kinases in mammalian tissues. Proc Natl Acad Sci U S A 71: 4037–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gill GN, Holdy KE, Walton GM, Kanstein CB (1976) Purification and characterization of 3′:5′-cyclic GMP-dependent protein kinase. Proc Natl Acad Sci U S A 73: 3918–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gudi T, Lohmann SM, Pilz RB (1997) Regulation of gene expression by cyclic GMP-dependent protein kinase requires nuclear translocation of the kinase: identification of a nuclear localization signal. Mol Cell Biol 17: 5244–5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Francis SH, Corbin JD (1994) Structure and function of cyclic nucleotide-dependent protein kinases. Annu Rev Physiol 56: 237–272. [DOI] [PubMed] [Google Scholar]
- 57. O'Halloran DM, Hamilton OS, Lee JI, Gallegos M, L'Etoile ND (2012) Changes in cGMP levels affect the localization of EGL-4 in AWC in Caenorhabditis elegans . PLoS One 7: e31614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hao Y, Xu N, Box AC, Schaefer L, Kannan K, et al. (2011) Nuclear cGMP-dependent kinase regulates gene expression via activity-dependent recruitment of a conserved histone deacetylase complex. PLoS Genet 7: e1002065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ortiz CO, Etchberger JF, Posy SL, Frokjaer-Jensen C, Lockery S, et al. (2006) Searching for neuronal left/right asymmetry: genomewide analysis of nematode receptor-type guanylyl cyclases. Genetics 173: 131–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hirose T, Nakano Y, Nagamatsu Y, Misumi T, Ohta H, et al. (2003) Cyclic GMP-dependent protein kinase EGL-4 controls body size and lifespan in C. elegans . Development 130: 1089–1099. [DOI] [PubMed] [Google Scholar]
- 61. Troemel ER, Chou JH, Dwyer ND, Colbert HA, Bargmann CI (1995) Divergent seven transmembrane receptors are candidate chemosensory receptors in C. elegans . Cell 83: 207–218. [DOI] [PubMed] [Google Scholar]
- 62. Kim K, Sato K, Shibuya M, Zeiger DM, Butcher RA, et al. (2009) Two chemoreceptors mediate developmental effects of dauer pheromone in C. elegans . Science 326: 994–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Esposito G, Di Schiavi E, Bergamasco C, Bazzicalupo P (2007) Efficient and cell specific knock-down of gene function in targeted C. elegans neurons. Gene 395: 170–176. [DOI] [PubMed] [Google Scholar]
- 64. Chao MY, Komatsu H, Fukuto HS, Dionne HM, Hart AC (2004) Feeding status and serotonin rapidly and reversibly modulate a Caenorhabditis elegans chemosensory circuit. Proc Natl Acad Sci U S A 101: 15512–15517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fukuto HS, Ferkey DM, Apicella AJ, Lans H, Sharmeen T, et al. (2004) G protein-coupled receptor kinase function is essential for chemosensation in C. elegans . Neuron 42: 581–593. [DOI] [PubMed] [Google Scholar]
- 66. O'Halloran DM, Altshuler-Keylin S, Lee JI, L'Etoile ND (2009) Regulators of AWC-mediated olfactory plasticity in Caenorhabditis elegans . PLoS Genet 5: e1000761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nakai J, Ohkura M, Imoto K (2001) A high signal-to-noise Ca2+ probe composed of a single green fluorescent protein. Nat Biotechnol 19: 137–141. [DOI] [PubMed] [Google Scholar]
- 68. Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, et al. (1997) Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388: 882–887. [DOI] [PubMed] [Google Scholar]
- 69. Tian L, Hires SA, Mao T, Huber D, Chiappe ME, et al. (2009) Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods 6: 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. McGrath PT, Xu Y, Ailion M, Garrison JL, Butcher RA, et al. (2011) Parallel evolution of domesticated Caenorhabditis species targets pheromone receptor genes. Nature 477: 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hess HA, Roper JC, Grill SW, Koelle MR (2004) RGS-7 completes a receptor-independent heterotrimeric G protein cycle to asymmetrically regulate mitotic spindle positioning in C. elegans . Cell 119: 209–218. [DOI] [PubMed] [Google Scholar]
- 72. Dong MQ, Chase D, Patikoglou GA, Koelle MR (2000) Multiple RGS proteins alter neural G protein signaling to allow C. elegans to rapidly change behavior when fed. Genes Dev 14: 2003–2014. [PMC free article] [PubMed] [Google Scholar]
- 73. Kennelly PJ, Krebs EG (1991) Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J Biol Chem 266: 15555–15558. [PubMed] [Google Scholar]
- 74. Aitken A, Bilham T, Cohen P, Aswad D, Greengard P (1981) A specific substrate from rabbit cerebellum for guanosine-3′:5′-monophosphate-dependent protein kinase. III. Amino acid sequences at the two phosphorylation sites. J Biol Chem 256: 3501–3506. [PubMed] [Google Scholar]
- 75. Thomas MK, Francis SH, Corbin JD (1990) Substrate- and kinase-directed regulation of phosphorylation of a cGMP-binding phosphodiesterase by cGMP. J Biol Chem 265: 14971–14978. [PubMed] [Google Scholar]
- 76. Edlund B, Zetterqvist O, Ragnarsson U, Engström L (1977) Phosphorylation of synthetic peptides by (32P)ATP and cyclic GMP-stimulated protein kinase. Biochem Biophys Res Commun 79: 139–144. [DOI] [PubMed] [Google Scholar]
- 77. Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4: 1633–1649. [DOI] [PubMed] [Google Scholar]
- 78. Hofmann F, Feil R, Kleppisch T, Schlossmann J (2006) Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev 86: 1–23. [DOI] [PubMed] [Google Scholar]
- 79. Burns ME, Arshavsky VY (2005) Beyond counting photons: trials and trends in vertebrate visual transduction. Neuron 48: 387–401. [DOI] [PubMed] [Google Scholar]
- 80. Soh JW, Mao Y, Liu L, Thompson WJ, Pamukcu R, et al. (2001) Protein kinase G activates the JNK1 pathway via phosphorylation of MEKK1. J Biol Chem 276: 16406–16410. [DOI] [PubMed] [Google Scholar]
- 81. Johlfs MG, Fiscus RR (2010) Protein kinase G type-Iα phosphorylates the apoptosis-regulating protein Bad at serine 155 and protects against apoptosis in N1E-115 cells. Neurochemistry International 56: 546–553. [DOI] [PubMed] [Google Scholar]
- 82. Singh K, Chao MY, Somers GA, Komatsu H, Corkins ME, et al. (2011) C. elegans Notch signaling regulates adult chemosensory response and larval molting quiescence. Curr Biol 21: 825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Neher E, Sakaba T (2008) Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 59: 861–872. [DOI] [PubMed] [Google Scholar]
- 84. Komalavilas P, Lincoln TM (1994) Phosphorylation of the inositol 1,4,5-trisphosphate receptor by cyclic GMP-dependent protein kinase. J Biol Chem 269: 8701–8707. [PubMed] [Google Scholar]
- 85. Wagner LE 2nd, Li WH, Yule DI (2003) Phosphorylation of type-1 inositol 1,4,5-trisphosphate receptors by cyclic nucleotide-dependent protein kinases: a mutational analysis of the functionally important sites in the S2+ and S2- splice variants. J Biol Chem 278: 45811–45817. [DOI] [PubMed] [Google Scholar]
- 86. Luo C, Gangadharan V, Bali KK, Xie RG, Agarwal N, et al. (2012) Presynaptically localized cyclic GMP-dependent protein kinase 1 is a key determinant of spinal synaptic potentiation and pain hypersensitivity. PloS Biology 10: e1001283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, et al. (2006) Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science 312: 1659–1662. [DOI] [PubMed] [Google Scholar]
- 88. Kolhekar R, Meller ST, Gebhart GF (1993) Characterization of the role of spinal N-methyl-D-aspartate receptors in thermal nociception in the rat. Neuroscience 57: 385–395. [DOI] [PubMed] [Google Scholar]
- 89. Trent C, Tsuing N, Horvitz HR (1983) Egg-laying defective mutants of the nematode Caenorhabditis elegans . Genetics 104: 619–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chase DL, Patikoglou GA, Koelle MR (2001) Two RGS proteins that inhibit Gαo and Gαq signaling in C. elegans neurons require a Gβ5-like subunit for function. Curr Biol 11: 222–231. [DOI] [PubMed] [Google Scholar]
- 91. Robatzek M, Niacaris T, Steger K, Avery L, Thomas JH (2001) eat-11 encodes GPB-2, a Gβ5 ortholog that interacts with Goα and Gqα to regulate C. elegans behavior. Curr Biol 11: 288–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. van der Linden AM, Simmer F, Cuppen E, Plasterk RH (2001) The G-protein β-subunit GPB-2 in Caenorhabditis elegans regulates the Goα-Gqα signaling network through interactions with the regulator of G-protein signaling proteins EGL-10 and EAT-16. Genetics 158: 221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chase DL, Koelle MR (2007) Biogenic amine neurotransmitters in C. elegans. In: Community TCeR, editor. WormBook: WormBook. [DOI] [PMC free article] [PubMed]
- 94. Wragg RT, Hapiak V, Miller SB, Harris GP, Gray J, et al. (2007) Tyramine and octopamine independently inhibit serotonin-stimulated aversive behaviors in Caenorhabditis elegans through two novel amine receptors. J Neurosci 27: 13402–13412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Harris GP, Hapiak VM, Wragg RT, Miller SB, Hughes LJ, et al. (2009) Three distinct amine receptors operating at different levels within the locomotory circuit are each essential for the serotonergic modulation of chemosensation in Caenorhabditis elegans . J Neurosci 29: 1446–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ezcurra M, Tanizawa Y, Swoboda P, Schafer WR (2011) Food sensitizes C. elegans avoidance behaviours through acute dopamine signalling. EMBO J 30: 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Potter LR (2011) Guanylyl cyclase structure, function and regulation. Cell Signal 23: 1921–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lincoln TM, Wu X, Sellak H, Dey N, Choi CS (2006) Regulation of vascular smooth muscle cell phenotype by cyclic GMP and cyclic GMP-dependent protein kinase. Front Biosci 11: 356–367. [DOI] [PubMed] [Google Scholar]
- 99. Hofmann F, Bernhard D, Lukowski R, Weinmeister P (2009) cGMP regulated protein kinases (cGK). Handb Exp Pharmacol 137–162. [DOI] [PubMed] [Google Scholar]
- 100. Arshavsky VY, Burns ME (2012) Photoreceptor signaling: supporting vision across a wide range of light intensities. J Biol Chem 287: 1620–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Garbers DL, Chrisman TD, Wiegn P, Katafuchi T, Albanesi JP, et al. (2006) Membrane guanylyl cyclase receptors: an update. Trends Endocrinol Metab 17: 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Brenner S (1974) The genetics of Caenorhabditis elegans . Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Mello CC, Kramer JM, Stinchcomb D, Ambros V (1991) Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. Embo J 10: 3959–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Fukushige T, Hawkins MG, McGhee JD (1998) The GATA-factor elt-2 is essential for formation of the Caenorhabditis elegans intestine. Dev Biol 198: 286–302. [PubMed] [Google Scholar]
- 105. Rongo C, Whitfield CW, Rodal A, Kim SK, Kaplan JM (1998) LIN-10 is a shared component of the polarized protein localization pathways in neurons and epithelia. Cell 94: 751–759. [DOI] [PubMed] [Google Scholar]
- 106. Ezak MJ, Hong E, Chaparro-Garcia A, Ferkey DM (2010) Caenorhabditis elegans TRPV channels function in a modality-specific pathway to regulate response to aberrant sensory signaling. Genetics 185: 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zimmer M, Gray JM, Pokala N, Chang AJ, Karow DS, et al. (2009) Neurons detect increases and decreases in oxygen levels using distinct guanylate cyclases. Neuron 61: 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. L'Etoile ND, Bargmann CI (2000) Olfaction and odor discrimination are mediated by the C. elegans guanylyl cyclase ODR-1. Neuron 25: 575–586. [DOI] [PubMed] [Google Scholar]
- 109. Chalasani SH, Kato S, Albrecht DR, Nakagawa T, Abbott LF, et al. (2010) Neuropeptide feedback modifies odor-evoked dynamics in Caenorhabditis elegans olfactory neurons. Nat Neurosci 13: 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Chronis N, Zimmer M, Bargmann CI (2007) Microfluidics for in vivo imaging of neuronal and behavioral activity in Caenorhabditis elegans . Nat Methods 4: 727–731. [DOI] [PubMed] [Google Scholar]
- 111. Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N (2010) Computer control of microscopes using µManager. Curr Protoc Mol Biol Chapter 14: Unit 14.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wood WB (1988) The Nematode Caenorhabditis elegans; researchers TcoCe, editor. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Length of starvation alters C. elegans sensitivity to quinine. Animals lacking EGL-4, RGS-2, RGS-3 or RGS-2 and RGS-3 function were tested for response to both (A) 10 mM quinine and (B) 1 mM quinine when starved for increasing lengths of time. (A) rgs-2(lof) and rgs-3(lof) animals responded to 10 mM similarly to wild-type animals when assayed 30 minutes after removal from their bacterial food source (p>0.5 and p>0.1, respectively). rgs-2(lof);rgs-3(lof) double mutant animals did not respond similarly to wild-type animals until 60 minutes after removal from food (p>0.5). (B) The behavioral sensitivity of rgs-2(lof), rgs-3(lof) and rgs-2(lof);rgs-3(lof) animals to dilute quinine increased with longer periods off food. The percentage of animals responding is shown. The combined data of n≥40 animals is shown. Error bars represent the standard error of the mean (SEM). Alleles used: egl-4(n479), rgs-2(vs17), and rgs-3(vs19) loss-of-function. WT = the N2 wild-type strain. lof = loss-of-function, min = minutes.
(PDF)